

Abstract
Antibiotics that operate via multiple mechanisms of action are a promising strategy to combat growing resistance. Previous studies have shown that dual action antifolates formed from a pyrroloquinazolinediamine core can inhibit the growth of bacterial pathogens without developing resistance. In this work, we expand the scope of dual action antifolates by repurposing the 2,4-diamino-1,6-dihydro-1,3,5-triazine (DADHT) cycloguanil scaffold to a variety of derivatives designed to inhibit dihydrofolate reductase (DHFR) and disrupt bacterial membranes. Dual mechanism DADHTs have activity against a variety of target pathogens, including M. tuberculosis, M. abscessus, and P. aeruginosa, among other ESKAPEE organisms. Through X-ray crystallography, we confirmed engagement of the E. coli DHFR target and found that some DADHTs stabilize a previously unobserved conformation of the enzyme but, broadly, bind in the occluded conformation. Using in vitro inhibition of purified E. coli and S. aureus DHFR and disruption of E. coli membranes, we determined that alkyl substitution of the dihydrotriazine at the 6-position best optimizes the DADHTs two mechanisms of action. By employing both mechanisms, the DADHT spectrum of activity was extended beyond the scope of traditional antifolates. We are optimistic that the dual mechanism approach, particularly through the action of antifolates, offers a unique means of combatting hard-to-treat bacterial infections.
Keywords: Antibiotic, antifolate, dihydrofolate reductase, dual mechanism, membrane disruption, triazine
Graphical Abstract
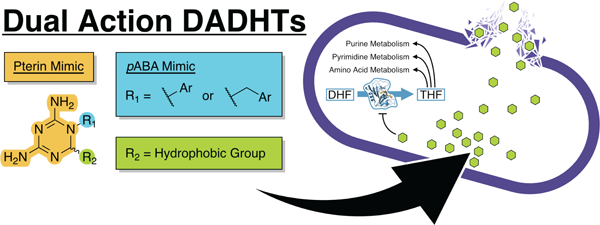
Antibiotics that act via multiple mechanisms of action (MoAs) have gained significant traction and have emerged as strong candidates to fight resistance.1 Molecules with multiple MoAs decrease the likelihood of escape mutations leading to resistance phenotypes. Compared to combination treatments, where each drug has its own pharmacokinetic and pharmacodynamic properties, single-agent, multi-MoA drugs gain the benefits of combination therapy and simplify dosing to ensure pressure is applied to multiple pathways simultaneously at the site of infection.2
Nature has provided a template for the multi-functional antibiotic strategy (Figure S1). Nisin is a ribosomally-synthesized antimicrobial peptide that interrupts peptidoglycan biosynthesis by binding to and sequestering lipid II, while simultaneously forming pores that induce membrane disruption in Gram-positive pathogens.3 Everninomicin P is a naturally occurring, nonenzymatic conjugate between the macrolide rosamicin and everninomicin F that non-simultaneously binds the peptidyl transferase center and the A site of the ribosome.4 Simocyclinone D8 is a bifunctional antibiotic containing aminocoumarin and polyketide moieties that allow the two weak-binding poles of the molecule to bind simultaneously at separate sites in the DNA-binding saddle of the E. coli DNA gyrase GyrA, achieve greater inhibition than either structural component alone.5 Nature was humanity’s first source of antibiotics and led us into a golden age of medicine. Now, nature is directing us toward strategies that engage multiple MoAs.
Synthetic chemists have responded by adopting this strategy in the design of new molecules (Figure S2). Oritavancin, a clinically used 4th generation aminoglycoside, employs three mechanisms of action: inhibition of peptidoglycan assembly by binding ᴅ-Ala-ᴅ-Ala/ᴅ-Ala-ᴅ-Lac, blocking transpeptidation at a secondary binding site, and membrane disruption.6,7 Cadazolid is another example of a bifunctional antibiotic that, combining the fluoroquinolone pharmacophore of ciprofloxacin and the oxazolidinone pharmacophore of tedizolid, inhibits both DNA gyrase and the bacterial ribosome.1 For both natural product and synthetic/semisynthetic multi-mechanism antibacterials, the addition of membrane disrupting functionality provides a robust secondary MoA.
The introduction of dual mechanism antifolates that inhibit dihydrofolate reductase (DHFR) and disrupt bacterial membranes is an excellent example of how synthetic bifunctional antibiotics can be developed.8,9 These compounds, such as IRS-16 (Figure 1) based on a pyrroloquinazolinediamine core elaborated with hydrophobic aryl substituents, have broad-spectrum activity in vivo with the development of minimal resistance. Antifolates are mainstream antibiotics, with the DHFR inhibitor trimethoprim (TMP), alone and in combination with sulfamethoxazole, present on the WHO List of Essential Medicines for the treatment of urinary tract infections.10 Classical antifolates, such as aminopterin and methotrexate, are folate-derivatives that contain an amine in place of a carbonyl on the pterin and/or a methyl on the para-aminobenzoic acid (pABA)-derived nitrogen. Non-classical antifolates, such as TMP, cycloguanil (CYC, the active metabolite of the antimalarial proguanil), and pyrimethamine (PYR, another antimalarial), mimic the pterin of folate with a single heterocycle and include an aryl substituent to mimic the p-aminobenzoyl group.11 Ultimately, by cutting off the production of 5,6,7,8-tetrahydrofolate (THF), these antibiotics bottleneck folate-based single-carbon metabolism including, most importantly, the production of deoxythymidylate.12
Figure 1.

Chemical structures of current antimicrobial antifolates and general structure and mechanism for dual action antifolates.
In this work, we use scaffold repurposing to develop a series of dual action 2,4-diamino-1,6-dihydro-1,3,5-triazines (DADHTs) that inhibit DHFR and disrupt bacterial membranes (Figure 1). The DADHTs, adapting the heterocyclic core of CYC, present an alternative scaffold to the IRS-16 pyrroloquinazolinediamine and are easy to functionalize through multicomponent synthesis. We report the synthesis, in vitro characterization of DHFR inhibition, co-crystallization with Escherichia coli DHFR, and cell-based activity assays for 32 DADHT derivatives. This study reveals the structure-activity relationships (SAR) for DHFR inhibition and membrane disruption, leading to the discovery of compound 3d with potent activity against difficult-to-treat human pathogens, including Mycobacterium tuberculosis, M. abscessus, Staphylococcus aureus, and Pseudomonas aeruginosa. Our new dual action antifolates may play a central role in combatting resistance to traditional antifolate medications to keep these essential medicines in the clinic.
Results
Design and synthesis of 2,4-diamino-1,6-dihydro-1,3,5-triazine (DADHT) derivatives.
We designed a panel of DADHTs based on the known SAR of cycloguanil (CYC), a common antimalarial inhibitor of DHFR.11 The basic compound design features a variety of substitutions on the DADHT core, including substituted phenyl derivatives attached at N1 and alkyl derivatives attached at C6. The N1-aryl derivatives (compounds 1a-g and 2b-l) were derived from a one-pot cyclodehydration reaction between cyanoguanidine, acetone, and variable aniline analogs under acidic HCl refluxing conditions.13 The products were recrystallized and obtained in 9–63 percent yield as the corresponding HCl salts (Scheme 1).
