

Abstract
Cockayne syndrome (CS) is a rare genetic disorder characterized as a segmental premature-aging syndrome. The CS group B (CSB) protein has previously been implicated in transcription-coupled repair, transcriptional elongation, and restoration of RNA synthesis after DNA damage. Recently, evidence for a role of CSB in base excision repair of oxidative DNA lesions has accumulated. In our search to understand the molecular function of CSB in this process, we identify a physical and functional interaction between CSB and poly(ADP-ribose) polymerase-1 (PARP-1). PARP-1 is a nuclear enzyme that protects the integrity of the genome by responding to oxidative DNA damage and facilitating DNA repair. PARP-1 binds to single-strand DNA breaks which activate the catalytic ability of PARP-1 to add polymers of ADP-ribose to various proteins. We find that CSB is present at sites of activated PARP-1 after oxidative stress, identify CSB as a new substrate of PARP-1, and demonstrate that poly(ADP-ribosyl)ation of CSB inhibits its DNA-dependent ATPase activity. Furthermore, we find that CSB-deficient cell lines are hypersensitive to inhibition of PARP. Our results implicate CSB in the PARP-1 poly(ADP-ribosyl)ation response after oxidative stress and thus suggest a novel role of CSB in the cellular response to oxidative damage.
Cockayne syndrome (CS) is a rare autosomal recessive disorder characterized as a segmental premature-aging syndrome. One of the major clinical hallmarks of CS is severe neurological abnormalities (reviewed in reference 27). Approximately 80% of all cases of CS are caused by mutations in the CSB gene. The CS group B (CSB) protein belongs to the SWI/SNF2 protein family and thus contains seven characteristic ATPase motifs which have only recently been successfully crystallized (12, 42). Accordingly, CSB is a DNA-dependent ATPase, and while no helicase activity has been reported for CSB (35), it is able to remodel chromatin in vitro (6).
Cells from patients with CS are sensitive to UV light and deficient in transcription-coupled repair (TCR) of UV-induced and other helix-distorting lesions (46). TCR is a subpathway of nucleotide excision repair, and TCR preferentially removes lesions from the transcribed strand of RNA polymerase II-transcribed genes. TCR requires active transcription and is likely to be initiated by stalling of RNA polymerase II at the site of a DNA lesion (reviewed in reference 41). Failure to remove RNA polymerase II and repair the lesion is believed to be a strong apoptotic signal (reviewed in reference 18).
Oxidative DNA lesions are produced either endogenously or exogenously by reactive oxygen species. Most of these lesions are repaired by the base excision repair (BER) pathway. Lesion-specific DNA glycosylases initiate the repair by removing the aberrant bases. Subsequently, AP endonuclease 1, or alternatively, polynucleotide kinase, generates substrates for a DNA polymerase to insert new and correct nucleotides, and a DNA ligase completes repair (37, 48).
Recently, the CSB protein has been implicated in the repair of oxidative DNA lesions. Besides a potential role for CSB in TCR of certain oxidative DNA lesions, CSB has also been implicated in general genome BER (reviewed in reference 27). The in vitro incision of oligonucleotides containing 7,8-dihydro-8-oxoguanine (8-oxoG) residues is impaired in extracts from CSB-deficient cells compared to wild-type (WT) cells (10, 40, 45). The in vivo repair of photosensitizer-induced oxidative DNA lesions in both the DHFR gene and mitochondrial DNA is also impaired (39, 40). Importantly, after exposure to γ rays, CSB-deficient cells accumulate oxidative lesions in the DNA to a greater extent than WT cells (45). Additionally, cells from csb and ogg1 (8-oxoG DNA glycosylase 1) double-knockout mice accumulate more oxidative lesions in genomic DNA than cells from the ogg1 single-knockout mice (32). Altogether, the results suggest that CSB plays a role in BER. However, the molecular role of CSB in the repair of oxidative lesions in the overall genome is unknown.
In our search for a more precise characterization of the function of CSB in BER, we have searched for protein partners of CSB among known BER proteins. Hence, identifying interactions between CSB and established repair proteins will elucidate at which molecular stages in the repair pathway that CSB participates. In this study, we identify poly(ADP-ribose) (PAR) polymerase 1 (PARP-1) as a protein that interacts both physically and functionally with CSB.
PARP-1 is an abundant nuclear DNA damage surveillance protein that can be characterized as a “molecular nick sensor.” PARP-1 binds with high affinity to and is activated by DNA single-strand breaks (SSBs). When activated, PARP-1 adds polymers of ADP-ribose to various proteins using NAD+ as a substrate (reviewed in reference 17). The acceptor proteins shown to be poly(ADP-ribosyl)ated by PARP-1 include histones, transcription factors, and PARP-1 itself. PARP-1 was very recently shown to be a structural component of chromatin (21), and it is involved in opening the chromatin structure around SSBs by poly(ADP-ribosyl)ating histones (43). BER has been shown to be greatly stimulated by PARP-1 (9, 11, 34). PARP-1 is believed to recruit the DNA repair apparatus to an SSB and is found in complex with the BER protein X-ray repair cross-complementing group 1 (XRCC1), DNA ligase III, and DNA polymerase β (4, 22, 28). Importantly, the PARP-1 recruitment of BER proteins requires the presence of SSBs, since the scaffold protein XRCC1 preferentially binds auto-poly(ADP-ribosyl)ated PARP-1 (13, 26, 31). Furthermore, an additional role for PARP-1 and poly(ADP-ribosyl)ation in polymerase β-independent long-patch BER of 8-oxoG has been proposed (25).
In this study, we demonstrate that CSB and PARP-1 physically interact. CSB binds to both unmodified and poly(ADP-ribosyl)ated PARP-1 in vitro, and CSB interacts with PARP-1 in vivo in both the absence and presence of oxidative stress. Interestingly, we show that after oxidative stress, the CSB/PARP-1 complex relocates to sites of DNA damage in the cell. We find that CSB is a novel substrate for PARP-1 poly(ADP-ribosyl)ation in vitro and that this modification inhibits the catalytic ATPase activity of CSB. Importantly, CSB also is poly(ADP-ribosyl)ated in vivo after oxidative stress. Furthermore, we find that CSB-deficient cells are significantly more sensitive to PARP inhibitors than CSB-complemented cells, and this sensitivity cannot be rescued by complementing with CSB protein containing site-directed mutations in the ATPase domain. Finally, we discuss the importance of these results for implicating CSB in the PARP-1-mediated response to oxidative DNA damage.
MATERIALS AND METHODS
Cell lines and culture conditions.
CS1AN.S3.G2/pc3.1-CSBwt, CS1AN.S3.G2/pc3.1-CSB-E646Q, CS1AN.S3.G2/pc3.1-CSB-T912/913V, CS1AN.S3.G2/pc3.1-CSB-Q942E, CS1AN.S3.G2/pc3.1-CSB-R946A, and CS1AN.S3.G2/pc3.1 cells were cultured as described previously (30, 36). CS1AN.S3.G2/pCMV-dtCSB cells were cultured as the rest of the CS1AN/S3.G2 stable transfected cells. HeLa cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Activation of PARP poly(ADP-ribosyl)ation activity in vivo was obtained by incubating proliferating cells with 250 μM H2O2 in phosphate-buffered saline (PBS) for 10 min before cell extract preparation and Western blot analysis.
Generation of stable transfectant cell line expressing double-tagged CSB protein.
CSBwt containing an N-terminal hemagglutinin antigen (HA) epitope and a C-terminal His6 from the pSLME6-dtCSB vector (kindly provided by W. Vermeulen, Rotterdam, The Netherlands) was subcloned into the SacI-BamHI sites of the mammalian expression vector pCMV-Script (Stratagene), creating pCMV-dtCSB. CS1AN.S3.G2 cells were transfected with pCMV-dtCSB using Lipofectamine (Invitrogen) according to the manufacturer's instructions. Stably transfected cell lines were selected and maintained in medium containing 400 μg/ml geneticin. After 14 days of selection, surviving cells were trypsinized and seeded for isolation of clones. Individual clones were screened for expression of CSB protein.
Recombinant proteins.
PARP-1 (generously provided by Gilbert de Murcia, Strasbourg, France) and HA- and His-double-tagged CSB were purified from insect cells as described previously (references 16 and 5, respectively). The cloning, expression, and purification of the CSB fragments will be described in detail elsewhere (T. Thorslund et al., unpublished data). Briefly, the designated CSB fragments (CSB2-341, CSB310-520, CSB465-1056, CSB953-1204, and CSB1187-1493) were amplified by PCR and cloned into the pTriEx-4 Neo vector (Novagen). The vector encodes N-terminal His and S tags and C-terminal herpes simplex virus (HSV) and His tags. The five CSB fragments were overexpressed in Escherichia coli and purified using Ni-nitrilotriacetic acid agarose (QIAGEN).
ELISA.
