

Abstract
Increasing evidence has indicated the intimate relationship between the gastrointestinal tract and respiratory tract. The microbial ecosystem has been confirmed to share key conceptual features with gut‐lung microbiome disorder and dysregulation during chronic obstructive pulmonary disease (COPD) exacerbations. However, the dynamic changes of the gut‐lung microbiome during COPD exacerbations and its potential role in disease etiology remain poorly understood. The present study investigated the dynamic changes of gut and lung microorganisms during acute exacerbation of chronic obstructive pulmonary disease (AECOPD). A longitudinal 16S ribosomal DNA survey of the gut and lung microbiome was completed on 90 feces and sputum samples collected from 15 subjects with AECOPD at three visits, which were defined as exacerbation, seven‐day stable state. The present analysis revealed a dynamic gut‐lung microbiota, where changes appeared to be associated with exacerbation events indicative of specific exacerbation phenotypes. Antibiotic and steroid treatments appeared to have differential effects on the gut‐lung microbiome, and the microbiome was associated with disease progression, but not with severity. The abundance and diversity of the microbiome was strongly influenced by the disease progression and therapy. Using culture‐independent methods to impact the gut and lung microbiota on AECOPD may be the key to understanding the interactions between the gut and lung, highlighting its potential as a biomarker, and possibly a target for future respiratory therapeutics.
Keywords: acute exacerbations of chronic obstructive pulmonary disease, dynamic changes, gut‐lung, microbiome
1. INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is one of the most prevalent respiratory diseases. It is characterized by persistent symptoms and impaired lung function as a consequence of airway inflammation, small airway obliteration, and alveolar destruction.1 Acute exacerbation of COPD (AECOPD) is the sudden worsening of symptoms, in which bacterial colonization is one major etiological factor.2 However, the dynamics of bacterial ecology during exacerbations and its role in disease pathogenesis remain poorly understood. Studies that have used culture‐independent techniques, such as PCR amplification and sequencing of the 16S ribosomal (r) RNA gene, have characterized a distinct bacterial community in the airway of COPD patients, when compared to healthy subjects, suggesting that changes in the lung microbiota could be associated with enhanced airway inflammation and disease progression.3, 4 However, at present, most lung microbiome studies have involved relatively small cohorts of subjects with limited longitudinal sampling and concurrent clinical information.
Increasing evidence has indicated the intimate relationship between the gastrointestinal tract (GIT) and respiratory tract. The gut microbiota interacts with the host immune system in ways that influence disease development. The human GIT is colonized by complex and diverse communities of commensal microorganisms. Through downstream signaling pathways, these microorganisms contribute to the regulation of cell proliferation, differentiation and gene expression in host epithelial cells. Chronic lung diseases, such as COPD, are common and often occur together with chronic GIT diseases, such as inflammatory bowel disease (IBD), or irritable bowel syndrome (IBS).5, 6 Up to 50% of adults with IBD and 33% of patients with IBS have pulmonary involvement, such as inflammation or impaired lung function, although many patients have no history of acute or chronic respiratory disease.7 Furthermore, patients with COPD are two to three times more likely to be diagnosed with IBD.8 In addition, individuals with COPD have functional and structural alterations in their intestinal mucosa, and patients with COPD typically have increased intestinal permeability.8, 9 Although the mature GIT and respiratory tract have different environments and functions, these have the same embryonic origin and, consequently, have structural similarities. Thus, it is not surprising that these two sites might interact in health and disease. However, the underlying mechanisms are not well understood.
In the present study, the 16S ribosomal DNA gene sequencing from a cohort of AECOPD patients requiring antibiotic and bronchodilator therapy was used to profile gut and airway bacterial communities. This was an observational study with time continuity. The bacterial microecology disordered in the lung and intestines may be keys to the exacerbation events, and it is possible that a gut target is the precise treatment for COPD.