Scheme 1.

Synthesis of N1-aryl-substituted DADHTs.
The C6-alkyl DADHTs (compounds 3a-h) were derived from a two-step dehydrative cyclization reaction sequence.14 First, p-Cl-aniline was condensed with cyanoguanidine under refluxing EtOH/HCl conditions to provide the corresponding p-chlorophenylbiguanide HCl salt. Second, the biguanide HCl salt was separately condensed with a variety of aldehydes under refluxing EtOH to provide final 6-alkyl-DADHT products in 23–96 percent yield as the corresponding HCl salts (Scheme 2). We chose not to chemically separate the enantiomers produced through this synthetic scheme, as chiral dihydrotriazine-containing compounds are known to quickly undergo electrocyclic racemization at biologically relevant temperatures.15 We reasoned that if there is a difference in binding affinity between epimers, the chirality of the active site would select for the best binding epimer.
Scheme 2.

Synthesis of C6-substituted DADHTs.
The synthesis of compounds 4–8 proceeded similarly to above using the corresponding anilines, aldehydes, and/or ketones as specified in the experimental section of the Supporting Information to provide DADHT analogs with unique combinations of substitutions (Scheme 3). The advantage of this overarching synthetic strategy to the DADHTs is the ready incorporation of hydrophobic alkyl groups at distinct positions (i.e., C6 and N1-aryl groups) to assemble the dual action antifolates in rapid and combinatorial fashion with high purity and no need for chromatographic purification.
Scheme 3.

Structures of additional compounds synthesized.
Antibacterial activity of DADHTs.
We evaluated the biological activity of the 32 DADHTs against a panel of pathogenic microorganisms using a Kirby-Bauer16 agar diffusion assay (Table S5, Figures S3–S16). Of the Gram-positive bacteria, Micrococcus luteus and Bacillus subtilis showed susceptibility to compounds 1a–g, 2e, 2f, 2h–l, 3c–f, 3h, and 5 (B. subtilis was also susceptible to CYC, 2b, 2d, and 8). In both tested strains of S. aureus, ethers 1c and 1d with terminal phenyl groups showed moderate activity with the longer, 3-carbon chain of 1d providing additional inhibition over 1c. Compound 2e, which places the ether substitution para to the dihydrotriazine, maintained similar activity to its isomer 1d. Shortening the phenyl substituent of 2l as in 2k and replacing the phenyl of 2k with a cyclohexyl in 2i did not have a significant effect while replacing the phenyl with an butyl group as in 2j reduced inhibitory activity. Compounds 1b, 1g, 2b–d, 2g, 3a, 3b, 3f, and 3g showed low-to-no activity. Increasing the three-carbon chain of 2g to the ten-carbon chain in 2h was sufficient to achieve activity. Incrementally increasing the length of the C6 substituent in compound series 3 similarly improved activity as seen through compounds 3c–e and 3h. The substitution of the para-chlorophenyl of 3d with an ortho-methyl group in compound 5 did not have a significant effect on activity. By comparison to CYC, which showed no activity in M. luteus and S. aureus and low activity in B. subtilis, the general trend is that long, lipophilic substituents increase inhibitory activity. This trend was also observed in Mycobacterium vaccae.
In Gram-negative organisms, far fewer compounds inhibited growth. In all cases, longer substituents increased the activity through the same trends as observed in Gram-positive strains. Surprisingly, even some strains of P. aeruginosa were susceptible to a selection of these molecules. In most cases, P. aeruginosa is resistant to antifolates, making this a target pathogen of interest for the dual action antifolate field.8,17 Although very slight growth inhibition was seen by 3c and 3g, P. aeruginosa required the long alkyl chains of 3d, 3e, and 5 for clear growth inhibition.
Based on the Kirby-Bauer assay results, the most active compounds were 1f, 2l, 2h, 3d, and 3e, with compounds 2h, 3d, and 3e achieving broad-spectrum activity. Although we observed the broad trend that greater substituent length lends to greater growth inhibition, 3e maintained lower activity than 3d consistently across all organisms studied. This may represent a maximum length of substituents at the 6-position on the dihydrotriazine, or it may simply be an artifact of reduced solubility of such a lipophilic molecule.
Motivated by the broad-spectrum activity and to better characterize the most active DADHTs, we performed broth microdilution susceptibility testing in S. aureus, E. coli, and P. aeruginosa (Table 1). In S. aureus, compounds 1e, 2f, 2h, 3d, 3e, and 5 all showed the greatest potency (MIC90 = 2–16 μM). These compounds all contain long, lipophilic substituents. Compounds 1c, 1d, 1f, 2e, 2i, 2k, and 2l containing a phenyl group at the end of an ether or thioether linkage to the N-phenyl group showed reduced activity (MIC90 = 32–128 μM). In E. coli, compounds 3d, 3e, and 5 showed the greatest activity with MIC90 values of 8–64 μM. These were also the only compounds to show growth inhibition against P. aeruginosa with MIC90 of 32–128 μM. Additional testing against a panel of ESKAPEE organisms revealed that compound 3d extends its activity to Enterococcus faecium, Acinetobacter baumannii, and Enterobacter aerogenes (Table S6). Taken together, these trends of antibacterial activity indicate that alkyl-substitution at the 6-position on the dihydrotriazine provides broad-spectrum bacterial growth inhibition.
Table 1.
Minimum inhibitory concentrations (MIC90) of DADHTs by broth microdilution.
| Compound | Minimum Inhibitory Concentration (MIC90) (μM) | ||||
|---|---|---|---|---|---|
| S. aureus ATCC 11632 | E. coli ATCC 25922 | P. aeruginosa PAO1 | |||
| − Thya | + Thy | − Thy | + Thy | − Thy | |
| CYC | >128 | ntb | >128 | nt | >128 |
| 1a | 128 | nt | >128 | nt | >128 |
| 1b | >128 | nt | >128 | nt | >128 |
| 1c | 64 | nt | >128 | nt | >128 |
| 1d | 32 | nt | >128 | nt | >128 |
| 1e | 4 | 4 | 128 | nt | >128 |
| 1f | 64 | nt | >128 | nt | >128 |
| 1g | 128 | nt | >128 | nt | >128 |
| 2b | >128 | nt | >128 | nt | >128 |
| 2c | >128 | nt | >128 | nt | >128 |
| 2d | >128 | nt | >128 | nt | >128 |
| 2e | 64 | nt | >128 | nt | >128 |
| 2f | 4 | 4 | >128 | nt | >128 |
| 2g | 128 | nt | >128 | nt | >128 |
| 2h | 16 | 32 | >128 | nt | >128 |
| 2i | 64 | nt | >128 | nt | >128 |
| 2j | 128 | nt | >128 | nt | >128 |
| 2k | 128 | nt | >128 | nt | >128 |
| 2l | 64 | nt | >128 | nt | >128 |
| 3a | >128 | nt | >128 | nt | >128 |
| 3b | >128 | nt | >128 | nt | >128 |
| 3c | 64 | nt | >128 | nt | >128 |
| 3d | 2 | 4 | 32 | 64 | 64 |
| 3e | 4 | 2 | 8 | 8 | 32 |
| 3f | >128 | nt | >128 | nt | >128 |
| 3g | >128 | nt | >128 | nt | >128 |
| 3h | 128 | nt | >128 | nt | >128 |
| 4 | >128 | nt | >128 | nt | >128 |
| 5 | 4 | 8 | 64 | 64 | 128 |
| 6 | >128 | nt | >128 | nt | >128 |
| 7 | >128 | nt | >128 | nt | >128 |
| 8 | >128 | nt | >128 | nt | >128 |
| TMP | 4 | 8c | 2 | >128 | nt |
−/+ Thy indicates absence or presence of 7.4 mg/mL thymidine.
nt = not tested.
weak growth observed as high as 128 μM.