Enzyme-linked immunosorbent assay (ELISA) was performed essentially as described previously (3). Briefly, for analysis of the in vitro interaction of CSB and PARP-1, the wells in a microtiter plate were coated with 8 nM bovine serum albumin (BSA), CSB, or PARP-1, as indicated. For the binding step, 8 nM PARP-1 or CSB was added to the indicated wells, and the binding reaction was done in either the absence or presence of ethidium bromide (30 μg/ml) or DNase I (5 μg/ml). Bound PARP-1 was detected with rabbit anti-PARP-1 antibodies (Alexis Biochemicals), while bound CSB was detected with rabbit anti-CSB antibodies (kindly provided by Jean-Marc Egly, Strasbourg, France). For determination of the Kd values for CSB interaction with unmodified or poly(ADP-ribosyl)ated PARP-1, PARP-1 was either mock treated or poly(ADP-ribosyl)ated in vitro (see below) before the wells were coated (8 nM PARP-1 per well). No difference in coating efficiency of unmodified compared to poly(ADP-ribosyl)ated PARP-1 was found. For the binding step, serial dilutions of CSB (ranging between 0.25 and 16 nM/well) were added to the corresponding wells in the presence of ethidium bromide (30 μg/ml), and subsequently, all steps were performed as described above. The fraction of the immobilized PARP-1 bound to the microtiter well that was specifically bound by CSB was analyzed by a Hill plot as described previously (3).
In vitro coimmunoprecipitation of purified CSB and PARP-1.
Protein A magnetic beads (New England Biolabs) were incubated with polyclonal rabbit PARP-1 antibodies (Alexis Biochemicals) in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.4], 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4, 1 mM NaF, and 1 tablet Complete protease inhibitor cocktail [Roche] per 50 ml). HA- and His-tagged CSB (250 ng) and 250 ng PARP-1 were mixed as indicated in either the absence or presence of DNase I (5 μg/ml) in RIPA buffer containing 1 μg/μl BSA and added to the antibody-bound beads. Subsequently, the beads were washed extensively with RIPA buffer and finally dissolved in 2× sodium dodecyl sulfate (SDS) loading buffer, boiled, and analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and Western blot. PARP-1 and CSB were detected by monoclonal PARP-1 and HA antibodies (Santa Cruz), respectively. Secondary ECL horseradish peroxidase-conjugated anti-mouse antibodies (Amersham Biosciences) were used to visualize the immunocomplexes. When the in vitro interaction of CSB and unmodified or poly(ADP-ribosyl)ated PARP-1 was investigated, 1,000 ng CSB was mixed with 500 ng PARP-1, which was either mock treated or poly(ADP-ribosyl)ated, in RIPA buffer with BSA (1 μg/ml) in either the absence or presence of DNase I (5 μg/ml). Subsequently, CSB was immunoprecipitated using polyclonal rabbit CSB antibodies (Santa Cruz). The precipitate was analyzed by SDS-PAGE and Western blot using monoclonal PAR antibodies (Alexis), monoclonal PARP-1 antibodies (Santa Cruz), or rabbit polyclonal CSB antibodies (Santa Cruz).
In vivo CSB immunoprecipitation.
HeLa whole-cell extract (5 mg) was incubated with protein A magnetic beads and either rabbit immunoglobulin G or polyclonal rabbit CSB antibody (Santa Cruz). After extensive washing in RIPA buffer, the precipitate was analyzed by SDS-PAGE and Western blot. Endogenous PARP-1 and CSB were visualized as described above. When investigating the coimmunoprecipitation of PARP-1 with CSB after oxidative stress, we used nuclear extracts from mock-treated or H2O2-treated CS1AN.S3.G2/pCMV-dtCSB cells. The H2O2-treated cells were incubated with 250 μM H2O2 for 10 min in PBS before extract preparation. For the study of in vivo poly(ADP-ribosyl)ation of CSB after oxidative stress, HeLa or CS1AN.S3.G2 cells were either mock treated or incubated with H2O2 before preparation of whole-cell extracts. CSB was immunoprecipitated using polyclonal CSB-specific antibodies (Santa Cruz), and the precipitate was washed extensively using a high-salt RIPA buffer (0.4 M NaCl) and subsequently with a low-salt RIPA buffer (50 mM NaCl). Finally, the precipitated CSB was analyzed by SDS-PAGE and Western blot using rabbit polyclonal CSB antibodies (Santa Cruz) and mouse monoclonal PAR antibodies (Alexis).
In vitro CSB fragment pull-down assay.
S-protein agarose (Novagen) was incubated with 1 μg of each of the five purified CSB fragments (described above) and washed in PBS-0.1% Tween 20, and then 0.5 μg PARP-1 (in PBS-0.1% Tween 20 with BSA [1 μg/μl]) was added. The beads were washed extensively in PBS-0.1% Tween 20, and the precipitate was analyzed by SDS-PAGE and Western blot. PARP-1 was visualized as described above, while the tagged CSB fragments were visualized by monoclonal HSV antibodies (Novagen).
In vitro poly(ADP-ribosyl)ation of PARP-1, CSB, and CSB fragments.
Recombinant PARP-1, CSB, and/or each of the five CSB fragments (all 20 ng/μl), were mixed as indicated in ribosylation buffer (10 mM Tris-HCl [pH 8], 1 mM MgCl2, 1 mM dithiothreitol). The poly(ADP-ribosyl)ation reaction was initiated by addition of NAD+ (1 mM) and activated (sonicated) DNA (0.1 mg/ml) in either the absence or presence of 3-aminobenzamide (3-AB) (10 mM). The reactions were terminated after incubation at room temperature for up to 15 min by addition of 10 mM 3-AB to the tubes not containing the PARP inhibitor or an equal volume of water to the control tubes that already contained 3-AB. The reactions were used either for in vitro coimmunoprecipitation or in ATP hydrolysis assays as described below or analyzed by Western blot using PAR, PARP-1, or CSB antibodies as described above.
Immunofluorescence.
To investigate colocalization of CSB, PARP-1, and PAR, HeLa cells grown on coverslips were either mock treated or treated with 250 μM H2O2 for 10 min in PBS before the cells were fixed according to the procedure recently described by Horibata et al. for CSB immunofluorescence using the rabbit polyclonal CSB antibody (Santa Cruz) described in this paper (19). For visualization of PARP-1 and PAR, we used a 1:100 dilution of mouse monoclonal PARP-1 antibody from Santa Cruz (F-2) and a 1:400 dilution of mouse monoclonal PAR antibodies from Alexis (10H). We used 1:400 dilutions of secondary antibodies, goat anti-mouse antibody conjugated to Alexa Fluor 488 (Molecular Probes) and goat anti-rabbit conjugated to Cy3 (Jackson Laboratories). Slides were analyzed with an Axiovert 200 M fluorescence microscope, and pictures were processed using Metamorph imaging system 4.1 (Universal Imaging Corporation) using deconvolution. When CSB focus formation after oxidative stress was analyzed, HeLa cells were either mock treated or incubated with 33 μM of the PARP inhibitor 3,4-dihydro-5-[4-(piperidinyl)butoxyl]-1(2H)-isoquinolinone (DPQ) for 1 h in serum-free medium before incubation with 500 μM H2O2 (still in the presence of 33 μM DPQ) in serum-free medium at 37°C for 30 min. Cells were fixed and CSB visualized as described above, and images were acquired with a ×63 objective with a Zeiss LSM 410 confocal system.
Single-cell images are representatives of at least 50 randomly selected cells.
In vitro PARP-1 poly(ADP-ribosyl)ation of histone H1.
A combined ribosylation and ELISA method to detect poly(ADP-ribosyl)ation of histone H1 was performed essentially as described previously (47). Briefly, ELISA plates were coated with histone H1 protein (10 μg/well). After blocking, PARP-1 (10 nM) and CSB (7, 14, and 22 nM) were added as indicated, and the poly(ADP-ribosyl)ation reaction was started in either the presence or absence of 3-AB. The reactions were stopped after 5 min, and subsequently, the wells were washed extensively before formation of the PAR polymer was analyzed with PAR-specific antibodies and colorimetric detection.
ATPase activity assays.
Standard CSB ATPase activity assays were performed essentially as described previously (5), with minor modifications. Twelve picomoles of CSB [either mock treated or poly(ADP-ribosyl)ated] and 17 pmol of PARP-1 [either mock treated or poly(ADP-ribosyl)ated] were used in each reaction. All the reactions contained 150 ng pUC19 plasmid DNA, and the buffer conditions were the same in all the reactions. Thin-layer chromatography analysis was performed in 0.8 M LiCl in 1 M formic acid.
Clonogenic survival.
Cells (CS1AN.S3.G2/pc3.1-CSBwt and CS1AN.S3.G2/pc3.1) were trypsinized and 500 cells were seeded per 10-cm2 dish and allowed to attach overnight before the medium was substituted with growth medium containing the indicated concentration of 3-AB (0 to 10 mM). After 3 days of incubation in the presence of 3-AB, the cells were washed with PBS three times and returned to normal growth medium. The cells were subsequently grown for 10 days, washed once with PBS, fixed with methanol, and stained with methylene blue. Blue colonies were counted to determine the clonogenic survival of cells. The assay was performed four times in triplicate.
MTT proliferation assays.