2. METHODS
2.1. Subjects and samples
Available endotracheal aspirates (ETAs) and feces samples from 15 patients with AECOPD were collected between August 2015 and April 2016 from the emergency intensive care units at Shanghai General Hospital. A longitudinal 16S rRNA‐based microbiome survey was performed on 90 samples (45/sputum; 45/feces) collected from 15 subjects. The samples were longitudinally collected at exacerbation (defined according to CDER10) as day 1, day 7, and day 14. Exacerbations were treated with inhaled corticosteroids and antibiotics according to guidelines.10
2.2. Microbiome analysis
Bacterial genomic DNA was extracted from sputum samples using proteinase K digestion and feces samples using the Qiagen DNA Mini kit (Qiagen, Valencia, California), according to manufacturer's protocols, and the V4‐V5 hypervariable regions of the 16S rRNA gene were PCR‐amplified with the appropriate controls against reagent contamination. Amplified DNA fragments were pyrosequenced using the 454 Genome Sequencer FLX platform (454 Life Sciences, Roche Diagnostics, Burgess Hill, United Kingdom). The sequencing reads were processed using quantitative insights into microbial ecology (QIIME) pipeline version 1.7.11 Stringent criteria were used to remove low‐quality and chimeric reads. The remaining rarified reads were subject to open reference operational taxonomic unit (OTU) picking (97% identity cutoff). The mean sequencing depth used for analysis was 31 615. No sample drop‐off. Then, the sequence data were deposited to NCBI (the National Center for Biotechnology Information), SRA (Sequence Read Archive) (SRP065072). http://www.ncbi.nlm.nih.gov/Traces/sra
2.3. Statistical analyses
Briefly, the exacerbation phenotypes were defined using the microbiological and clinical criteria, as previously established.12 Partial least squares discriminant analysis, receiver operating characteristic curve reconstruction, and network analysis were performed on exacerbation phenotypes and microbiota and/or clinical data. A general linear mixed model was constructed between clinical variables and four measures of α diversity (microbial diversity within a sample): OTU richness, Shannon's H, chao1, and Faith's phylogenetic diversity. In order to identify the clinical predictors of β diversity (microbial composition dissimilarity between samples), canonical correspondence analysis (CCA) was performed on clinical variables, and the relative abundance of taxa at the phylum, genus, and OTU levels. Biomarker factors were identified using principal component analysis (PCA). The false discovery rate method was used to adjust (adj.) the P values for multiple test.
3. RESULTS
3.1. Characteristics of pyrosequencing results
A total of 2 916 257 usable sequences were obtained from 90 samples by pyrosequencing. Among these, 2 583 719 high‐quality sequences were selected, with an average of 33 124 sequences per sample. Then, 126 OTUs were delineated at a 97% similarity level. The values of Good's coverage of all libraries were above 99%. The rarefaction curves approached a plateau with the present sequencing, and the Shannon and Simpson index curve estimates of all samples were stable, suggesting that although new phylotypes would be expected with additional sequencing, most of the diversity had already been captured (Figures 1 and 2). Diversity and abundance estimators of the community are shown in Table 1. Chao and abundance‐based coverage estimation (ACE) index reflecting community abundance, the results showed differences in different stages (P < .0010).There were statistically significant differences in Shannon and Simpson indices, Chao I, ACE indices, and OTU numbers in day 1, day 7, and day 14 of the ETA samples (P < .05), demonstrating a significantly higher diversity in patients in different periods. The characteristics of each group are presented in Table 1.
Figure 1.
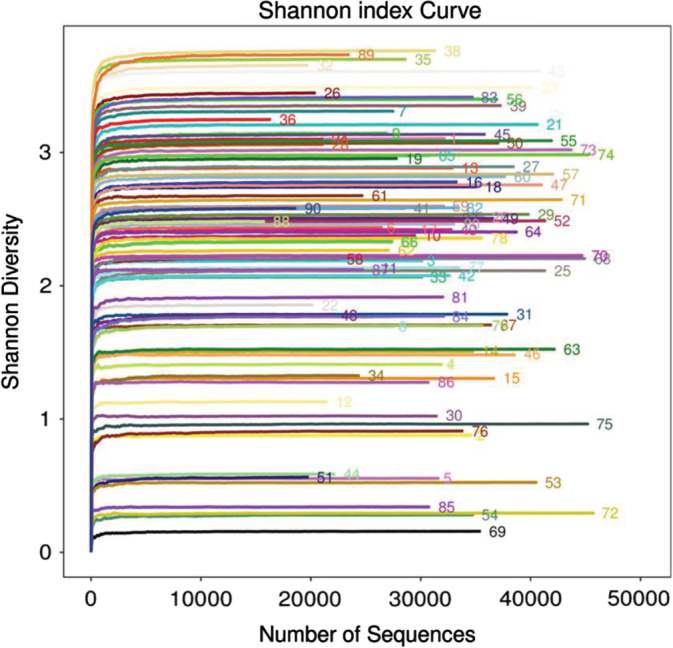
Alpha diversity analysis/Shannon‐Wiener of 90 samples. OTU with 97% similarity was used to calculate Shannon and Simpson values under different random samples with mothur, and curve curves were made with R language tools. The x‐coordinate is the number of sequences per sample, and the y‐coordinate is rarefaction measure. When the curve tends to be flat, it indicates that the sequencing data volume is large enough to reflect the majority of microbial information in the sample. The higher the y value is, the higher the community diversity is. The sequence number in the figure represents the sequence number of the sample. OTU, operational taxonomic unit
Figure 2.