In vitro inhibition of DHFR by DADHTs.
Guided by the bacterial growth inhibition studies, we selected nine of the DADHTs for in vitro studies to test the compounds as inhibitors of bacterial DHFR. We selected compounds 1d-f, 2f, 3c–e, and 5 to explore the effect of C6 and N-aryl substituents through the determination of apparent half-maximal inhibitory concentrations (IC50s, Table 2, Figures S17–S19). We cloned, heterologously expressed, and purified His6-tagged variants of DHFR from E. coli (EcDHFR) and S. aureus (SaDHFR and SaDHFR-F98Y) representing relevant targets from Gram-negative and Gram-positive bacterial pathogens, respectively.
Table 2.
In vitro inhibition (apparent IC50) of bacterial DHFR by select DADHTs.
| Compound |
EcDHFR IC50 (μM ± SD) |
SaDHFR IC50 (μM ± SD) |
|---|---|---|
| CYC | 23 ± 16 | nfa |
| 1d | 3 ± 1 | ntb |
| 1e | 1.0 ± 0.3 | 2.5 ± 0.5 |
| 1f | 5 ± 2 | nt |
| 2f | 26 ± 9 | 14 ± 5 |
| 3c | 3 ± 1 | nt |
| 3d | 5 ± 3 | 1.7 ± 0.3 |
| 3e | 2.1 ± 0.2 | 0.9 ± 0.3 |
| 5 | 7 ± 5 | 23 ± 14 |
| TMP | 9.9 × 10−3 ± 7 × 10−3 | 3.3 × 10−3 ± 7 × 10−4 |
nf = no fit to the data due to partial inhibition, see Figure S17 and S18.
nt = not tested.
CYC, the parent compound for these DADHTs, was up to four orders of magnitude less potent (IC50 = 23 μM) than TMP (IC50 = 9.9 nM) against EcDHFR and did not inhibit SaDHFR (Table 2; Figures S17–S19). This is not surprising, as CYC is optimized for plasmodial DHFR,11 while TMP is specific for bacterial DHFR.18 Compared to CYC, compounds 1d-f, 3c–e, and 5 showed the greatest activity against EcDHFR (IC50 = 0.7–12 μM), while compounds 1e, 3d, and 3e were most effective against SaDHFR (IC50 = 0.6–3 μM). Between the aryl-substituted ethers 1e and 2f, the meta-substitution of 1e provides greater inhibition of both EcDHFR (IC50 = 1.0 μM) and SaDHFR (IC50 = 2.5 μM) compared to the para-substituted 2f (EcDHFR IC50 = 26 μM, SaDHFR IC50 = 14 μM) (Figures S17–S18). This may explain the marginally higher whole cell activity of 1e compared to 2f (Table 1). Compounds 1d and 1f, which contain a phenyl substituent but different linkers, were comparable versus EcDHFR (IC50 = 2–7 μM). Although a difference was observed in the antibacterial activity of compounds 3c–e and 5, inhibition of EcDHFR by these compounds showed significant differences (IC50 = 1.9–12 μM). In SaDHFR, compound 5 (IC50 = 23 μM), which replaces the p-chlorophenyl substituent of 3d for an o-tolyl substituent, showed reduced inhibition compared to 3d and 3e (IC50 = 0.6–1.7 μM), consistent with the known SAR of ortho substitution on the N-phenyl ring.19 Aside from CYC and compound 2f, all tested compounds displayed statistically significant increases in IC50 versus SaDHFR-F98Y, a clinically relevant mutant,20 relative to SaDHFR (Figure S18 and S19). These data indicate that bulky hydrophobic substituents on either the N1-aryl group or the 6-position of the dihydrotriazine were tolerated compared to CYC for inhibiting bacterial DHFR. There is also a preference for para-chlorophenyl substitution at N1 in 6-substituted DADHTs. For the N1-aryl-substituted compounds (1d–f and 2f), this is consistent with previous work done to expand the cycloguanil scaffold through the functionalization of this ring.21
Structural analysis of EcDHFR bound to CYC and 3d.
To understand how DADHTs containing bulky hydrophobic groups are accommodated in the DHFR active site, we determined the X-ray crystal structures of EcDHFR bound to CYC (Figure 2abc) and compound 3d (Figure 2def) at 2.17 and 2.35 Å resolutions, respectively (Table S7). Due to the flexibility of the C6-nonyl moiety of 3d, only an ethyl substituent could be accurately fit to the observed electron density of this ligand (Figure S20). We believed the (S)-form of 3d was the best model, although we could not definitively rule out the potential for the (R)-form based on the observed density.
Figure 2. X-ray crystal structures of EcDHFR bound to cycloguanil (CYC) and compound 3d.

a and d show the cartoon depiction of EcDHFR with the respective ligands (CYC and 3d) bound. b and e show the surface depiction of EcDHFR with the respective ligands (CYC and 3d) bound. c shows the polar contacts between compound CYC and the active site of EcDHFR. f shows the polar contacts between 3d and the active site of EcDHFR.
Comparison of the EcDHFR•CYC and EcDHFR•3d structures reveals similar interactions between binding pocket residues and the respective ligand (Figure 2cf). Both ligands form hydrogen bonding interactions with the main-chains of Ile5 and Ile94 and side-chains of Tyr100 and Asp27, as well as hydrophobic interactions with Ala7 and the side-chains of Met16 and Phe31. Moreover, both structures reveal EcDHFR with the conformationally mobile Met20 loop (Ala9– Leu24) occluding the interface between the cofactor (NADPH) binding site and the substrate binding site, although there is some variation between the two structures. In previously reported EcDHFR structures where NADPH is bound to the cofactor binding site, the occluded conformation prevents the nicotinamide moiety of NADPH from accessing the substrate binding site.22 Although we do not report an EcDHFR structure with NADPH bound, we hypothesize that the occluded conformations accessed by EcDHFR•CYC and EcDHFR•3d would act similarly.
