

Abstract
Amino acids are essential building blocks in biology and chemistry. Whereas nature relies on a small number of amino acid structures, chemists desire access to a vast range of structurally diverse analogues1-3. The selective modification of amino acid side-chain residues represents an efficient strategy to access non-canonical derivatives of value in chemistry and biology. While semisynthetic methods leveraging the functional groups found in polar and aromatic amino acids have been extensively explored, highly selective and general approaches to transform unactivated C─H bonds in aliphatic amino acids remain less developed4,5. Here we disclose a stepwise dehydrogenative method to convert aliphatic amino acids into structurally diverse analogues. The key to the success of this approach lies in the development of a selective catalytic acceptorless dehydrogenation method driven by photochemical irradiation, which provides access to terminal alkene intermediates for downstream functionalization. Overall, this strategy enables the rapid synthesis of new amino acid building blocks and suggests possibilities for the late-stage modification of more complex oligopeptides.
Natural and unnatural non-canonical amino acids (ncAAs) find applications as starting materials in the synthesis of functional molecules6-8, as a source of chiral information in enantioselective synthesis and catalysis9 and as tools to expand and explore the function of native biological machinery (Fig. 1a)10-12. Proteinogenic amino acids are an ideal starting point to access ncAA scaffolds, provided that selective side-chain modification can be achieved in the presence of Lewis basic backbone functional groups and without loss of enantiopurity at the α-stereogenic centre. A small number of amino acids possessing orthogonally reactive functional groups (for example, cysteine, serine, lysine, glutamic acid, tryptophan, inter alia) have powered ncAA semisynthesis5,13 and enabled residue-specific bioconjugation4,14. By contrast, the absence of readily functionalizable chemical handles in the aliphatic amino acids (for example, leucine, isoleucine, valine) has constrained synthetic access to derivatives in proximal chemical space and limited opportunities to leverage these residues in more complex settings (Fig. 1b)15.
Fig. 1 ∣. Strategies to access non-canonical amino acids.

a, Non-canonical amino acids are essential building blocks in bioactive molecules and chiral reagents. b, Aliphatic amino acids are abundant but underutilized feedstocks in organic synthesis. c, This work: terminal-selective dehydrogenation enables access to diverse non-canonical amino acid scaffolds. d, High terminal selectivity is eroded under thermal catalytic acceptorless dehydrogenation conditions. e, Directed, intramolecular HAT provides access to terminal olefins, but requires auxiliary installation.
We sought to devise a general platform to enable more effective utilization of aliphatic amino acid building blocks. We reasoned that a terminal alkene would provide an ideal intermediate functional handle to introduce downstream functionality (Fig. 1c). Ideally, the desired alkene-containing intermediates could be accessed directly through a selective, catalytic, dehydrogenation of the saturated precursor. However, terminal-selective dehydrogenation remains an unsolved synthetic challenge in nearly every synthetic context, and only isolated examples of selective amino acid dehydrogenation exist16-18. Impressive terminal selectivities have been achieved in alkane dehydrogenation using noble metal catalysts operating under acceptorless conditions19-21. However, metal hydride species formed in situ can promote undesired thermal olefin isomerization, compromising terminal selectivity and leading to complex product mixtures at higher reaction conversions (Fig. 1d). A more common strategy to control positional selectivity in olefination reactions has been to use an internal oxidant and/or directing group (Fig. 1e)16,17,22-24. However, the requirement for pre-installation of a stoichiometric auxiliary limits the generality of such approaches in native and more complex settings.