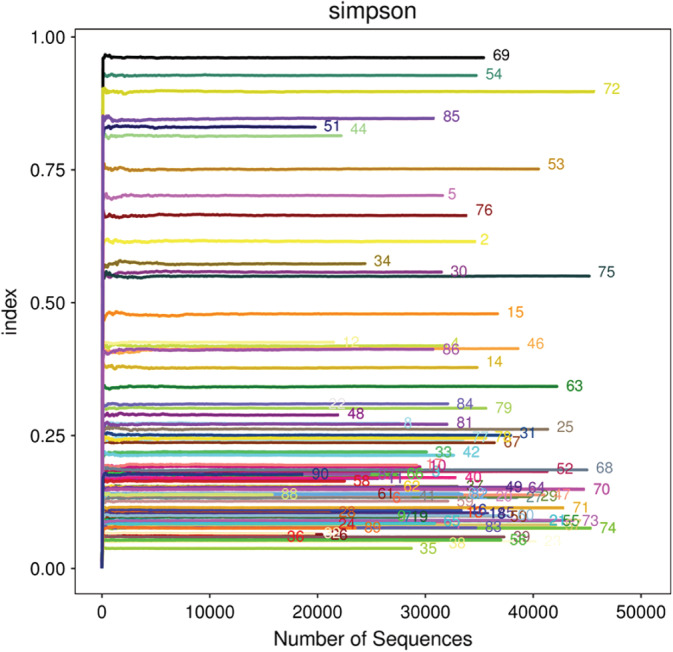
Alpha diversity analysis/Simpson of 90 samples. OTU with 97% similarity was used to calculate Simpson values under different random samples with mothur, and curve curves were made with R language tools. The x‐coordinate is the number of sequences per sample, and the y‐coordinate is rarefaction measure. When the curve tends to be flat, it indicates that the sequencing data volume is large enough to reflect the majority of microbial information in the sample. The higher the y value is, the less the community diversity is. The sequence number in the figure represents the sequence number of the sample. OTU, operational taxonomic unit
Table 1.
Comparisons of microbiota between exacerbation (day 1), treatment (day 7), and stable (day 14)
| Group | Samples (n = 45) | Reads | OTUs | ACE | Chao I | Shannon | Simpson |
|---|---|---|---|---|---|---|---|
| Feces | |||||||
| Day 1 | 35 776 ± 7361 | 177 (166 184) | 233 (217, 317) | 237 (220, 272) | 2.683 ± 0.803 | 0.097 (0.076, 0.262) | |
| Day 7 | 31 045 ± 7599 | 142 (137 147) | 180 (174, 230) | 181 (172, 198) | 2.387 ± 0.874 | 0.136 (0.085, 0.245) | |
| Day 14 | 30 013 ± 7643 | 229 (202 266) | 314 (264, 373) | 324 (260, 340) | 2.922 ± 0.874 | 0.099 (0.064, 0.177) | |
| X 2 | 4.190a | 39.156 | 23.555 | 31.086 | 1.485a | 2.043 | |
| P | .022 | <.001 | <.001 | <.001 | .238 | .36 | |
| Sputum | |||||||
| Day 1 | 31 615 ± 8177 | 96 (92, 100) | 190.667 ± 72.007 | 151.667 ± 25.176 | 2.33 (1.48, 2.40) | 0.298 ± 0.266 | |
| Day 7 | 32 272 ± 7465 | 73 (61, 77) | 128.333 ± 37.498 | 103.533 ± 15.278 | 1.41 (0.88, 1.86) | 0.433 ± 0.245 | |
| Day 14 | 33 434 ± 5708 | 114 (106,12) | 162.333 ± 41.086 | 154.733 ± 34.108 | 2.40 (2.12, 2.65) | 0.192 ± 0.160 | |
| X 2 | 0.247a | 38.906 | 5.294a | 18.274a | 11.661 | 4.190a | |
| P | .782 | <.001 | .009 | <.001 | .003 | .022 | |
Note: The 90 samples were divided into two groups and three stages, analyzed for diversity through Reads, OTUs, ACE, Chao I, Shannon and Simpson. day 1: exacerbation stage; day 7: treatment stage; day 14: stable stage.
Abbreviation: OTU, operational taxonomic unit.
The data was not normally distributed, statistical analysis used one‐way ANOVA method.