For survival after 3-AB and DPQ treatment, the indicated cell lines were trypsinized and 1,000 cells were seeded in each well in a 96-well plate and allowed to attach overnight before substitution of the medium with medium containing the indicated amounts of 3-AB (0 to 10 mM) or DPQ (0 to 1 mM). After 3 (3-AB) or 5 (DPQ) days of incubation with the drug, 100 μg/μl 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) was added to each well, and the plate was incubated for an additional 5 h. The MTT-containing medium was subtly removed, and the crystals were dissolved by the addition of 100 μl of dimethyl sulfoxide. The absorbance was analyzed at 562 nM, and the percent survival was calculated. The assays were performed at least five times in triplicate for each drug, cell line, and time point.
RESULTS
CSB interacts with PARP-1 in vitro and in vivo.
To elucidate the function of CSB in BER, we screened for protein partners of CSB among known BER proteins and identified PARP-1 as a CSB-interacting protein. Direct binding of CSB and PARP-1 was first demonstrated by in vitro coimmunoprecipitation (Fig. 1A). Purified CSB was incubated either alone or in the presence of PARP-1 or PARP-1 plus DNase I. Subsequently, PARP-1 was immunoprecipitated, and the precipitate was analyzed for the presence of CSB. As shown in Fig. 1A (left), CSB coimmunoprecipitated with purified PARP-1 in both the presence and absence of DNase I, demonstrating an in vitro interaction between CSB and PARP-1 which is not DNA mediated. Furthermore, CSB was not precipitated in the absence of PARP-1, demonstrating specificity of the PARP-1 antibody. The membrane was then reprobed with an antibody against PARP-1 (Fig. 1A, right) to show PARP-1 immunoprecipitation, and since the membrane had not been stripped, the strong signal from recombinant CSB reappeared. The direct interaction between CSB and PARP-1 was confirmed by ELISA, coating with either PARP-1 (Fig. 1B, top) or CSB (Fig. 1B, bottom) and, subsequently, binding with CSB or PARP-1, respectively. Incubation with either ethidium bromide or DNase I did not affect the interaction, and neither CSB nor PARP-1 bound to wells coated with BSA, and the antibodies did not cross-react. This confirms the direct interaction between CSB and PARP-1 and demonstrates that the interaction is not DNA mediated. Next, we analyzed whether CSB and PARP-1 exist in complex in vivo. Whole-cell extracts from HeLa cells were prepared, and endogenous CSB was immunoprecipitated. As shown in Fig. 1C (left), approximately 50% of the endogenous CSB protein was immunoprecipitated, while no CSB was precipitated using control immunoglobulin G. The precipitate was subsequently analyzed for the presence of PARP-1 (Fig. 1C, right), and we found that endogenous PARP-1 coimmunoprecipitated with CSB; hence, a CSB/PARP-1 complex exists in vivo.
FIG. 1.

CSB interacts with PARP-1 in vitro and in vivo. (A) CSB (250 ng), PARP-1 (250 ng), and DNase I (5 μg/ml) were mixed as indicated. PARP-1 was immunoprecipitated (IP) with polyclonal PARP-1 antibodies, and the precipitate was analyzed by SDS-PAGE and Western blot (W). Left panel, CSB visualized by monoclonal HA antibody. Right panel, the same membrane subsequently probed for PARP-1. A molecular weight marker is indicated on the left side of the membrane. (B) Wells in a microtiter plate were coated with either 8 nM PARP-1 or 8 nM CSB, as indicated. After blocking with BSA, the wells were incubated with 8 nM CSB or 8 nM PARP-1 or no protein (−) as indicated, either alone or in the presence of ethidium bromide (EtBr) (30 μg/ml) or DNase I (5 μg/ml). Top, PARP-1-bound CSB detected with CSB antibodies followed by colorimetric analysis. Bottom, CSB-bound PARP-1 detected with PARP-1 antibodies followed by colorimetric analysis. OD490, optical density at 490 nm. (C) Endogenous CSB was immunoprecipitated from HeLa whole-cell extract using CSB-specific antibodies. The precipitate was analyzed by Western blotting for the presence of CSB (left) and PARP-1 (right). IgG, immunoglobulin G.
To map the region of CSB that mediates the direct interaction to PARP-1, we cloned five tagged CSB fragments covering the entire region of the protein: CSB2-341, CSB310-520, CSB465-1056, CSB953-1204, and CSB1187-1493 (Fig. 2A). The fragments were expressed and purified from E. coli and mixed with purified PARP-1, and in vitro coimmunoprecipitation was performed using S-protein agarose specific for the N-terminal tag on the CSB fragments. As shown in Fig. 2B, recombinant PARP-1 bound strongly to the N terminus of CSB (amino acids 2 to 341), whereas the other four fragments bound very little or no PARP-1. The fragments were all present in similar amounts in the pull-down experiment as shown in Fig. 2B (bottom). Thus, the direct interaction of CSB and PARP-1 in vitro is mediated by the N terminus of CSB. This part of the CSB protein has no conserved domains and has not previously been assigned a function.
FIG. 2.

The N terminus of CSB mediates the interaction with PARP-1. (A) Schematic representation of full-length CSB and the CSB fragments used to map the interaction with PARP-1. Full-length CSB contains an acidic domain (Ac), two bipartite nuclear localization signals (NLS), and the seven conserved ATPase motifs (I, IA, and II to VI). The five CSB fragments cover amino acids 2 to 341, 310 to 520, 465 to 1056, 953 to 1204, and 1187 to 1493 of CSB and are all His and S tagged in the N terminus and HSV and His tagged in the C terminus. (B) The five CSB fragments were expressed and purified from E. coli, bound to S-protein agarose, and incubated with 0.5 μg PARP-1. The beads were washed extensively and analyzed by SDS-PAGE and Western blot. First, precipitated PARP-1 was visualized with monoclonal PARP-1 antibodies, and after stripping the membrane, the tagged CSB fragments were visualized by monoclonal HSV antibodies. Purified PARP-1 was loaded in the first lane, and the migration of the molecular weight marker is illustrated on the left side.
CSB also interacts with poly(ADP-ribosyl)ated PARP-1 in vitro and in vivo.
Following exposure of cells to oxidative stress and the formation of SSBs, PARP-1 binds to nicks in the DNA, is activated, and adds polymers of ADP-ribose to various proteins as well as to itself. We therefore investigated the effect of this PARP-1 modification on the interaction with CSB. First, in vitro coimmunoprecipitation experiments with purified CSB and either unmodified PARP-1 or PARP-1 that had been poly(ADP-ribosyl)ated in vitro were performed, and the coimmunoprecipitations were performed both in the absence and in the presence of DNase I. As shown in Fig. 3A (middle), unribosylated PARP-1 coimmunoprecipitated with CSB, confirming the direct interaction between the two proteins (also demonstrated in Fig. 1). The in vitro poly(ADP-ribosyl)ation reaction of PARP-1 is very efficient and adds long chains of ADP-ribose units to PARP-1, and after this dramatic in vitro modification, the monoclonal PARP-1 antibody does not recognize PARP-1. However, when analyzing the CSB precipitate using antibodies against PAR (Fig. 3A, bottom), we found that CSB also immunoprecipitated poly(ADP-ribosyl)ated PARP-1, which shows that CSB can interact with the activated form of PARP-1. Furthermore, CSB and poly(ADP-ribosyl)ated PARP-1 also interacted in the presence of DNase I; thus, the interaction is not DNA dependent. To address whether CSB has higher affinity for one of the two forms of PARP-1, that is, the unmodified PARP-1 and the activated auto-poly(ADP-ribosyl)ated PARP-1, we performed quantitative ELISA experiments to determine the Kd of the interactions. The quantitative ELISA studies were carried out in the presence of ethidium bromide to eliminate any putative effects of DNA. As seen in Fig. 3B, the kinetics of the interaction between CSB and either unmodified (dotted line) or poly(ADP-ribosyl)ated (solid line) PARP-1 are very similar. The Kd values were approximately 0.8 nM and 0.7 nM, respectively, and this indicates that CSB is not likely to interact with the automodification domain in PARP-1. Since other cellular factors could influence the complex formation of CSB and PARP-1 after damage, we investigated the interaction between CSB and unmodified or poly(ADP-ribosyl)ated PARP-1 in vivo. Nuclear extracts from untreated and H2O2-treated cells stably expressing HA- and His-tagged CSB (CS1AN.S3.G2/pCMV-dtCSB) were prepared, and in vivo coimmunoprecipitation experiments were performed. The H2O2 treatment used is known to activate the poly(ADP-ribosyl)ation response (47) (see Fig. 6C). As shown in Fig. 3C, PARP-1 coimmunoprecipitated with CSB in both untreated cells and cells that had been challenged with oxidative stress. To summarize, these results show that CSB interacts in vitro with PARP-1 when PARP-1 is both unmodified and auto-poly(ADP-ribosyl)ated and that the CSB/PARP-1 complex exists in vivo both before and after oxidative stress.
FIG. 3.