We report photochemical conditions that promote terminal-selective catalytic acceptorless dehydrogenation of aliphatic amino acid residues. Our approach was inspired by seminal studies from Sorensen and West, who reported that a dual catalytic system composed of tetra-n-butylammonium decatungstate (TBADT) and a cobaloxime complex (Co(dmgH)2(Py)Cl) (dmg, dimethylglyoximate) can promote the dehydrogenation of unactivated aliphatic substrates25. These proof-of-concept studies established the basic mechanistic framework for photocatalytic dehydrogenation; however, the functional group compatibility, site- and chemoselectivity and overall synthetic versatility of this method remain underexplored26. We reasoned that the combination of decatungstate (DT) and cobaloxime cocatalysts might enable terminal-selective dehydrogenation due to the well-established steric preference of DT-mediated H atom abstraction (HAA)27 and Co-based metal-hydride hydrogen atom transfer (MHAT) steps28. Importantly, we have previously reported that this same combination of catalysts can promote contra-thermodynamic internal-to-terminal olefin isomerization29. We thus envisioned that any background isomerization process might enrich, rather than erode, the terminal selectivity of the proposed dehydrogenation step.
We selected (l)-Bz-Leu-OtBu (1a) as a model substrate for the proposed dehydrogenation reaction (Fig. 2a). The formation of terminal γ,δ-dehydrogenated product 2a was observed in 86% yield with a >20:1 ratio of terminal/internal isomers and 94% enantiospecificity (e.s.) under optimal reaction conditions using 6 mol% Na4W10O32 (NaDT) and 5 mol% Co(dmgH)(dmgH2)Br2 (Co-H) in MeCN at room temperature under 390 nm light-emitting diode (LED) irradiation (Fig. 2a). The yield of2a was decreased to 46% when TBADT was used in place of NaDT (Fig. 2a, entry 1). The lower yield observed using TBADT can be attributed to significant background catalyst deactivation, which may arise from a light-driven elimination pathway (Supplementary Information)25,30,31. Other synthetically useful thermal and photochemical dehydrogenation methods were evaluated for their ability to convert 1a into 2a; however, none of the conditions we tested yielded the desired product (Supplementary Information)32,33. No reaction was observed without DT, cobaloxime or LED irradiation (Fig. 2a, entry 4). The optimal reaction conditions were found to be amenable to scale (Fig. 2a, entry 5), enabling the isolation of 900 mg of 2a (4 mmol scale, 79% yield, 94% e.s.).
Fig. 2 ∣. Development of the dehydrogenation method.

a, Reaction optimization. See Section 5 of the Supplementary Information for full details. b, Proposed mechanism and mechanistic studies. See Section 9 of the Supplementary Information for full experimental details. c, Substrate scope using amino acid substrates. Reactions were performed on a 0.4 mmol scale with 6 mol% NaDT and 5 mol% Co(dmgH)(dmgH2)Br2 under 390 nm LED irradiation at room temperature (RT) in acetonitrile (0.1 M) for 24 h. Isolated yields are the average of two runs. See Section 6 of the Supplementary Information for full experimental details. aDebrominative olefination reaction conditions: 2 mol% 4CzIPN, 5 mol% Co(dmgH2)(dmgH)Br2, 20 mol% DIPEA, 1.0 equiv. K2CO3, MeCN (0.1 M). bReaction was performed with 6 mol% NaDT and 15 mol% Co(dmgH)(dmgH2)Br2 in 9:1 MeCN:H2O (0.1 M). cNMR yields. dReaction was performed with 1g-Cl as the substrate. eReaction was performed with 6 mol% NaDT and 25 mol% Co(dmgH)(dmgH2)Br2 in 3:1 MeCN:H2O (0.1 M). TCP, N-tetrachlorophthaloyl.