3.2. Airway microbiome profiles
Based on the overall bacterial phyla composition, the core bacterial floras in the ETA samples comprised of five bacteria phylas: Firmicutes phyla, Proteobacteria phyla, Bacteroidetes phyla, Actinobacteria phyla, and Fusobacteria phyla (Figure 3). Approximately 98.4% of the sequences belonged to one of five bacterial phyla: Firmicutes (51.8%), Proteobacteria (30.1%), Bacteroidetes (14.2%), Actinobacteria (2.4%), and Fusobacteria (1.2%). A total of more than 179 bacterial species from 16 phyla were identified in the lungs, the most abundant were Streptococcus (26.1%), Haemophilus (4.1%), Moraxella (5.6%), Pseudomonas (4.4%), Prevotella (8.15%), Neisseria (7.75%), Gemella (6.12%), and Veillonella(5.5%), and all of which are typical members of the lung microbiota.13 However, respiratory pathogens were also detected, which included Acinetobacter (4.3%) and Klebsiella (3.2%). Furthermore, there was a significantly greater inter‐subject variation in microbiome community at the same visit, when compared with the temporal variation within each subject, suggesting that for the present cohort, the lung microbiome was relatively unstable over time (Figure 4).
Figure 3.
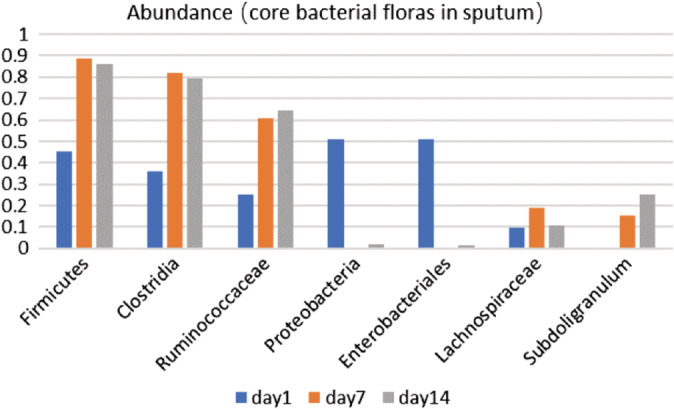
Relative abundance of core bacterial phyla in sputum at different periods day 1: acute phase, day 7: treatment phase; day 14: stability
Figure 4.
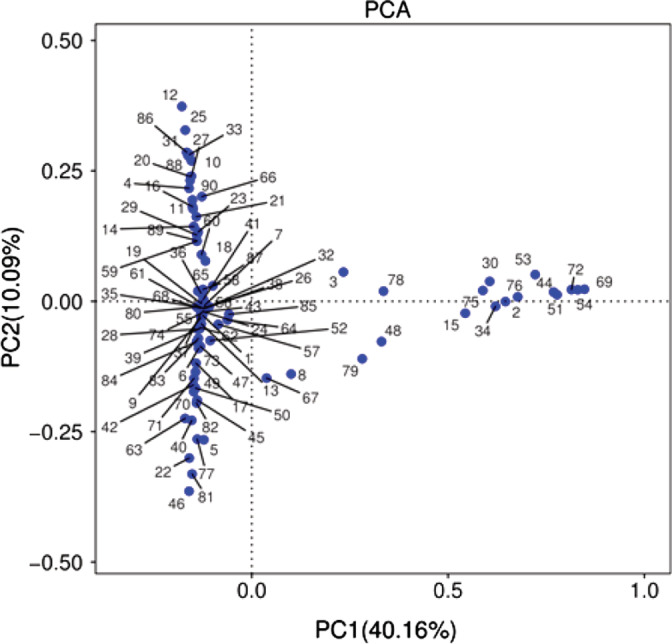
Principal component analysis (PCA) used OTU, pc‐ord, or CANOCO with 97% similarity. PC1 and PC2, respectively, represent the suspected influencing factors for the deviation of the microbial composition of the two groups of 90 samples. The more similar the composition of the sample is, the closer the distance is reflected in the PCA diagram, and the sample shows the distribution of aggregation, indicating that the flora in feces and sputum is relatively similar. The sequence number in the figure represents the sequence number of the sample. OTU, operational taxonomic unit
3.3. Microbiome shifts during exacerbations
In the present results, an overall reduced α diversity (microbial diversity within a sample) with a small and nonsignificant microbial composition shift toward an increase in the relative abundance of Proteobacteria (adj. P = .42 using paired t test) and a decrease in Firmicutes (adj. P = .73 using paired t test) during exacerbations (day 1), when compared to stable (day 14) samples. Moraxella exhibited the greatest change during exacerbations, with an average increase in relative abundance of 5% (adj. P = .22 using paired t test). This was followed by a decrease in Streptococcus (3.8%, adj. P = .58 using paired t test) and an increase in Haemophilus (3.0%, adj. P = .57 using paired t test). Although these changes were not statistically significant, when compared to paired stable and exacerbation samples, a significant increase in Acinetobacter was observed when comparing exacerbation vs all stable samples (adj. P = .022 using t test).