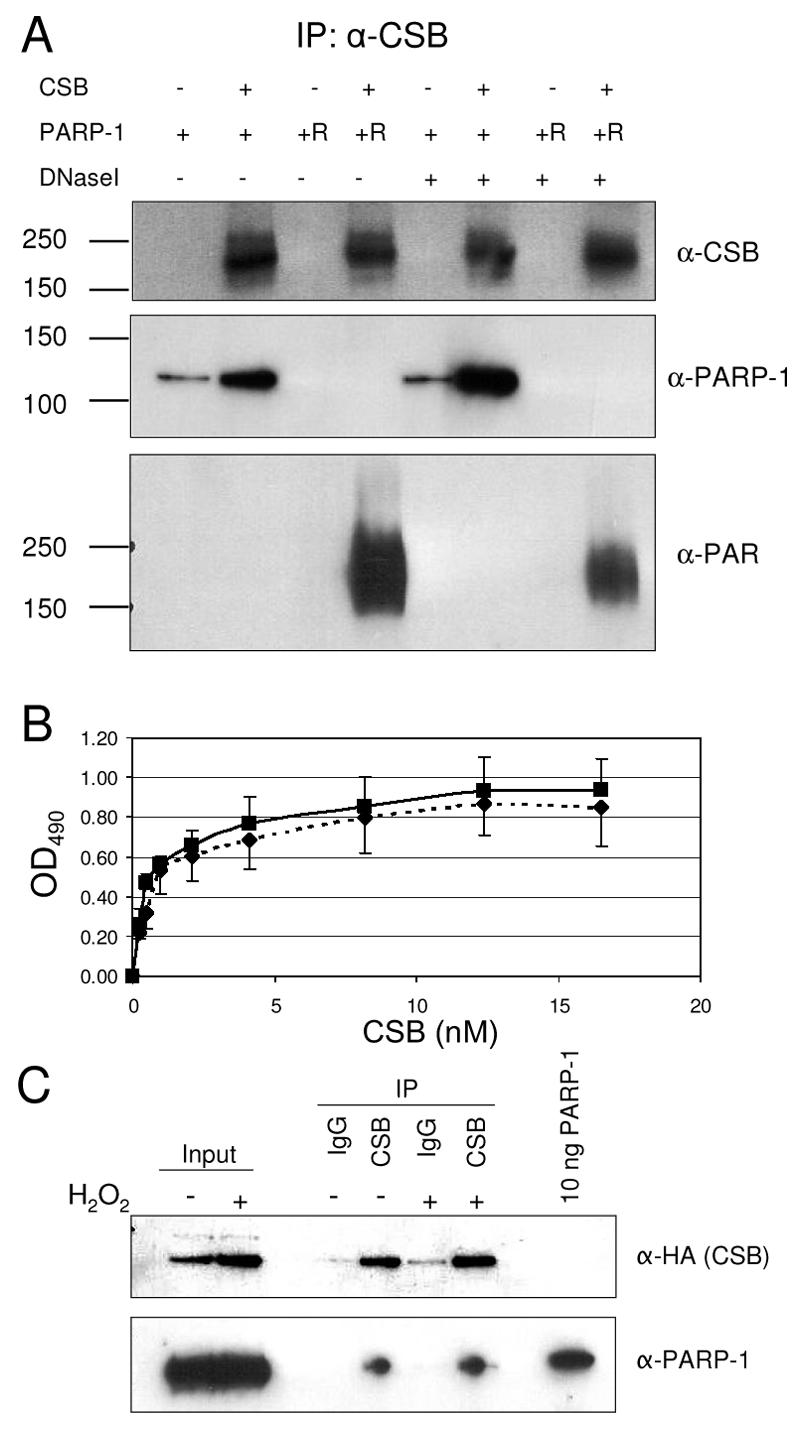
CSB interacts with both unmodified PARP-1 and poly(ADP-ribosyl)ated PARP-1. (A) Recombinant PARP-1 was either mock treated or poly(ADP-ribosyl)ated in ribosylation mix in vitro. Subsequently, 500 ng unmodified (+) or poly(ADP-ribosyl)ated (+R) PARP-1 was mixed with 1,000 ng CSB (+) (or equimolar amounts of BSA [−]) and DNase I, as indicated. CSB was immunoprecipitated using rabbit polyclonal CSB antibodies, and the precipitate was washed extensively and analyzed by Western blot. The top panel shows the immunoprecipitation (IP) of CSB, the middle panel shows the coimmunoprecipitated unmodified PARP-1 [this antibody does not recognize the heavily modified poly(ADP-ribosyl)ated PARP-1], and the bottom panel shows coimmunoprecipitated heavily poly(ADP-ribosyl)ated PARP-1, visualized by PAR-specific antibodies. All the blots shown are the same membrane probed, stripped, and reprobed with the different antibodies. (B) Recombinant PARP-1 was either mock treated or poly(ADP-ribosyl)ated in vitro. Wells in a microtiter plate were coated with 8 nM unmodified PARP-1 or poly(ADP-ribosyl)ated PARP-1 and blocked with BSA. Subsequently, the wells were incubated with serial dilutions of CSB (0.25 nM to 16.5 nM). Bound CSB was detected with polyclonal CSB antibodies. Filled squares and solid lines represent CSB binding to poly(ADP-ribosyl)ated PARP-1, while filled diamonds and dotted lines represent CSB binding to unmodified PARP-1. OD490, optical density at 490 nm. (C) CS1AN cells stably transfected with HA- and His-double-tagged CSB were either mock treated or incubated with H2O2. Subsequently, nuclear extracts were prepared from these cells and CSB was immunoprecipitated using CSB-specific antibodies. The precipitate was analyzed by Western blotting for the presence of HA-tagged CSB (top) and PARP-1 (bottom).
FIG. 6.

CSB does not regulate the PARP-1 poly(ADP-ribosyl)ation activity. (A) ELISA plates were coated with histone H1, and after blocking, CSB (125 to 375 ng) and PARP-1 (125 ng) were added to the indicated wells in 50 μl poly(ADP-ribosyl)ation reaction mix in either the presence or absence of 3-AB. The poly(ADP-ribosyl)ation reaction was stopped after 5 min, and the PAR polymer was detected with polyclonal PAR antibodies and colorimetric analysis. (B) Recombinant PARP-1 (20 ng/μl) and CSB (20 ng/μl) were mixed in ribosylation reaction mix, as indicated, in either the presence or absence of 10 mM 3-AB. After incubation for 5 or 10 min, the reactions were stopped and analyzed by SDS-PAGE and Western blot. The same membrane was probed, stripped, and reprobed with the indicated antibodies. Top panel, monoclonal PAR antibodies. Middle panel, monoclonal HA antibodies (recognizes the N-terminal tag on CSB). Bottom panel, monoclonal PARP-1 antibodies. The migration of the molecular weight marker is illustrated on the left side. (C) CS1AN/CSBwt (WT) and CS1AN/vector (CS-B) cells were either mock treated or incubated with 250 μM H2O2 in PBS for 10 min to activate PARP poly(ADP-ribosyl)ation activity in vivo and subsequently analyzed by SDS-PAGE and Western blot. Top panel, PAR antibodies. Middle panel, monoclonal PARP-1 antibodies. Bottom panel, polyclonal goat lamin B antibodies. The same membrane was probed with the three different antibodies. The migration of the molecular weight marker is illustrated on the left side.
The CSB/PARP-1 complex relocates in the nucleus after oxidative stress.
Since CSB was found to interact with both unmodified and auto-poly(ADP-ribosyl)ated PARP-1 and exist in complex in vivo both before and after oxidative stress, we next examined the colocalization of endogenous CSB and PARP-1/PAR in HeLa cells before and after H2O2 treatment. We observed that PARP-1 was highly concentrated in the nucleolus but also present throughout the nucleus, and as reported previously (29), this subcellular localization of PARP-1 was not significantly affected by low doses (250 μM) of H2O2 (Fig. 4A). PARP-1 poly(ADP-ribosyl)ation activity is activated by PARP-1 binding to SSBs, and thus, PAR is primarily observed after DNA damage (13). Accordingly, we observed distinct PAR nuclear foci after treatment with H2O2 (Fig. 4B). PARP-1 itself is a primary substrate for poly(ADP-ribosyl)ation in vivo, and thus, extensive colocalization between PARP-1 and PAR was observed after H2O2 treatment (data not shown). Immunofluorescence studies of endogenous CSB have recently been reported (19), and this procedure was used to visualize CSB. As also reported previously (2), we observed that CSB was heterogeneously distributed in the nucleoplasm and present in larger foci in nucleoli (Fig. 4). The overall subcellular localization of CSB did not change significantly after the relatively low dose of H2O2 used in these experiments; however, we did observe that CSB foci in the nucleoplasm of the majority of the cells appeared more distinct and confined after damage induction (Fig. 4). As shown later, treatment with higher doses of H2O2 caused CSB to form distinct foci (Fig. 5D). Interestingly, in undamaged cells, CSB and PARP-1 were only found to colocalize in foci in the nucleolus, but after oxidative stress, CSB and PARP-1 also colocalized in the nucleoplasm (Fig. 4A). Furthermore, after H2O2 treatment, we observed significant colocalization of CSB and PAR in clearly visible foci both in the nucleolus and outside the nucleolus (Fig. 4). A visual estimation of 25 cells indicated that CSB was present at approximately 50% of all nuclear PAR foci. Taken together, these results demonstrate that the CSB/PARP-1 complex partially relocates in the nucleus after oxidative stress and, in particular, that CSB is present at sites of active PARP-1. Thus, the CSB/PARP-1 complex seems to relocate to sites of H2O2-induced DNA damage.