We conducted a series of experiments to probe the basis for terminal selectivity, based on a proposed reaction mechanism involving sequential H atom abstraction and MHAT steps (Fig. 2b). To assess the site selectivity of initial radical generation, we subjected 1a to ‘DT-only’ reaction conditions consisting of 1% NaDT and 5% (4-Cl-C6H4S)2 in MeCN/D2O under 390 nm LED irradiation34. The pattern of isotope exchange observed in recovered 1a reveals the initial HAA step to be highly selective for the methine γ-position (98% D-incorporation), consistent with previously observed selectivity patterns in DT-mediated C─H fluorination35,36 and other functionalization reactions27. Only minor isotope exchange (6% D-incorporation) was observed at the terminal δ-position, and no isotope exchange occurred at the α or β positions of the leucine side chain (Fig. 2b, HAA selectivity). To assess the site selectivity of cobaloxime-mediated HAA/MHAT, we subjected independently prepared 1a-Br to debrominative olefination conditions proposed to proceed through a similar Co-mediated MHAT step following reductive generation of the γ-position radical (Fig. 2b, MHAT selectivity)28. Under these conditions, the reaction of 1a-Br led to the formation of 39% 2a and 5% Int-2a (8:1 terminal/internal). Finally, independently prepared internal olefin Int-2a was subjected to the standard dehydrogenation conditions. After irradiating for 24 h, 68% 2a was obtained, with only 4% Int-2a remaining (17:1 terminal/internal). By contrast, no Int-2a was observed to form when 2a was resubjected to the standard conditions. Collectively, these findings suggest that high terminal selectivity arises from selective HAA and MHAT steps, as well as background internal-to-terminal isomerization.
A range of amino acids was evaluated as substrates under the optimal dehydrogenation conditions to assess the scope and selectivity of the reaction (Fig. 2c). Leucine derivatives bearing common N-protecting groups, such as -Boc, -TCP, -Cbz and -CO2Ph, reacted to form the corresponding dehydrogenated products 2b–2e without significant loss of enantioenrichment (96–99% e.s.). In general, improved product yields can be obtained by recharging the reaction mixture with fresh reagents after 24 h: for example, the yield of product 2b increased from 51% (45% RSM, 96% e.s.) to 74% yield (0% RSM) with minimal erosion of enantiomeric excess (e.e.) (91% e.s.) after recharging. The reaction of N-unprotected substrate, H-Leu-OtBu·HCl, led to the formation of dehydrogenation products 2g and 2g′ in 57% yield (3.8:1 regioisomeric ratio (r.r.)), whereas C-unprotected and fully unprotected substrates reacted to form 2h and 2i in 23% and 37% yields, respectively.
Other substrates derived from (l)-valine, (l)-homoleucine and (l)-β-amino acids also reacted to form terminal alkene-containing products (2j–2o) in good yields and high enantiospecificities. A 3:1 mixture of regioisomers 2p and 2p′ was obtained from the reaction of isoleucine derivative 1p. Protected (l)-2-aminobutyric acid (homoalanine) reacted to form vinylglycine derivative 2q in 50% yield, however erosion of the product enantioselectivity (71% e.s.) was observed. Similar erosion of e.e. was observed when (l)-homoglutamic acid was used as a substrate; in this case, internal dehydrogenation product 2r was obtained in 55% yield (49% e.s.).
Whereas conjugated α,β-dehydroamino acids are well-established and versatile intermediates37, the corresponding terminal β,γ- and γ,δ-dehydroamino acids have not been systematically studied38,39. We envisioned that the complementary polarity and reactivity profile of the terminal olefin functional group and pre-established α-position stereogenicity could present new opportunities for ncAA synthesis (Fig. 3). For example, dehydrogenated building blocks 2a, 2j and 2m were converted to azetidine- (3a) and aziridine-containing (3j, 3m) amino acids through a cyclization/dehalogenation sequence mediated by N-iodosuccinimide and tributyltin hydride, respectively. Aziridine 3j is a protected derivative of pleurocybellaziridine, a cytotoxic ncAA produced by the angel’s wing mushroom, Pleurocybella porrigens40. Our approach enables synthetically useful access to this target in four steps overall from commercial H-Val-OtBu.
Fig. 3 ∣. Product derivatization and elaboration.

See Section 7 of the Supplementary Information for full experimental details. aNMR yield.
The direct functionalization of the activated methine C─H bonds of valine and leucine analogues has previously been achieved under chemoenzymatic41-43, transition metal-mediated and radical-based conditions13,44,45. However, direct functionalization of the terminal methyl C─H bonds remains challenging46-48. The olefin intermediates obtained here enable straightforward access to terminal-modified ncAAs using anti-Markovnikov alkene hydrofunctionalization conditions. For example, dehydrogenation followed by radical trifluoromethylation provided access to homo-isoleucine analogue 4a in 90% yield from 2a; thiol-ene and radical hydroazidation reactions led to thioglucoside 5a and azide-containing amino acid 6a, respectively. In addition to radical-mediated transformations, 2a also reacted under established hydroboration conditions to afford terminal alcohol-containing product 7a in 62% yield (2:1 diastereomeric ratio (d.r.)) after oxidative workup. For comparison, a previously reported route to prepare an analogous linear alcohol (possessing a different protecting group pattern) required six steps and involved an enzymatic kinetic resolution49.