During the three time points in the course of the disease, the abundance and diversity of the flora in the acute phase significantly changed, when compared with the stable phase (P = .001).
3.4. Gut microbiome profiles
In the gut samples, these contained more OTU, and Firmicutes and Bacteroidetes (P < .0001), when compared to lung samples, while Acidobacteria and Cyanobacteria were very low (1.180/0.269). The Phyla Bacteroidetes (21.2%) appeared to be the second most abundant after the first genus Firmicutes (60.6%). The other phylums with obvious abundance were Proteobacteria (15.6%), Actinobacteria (2.3%), and Verrucomicrobia (1.8%) (Figure 5). More than 256 species of 20 bacterial phyla were found in the gut samples. Excluding the unclassified genera, the second genus was Bifidobacterium (14.7%). During the course of AECOPD COPD (acute stage, treatment stage and stable stage), the OTUs significantly decreased and the abundance of bacteria varied, but the diversity did not significantly change.
Figure 5.
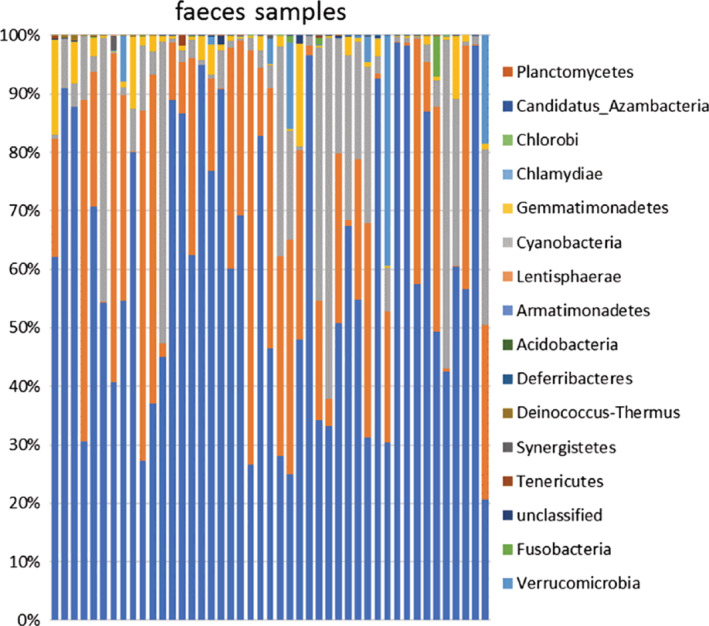
Relative abundance of bacterial phyla in microbiota of faces samples (n = 45)
4. DISCUSSION
COPD is one of the most prevalent respiratory diseases, and is characterized by persistent symptoms and impaired lung function as a consequence of airway inflammation, small airway obliteration and alveolar destruction.14 AECOPD presents with the sudden worsening of symptoms, in which bacterial colonization is one major inducing factor.15 Although nearly half of AECOPDs is associated with bacterial infections, the present knowledge of microbial species associated with these events remains limited through culture‐dependent methods. Studies of the lung microbiome using culture independent techniques and its impact on lung immunity is a relatively new field, and this may contribute to new advances in understanding respiratory diseases.16 At present, new evidence has identified microbial communities both in healthy humans and in individuals with the disease.17 The lung has a distinct bacterial community, and there is a possibility that the core lung microbiome is established in utero during and after birth in very early life, and it has been suggested to be with gut microbiota in human and animal studies.18, 19, 20, 21 There are different bacterial floras in the airway of COPDs, and the bacteria are either colonizing or pathogenic. However, the potential impact on the development of the disease has been previously unconfirmed. Furthermore, as the disease progresses, the diversity of bacteria in the respiratory sample changes, indicating that the pathogenesis of these events may involve a variety of microbial changes. In the present study, the 16 seconds rRNA analysis revealed that there was a dynamic change in bacterial population in the lung of 15 patients treated for AECOPD. Furthermore, there were significant differences in the longitudinal timeline of OTUs (day 1/day 7 P = .002; day 7/day 14 P = .002; day/day 4 P < .001). The OTUs of acute onset were significantly reduced, when compared to the stable state, but the tough was in day 7 after treatment. Most patients had no complete remission, and antibacterial drugs and inhaled hormones were continued, but intravenous hormones were stopped. The core floras of phylum levels, such as Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria, and Fusobacteria also decreased, and the unconventional colonization members of pseudomonas and enterobacteriaceae slightly increased, but there was no statistical difference. The positive samples were tested by culture‐dependent methods, and positive rate was less than 10% for pathogenic bacteria, such as Klebsiella.