Despite the Met20 loop occluding the nicotinamide binding region in both structures, Met16 faces opposite directions in the two structures and the backbone of residues Ile14–Met16 are 2 Å closer to the binding pocket of CYC than that of 3d (Figure S21a). As a result of this proximity, the CYC chlorophenyl ring is tilted away from Ile14–Met16, differing from the chlorophenyl ring of 3d by an angle of ~18°, while the dihydrotriazine rings of both ligands remain well aligned. The conformation of Met16 brings its side-chain within 4 Å of the CYC p-chlorophenyl ring, allowing for hydrophobic interaction. Another significant difference is that the Glu18–Leu23 region of the Met20 loop is not resolved in the 3d structure, likely due to the destabilizing nature of the disordered C6-nonyl extended alkyl tail of 3d in contrast to the compact 6,6-dimethyl group of CYC, which is only 5 Å removed from Pro21.
Comparison of the EcDHFR•CYC structure to structures of DHFR•CYC complexes from M. tuberculosis (PDB: 6NNH) and Candida auris (PDB: 8CRH) reveals largely similar interactions between binding pocket residues and CYC (Figure S21b). However, in contrast to the occluded conformation observed in EcDHFR•CYC, the structures of MtDHFR•CYC and CaDHFR•CYC depict the protein in either its open or closed conformation, in which residues Ile14–Glu17 are swung away from the substrate binding pocket by as much as 9 Å, allowing access to NADPH. We also observed that the angle between the planes of the p-chlorophenyl of CYC and the aromatic side-chain of Phe31 varies 55° to 75° in MtDHFR•CYC and CaDHFR•CYC, whereas the same angle measures ~40° in EcDHFR•CYC, indicating a less significant face-to-edge π-π stacking interaction in EcDHFR•CYC. Loss of a favorable π-π stacking orientation between CYC and Phe31 in the EcDHFR complex may be explained by the interaction of CYC with Met16, as described above.
While there are no published structures of DHFR in complex with 3d, several structures of DHFR from various species have been solved in complex with PYR, which only differs from the ethyl-tail model of 3d in its pyrimidine ring in place of a dihydrotriazine ring. As in the CYC complexes, the major difference observed in the binding pocket between the EcDHFR•3d structure and DHFR•PYR complexes from M. tuberculosis (PDB: 6NNI) and Trypanosoma brucei (PDB: 3QFX) is the proximity of the Ile14–Glu17 loop to 3d (Figure S21c). The side-chain of Met16 in the 3d complex is oriented away from the binding pocket and is thus ~1 Å farther from 3d than CYC. As a result, the p-chlorophenyl moiety of 3d achieves an orientation that is more favorable for a face-to-edge π-π stacking interaction with the aromatic side-chain of Phe31 (55° angle between rings). Additionally, the side-chain of Ile50 in the EcDHFR•3d structure is positioned 1–1.5 Å closer to the binding pocket than the side-chains of corresponding Leu50 and Leu90 in the MtDHFR•PYR and TbDHFR•PYR complexes, respectively, inducing a tilt in the chlorophenyl ring of 3d that separates it from that of PYR by an angle of ~11°.
Comparison with EcDHFR structures reveals distinct conformation of the Met20 loop in EcDHFR•CYC and EcDHFR•3d.
To better understand the conformation of the Met20 loop in the EcDHFR•CYC and EcDHFR•3d structures, we overlayed 184 unique EcDHFR structures from PDB entries associated with UniProtKB accession number P0ABQ4 (Table S8). We categorized each structure into closed, open, occluded, and disordered forms22 and observed variation that led us to further sub-group the occluded conformation into occluded conformers A, B, and C with 2 ungrouped “unique” occluded conformers (Table S8, Figure S22–S23). All structures in occluded subgroup A have folate, DHF, THF, or 5-formyl-THF bound, while subgroup B includes a mixture of folates and inhibitors, and subgroup C includes exclusively inhibitor-bound EcDHFR. Interestingly, all structures in occluded subgroup B are in space group P61 and represent chain A, with the corresponding B chains in the open conformation. Occluded subgroup A represents a mixture of space groups and chains, while structures in occluded subgroup C are exclusively in space group P6122. We also sub-grouped the disordered conformation into disordered conformers A and B and 1 ungrouped “unique” disordered conformer. We then aligned and overlayed the structures of EcDHFR•CYC and EcDHFR•3d onto the previously described alignment (Figures S24–S27). Compared to each closed, open, occluded, and disordered group, both EcDHFR•CYC and EcDHFR•3d reveal newly observed occluded conformational states, while EcDHFR•3d also represents a unique disordered conformer. Analysis using the DALI Protein Structure Comparison Server23 further substantiated this observation, revealing that structures with the highest alignment are almost entirely in an occluded conformation (Figure S28, Tables S9–S10). We also noted that all but one of the structures in the same space group as EcDHFR•CYC and EcDHFR•3d exhibit an occluded conformation (Figures S29–S30).
Although the position of the Met20 loop in EcDHFR•3d is similar to other DHFRs adopting the occluded conformation (Figures S26–S27), hierarchical clustering analysis based on global and Met20 loop Cα RMSD differences indicated that EcDHFR•CYC adopts a conformation entirely unique amongst EcDHFR structures (Figures S31–32). The N-terminal end of the Met20 loop overlays very tightly in a turn from Ala9 to Arg12 in all previously solved EcDHFR structures. In the EcDHFR•CYC structure, this region is constricted from Ala9 to Asp11, resulting in a single residue shift in the Met20 loop. At the C-terminal end of the Met20 loop, Asn23 adopts an altered backbone conformation that orients the side-chain toward Ser148. This exchanges the hydrogen bond between the Asn23 backbone amide and Ser148 Oγ that is characteristic of the occluded conformation22 for interaction between Asn23 HNδ and Ser148 Oγ (Figure S33a). The only other EcDHFR structure with a similar change is the EcDHFR•TMP complex (PDB: 7NAE).18 In 7NAE, the backbone conformational change in Asn23 is accommodated by an additional backbone change in Ile24 that shifts the Cα of Asn23 by 3.2 Å compared to EcDHFR•CYC, positioning Trp22 in the same location as the rest of the occluded conformers. All previously determined EcDHFR structures, including 7NAE, show Trp22 buried in a hydrophobic pocket formed by the inward-facing side-chains of the Met20 loop (Figures S34–S35). Due to the conformation of Asn23 in EcDHFR•CYC, Trp22 becomes solvent-facing above the substrate binding site (Figure S34). As a result, the side-chains of Ile14 and Met20 reorient to fill the hydrophobic pocket that Trp22 would otherwise inhabit (Figures S36, S37). These changes to the Met20 loop result in an occluded conformation that places Met16 directly over top of the substrate binding site sandwiched between the aryl groups of CYC and Trp22 (Figure S33b, S38).