In addition to enabling terminal-selective side-chain modifications, olefin intermediates provide opportunities for functionalization at other side-chain positions. For example, ketone 7a was obtained in 79% yield by subjecting intermediate 2a to standard ozonolysis conditions, and product 8a was obtained in 29% yield under oxidative allylic amination conditions. Finally, N-allylation followed by ring-closing metathesis provided access to enantioenriched pipecolic acid derivative 9a that could serve as an intermediate for the synthesis of an FDA-approved thrombin inhibitor, argatroban. In comparison, previous enantioselective syntheses of structurally related compounds relied on the use of stoichiometric chiral auxiliaries to establish the desired α-position stereochemistry50,51.
The synthetic efficiency of dehydrogenative tailoring is further illustrated by the synthesis of ncAA 11, which has been proposed to serve as the surrogate for γ,δ-dihydroxy-leucine, a key residue that occurs in the natural product alloviroidin. This intermediate can be obtained in just two steps from commercially available l-leucine derivative, Cbz-Leu-OMe. By contrast, a previously established route to 11 required a six-step sequence starting from achiral precursors38.
In addition to derivatives arising from the modification of l-leucine, we examined the feasibility of accessing β-branched amino acids through dehydrogenative tailoring of L-isoleucine. Following dehydrogenation, intermediate 2p was subjected to dihydroxylation, oxytrifluoromethylation and olefin cross-metathesis conditions to provide highly functionalized ncAAs 3p, 4p and 5p, respectively, in workable yields. The terminal alkene within 2p not only serves as a reactive handle in thiol-ene ‘click’ reactions, but also can be leveraged to introduce diverse ‘clickable’ thiol, azide and alkyne functional groups (for example, products 6p, 7p and 8p) in just one or two steps.
Next, we subjected a series of dipeptide substrates (12a–12l) to the standard dehydrogenation conditions, to assess the feasibility of dehydrogenation on more complex substrates (Fig. 4). In general, protected amino acid side chains were well tolerated under the reaction conditions, with slightly diminished yields observed in the presence of histidine, tryptophan and lysine residues. Several unprotected dipeptides were also tested as substrates (Supplementary Information); however, low reaction yields (<10%) were observed under the standard reaction conditions. This series also reveals efficient dehydrogenation to be possible irrespective of whether the aliphatic residue was placed in the N-terminal or C-terminal position (for example, 13e and 13f): a potential advantage of using an undirected dehydrogenation protocol that does not necessitate installation of a directing auxiliary in a specific position.
Fig. 4 ∣. Dehydrogenation of oligopeptide substrates.