In the past, antimicrobial drugs have been considered to have inhibitory effects on microorganisms, but the inhibitory effect intensity was correlated with the type and use time of drugs, and similar phenomena were found in the present study. Furthermore, the abundance and diversity of microorganisms in the lungs dynamically fluctuated with disease progression, treatment and stability, with statistically significant differences in values over the three periods. However, the abundance and diversity of bacteria and microorganisms were recovered at the stable period of the disease, and there was no statistical difference between day 1 and day 14 (Table 1).
In addition to observe the dynamics of the bacteria microorganisms in the lung, feces samples in AECOPD were also analyzed. The human intestine harbors nearly 100 trillion microorganisms, which comprises more than 1000 distinct bacterial species.22 Dysbiosis of the gut microbiota plays a pivotal role in the pathogenesis of chronic bowel diseases, such as chronic IBD, Crohn's disease (CD), and ulcerative colitis (UC) (Clemente). Notably, the total number of bacteria in lung tissue, which has a density of 10 to 100 bacterial cells per 1000 human cells, is relatively small, when compared with that of the gut microbiota. In the present study, it was also observed that the lung microbiome was significantly lower than the gut. The gut and respiratory tract epithelia retain some anatomical similarities. Both are derived from the endoderm and consist of columnar epithelial cells with projections of microvilli (gut) or cilia (respiratory tract) that function as a physical barrier, and as sentinels for the immune system in conjunction with associated lymphoid tissues. Both secrete mucus through goblet cells, as well as secretory immunoglobulin A (sIgA; although this was less in the lungs).23 Bacteria from the gut can travel to the lungs through aspiration of vomit or oesophageal reflux, and a disturbance in epithelial integrity may enable bacterial translocation and their components and metabolites to enter the circulatory system, which can cause systemic inflammation.24 These evidences have demonstrated that the gut microbiota contributes to the development and differentiation of the mammalian immune system. This indicates that the GIT is a reservoir of airway microbial transmission for vulnerable patients. The bacteria flora of feces and sputum in the same patient were compared, and it was found that the core flora was basically similar, with only differences in quantity.25 At different stages of the disease, the change in microflora abundance and diversity in these two kinds of samples were also similar.
In the present study, all patients were given intravenous antibiotics (moxifloxacin), inhaled hormones (budesonide + Ipratropine) and bronchodilators (Terbutaline), and small doses of intravenous hormones (Methylprednisolone) in the early treatment phase, which lasted for 3 to 5 days. The abundance and diversity of sputum bacterial flora varied, and revealed a trend of first falling, and subsequently rising. There was no difference between day 1 and day 14. Interestingly, the bacteria of these feces samples differed from the sputum, and the abundance decreased in day 7 and became higher in day 14. However, the diversity of bacteria did not significantly change from the beginning of the disease progression to the stable period after treatment, and there was no statistical difference, which was not consistent with previous research results. It has been well‐documented that despite methodological differences, antibiotic treatment, especially amoxycillin, cephalosporins, clindamycin, or broad‐spectrum antibiotics, has a marked effect on the composition of the intestinal microbiota,26, 27 particularly long‐term disturbances in (specific) bacterial populations.25 Furthermore, it has been widely accepted that antibiotic treatment profoundly affects the intestinal microbiota. However, these present findings revealed that the dominant fecal microbiota was not affected upon antibiotic treatment in COPD patients with a history of frequent antibiotic use. This prolonged antibiotic pressure appears to have caused a long‐term imbalance in the microbiota, with a shift toward populations resistant to antibiotics.
The present data samples, as well as the representative group of patients with severe COPD, were small. Therefore, it would be prudent to extrapolate these findings. Although the intubation was closely correlated to bacterial community abundance and diversity, antimicrobial agents may be more obvious to the bacterial microecology. The role of bacterial community diversity remains in respiratory failure.
The results of the present study emphasizes the advantages of providing more and higher resolution methods, when compared to traditional bacterial culture methods, in comprehensively evaluating the gut and airway microbiota of chronic CODP patients.