Based on an overlay of NADP(H) bound in the cofactor binding site of EcDHFR in the closed conformation, the backbone of Ile14 and Gly15 and the side-chain of Glu17 are responsible for occlusion of the cofactor binding site in EcDHFR:CYC (Figure S33c). This contrasts with occluded subgroups A and C which feature Met16 oriented toward the cofactor binding site to occlude the connection between the two binding sites. In these structures, such as 7NAE, a hydrogen bond between Met16 and Ser49 positions the methionine in the canonical occluded position. An overlay of 7NAE and EcDHFR•CYC, however, shows a steric clash between the p-Cl on the CYC aryl group and the side-chain of Ile50, pushing the isoleucine away by 2 Å (Figure S33d). This shifts the position of helix α2 to break the hydrogen bond between Met16 and Ser49, releasing Met16 and the rest of the Met20 loop to adopt a new conformation. Notably, structures in occluded conformation subgroup B also lack this interaction and place Met16 above the cofactor binding site, away from the canonical occluding position (Figure S38e). Together, these features provide structural insights into the unique EcDHFR•CYC Met20 loop conformation and provides a guide for future structure-based drug design.
Membrane Disruption Contributes to DADHT Antibacterial Activity
To determine if the DADHTs operate solely through an antifolate mechanism in E. coli and S. aureus, we determined MIC90 values in the presence of thymidine for the compounds with the greatest antibacterial potency (Table 1). Inclusion of thymidine in antibacterial susceptibility assays antagonizes the activity of single-mechanism DHFR inhibitors.24 If DADHTs act uniquely as antifolates, the introduction of thymidine to the media would rescue bacterial growth. In the presence of thymidine, we observed the bacterial DHFR inhibitor TMP suffer complete loss of growth inhibition in E. coli and an increase from MIC90 = 4–8 μM in S. aureus, although weak growth of S. aureus was observed as high as 128 μM. By contrast, the addition of thymidine did not change the observed MIC90 values beyond the margin of error for compounds 1e, 2f, 2h, 3d, 3e, and 5 in both organisms. The maintenance of growth inhibition in the presence of thymidine led us to believe that DADHTs act by a second mechanism, in addition to DHFR inhibition, presumably membrane disruption.
To evaluate the potential for membrane disruption as this second mode of action, we performed a SYTOX Green uptake assay in E. coli ATCC 25922. Compared to the membrane disrupting peptide melittin as a positive control, CYC and TMP showed no uptake of SYTOX Green at any of the concentrations tested (Figure S39ab). In contrast, compounds 3d (Figures 3a and S39d) and 3e (Figures 3b and S39e) containing 6-(nonyl) and 6-(undecyl) substitutions, respectively, caused significant SYTOX Green uptake relative to the controls. Compound 3c, which has a shorter 6-(hexyl) alkyl group, showed reduced SYTOX Green uptake relative to compounds 3d and 3e (Figures S39cde). The length of the alkyl substituent correlated to the half maximal effective concentration (EC50) of endpoint SYTOX Green uptake in treated cells: 3c (hexyl) EC50 = 660 μM; 3d (nonyl) EC50 = 150 μM; 3e (undecyl) EC50 = 16 μM (Figures S39fgh).
Figure 3. 6-(nonyl) and 6-(undecyl) substitutions show significant membrane disruption in E. coli.

a and b show the mean of three independent trials measuring fluorescence intensity at 520 nm upon treatment of E. coli ATCC 25922 cells with compounds 3d and 3e, respectively, at 0 min. Data points above the solubility limits of each compound in PBS are not shown. Full datasets can be found in Figure S39. c shows a plot of 1/(MIC90) vs clogP calculated using ChemDraw Professional v21. MIC90 data for IRS-16 was sourced from the literature.8 MIC90 data for all other compounds was sourced from Table 1. Compounds with MIC90 > 128 μM were excluded.
Previous reports on dual action antifolates linked membrane-disrupting activity in E. coli with activation of the mechanosensitive channel of large conductance, MscL (but not the small mechanosensitive channel, MscS) by cumene or biphenyl structural motifs.25 We obtained E. coli knock-out strains of the small (MscS) and large (MscL) mechanosensitive channels from the Keio collection, a library of W3110 lineage E. coli K12 cells containing single genes substituted with a kanamycin-resistance cassette.26,27 We determined MIC values against E. coli W3310ΔmscL::Kan (Keio strain JW3252), E. coli K12 W3310ΔmscS (Keio strain JW2891), and the WT parent strain E. coli W3310 (Table S11). We observed no significant differences in the MIC values of CYC, TMP, or compounds 1e, 2f, 3d, 3e, or 5 against this strain set. Hence, it appears that the membrane-disrupting activity of DADHTs is due to a nonspecific mechanism.
Anticancer activity, cytotoxicity, and hemolytic activity of DADHTs.
Some antifolates can be cytotoxic and selectively inhibit the growth of rapidly dividing cancer cells.21,28 To test for anticancer activity and probe the general cytotoxicity of DADHTs, additional biological testing was performed against MCF-7 breast cancer cells and PC3 prostate cancer cells (Table S12). Compounds that showed high activity in the antibacterial testing, particularly those with clogP > 2.5 (Table S13), also showed a reduction in MCF-7 and PC3 cell viability. Compounds 2e, 3d, and 3e showed the greatest cytotoxicity against both cell types with up to 100% growth inhibition and IC50 values ranging from 1–4 μM for MCF-7 cells and 5–10 μM for PC3 cells. The remaining compounds displayed a range of activities with a trend of lower activity correlating to lower hydrophobicity. In addition to MCF-7 and PC3 cells, we also determined the IC50 in VERO cells to be <6.25 μM. This observed mammalian cytotoxicity poses a challenge in the development of these compounds as antibacterial agents but could indicate promise as anticancer agents.
Based on data indicating that compounds 3d and 3e act as potent membrane disruptors and show mammalian cytotoxicity, we tested for non-specific membrane disruption using an in vitro hemolysis assay. We determined the hemolytic activity (HA50) of these compounds by measuring OD650nm of human erythrocytes after treatment with CYC, 3d, or 3e. While CYC showed no detectable hemolysis over all tested concentrations, both 3d and 3e began to lyse cells between 32 μM and 64 μM with more significant hemolysis at higher concentrations (Figure S40a). Compound 3d showed an HA50 of 84 μM (Figure S40b), and compound 3e showed an HA50 of 63 μM (Figure S40c). Similar to the results from bacterial membrane disruption (Figures 3, S39), increasing the length of the 6-alkyl substituent increases the degree of hemolysis, suggesting that the antibacterial and anticancer activity of DADHTs is due to a combination of DHFR inhibition and general membrane disruption.
DADHT anti-bacterial activity extends to anti-mycobacterial activity.