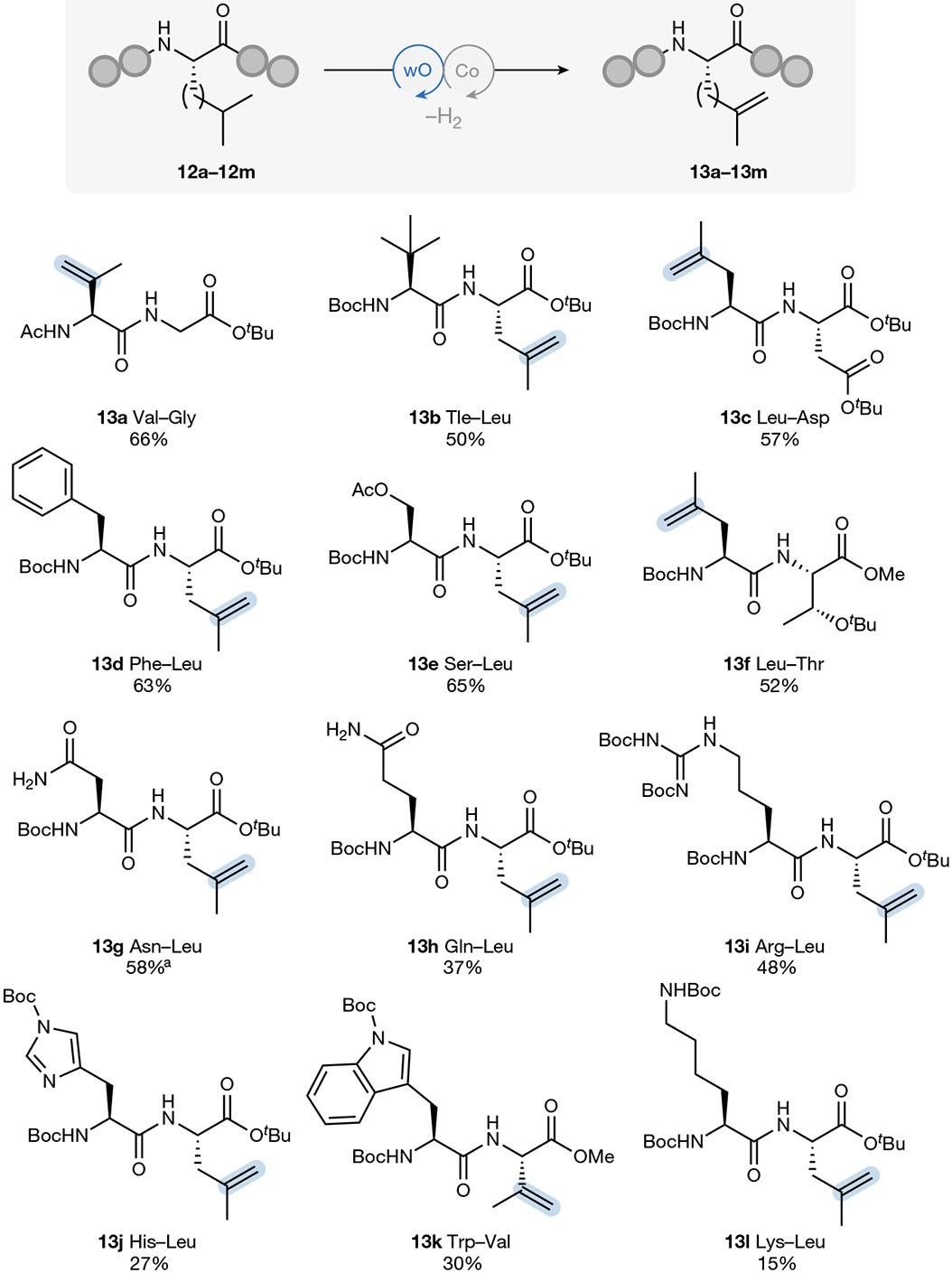
Reactions were performed on a 0.2 mmol scale with 6 mol% NaDT and 5 mol% Co(dmgH) (dmgH2)Br2 under 390 nm LED irradiation at room temperature in acetonitrile (0.1 M) for 18 h. Isolated yields are the average of two runs. See Sections 8.1-8.12 of the Supplementary Information for full experimental details. aReaction performed with 24 mol% NaDT and 20 mol% Co(dmgH)(dmgH2)Br2 in MeCN (0.1 M).
Finally, we sought to illuminate potential applications of dehydrogenative tailoring at a later synthetic stage (Fig. 5). We envisioned that the ability to interconvert side-chain residues within an oligopeptide in a targeted manner could prove enabling, and selected tripeptide 12m as a model substrate (Fig. 5a). Following dehydrogenation, 13m was transformed into the corresponding alanine-containing peptide 14m under Kwon hydrodealkenylation conditions52, in what amounts to an otherwise highly challenging ‘residue editing’ operation. Conversely, under standard ozonolysis conditions, 13m was transformed into ketone-containing peptide 15m.