Bacterial OTUs, particularly OTUs that belong to Haemophilus, were identified as microbial “hubs” that had a disproportionately large number of negative connections with other OTUs. These correlations were highly robust across all samples, indicating that this might represent a general pattern of the COPD lung microbiome. The overgrowth of these bacterial OTUs could thereby drive respiratory tract dysbiosis, which has been suggested to be a potential cause of lung disease exacerbations.28 Recent studies have highlighted the importance of ecological interactions in multiple human body habitats. An emerging paradigm is the “keystone species” hypothesis, where even marginal changes in the abundances of relatively few bacterial species could have profound effects on the overall microbial community structure, and consequently alter human disease states.24, 28, 29 Based on the present network analysis, it was speculated that the increased abundance of Haemophilus spp. and the possibly of other Proteobacteria might remodel the normal lung microbial ecosystem into a state of dysbiosis, which could elicit a host pro‐inflammatory response. These present results suggest that the “keystone species” hypothesis in the context of the lung microbiome warrants further study, since it might provide a conceptual basis for novel therapeutic strategies that target a few key bacterial targets to counteract a dysbiotic microbial community in COPD.
As the disease progressed, airway colonization bacteria, such as streptococcus, were reduced, while pseudomonas and enterobacteriaceae, such as klebsiella, were increased. Therefore, it was speculated that the lung microbiome could serve as an additional line of defense that shapes the lung inflammatory response induced by respiratory pathogens.16
The investigators acknowledge that the study has some limitations. Inherent to the nature and severity of the disease, diverse and extensive medication was used in the AECOPD population, in which patients had to be on stable medication for corticosteroids, receive bronchodilators therapy. These medications had an impact on the composition of the microbiota.
5. CONCLUSION
The dynamic changes of the bacterial microbiome in the gut and lung with AECOPD were observed in this study. The lung and intestinal bacterial communities have their unique characteristics and diversity. The composition and diversity of dominant bacterial communities in the gut was more similar than that in the intestines, but the gut was more pronounced. The fluctuation of disease was related to the abundance and diversity of the bacterial community. Finally, in our study, patients were given corticosteroids and antibiotics as part of treatment. These would affect microbiota in the lungs and the gut. In order to avoid the confusion of these variables, we selected using self‐control. At the same time, in terms of the treatment plan, the treatment drugs and doses of all patients were the same or similar, and no difference between the course of treatment and the severity of the disease, so as to avoid the deviation of data as far as possible. The necessity and rationality of antibiotics treatment in AECOPD should be further investigated, and the intensity and duration of antibiotic treatment also needs to be observed. The correction of microbial imbalance in the lung‐gut may lead to potential avenues for respiratory therapeutics.
CONFLICT OF INTEREST
All authors declare no conflict of interest.
Sun Z, Zhu Q‐L, Shen Y, Yan T, Zhou X. Dynamic changes of gut and lung microorganisms during chronic obstructive pulmonary disease exacerbations. Kaohsiung J Med Sci. 2020;36:107–113. 10.1002/kjm2.12147
REFERENCES
- 1. Vestbo J, Hurd SS, Agustí AG, Jones PW, Vogelmeier C, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187:347–365. [DOI] [PubMed] [Google Scholar]
- 2. Leung JM, Tiew PY, Mac Aogáin M, Budden KF, Yong VF, Thomas SS, et al. The role of acute and chronic respiratory colonization and infections in the pathogenesis of COPD. Respirology. 2017;22:634–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stokholm J, Schjørring S, Pedersen L, Bischoff AL, Følsgaard N, Carson CG, et al. Living with cat and dog increases vaginal colonization with E. coli in pregnant women. PLoS One. 2012;7:e46226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smith B, Li N, Andersen AS, Slotved HC, Krogfelt KA. Optimising bacterial DNA extraction from faecal samples: Comparison of three methods. Open Microbiol J. 2011;5:14–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rodriguez‐Roisin R, Bartolome SD, Huchon G, Krowka MJ. Inflammatory bowel diseases, chronic liver diseases and the lung. Eur Respir J. 2016;47:638–650. [DOI] [PubMed] [Google Scholar]
- 6. Papanikolaou I, Kagouridis K, Papiris SA. Patterns of airway involvement in inflammatory bowel diseases. World J Gastrointest Pathophysiol. 2014;5:560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vutcovici M, Brassard P, Bitton A. Inflammatory bowel disease and airway diseases. World J Gastroenterol. 2016;22:7735–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Virta LJ, Ashorn M, Kolho KL. Cow's milk allergy, asthma, and pediatric IBD. J Pediatr Gastroenterol Nutr. 2013;56:649–651. [DOI] [PubMed] [Google Scholar]
- 9. Rutten EPA, Lenaerts K, Buurman WA, WOTUers EFM. Disturbed intestinal integrity in patients with COPD: Effects of activities of daily living. Chest. 2014;145:245–252. [DOI] [PubMed] [Google Scholar]
- 10.U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). Regulatory Information Guidances (Drugs) Acute Bacterial Exacerbations of Chronic Bronchitis in Patients With Chronic Obstructive Pulmonary Disease: Developing Antimicrobial Drugs for Treatment; 2012 Sep.