FIG. 4.

Immunofluorescence staining of endogenous CSB (A and B), PARP-1 (A), and PAR (B) in HeLa cells that have been either mock treated or treated with 250 μM H2O2 just prior to fixation. DNA was stained by DAPI (4′,6′-diamidino-2-phenylindole). Merged images of CSB and PARP-1 (A) and CSB and PAR (B) are shown, and colocalization is indicated by yellow foci.
FIG. 5.

CSB is a substrate for PARP-1 poly(ADP-ribosyl)ation. (A) PARP-1 (20 ng/μl) was incubated either alone (−) or with each of the five different purified CSB fragments as indicated, each (20 ng/μl) in ribosylation reaction mix. After 15 min of incubation, the reactions were stopped and the fragments were affinity purified using S-protein agarose specific for the N-terminal tags on the CSB fragments. The precipitates were then analyzed by SDS-PAGE and Western blot. Left panel, monoclonal poly(ADP-ribose) antibodies. Right panel, monoclonal HSV antibodies (recognizes the C-terminal tag on the CSB fragments). The two Western blots shown are the same membrane probed, stripped, and reprobed with the two different antibodies, re-spectively. The migration of the molecular weight marker is illustrated on the left side. (B) CSB and PARP-1 were either mock treated or poly(ADP-ribosyl)ated. Subsequently, [γ-32P]ATP was incubated either in the absence (−) or in the presence of mock-treated (+) or poly(ADP-ribosyl)ated (+R) CSB (12 pmol) and PARP-1 (17 pmol) as indicated. The reactions were analyzed by thin-layer chromatography, radioactivity was detected by phosphorimaging, and ATP hydrolysis was quantified using Quantity One software (Bio-Rad). In each single experiment, the percentage of ATP hydrolysis for each reaction was then calculated relative to the activity observed with unmodified CSB alone. Data represent the means and standard deviations of six duplicate experiments. (C) CS1AN and HeLa cells were either mock treated or incubated with H2O2, and subsequently, whole-cell extracts were prepared from these cells. CSB was immunoprecipitated (IP) using CSB-specific antibodies, and the precipitate was analyzed by Western blotting for the presence of CSB (top) and poly(ADP-ribosyl)ated protein using PAR-specific antibodies (bottom). The migration of the molecular weight marker is illustrated on the left side. (D) Confocal images of HeLa cells showing the localization of CSB in undamaged (mock) or H2O2-treated cells. To inhibit the PARP poly(ADP-ribosyl)ation response induced by H2O2, cells were incubated with DPQ during damage induction or without H2O2 treatment as a control.
CSB is a substrate for poly(ADP-ribosyl)ation, and the modification alters the CSB activity in vitro.
It has been shown that PARP-1 can poly(ADP-ribosyl)ate a variety of proteins, and some of these posttranslational modification substrates have also been demonstrated to directly interact with PARP-1 (reviewed in reference 8). Therefore, we next investigated if CSB is a substrate for PARP-1 poly(ADP-ribosyl)ation. First, we incubated PARP-1 with each of the five purified and recombinant CSB fragments (described in Fig. 2) in a ribosylation reaction. Subsequently, the CSB fragments were affinity purified and analyzed for PAR modification by Western blot. As shown in Fig. 5A, we observed a PAR signal in the reaction incubated with the CSB fragment CSB2-341 and a weaker PAR signal in the reaction incubated with CSB1187-1493 (Fig. 5A, left). The bands appeared at the exact positions of CSB fragments, as demonstrated in Fig. 5A (right), where the membrane had been stripped and reprobed for the C-terminal HSV tag on the fragments. The CSB2-341 fragment, which was found to be significantly ribosylated, was also the fragment that mediated the direct interaction with PARP-1 (Fig. 2). Importantly, the PAR-specific antibody did not recognize CSB2-341 or CSB1187-1493 when the fragments were not poly(ADP-ribosyl)ated in vitro (data not shown). Hence, CSB is a substrate for PARP-1 poly(ADP-ribosyl)ation in vitro; however, the extent of PAR chains added to the CSB fragments appears to be limited, since the mobility of the fragments did not change significantly. Next, we wished to investigate whether the poly(ADP-ribosyl)ation of CSB affected its catalytic activity. CSB is a DNA-dependent ATPase, and therefore, we performed ATP hydrolysis experiments with either unmodified or poly(ADP-ribosyl)ated CSB. Interestingly, poly(ADP-ribosyl)ation of CSB in vitro resulted in a ∼40% reduction in ATPase activity (Fig. 5B). Since we did not separate CSB and PARP-1 after the in vitro poly(ADP-ribosyl)ation reaction, we included several controls. PARP-1 by itself, either unmodified or poly(ADP-ribosyl)ated, did not show any ATP hydrolysis activity. Furthermore, addition of either unmodified PARP-1 or PARP-1 that had been pre-poly(ADP-ribosyl)ated to the CSB ATPase reaction did not cause a decrease in ATPase activity. Only the specific PARP-1 poly(ADP-ribosyl)ation of CSB in vitro inhibited the ATPase activity of CSB. Finally, we investigated whether CSB is poly(ADP-ribosyl)ated in vivo after oxidative stress. Whole-cell extracts were prepared from untreated or H2O2-treated HeLa cells and CSB-deficient CS1AN cells. CSB was immunoprecipitated with CSB-specific antibodies from these extracts using very stringent conditions and subsequently analyzed by Western blot, probing for CSB and PAR. As seen in Fig. 5C (top), CSB was precipitated from the HeLa cells under these stringent conditions, and similar amounts of CSB were precipitated from both the untreated extract and the extract prepared from H2O2-treated HeLa cells. Using PAR-specific antibodies (Fig. 5C, bottom), a band of the same size as CSB appeared only in the immunoprecipitate from HeLa cells, suggesting that this band is indeed ribosylated CSB. Note that this PAR band is very unlikely to be poly(ADP-ribosyl)ated PARP-1, since the bands appearing when the membrane was reprobed with PARP-1-specific antibodies were of lower molecular weight (data not shown). Interestingly, the signal from poly(ADP-ribosyl)ated CSB increased after treatment with H2O2, indicating that CSB is modified by PARP after oxidative stress. Thus, the results presented here demonstrate that the N terminus of CSB is ribosylated by PARP-1 in vitro and that this modification inhibits the catalytic activity of CSB. Furthermore, after oxidative stress, CSB is poly(ADP-ribosyl)ated in vivo. To investigate the importance of the poly(ADP-ribosyl)ation of CSB in vivo, we further analyzed the localization of CSB after oxidative stress and the influence of poly(ADP-ribosyl)ation. As stated above, CSB was found to be diffusely distributed throughout the nucleoplasm in untreated HeLa cells (Fig. 4 and Fig. 5D, top). We observed that when HeLa cells were exposed to high doses of H2O2 (500 μM) for 30 min, distinct CSB foci could be observed in the nucleus (Fig. 5D, second panel), and more than half of these foci colocalized with PAR (data not shown). Interestingly, when PARP poly(ADP-ribosyl)ation activity was inhibited by the drug DPQ during exposure of HeLa cells to H2O2, no significant CSB foci were observed (Fig. 5D, bottom). The DPQ drug by itself did not influence CSB localization (Fig. 5D, third panel). Thus, the PARP-1 poly(ADP-ribosyl)ation activity and not the DNA damage induced by H2O2 is essential for CSB to form foci after oxidative stress.
PARP-1 poly(ADP-ribosyl)ation activity is not dependent on CSB.
PARP-1 poly(ADP-ribosyl)ation of CSB affected the catalytic activity of CSB, and next, we analyzed if CSB reversely affected PARP-1 poly(ADP-ribosyl)ation activity. First, we used an ELISA-based assay with purified proteins, where the effect of CSB on PARP-1 poly(ADP-ribosyl)ation of histone H1 was investigated. As shown in Fig. 6A, there was no significant change in poly(ADP-ribosyl)ation of histone H1 when incubated with increasing amounts of CSB. We also analyzed the effect of CSB on PARP-1 auto-poly(ADP-ribosyl)ation in vitro. As shown in Fig. 6B (top), CSB had no effect on PARP-1 auto-poly(ADP-ribosyl)ation, since the Western signal for PAR was very similar in the lanes incubated without or with CSB, respectively. As a control, we show the same membrane probed for the HA tag on CSB and for PARP-1 protein (Fig. 6B, middle and bottom, respectively). As mentioned previously, the monoclonal PARP-1-specific antibody is not able to recognize the heavily in vitro-poly(ADP-ribosyl)ated form of PARP-1, and the decrease in PARP-1 signal (Fig. 6B, bottom) corresponds to the increased PAR signal (Fig. 6B, top). Incubation with the PARP-1 inhibitor 3-AB completely inhibited the PAR reaction. Finally, we examined the effect of CSB on poly(ADP-ribosyl)ation in vivo. We incubated isogenic CSB-deficient cells (CS-B cells) and CSB-complemented cells (WT cells) with H2O2 and subsequently analyzed them for PAR formation in vivo by Western blot. As shown in Fig. 6C (top), we observed no obvious difference in PAR formation after H2O2 treatment between CS-B and WT cells. Importantly, we did not observe any difference in the amount of PARP-1 protein in CS-B and WT cells (Fig. 6C, middle). As a control for loading, we probed for the nuclear structural protein lamin B (Fig. 6C, bottom). We also analyzed the PAR formation in vivo in CS-B and WT cells after H2O2 treatment by immunofluorescence with PAR-specific antibodies and also did not observe any difference in poly(ADP-ribosyl)ation activity between CS-B and WT cells (data not shown). Thus, PARP-1 poly(ADP-ribosyl)ation activity is not dependent on the CSB protein.