Fig. 5 ∣. Peptide modification enabled by dehydrogenative tailoring.

a, Peptide tailoring. Reactions were conducted on a 0.2 mmol scale with 6 mol% NaDT and 5 mol% Co(dmgH)(dmgH2)Br2 under 390 nm LED irradiation at room temperature in acetonitrile (0.1 M) for 18 h. Isolated yields are reported (average of two runs). b, Late-stage peptide–peptide conjugation. aReaction was performed with 5 mol% NaDT and 25 mol% Co(dmgH)(dmgH2)Br2 in 3:1 MeCN:H2O (0.1 M). See Sections 8.13-8.17 of the Supplementary Information for full experimental details.
Terminal alkene functional groups installed at a late synthetic stage can also serve as useful bioorthogonal handles for thiol-ene click reactions. To illustrate this, we subjected a derivative of the opioid peptide neurotransmitter Leu-enkephalin 16 to modified dehydrogenation conditions and obtained alkene-containing product 17 in 24% yield. Using reduced glutathione, 18, as the thiol-ene coupling partner, 17 was readily converted to thioether-linked peptide-coupling product 19 in 37% yield (Fig. 5b).
Supplementary Material
Acknowledgements
We thank J. Yang (MIT) for HPLC separation of product 2p and S. Garhwal (MIT) for supercritical fluid chromatography data collection. Financial support for this work was provided by the National Institutes of Health (GM146248) and the National Science Foundation (NSF) through a Graduate Research Fellowship to G.O. (DGE1745303).
Footnotes
Competing interests The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-024-07988-8.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-024-07988-8.
Data availability
All data supporting the findings of this paper are available in the main text or the Supplementary Information.
References
- 1.Hruby VJ & Qian X in Peptide Synthesis Protocols (eds Pennington MW & Dunn BM) 249–286 (Humana, 1995). [Google Scholar]
- 2.Nájera C & Sansano JM Catalytic asymmetric synthesis of α-amino acids. Chem. Rev 107, 4584–4671 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Almhjell PJ, Boville CE & Arnold FH Engineering enzymes for noncanonical amino acid synthesis. Chem. Soc. Rev 47, 8980–8997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.deGruyter JN, Malins LR & Baran PS Residue-specific peptide modification: a chemist’s guide. Biochemistry 56, 3863–3873 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noisier AFM & Brimble MA C─H functionalization in the synthesis of amino acids and peptides. Chem. Rev 114, 8775–8806 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Walsh CT, O’Brien RV & Khosla C Nonproteinogenic amino acid building blocks for nonribosomal peptide and hybrid polyketide scaffolds. Angew. Chem. Int. Ed. Engl 52, 7098–7124 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blaskovich MAT Unusual amino acids in medicinal chemistry. J. Med. Chem 59, 10807–10836 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Hickey JL, Sindhikara D, Zultanski SL & Schultz DM Beyond 20 in the 21st century: prospects and challenges of non-canonical amino acids in peptide drug discovery. ACS Med. Chem. Lett 14, 557–565 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reetz MT New approaches to the use of amino acids as chiral building blocks in organic synthesis. Angew. Chem. Int. Ed. Engl 30, 1531–1546 (1991). [Google Scholar]
- 10.Rezhdo A, Islam M, Huang M & Van Deventer JA Future prospects for noncanonical amino acids in biological therapeutics. Curr. Opin. Biotechnol 60, 168–178 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saleh AM, Wilding KM, Calve S, Bundy BC & Kinzer-Ursem TL Non-canonical amino acid labeling in proteomics and biotechnology. J. Biol. Eng 13, 43 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lugtenburg T, Gran-Scheuch A & Drienovská I Non-canonical amino acids as a tool for the thermal stabilization of enzymes. Protein Eng. Des. Sel 36, gzad003 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aguilar Troyano FJ, Merkens K, Anwar K & Gómez-Suárez A Radical-based synthesis and modification of amino acids. Angew. Chem. Int. Ed. Engl 60, 1098–1115 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boutureira O & Bernardes GJL Advances in chemical protein modification. Chem. Rev 115, 2174–2195 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Capecchi A & Reymond J-L Peptides in chemical space. Med. Drug Discov 9, 100081 (2021). [Google Scholar]