Given the unique cell envelope composition of mycobacteria and the need for the development of new anti-tuberculosis drugs, we next aimed to determine if the DADHTs have activity against M. tuberculosis and M. abscessus. To assess activity against M. tuberculosis, we used a standardized microplate alamar blue assay (MABA)29 to determine MIC90 values, assaying in GAST (glycerol carbon source) and 7H12 media to evaluate dependence of activity on glycerol metabolism which is known to generate false positives in antibacterial screens30 (Table 3). Briefly, we found that compounds 3d and 3e, including long alkyl substitution at the 6-position, showed the greatest activity (MIC90 = 0.7–1.5 μM) while compounds 1d–f, 2e, 2f, 2h–k, and 3c displayed moderate activity (MIC90 = 2.5–7.5 μM). No significant differences were seen between GAST and 7H12 media. Notably, the SAR in M. tuberculosis follows the same trend observed in M. vaccae via the Kirby-Bauer agar diffusion method (Table S5). We further probed the activity of compound 3d against M. tuberculosis through the low-oxygen recovery assay (LORA), which simulates the non-replicating conditions of latent tuberculosis (Table S14).29 Although it is uncommon for antibacterials to have any activity in the LORA assay, compound 3d showed an MIC90 of 1.12 μM.
Table 3.
Antimycobacterial activity of dual action DADHTs.
| Compound | Minimum Inhibitory Concentration (MIC90, μM) | Half-Maximal Inhibitory Concentration (IC50, μM) |
|||
|---|---|---|---|---|---|
| M. tuberculosis H37Rv | M. abscessus L948 | ||||
| MABA: GAST Media | MABA: 7H12 Media |
MABA | Biofilm: Formationa | Biofilm: Preformedb | |
| CYC | >100 | 74.8 | > 200 | > 200 | > 80 |
| 1b | >100 | 48.3 | ntc | nt | nt |
| 1c | 9.2 | 10 | nt | nt | nt |
| 1d | 3.1 | 5.8 | nt | nt | nt |
| 1e | 2.9 | 3 | > 200 | > 200 | > 80 |
| 1f | 6.2 | 4.7 | nt | nt | nt |
| 1g | 10 | 7.7 | nt | nt | nt |
| 2b | >100 | 71.9 | nt | nt | nt |
| 2c | >100 | >100 | nt | nt | nt |
| 2d | >100 | >100 | nt | nt | nt |
| 2e | 5 | 4.6 | nt | nt | nt |
| 2f | 5 | 5.6 | > 200 | > 200 | > 80 |
| 2h | 2.5 | 2.9 | nt | nt | nt |
| 2i | 3.7 | 6.2 | nt | nt | nt |
| 2j | 5.4 | 6.2 | nt | nt | nt |
| 2k | 5.4 | 7.5 | nt | nt | nt |
| 2l | 8.1 | 8.6 | nt | nt | nt |
| 3a | >100 | >100 | nt | nt | nt |
| 3b | 69 | 48.6 | nt | nt | nt |
| 3c | 5.7 | 5.5 | > 200 | > 200 | > 80 |
| 3d | 1.5 | 1.4 | 54.5 | 34.4 | > 80 |
| 3e | 1.3 | 0.7 | 85.2 | 167.3 | > 80 |
| 3f | 28 | 23.3 | nt | nt | nt |
| 3g | 60.4 | 45.4 | nt | nt | nt |
| 3h | 10.2 | 9.7 | nt | nt | nt |
| 4 | 50.8 | 71.3 | nt | nt | nt |
| 5 | nt | nt | 61.6 | 93.1 | > 80 |
| Rifampicin | <0.016 | 0.05 | nt | nt | nt |
| Isoniazid | 0.12 | 0.24 | nt | nt | nt |
| Moxifloxacin | 0.042 | 0.43 | nt | nt | nt |
| Streptomycin | 0.27 | 0.21 | nt | nt | nt |
| Pretomanid | 0.51 | 0.38 | nt | nt | nt |
| Clarithromycin | nt | nt | 15.4 | 11.7 | > 80 |
Test compounds were added before biofilms had formed.
Test compounds were added after biofilms had formed.
nt = not tested.
In M. abscessus, we determined the whole cell growth inhibitory activity at 50 μM of each compound (Figure S41). Compound 3d was the most potent, causing complete growth inhibition in liquid 7H9 medium. Compound 5 nearly reached this level of activity and reduced growth 40-fold compared to the untreated control and to CYC. Compounds 1d, 2e, 2i, 2l, and 3e also showed strong activity against M. abscessus, reducing the OD600 between 3- and 5-fold versus the untreated control. To better resolve the growth inhibitory activity of these compounds against M. abscesssus, we performed the MABA assay (Table 3, Figure S42), which revealed that compounds 3d (IC50 = 54.5 μM), 3e (IC50 = 85.2 μM), and 5 (IC50 = 61.6 μM) show the greatest activity relative to CYC (no inhibition) and clarithromycin (IC50 = 15.4 μM). Notably, while clarithromycin plateaus at ~70% inhibition from 12.5–200 μM, compounds 3d and 5 reach 100% inhibition at 100 μM. These data demonstrate the advantage of the 6-(nonyl) substituent, found in both compounds 3d and 5, in combatting mycobacteria and may suggest a bactericidal phenotype.
Due to the clinical relevance of M. abscessus biofilms in pulmonary infections experienced by cystic fibrosis patients,31 we also screened the ability of these compounds, at 50 μM, to inhibit growth of M. abscessus biofilms in vitro (Figure S43). Compounds 3d, 3e, and 5 showed near complete inhibition of biofilm growth after incubating for 1 week under low-oxygen conditions, with compound 1e showing partial growth inhibition. Biofilm growth was recovered in wells containing 1e, 3e, and 5 upon reintroducing oxygen to assay plates and incubating for an additional week. By contrast, 3d maintained partial growth inhibition after the 2-week time point. We then selected a subset of these compounds to determine IC50 values for this biofilm growth inhibition (Table 3, Figure S44). Compound 3d best inhibited the formation of M. abscessus (IC50 = 34.4 μM), while compounds 5 (IC50 = 93.1 μM) and 3e (IC50 = 167.3 μM) showed moderate biofilm growth inhibition. Compounds 1e, 2f, and 3c, all of which showed activity against M. tuberculosis, did not inhibit the formation of M. abscessus biofilms. This SAR is consistent with the trends observed in other organisms where the 6-nonyl substitution offers optimal antibacterial potential. Neither clarithromycin nor the DADHTs were able to disrupt preformed M. abscessus biofilms (Table 3, Figure S45). While the inability to disrupt preformed biofilms was not surprising, the ability of select DADHTs to inhibit the formation of M. abscessus biofilms is quite uncommon.
Motivated by this antimycobacterial activity that distinguishes DADHTs from clinically used antifolates, such as TMP, CYC, and PYR, which lack whole cell activity against mycobacteria due to low cell permeability,32 we performed molecular docking of ligands into MtDHFR•CYC (PDB: 6NNH). Computational docking suggests that 3d and 3e can be accommodated in the active site of MtDHFR and likely bind in similar modes to that observed in the EcDHFR•3d structure, with the 6-substitution oriented to exit the active site through the space that accommodates the glutamate tail of bound folates (Figure S46). These studies demonstrate that dual action DADHTs are potent antimycobacterial agents and highlights mycobacteria as flagship target pathogens for the future development of dual action antifolates more broadly.