CSB-deficient cells are sensitive to inhibition of PARP.
To further elucidate the cellular function of the CSB/PARP-1 complex, we investigated the sensitivity of CSB-proficient and -deficient cells to inhibition of PARP-1 poly(ADP-ribosyl)ation activity using specific PARP inhibitors. First, we used a clonogenic survival assay to analyze the sensitivity of isogenic CSB-deficient (vector) and CSB-complemented cells (WT) to inhibition of PARP activity by 3-AB. Interestingly, we found that after incubating cells for 3 days with 3-AB, cells deficient in CSB were hypersensitive and displayed a significant reduction in survival compared to cells expressing functional CSB (Fig. 7A). We also analyzed the sensitivity of CSB-deficient and -complemented cells to a different PARP inhibitor, DPQ. DPQ is a more potent and specific PARP poly(ADP-ribosyl)ation inhibitor compared to 3-AB; thus, the use of DPQ eliminates some of the putative nonspecific actions observed with 3-AB (38). This survival analysis was done using the MTT assay and incubation with the drug for 5 days. As shown in Fig. 7B, CSB-deficient cells (vector) were also hypersensitive to inhibition of PARP activity by DPQ compared to the CSB-complemented cells (WT), thus confirming the 3-AB results.
FIG. 7.

CSB-deficient cells are sensitive to PARP inhibitors. (A) Clonogenic survival of CS1AN/CSBwt (WT) and CS1AN/vector (VECTOR) cells after treatment with the indicated amounts of the PARP inhibitor 3-AB. Solid triangles indicate survival of WT cells. Solid circles indicate survival of vector cells. Data represent the means and standard deviations of four triplicate experiments. (B) MTT survival of CS1AN/CSBwt (WT) cells, indicated by solid triangles, and CS1AN/vector (VECTOR) cells, indicated by solid circles, after treatment with the PARP inhibitor DPQ. Data represent the means and standard deviations of five triplicate experiments.
Previously, we established and described cell lines expressing CSB protein with single amino acid substitutions in different conserved motifs of the DNA-dependent ATPase domain (Fig. 8A). These cell lines and the corresponding CSB mutant proteins have previously been characterized by measuring the cellular sensitivity to UV light, oxidative stress, and biochemical ATPase activity; we have included these data in Table 1 for easy reference (5, 30, 36, 45). To further investigate the function of CSB in PARP poly(ADP-ribosyl)ation-inhibited cells, we analyzed the sensitivity of these mutant cell lines towards 3-AB using the MTT assay. Again, CSB-deficient cells (vector) were more sensitive to the PARP inhibitor 3-AB than the CSB-complemented cells (WT) (Fig. 8B). Two of the cell lines expressing CSB protein with site-specific point mutations (CS1AN/CSB-E646Q and CS1AN/CSB-R946A) behaved very similar to the vector cell line not expressing CSB protein, while the two cell lines CS1AN/CSB-T912-913V and CS1AN/CSB-Q942E demonstrated an intermediate sensitivity to 3-AB compared to WT and vector cells. Thus, the integrity of the ATPase domain and the catalytic activity of CSB seem to be important for the ability of the CSB protein to complement the 3-AB sensitivity of CSB-deficient cells.
FIG. 8.

Sensitivity of cells expressing mutant CSB protein to inhibition of PARP. (A) Schematic representation of the CSB protein and CSB amino acid sequence of conserved regions in ATPase motifs II, V, and VI. Amino acids mutated in CSB in the different cell lines used are indicated in red letters. The corresponding amino acid sequence of five other SWI/SNF2 proteins are indicated below to illustrate the conservation of the mutated amino acids (E646Q, T912-913V, E942Q, and R946A). (B) MTT survival of CS1AN/CSBwt (WT), CS1AN/CSB-E646Q (E646Q), CS1AN/CSB-T912/913V (T912/913V), CS1AN/CSB-Q942E (Q942E), CS1AN/CSB-R946A (R946A), and CS1AN/vector (VECTOR) cells after treatment with the indicated amounts of the PARP inhibitor 3-AB. Survival of WT cells is indicated by solid triangles, and survival of vector cells is indicated by solid circles. The four CSB mutant cell lines, E646Q, T912/913V, Q942E, and R946A, are indicated by open squares, +, ×, and open diamonds, respectively. Data represent the means and standard deviations of four triplicate experiments.
TABLE 1.
Activity of CSBa
| Characteristic | CSB activity
|
|||||
|---|---|---|---|---|---|---|
| CSBwt | Vector | CSBE8646Q | CSBT912-913V | CSBQ942E | CSBR946A | |
| Protein | ||||||
| ATPase activity | +++ | ND | ÷ | + | + | ND |
| ATP binding | +++ | ND | +++ | +++ | +++ | ND |
| DNA binding | +++ | ND | +++ | ++ | ++ | ND |
| Cellular | ||||||
| UV survival | +++ | ÷ | ÷ | ÷ | ÷ | ÷ |
| γ survival | +++ | ÷ | ND | ++ | + | + |
| 8-oxoG incision | +++ | ÷ | +++ | ++ | + | + |
| 3-AB survival | +++ | ÷ | ÷ | + | + | ÷ |
+++, same activity as CSBwt protein; ++, lower but still significant activity; +, deficient but residual activity; ÷, deficient; ND, not determined.
DISCUSSION
In this study, we identify a physical and functional interaction between CSB and PARP-1. Many reports have recently implicated CSB in BER of oxidative DNA lesions in the general genome, and our result is the first demonstration of a direct interaction between CSB and a protein involved in BER. We also show that CSB forms distinct foci and can be found at sites of activated PARP-1, i.e., DNA damage, in cells after H2O2 treatment. Furthermore, we find that CSB is a substrate for poly(ADP-ribosyl)ation by PARP-1 after oxidative stress, altogether suggesting a role for CSB in the PARP-1-mediated DNA damage response and repair. In support of these data, Flohr et al. recently suggested that PARP-1 stimulation of BER depends on CSB (15). Using the photosensitizer Ro 19-8022, which upon light activation primarily induces 8-oxoG lesions (49), and alkaline elution, they found that the presence of CSB was vital for PARP-1 stimulation of 8-oxoG repair. The finding in the present study that CSB resides in a physical complex with PARP-1 supports the hypothesis that PARP-1 stimulation of BER may depend on CSB. CSB has previously been implicated in general genome BER of 8-oxoG, since the repair of this lesion is impaired in CSB-deficient cells, and 8-oxoG lesions accumulate to a greater extent in these cells after exposure to γ rays (10, 32, 39, 40, 45). Thus, we speculate that the CSB/PARP-1 complex is important for repair of 8-oxoG.
Functional studies with the catalytically inactive E646Q CSB motif II mutant have suggested that the biochemical ATPase activity of CSB is not required for CSB to function in the BER process, as this mutant fully complements the 8-oxoG incision deficiency (Table 1). Although the molecular role of CSB in BER is unknown, we speculate that CSB may stimulate the incision of 8-oxoG either by directly stimulating the OGG1 protein, for which a functional but no physical interaction has been observed (44), or by stimulating other 8-oxoG-incising DNA glycosylases. Alternatively, CSB may alter the DNA structure around the 8-oxoG lesion, providing better access for the DNA glycosylase to the lesion. This last notion is supported by the observation that the E646Q mutant CSB protein binds DNA with the same affinity as the wild-type protein (5) and the finding by Citterio et al. that the ATPase activity of CSB is not required for the protein to induce local changes in DNA topology (6). In addition, our results could imply that CSB may also function as a scaffold protein, keeping PARP-1 in close proximity to the incised lesion, thus stimulating faster repair and preventing generation and accumulation of the cytotoxic base excision repair SSB intermediates.