- 16.Voica A-F, Mendoza A, Gutekunst WR, Fraga JO & Baran PS Guided desaturation of unactivated aliphatics. Nat. Chem 4, 629–635 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herbort JH, Bednar TN, Chen AD, RajanBabu TV & Nagib DA γ C─H functionalization of amines via triple H-atom transfer of a vinyl sulfonyl radical chaperone. J. Am. Chem. Soc 144, 13366–13373 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chuentragool P, Parasram M, Shi Y & Gevorgyan V General, mild, and selective method for desaturation of aliphatic amines. J. Am. Chem. Soc 140, 2465–2468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang K et al. Selective dehydrogenation of small and large molecules by a chloroiridium catalyst. Sci. Adv 8, eabo6586 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobereiner GE & Crabtree RH Dehydrogenation as a substrate-activating strategy in homogeneous transition-metal catalysis. Chem. Rev 110, 681–703 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Choi J, MacArthur AHR, Brookhart M & Goldman AS Dehydrogenation and related reactions catalyzed by iridium pincer complexes. Chem. Rev 111, 1761–1779 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Parasram M, Chuentragool P, Wang Y, Shi Y & Gevorgyan V General, auxiliary-enabled photoinduced Pd-catalyzed remote desaturation of aliphatic alcohols. J. Am. Chem. Soc 139, 14857–14860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stateman LM, Dare RM, Paneque AN & Nagib DA Aza-heterocycles via copper-catalyzed, remote C─H desaturation of amines. Chem 8, 210–224 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou S, Zhang Z-J & Yu J-Q Copper-catalysed dehydrogenation or lactonization of C(sp3)─H bonds. Nature 629, 363–369 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.West JG, Huang D & Sorensen EJ Acceptorless dehydrogenation of small molecules through cooperative base metal catalysis. Nat. Commun 6, 10093 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ritu Kolb D., Jain N & König B Synthesis of linear enamides and enecarbamates via photoredox acceptorless dehydrogenation. Adv. Synth. Catal 365, 605–611 (2023). [Google Scholar]
- 27.Ravelli D, Fagnoni M, Fukuyama T, Nishikawa T & Ryu I Site-selective C─H functionalization by decatungstate anion photocatalysis: synergistic control by polar and steric effects expands the reaction scope. ACS Catal. 8, 701–713 (2018). [Google Scholar]
- 28.Zhao H. et al. Merging halogen-atom transfer (XAT) and cobalt catalysis to override E2-selectivity in the elimination of alkyl halides: a mild route toward contra-thermodynamic olefins. J. Am. Chem. Soc 143, 14806–14813 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Occhialini G, Palani V & Wendlandt AE Catalytic, contra-thermodynamic positional alkene isomerization. J. Am. Chem. Soc 144, 145–152 (2022). [DOI] [PubMed] [Google Scholar]
- 30.Yamase T, Takabayashi N & Kaji M Solution photochemistry of tetrakis(tetrabutylammonium) decatungstate(VI) and catalytic hydrogen evolution from alcohols. J. Chem. Soc. Dalton Trans 10.1039/DT9840000793 (1984). [DOI] [Google Scholar]
- 31.Wrzyszczyński A. et al. Unexpected Hofmann elimination in the benzophenone–(phenylthio)acetic tetrabutylammonium salt photoredox system. J. Am. Chem. Soc 125, 11182–11183 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Fuse H, Kojima M, Mitsunuma H & Kanai M Acceptorless dehydrogenation of hydrocarbons by noble-metal-free hybrid catalyst system. Org. Lett 20, 2042–2045 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Zhou M-J, Zhang L, Liu G, Xu C & Huang Z Site-selective acceptorless dehydrogenation of aliphatics enabled by organophotoredox/cobalt dual catalysis. J. Am. Chem. Soc 143, 16470–16485 