Discussion
In this work, we reported the discovery of a new class of dual action antifolates based on the cycloguanil scaffold: the DADHTs. We demonstrated that many of these compounds have broad-spectrum antibacterial activity, including potency against several ESKAPEE pathogens, M. tuberculosis, and M. abscessus. Specifically, we learned that alkyl substitution at the 6-position on the dihydrotriazine provides the most potent, broad-spectrum antibacterial activity with compound 3d achieving an optimal combination of whole cell activity, enzyme inhibition, and membrane disruption within the limits of solubility. Some of these molecules, including 3c–e that range from 6-(hexyl) to 6-(undecyl), have been described in previous work to have potency against Enterococcus faecalis and Lactobacillus arabinosus grown in conditions that increase dependence on folate metabolism.33,34 The 6-alkyl substituted DADHTs achieve strong growth inhibition of P. aeruginosa, which is known to encode efflux pumps that render it intrinsically resistant to antifolates, including TMP and IRS-16,8 but not the IRS-16 derivative fluorofolin.17 To our knowledge, 3d and related dual action DADHTs represent just the second example of antifolates effective against P. aeruginosa.
We similarly showed that substitutions on the N-aryl ring at the meta and para positions achieve greater antibacterial activity than the parent compound CYC. Specifically, long alkyl chains that may contain heteroatoms or terminal phenyl groups achieved the greatest activity from this class of molecule. Compounds 1e and 2f have previously been described in SAR studies attempting to explore the role of hydrophobic groups in binding to DHFR, although the present work is the first to demonstrate their broad-spectrum antibacterial activity.35,36
The addition of bulky groups at all three tested positions (i.e., C6 on the dihydrotriazine or meta- and para- on the N1-aryl) provides membrane disruption and does not reduce inhibitory activity against either EcDHFR or SaDHFR in comparison to the parent compound CYC. Although this indicates that the CYC pharmacophore is sufficient to inhibit the enzyme, a significant reduction in potency relative to TMP reveals that further optimization is necessary to specifically target bacterial DHFR. Previous efforts at fighting resistance and improving activity of such nonclassical antifolates, such as iclaprim and the propargyl-linked antifolates, have focused on adapting the aryl ring, or its analogous structure, that mimics the para-aminobenzoyl group of DHF to optimize π-π stacking with Phe31.37 Such traditional, single mechanism antifolates, represented by TMP, are hydrophilic and maintain very high activity against E. coli and S. aureus strains (Figure 3c, Table S13). A plot of 1/MIC90 vs clogP shows that dual action antifolates, represented by DADHTs and IRS-16, achieve greater activity with increasing hydrophobicity, allowing them to access membrane disruption as a second mechanism of action. Although the tested compounds 3d and 3e began to induce hemolysis between 32 μM and 64 μM, 3d and 3e maintained activity against Gram-positive organisms at much lower concentrations. This window suggests that it may be worthwhile to optimize these compounds for bacterial-specific activity.
Both IRS-168 and DADHT 3d are antibacterial and cytotoxic, but vary significantly in clogP values, 1.940 and 4.608, respectively. The increased hydrophobicity of DADHT 3d relative to IRS-16 may be responsible for its hemolytic activity.9 Hence, we propose that optimizing DADHTs for clogP values between 1.940 and 4.608 might reveal analogs with the desired dual action antibacterial activity and reduced cytotoxicity and hemolytic activity. One of the challenges associated with engineering membrane disrupting functionality into a small molecule is the loss of solubility with increasing clogP—a trend that we observed in the DADHT series (Figure S47) and has been reported for IRS-16.9 More extensive optimization of the hydrophobic sidechain is required prior to in vivo studies. Cytotoxicity derived from the antifolate MoA is an additional facet that must be considered. Antimalarial DADHT antifolates such as WR99210 have cytotoxicity on the same order (low μM IC50) as dual action DADHTs, but gain potent inhibition (sub-nM IC50) of P. falciparum growth due to the selective induction of human DHFR expression.38 As previously mentioned, a primary goal of dual action DADHT development would center around achieving a similar discrimination of pathogen from host in the bacterial setting. The DADHT 2,4-diamino-1,6-dihydro-1,3,5-triazine scaffold allows for easy modification of the aryl group and substitution at the 6-position to simultaneously enhance potency against bacterial DHFR and optimize bacterial-specific membrane disruption (Figure 4).
Figure 4.

Summary structure-activity relationships of hydrophobic cycloguanil derivatives.
The X-ray crystal structures reported here enable a structure-guided approach to optimizing the DADHTs. Global alignment and hierarchical clustering of EcDHFR structures prompted us to subgroup the occluded conformation and disordered conformation to aid in further comparisons and allowed us to decisively conclude that the Met20 loop of EcDHFR•CYC exists in a conformational space of its own. Although the structures of EcDHFR•CYC and EcDHFR•3d show the Met20 loop occluding the NADPH binding site, they differ in conformation. The partially disordered EcDHFR•3d Met20 loop places Met16 in the canonical EcDHFR occluded conformation while EcDHFR•CYC uniquely orients Met16 to face the substrate binding site. We speculate that the long, flexible 6-(nonyl) substituent of 3d may play a role in stimulating the partially disordered region of the Met20 loop and stabilizing the occluded conformation of EcDHFR. This added stability may result in the 10-fold increase in inhibitory potency observed for 3d compared to CYC (Table 2, Figures S17–18). Cao and colleagues proposed that inhibitor binding to the occluded conformation of EcDHFR takes advantage of the same network of interactions that underlies the enzyme’s slow THF release kinetics.39 Compound 3d may employ a similar strategy, offering the DADHT scaffold as a means to drug of the occluded conformation of DHFR. The EcDHFR•CYC and EcDHFR•3d structures help to rationalize the in vitro DHFR inhibition data showing that bulky hydrophobic substituents at C6 on the dihydrotriazine or meta- and para-positions on the N1-aryl substituent can be accommodated by the solvent exposed opening to the active site. Based on this guidance, we propose that the inclusion of a methylene spacer at N1 of the dihydrotriazine core might enhance DHFR binding by better mimicking the natural DHF structure and the optimized bacterial DHFR inhibitor trimethoprim (Figure 4).
One of the most promising applications of dual action antifolates is the development of treatments for infections caused by bacterial pathogens that are notoriously difficult to treat due to biofilm formation and a lack of membrane permeability. Compared to known antifolates, we observed potent inhibition of M. tuberculosis and M. abscessus growth by DADHTs that, notably, halted the formation of M. abscessus biofilms. The potent activity of DADHTs against these high-priority pathogens highlights the utility of the dual mechanism approach and emphasizes the importance of engineering cell permeability into antifolates. Although TMP, CYC, and PYR show low activity against M. tuberculosis40 and M. abscessus,41 other antifolates such as PQD-1, trimetrexate, WR99210, and the triaza-coumerins have shown that inhibiting folate metabolism may be a viable strategy in targeting mycobacterial infections.41,42 Antifolates are known to disrupt the biosynthesis of mycolic acids that make up a major component of the mycobacterial cell envelope by blocking the methionine cycle responsible for the formation of S-adenosyl methionine.32 Therefore it is not difficult to imagine that compounds 1e, 2h, 3d, 3e, and 5 may derive their strong antimycobacterial activity through a two-pronged attack on mycobacterial cell envelope: first, by impairing the biosynthesis of its component mycolic acids and, second, by inducing membrane disruption through the action of their hydrophobic side-chains.