We establish that CSB is a novel substrate for poly(ADP-ribosyl)ation by PARP-1 after oxidative stress and, intriguingly, that this modification reduces the catalytic ATPase activity of CSB. As we have previously shown that the ATPase activity of CSB is not essential for the function of CSB in BER, we are now challenged to understand the biological function of this posttranslational modification of CSB. The CSB fragment CSB2-341 is not heavily ribosylated in vitro, and full-length CSB is not heavily ribosylated in vivo, since the migration of the proteins does not change significantly after ribosylation. Proteins that are poly(ADP-ribosyl)ated often bind DNA with less affinity because of the extra negative charge of the PAR units. Therefore, it is possible that the inhibition of the DNA-dependent CSB ATPase activity by poly(ADP-ribosyl)ation is a secondary effect caused by an alteration in DNA binding of poly(ADP-ribosyl)ated CSB. However, it was recently demonstrated that CSB binds DNA by wrapping the DNA around its surface and that the wrapping was stimulated by ATP binding to CSB. On the other hand, ATP hydrolysis by CSB caused unwrapping of the DNA (1). Thus, it may also be speculated that the limited ribosylation of CSB, which inhibits ATPase activity, actually changes the equilibrium of CSB in favor of more wrapping of DNA, but this remains to be established.
In this study, we also demonstrate that CSB dysfunction profoundly sensitizes cells to the inhibition of PARP-1 enzymatic activity. Importantly, we detected no difference in PARP-1 protein level and in the PARP-1 poly(ADP-ribosyl)ation response after oxidative stress between the CSB-deficient and -proficient cell lines. Since the PARP enzymatic activity was only inhibited for a few days and no exogenous DNA damage was induced in these experiments, we speculate that the sensitivity was due to endogenous cytotoxic SSBs which were not repaired efficiently when PARP was inhibited. Since cells expressing functional CSB survive this inhibition of PARP better than CSB-deficient cells, we speculate that CSB may mediate an alternative repair pathway. SSBs are very potent in stalling RNA polymerase II (20), and CSB may mediate transcription-coupled repair (TCR) of these lesions. In support of this, in vivo results suggesting a role for CSB in TCR of oxidative DNA lesions have previously been published (7, 23, 24). Additionally, the drugs used to inhibit PARP in our study only inhibit the poly(ADP-ribosyl)ation activity of the protein and not the binding of PARP-1 to SSBs. Thus, PARP-1 itself bound to DNA could also interfere with transcription and other aspects of DNA metabolism. Very recently, a similar sensitivity to PARP inhibition was described for BRCA1- and BRCA2-deficient cells, and the sensitivity was shown to be caused by persistence of DNA damage which, when PARP was inhibited, could be repaired by BCRA1- and BRCA2-dependent homologous recombination (14). Our results suggest that CSB may also mediate a backup repair pathway of SSBs when PARP is inhibited, and this further illustrates how different pathways may cooperate to deal with devastating DNA lesions.
The sensitivity of CSB-deficient cells to inhibition of PARP was not complemented by the ATPase-dead E646Q CSB mutant protein. Since the ATPase activity of CSB is not required for its function in BER of 8-oxoG but is required in TCR, this again suggests that TCR might serve as a backup mechanism for repair of cytotoxic SSBs in the PARP-inhibited cells. The two cell lines expressing the CSB mutant proteins CSB-T912/913V and CSB-Q942E were also sensitive to inhibition of PARP. However, we observed a small increase in survival compared to the CSB-null cell line. Biochemical analysis of these two mutant proteins has demonstrated a small but residual ATPase activity (5) which may explain this difference. The cells expressing CSB with a different site-directed mutation (CSB-R946A) behaved similarly to the vector and E646Q cell lines. The ATPase activity of this mutant protein has not yet been characterized.
It is also possible that the hypersensitivity of CSB-deficient cells to inhibition of PARP poly(ADP-ribosyl)ation activity may be due to the chromatin-remodeling activities of both proteins. Recently, PARP-1 was shown to bind to nucleosomes and modulate chromatin structure through NAD+-dependent automodification (21). Furthermore, it has also been demonstrated that PARP-1 poly(ADP-ribosyl)ates histones, which causes chromatin decondensation (33), and as demonstrated by Citterio et al., CSB can remodel chromatin in vitro in an ATP-dependent manner (6). Thus, CSB and PARP-1 may have complementary roles in remodeling chromatin structure.
In conclusion, these studies demonstrate that CSB resides in a physical and functional complex with PARP-1 that redistributes in the nucleus in response to DNA damage. We show that CSB is posttranslationally modified by PARP-1 after oxidative stress, thus implicating CSB in the PARP-1 poly(ADP-ribosyl)ation response to SSBs. The biological function of the poly(ADP-ribosyl)ation of CSB is not yet clear, but future studies of biochemical and cellular consequences of this posttranslational modification for CSB activity should provide further insight. Furthermore, we speculate that CSB also may be involved in a PARP-1-independent TCR of SSBs, further emphasizing the importance of CSB in maintaining the genome free of oxidative DNA lesions. As patients with CS suffer from dramatic neurodegeneration and a variety of clinical features associated with progeria, we speculate that the reduced capability to repair oxidative damage in the absence of CSB may contribute to these CS phenotypes.
Acknowledgments
We thank Gilbert de Murcia for providing purified PARP-1, Jean-Marc Egly for providing CSB antibodies, and Wim Vermeulen for providing the pSLME6-dtCSB vector. Ulla Birk Henriksen is thanked for supreme technical assistance. David M. Wilson III is thanked for critical reading of the manuscript. Wen-Hsing Cheng is thanked for help with the immunofluorescence studies.
This work was supported by the Danish Medical Research Council (grant no. 22-03-0253). T.T. was supported by the Danish Medical Research Council (grant no. 22-02-0104) and the Knud Højgaard Foundation. M.C. was supported by the Carlsberg Foundation.
REFERENCES
- 1.Beerens, N., J. H. Hoeijmakers, R. Kanaar, W. Vermeulen, and C. Wyman. 2004. The CSB protein actively wraps DNA. J. Biol. Chem. 280:4722-4729. [DOI] [PubMed] [Google Scholar]
- 2.Bradsher, J., J. Auriol, L. P. de Santis, S. Iben, J. L. Vonesch, I. Grummt, and J. M. Egly. 2002. CSB is a component of RNA pol I transcription. Mol. Cell 10:819-829. [DOI] [PubMed] [Google Scholar]
- 3.Brosh, R. M., Jr., J. L. Li, M. K. Kenny, J. K. Karow, M. P. Cooper, R. P. Kureekattil, I. D. Hickson, and V. A. Bohr. 2000. Replication protein A physically interacts with the Bloom's syndrome protein and stimulates its helicase activity. J. Biol. Chem. 275:23500-23508. [DOI] [PubMed] [Google Scholar]
- 4.Caldecott, K. W., S. Aoufouchi, P. Johnson, and S. Shall. 1996. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick-sensor’ in vitro. Nucleic Acids Res. 24:4387-4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christiansen, M., T. Stevnsner, C. Modin, P. M. Martensen, R. M. Brosh, Jr., and V. A. Bohr. 2003. Functional consequences of mutations in the conserved SF2 motifs and post-translational phosphorylation of the CSB protein. Nucleic Acids Res. 31:963-973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Citterio, E., V. Van Den Boom, G. Schnitzler, R. Kanaar, E. Bonte, R. E. Kingston, J. H. Hoeijmakers, and W. Vermeulen. 2000. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol. Cell. Biol. 20:7643-7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper, P. K., T. Nouspikel, S. G. Clarkson, and S. A. Leadon. 1997. Defective transcription-coupled repair of oxidative base damage in Cockayne syndrome patients from XP group G. Science 275:990-993. [DOI] [PubMed] [Google Scholar]
- 8.D'Amours, D., S. Desnoyers, I. D'Silva, and G. G. Poirier. 1999. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 342:249-268. [PMC free article] [PubMed] [Google Scholar]
- 9.Dantzer, F., G. de La Rubia, J. Menissier-De Murcia, Z. Hostomsky, G. de Murcia, and V. Schreiber. 2000. Base excision repair is impaired in mammalian cells lacking poly(ADP-ribose) polymerase-1. Biochemistry 39:7559-7569. [DOI] [PubMed] [Google Scholar]
- 10.Dianov, G., C. Bischoff, M. Sunesen, and V. A. Bohr. 1999. Repair of 8-oxoguanine in DNA is deficient in Cockayne syndrome group B cells. Nucleic Acids Res. 27:1365-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durkacz, B. W., O. Omidiji, D. A. Gray, and S. Shall. 1980. (ADP-ribose)n participates in DNA excision repair. Nature 283:593-596. [DOI] [PubMed] [Google Scholar]