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y-A et al. Stereochemical editing logic powered by the epimerization of unactivated tertiary stereocenters. Science 378, 383–390 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halperin SD, Fan H, Chang S, Martin RE & Britton R A convenient photocatalytic fluorination of unactivated C─H bonds. Angew. Chem. Int. Ed. Engl 53, 4690–4693 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Yuan Z. et al. Site-selective, late-stage C─H 18F-fluorination on unprotected peptides for positron emission tomography imaging. Angew. Chem. Int. Ed. Engl 57, 12733–12736 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Bogart JW & Bowers AA Dehydroamino acids: chemical multi-tools for late-stage diversification. Org. Biomol. Chem 17, 3653–3669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edagwa BJ & Taylor CM Peptides containing γ,δ-dihydroxy-l-leucine. J. Org. Chem 74, 4132–4136 (2009). [DOI] [PubMed] [Google Scholar]
- 39.McLean JT, Milbeo P, Lynch DM, McSweeney L & Scanlan EM Radical-mediated acyl thiol-ene reaction for rapid synthesis of biomolecular thioester derivatives. Eur. J. Org. Chem 2021, 4148–4160 (2021). [Google Scholar]
- 40.Wakimoto T. et al. Proof of the existence of an unstable amino acid: pleurocybellaziridine in Pleurocybella porrigens. Angew. Chem. Int. Ed. Engl 50, 1168–1170 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Zwick CR & Renata H Remote C─H hydroxylation by an α-ketoglutarate-dependent dioxygenase enables efficient chemoenzymatic synthesis of manzacidin C and proline analogs. J. Am. Chem. Soc 140, 1165–1169 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Tao H. et al. Stereoselectivity and substrate specificity of the FeII/α-ketoglutarate-dependent oxygenase TqaL. J. Am. Chem. Soc 144, 21512–21520 (2022). [DOI] [PubMed] [Google Scholar]
- 43.Gomez CA, Mondal D, Du Q, Chan N & Lewis JC Directed evolution of an iron(II)- and α-ketoglutarate-dependent dioxygenase for site-selective azidation of unactivated aliphatic C─H bonds. Angew. Chem. Int. Ed. Engl 62, e202301370 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nanjo T, De Lucca EC & White MC Remote, late-stage oxidation of aliphatic C─H bonds in amide-containing molecules. J. Am. Chem. Soc 139, 14586–14591 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarver PJ, Bissonnette NB & MacMillan DWC Decatungstate-catalyzed C(sp3)─H sulfinylation: rapid access to diverse organosulfur functionality. J. Am. Chem. Soc 143, 9737–9743 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galonić DP, Vaillancourt FH & Walsh CT Halogenation of unactivated carbon centers in natural product biosynthesis: trichlorination of leucine during barbamide biosynthesis. J. Am. Chem. Soc 128, 3900–3901 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Cudic M, Marí F & Fields GB Synthesis and solid-phase application of suitably protected γ-hydroxyvaline building blocks. J. Org. Chem 72, 5581–5586 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shu C, Noble A & Aggarwal VK Metal-free photoinduced C(sp3)─H borylation of alkanes. Nature 586, 714–719 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Barbie P & Kazmaier U Total synthesis of cyclomarins A, C and D, marine cyclic peptides with interesting anti-tuberculosis and anti-malaria activities. Org. Biomol. Chem 14, 6036–6054 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Agami C. et al. Asymmetric syntheses of enantiopure 4-substituted pipecolic acid derivatives. Eur. J. Org. Chem 2001, 2385–2389 (2001). [Google Scholar]
- 51.Ferraboschi P, Mieri MD, Grisenti P, Lotz M & Nettekoven U Diastereoselective synthesis of an argatroban intermediate, ethyl (2R,4R)-4-methylpipecolate, by means of a mandyphos/rhodium complex-catalyzed hydrogenation. Tetrahedron Asymmetry 22, 1626–1631 (2011). [Google Scholar]
- 52.Smaligo AJ et al. Hydrodealkenylative C(sp3)─C(sp2) bond fragmentation. Science 364, 681–685 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this paper are available in the main text or the Supplementary Information.