- 11. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high‐throughput community sequencing data. Nat Methods. 2010;7:335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Turato G, Zuin R, Saetta M. Pathogenesis and pathology of COPD. Respiration. 2001;68:117–128. [DOI] [PubMed] [Google Scholar]
- 13. Garzoni C, Brugger SD, Qi W, Wasmer S, Cusini A, Dumont P, et al. Microbial communities in the respiratory tract of patients with intersitial lung disease. Thorax. 2013;68:1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Global Initiative for Chronic Obstructive Lung Disease . Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease; 2016 Jun 10.
- 15. Persson LJ, Aanerud M, Hardie JA, Miodini Nilsen R, Bakke PS, Eagan TM, et al. Antimicrobial peptide levels are linked to airway inflammation, bacterial colonisation and exacerbations in chronic obstructive pulmonary disease. Eur Respir J. 2017;49:1601328. [DOI] [PubMed] [Google Scholar]
- 16. Wang L, Hao K, Yang T, Wang C. Role of the lung microbiome in the pathogenesis of chronic obstructive pulmonary disease. Chin Med J (Engl). 2017;130:2107–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Young VB. The role of the microbiome in human health and disease: An introduction for clinicians. BMJ. 2017;356:j831. [DOI] [PubMed] [Google Scholar]
- 18. Hansen CH, Nielsen DS, Kverka M, Zakostelska Z, Klimesova K, Hudcovic T, et al. Patterns of early gut colonization shape future immune responses of the host. PLoS One. 2012;7:e34043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Makino H, Kushiro A, Ishikawa E, Muylaert D, Kubota H, Sakai T, et al. Transmission of intestinal Bifidobacterium longum subsp. longum strains from mother to infant, determined by multilocus sequencing typing and amplified fragment length polymorphism. Appl Environ Microbiol. 2011;77:6788–6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ball P. Epidemiology and treatment of chronic bronchitis and its exacerbations. Chest. 1995;108:43S–52S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Soler N, Torres A, Ewig S, Gonzalez J, Celis R, El‐Ebiary M, et al. Bronchial microbial patterns in severe exacerbations of chronic obstructive pulmonary disease (COPD) requiring mechanical ventilation. Am J Respir Crit Care Med. 1998;157:1498–1505. [DOI] [PubMed] [Google Scholar]
- 22. Feng T, Elson CO. Adaptive immunity in the host microbiota dialog. Mucosal Immunol. 2011;4:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dumas A, Bernard L, Poquet Y, Lugo‐Villarino G, Neyrolles O. The role of the lung microbiota and the gut‐lung axis in respiratory infectious diseases. Cell Microbiol. 2018;20:e12966. [DOI] [PubMed] [Google Scholar]
- 24. Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, et al. Microbial co‐occurrence relationships in the human microbiome. PLoS Comput Biol. 2012;8:e1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koning CJ, Jonkers D, Smidt H, RombOTUs F, Pennings HJ, WOTUers E, et al. The effect of a multispecies probiotic on the composition of the faecal microbiota and bowel habits in chronic obstructive pulmonary disease patients treated with antibiotics. Br J Nutr. 2010;103:1452–1460. [DOI] [PubMed] [Google Scholar]
- 26. Becattini S, Taur Y, Pamer EG. Antibiotic‐induced changes in the intestinal microbiota and disease. Trends Mol Med. 2016;22:458–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ianiro G, Tilg H, Gasbarrini A. Antibiotics as deep modulators of gut microbiota: Between good and evil. Gut. 2016;65:1906–1915. [DOI] [PubMed] [Google Scholar]
- 28. Koning CJ, Jonkers DM, Stobberingh EE, Mulder L, Rombouts FM, Stockbrügger RW. The effect of a multispecies probiotic on the intestinal microbiota and bowel movements in healthy volunteers taking the antibiotic amoxycillin. Am J Gastroenterol. 2008;103(1):178–189. [DOI] [PubMed] [Google Scholar]
- 29. Sullivan A, Edlund C, Nord CE. Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect Dis. 2001;1:101–114. [DOI] [PubMed] [Google Scholar]