- 12.Durr, H., C. Korner, M. Muller, V. Hickmann, and K. P. Hopfner. 2005. X-ray structures of the Sulfolobus solfataricus SWI2/SNF2 ATPase core and its complex with DNA. Cell 121:363-373. [DOI] [PubMed] [Google Scholar]
- 13.El-Khamisy, S. F., M. Masutani, H. Suzuki, and K. W. Caldecott. 2003. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 31:5526-5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farmer, H., N. McCabe, C. J. Lord, A. N. Tutt, D. A. Johnson, T. B. Richardson, M. Santarosa, K. J. Dillon, I. Hickson, C. Knights, N. M. Martin, S. P. Jackson, G. C. Smith, and A. Ashworth. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434:917-921. [DOI] [PubMed] [Google Scholar]
- 15.Flohr, C., A. Burkle, J. P. Radicella, and B. Epe. 2003. Poly(ADP-ribosyl)ation accelerates DNA repair in a pathway dependent on Cockayne syndrome B protein. Nucleic Acids Res. 31:5332-5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giner, H., F. Simonin, G. de Murcia, and J. Menissier-de Murcia. 1992. Overproduction and large-scale purification of the human poly(ADP-ribose) polymerase using a baculovirus expression system. Gene 114:279-283. [DOI] [PubMed] [Google Scholar]
- 17.Herceg, Z., and Z. Q. Wang. 2001. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat. Res. 477:97-110. [DOI] [PubMed] [Google Scholar]
- 18.Hoeijmakers, J. H. 2001. Genome maintenance mechanisms for preventing cancer. Nature 411:366-374. [DOI] [PubMed] [Google Scholar]
- 19.Horibata, K., Y. Iwamoto, I. Kuraoka, N. G. Jaspers, A. Kurimasa, M. Oshimura, M. Ichihashi, and K. Tanaka. 2004. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc. Natl. Acad. Sci. USA 101:15410-15415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kathe, S. D., G. P. Shen, and S. S. Wallace. 2004. Single-stranded breaks in DNA but not oxidative DNA base damages block transcriptional elongation by RNA polymerase II in HeLa cell nuclear extracts. J. Biol. Chem. 279:18511-18520. [DOI] [PubMed] [Google Scholar]
- 21.Kim, M. Y., S. Mauro, N. Gevry, J. T. Lis, and W. L. Kraus. 2004. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 119:803-814. [DOI] [PubMed] [Google Scholar]
- 22.Kubota, Y., R. A. Nash, A. Klungland, P. Schar, D. E. Barnes, and T. Lindahl. 1996. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 15:6662-6670. [PMC free article] [PubMed] [Google Scholar]
- 23.Leadon, S. A., and P. K. Cooper. 1993. Preferential repair of ionizing radiation-induced damage in the transcribed strand of an active human gene is defective in Cockayne syndrome. Proc. Natl. Acad. Sci. USA 90:10499-10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le Page, F., E. E. Kwoh, A. Avrutskaya, A. Gentil, S. A. Leadon, A. Sarasin, and P. K. Cooper. 2000. Transcription-coupled repair of 8-oxoguanine: requirement for XPG, TFIIH, and CSB and implications for Cockayne syndrome. Cell 101:159-171. [DOI] [PubMed] [Google Scholar]
- 25.Le Page, F., V. Schreiber, C. Dherin, G. De Murcia, and S. Boiteux. 2003. Poly(ADP-ribose) polymerase-1 (PARP-1) is required in murine cell lines for base excision repair of oxidative DNA damage in the absence of DNA polymerase beta. J. Biol. Chem. 278:18471-18477. [DOI] [PubMed] [Google Scholar]
- 26.Leppard, J. B., Z. Dong, Z. B. Mackey, and A. E. Tomkinson. 2003. Physical and functional interaction between DNA ligase IIIα and poly(ADP-ribose) polymerase 1 in DNA single-strand break repair. Mol. Cell. Biol. 23:5919-5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Licht, C. L., T. Stevnsner, and V. A. Bohr. 2003. Cockayne syndrome group B cellular and biochemical functions. Am. J. Hum. Genet. 73:1217-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masson, M., C. Niedergang, V. Schreiber, S. Muller, J. Menissier-de Murcia, and G. de Murcia. 1998. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol. 18:3563-3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meder, V. S., M. Boeglin, G. de Murcia, and V. Schreiber. 2005. PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J. Cell Sci. 118:211-222. [DOI] [PubMed] [Google Scholar]
- 30.Muftuoglu, M., R. Selzer, J. Tuo, R. M. Brosh, Jr., and V. A. Bohr. 2002. Phenotypic consequences of mutations in the conserved motifs of the putative helicase domain of the human Cockayne syndrome group B gene. Gene 283:27-40. [DOI] [PubMed] [Google Scholar]
- 31.Okano, S., L. Lan, K. W. Caldecott, T. Mori, and A. Yasui. 2003. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 23:3974-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osterod, M., E. Larsen, F. Le Page, J. G. Hengstler, G. T. Van Der Horst, S. Boiteux, A. Klungland, and B. Epe. 2002. A global DNA repair mechanism involving the Cockayne syndrome B (CSB) gene product can prevent the in vivo accumulation of endogenous oxidative DNA base damage. Oncogene 21:8232-8239. [DOI] [PubMed] [Google Scholar]
- 33.Realini, C. A., and F. R. Althaus. 1992. Histone shuttling by poly(ADP-ribosylation). J. Biol. Chem. 267:18858-18865. [PubMed] [Google Scholar]
- 34.Satoh, M. S., and T. Lindahl. 1992. Role of poly(ADP-ribose) formation in DNA repair. Nature 356:356-358. [DOI] [PubMed] [Google Scholar]
- 35.Selby, C. P., and A. Sancar. 1997. Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem. 272:1885-1890. [DOI] [PubMed] [Google Scholar]
- 36.Selzer, R. R., S. Nyaga, J. Tuo, A. May, M. Muftuoglu, M. Christiansen, E. Citterio, R. M. Brosh, Jr., and V. A. Bohr. 2002. Differential requirement for the ATPase domain of the Cockayne syndrome group B gene in the processing of UV-induced DNA damage and 8-oxoguanine lesions in human cells. Nucleic Acids Res. 30:782-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slupphaug, G., B. Kavli, and H. E. Krokan. 2003. The interacting pathways for prevention and repair of oxidative DNA damage. Mutat. Res. 531:231-251. [DOI] [PubMed] [Google Scholar]
- 38.Southan, G. J., and C. Szabo. 2003. Poly(ADP-ribose) polymerase inhibitors. Curr. Med. Chem. 10:321-340. [DOI] [PubMed] [Google Scholar]
- 39.Stevnsner, T., S. Nyaga, N. C. de Souza-Pinto, G. T. van der Horst, T. G. Gorgels, B. A. Hogue, T. Thorslund, and V. A. Bohr. 2002. Mitochondrial repair of 8-oxoguanine is deficient in Cockayne syndrome group B. Oncogene 21:8675-8682. [DOI] [PubMed] [Google Scholar]
- 40.Sunesen, M., T. Stevnsner, R. M. Brosh, Jr., G. L. Dianov, and V. A. Bohr. 2002. Global genome repair of 8-oxoG in hamster cells requires a functional CSB gene product. Oncogene 21:3571-3578. [DOI] [PubMed] [Google Scholar]
- 41.Svejstrup, J. Q. 2002. Mechanisms of transcription-coupled DNA repair. Nat. Rev. Mol. Cell Biol. 3:21-29. [DOI] [PubMed] [Google Scholar]
- 42.Thoma, N. H., B. K. Czyzewski, A. A. Alexeev, A. V. Mazin, S. C. Kowalczykowski, and N. P. Pavletich. 2005. Structure of the SWI2/SNF2 chromatin-remodeling domain of eukaryotic Rad54. Nat. Struct. Mol. Biol. 12:350-356. [DOI] [PubMed] [Google Scholar]
- 43.Tulin, A., and A. Spradling. 2003. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science 299:560-562. [DOI] [PubMed] [Google Scholar]
- 44.Tuo, J., C. Chen, X. Zeng, M. Christiansen, and V. A. Bohr. 2002. Functional crosstalk between hOgg1 and the helicase domain of Cockayne syndrome group B protein. DNA Repair 1:913-927. [DOI] [PubMed] [Google Scholar]
- 45.Tuo, J., M. Muftuoglu, C. Chen, P. Jaruga, R. R. Selzer, R. M. Brosh, Jr., H. Rodriguez, M. Dizdaroglu, and V. A. Bohr. 2001. The Cockayne Syndrome group B gene product is involved in general genome base excision repair of 8-hydroxyguanine in DNA. J. Biol. Chem. 276:45772-45779. [DOI] [PubMed] [Google Scholar]
- 46.Venema, J., L. H. Mullenders, A. T. Natarajan, A. A. van Zeeland, and L. V.Mayne. 1990. The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc. Natl. Acad. Sci. USA 87:4707-4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.von Kobbe, C., J. A. Harrigan, A. May, P. L. Opresko, L. Dawut, W. H.-Cheng, and V. A. Bohr. 2003. Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol. Cell. Biol. 23:8601-8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wiederhold, L., J. B. Leppard, P. Kedar, F. Karimi-Busheri, A. Rasouli-Nia, M. Weinfeld, A. E. Tomkinson, T. Izumi, R. Prasad, S. H. Wilson, S. Mitra, and T. K. Hazra. 2004. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 15:209-220. [DOI] [PubMed] [Google Scholar]
- 49.Will, O., E. Gocke, I. Eckert, I. Schulz, M. Pflaum, H. C. Mahler, and B. Epe. 1999. Oxidative DNA damage and mutations induced by a polar photosensitizer, Ro19-8022. Mutat. Res. 435:89-101. [DOI] [PubMed] [Google Scholar]