Although dual action antibiotics engage multiple targets, there is usually a dominate mechanism that drives efficacy.1 For DADHTs, moderate DHFR inhibition was demonstrated but it was the membrane disrupting activity that produced the most dominate antibacterial activity. We hypothesize that membrane disruption mechanism of dual MoA DADHTs operate through a general mechanism by inserting into the membrane and aggregating into pores that allow flux of small molecules and ions. The length-dependent nature of membrane disruption we observed for compounds 3c–e is consistent with this model. Bacterial phospholipids range from 14 to 20 carbons in length.43 As the length of the alkyl substitution increases between 3c–e, the molecule approaches this length. Since we observed human erythrocyte hemolysis and cytotoxicity to MCF-7 and PC3 cells and found no engagement with mechanosensitive channels MscL or MscS, we posit that the mechanism of membrane disruption must be general enough to act across against both prokaryotic and eukaryotic cells. Future research should focus on optimizing dual action antifolates to better balance the contribution of antibacterial activity across both mechanisms, DHFR inhibition and membrane disruption, to improve efficacy and decrease toxicity. This could include screening for the optimal pterin mimic heterocyclic core and optimization of hydrophobic side chains.
Conclusion
We have demonstrated the repurposing of a malaria-specific antifolate scaffold, CYC, into antibacterial antifolates, the DADHTs, through the incorporation of hydrophobic substituents. The DADHTs display a dual mechanism of action through direct inhibition of bacterial DHFR and membrane disruption. The DADHTs bind DHFR in the substrate binding site and stabilize a novel occluded conformation. Their dual mechanism nature expanded the spectrum of antibacterial activity to difficult-to-treat Gram-negative (P. aeruginosa and A. baumannii) and mycobacterial pathogens (M. tuberculosis and M. abscessus). The present study suggests that other antifolate scaffolds can be adapted to apply the dual action strategy. The DADHT scaffold is easy to modify through a streamlined multicomponent synthetic process, enabling rapid SAR exploration. Future optimization of the DADHTs requires reducing the cytotoxicity and hemolytic activity while maintaining the dual action nature against target pathogens. Antibiotics that operate via multiple mechanisms of action mark a significant step forward in the fight against resistance. We hope this work will continue that momentum and open new doors of possibility for combatting hard-to-treat pathogens.
Supplementary Material
Acknowledgments
We thank Jeff Kao and Manmillan Singh (Department of Chemistry, Washington University in St. Louis) for expertise in acquiring NMR spectra; Shahriar Mobashery and Nuno T. Antunes for assistance in susceptibility testing against the ESKAPEE panel; and Baojie Wan and Yuehong Wang for susceptibility testing against M. tuberculosis. This research used beamline 17-ID-1 (AMX) of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.
Funding
Research was supported by the Arnold and Mabel Beckman Foundation through a Beckman Scholars Award to J.D.G (scholar) and T.A.W. (mentor), dx.doi.org/10.13039/100000997. Additional support was provided by the Children’s Discovery Institute at St. Louis Children’s Hospital (grant MI-PD-II-2018–748, T.A.W.), the Research Corporation for Science Advancement through a Cottrell Scholar award (T.A.W.), the Alfred P. Sloan Foundation through a Sloan Fellowship Award (T.A.W.), the Camille and Henry Dreyfus Foundation through a Camille Dreyfus Teacher-Scholar Award (T.A.W.), and the National Institutes of Health (NIH) through grants 2U01AI123394 (T.A.W.), R01-AI171514 (J.M.J.), R37AI054193 (M.J.M. and T.A.W.), the McNair Scholars Program (C.L. and T.A.M.), and the Beta Beta Beta Biological Honor Society (T.A.M.). The ALS-ENABLE beamlines are supported in part by the NIH through grant P30 GM124169–01.
Abbreviations
- CYC
cycloguanil
- DADHT
2,4-diamino-1,6-dihydro-1,3,5-triazine
- DHF
7,8-dihydrofolate
- DHFR
dihydrofolate reductase
- MoA
Mechanism of action
- pABA
para-aminobenzoic acid
- PYR
pyrimethamine
- THF
5,6,7,8-tetrahydrofolate
- TMP
trimethoprim
- TSA
trichostatin A
Footnotes
Supporting Information
The Supporting Information is available free of charge online.
• Experimental methods; supplementary tables of strains used in this study, sequences and profiles of DHFRs used in this study, whole-cell growth inhibition, X-ray crystallographic statistics, EcDHFR conformational groups, top DALI results, and clogP values; supplementary figures of multi-MoA antibiotic structures, whole cell growth inhibition, IC50 curves, X-ray structure electron density maps and structural analysis, hierarchical clustering dendrograms, membrane disruption, hemolysis, molecular docking, solubility, and enzyme purification; and compound characterization data, including NMR spectra and LC-MS traces, for CYC and compounds 1a–g, 2b–l, 3a–h, and 4–8 (.pdf).
• Datasets for comparative structural analysis and hierarchical clustering (.xlsx).
• EcDHFR•CYC DALI results (.xlsx)
• EcDHFR•3d DALI results (.xlsx)
Notes
The authors declare no competing financial interest.
In Memoriam
We dedicate this study to the life, friendship, and scientific contributions of Dr. Ute Möllmann. Her dedication to microbiology has made a lasting impact on the study of antibiotics and microbial infections. Her kindness to others will be forever remembered and cherished by those who knew her.
References
- (1).Feng J; Zheng Y; Ma W; Ihsan A; Hao H; Cheng G; Wang X Multitarget Antibacterial Drugs: An Effective Strategy to Combat Bacterial Resistance. Pharmacol. Ther 2023, 252, 108550. 10.1016/j.pharmthera.2023.108550. [DOI] [PubMed] [Google Scholar]
- (2).Theuretzbacher Ursula. Dual-Mechanism Antibiotics. Nat Microbiol 2020, 5, 984–985. 10.1038/s41564-020-0767-0. [DOI] [PubMed] [Google Scholar]
- (3).Wiedemann I; Breukink E; Van Kraaij C; Kuipers OP; Bierbaum G; De Kruijff B; Sahl H-G Specific Binding of Nisin to the Peptidoglycan Precursor Lipid II Combines Pore Formation and Inhibition of Cell Wall Biosynthesis for Potent Antibiotic Activity. J. Biol. Chem 2001, 276 (3), 1772–1779. 10.1074/jbc.M006770200. [DOI] [PubMed] [Google Scholar]
- (4).Limbrick EM; Graf M; Derewacz DK; Nguyen F; Spraggins JM; Wieland M; Ynigez-Gutierrez AE; Reisman BJ; Zinshteyn B; McCulloch KM; Iverson TM; Green R; Wilson DN; Bachmann BO Bifunctional Nitrone-Conjugated Secondary Metabolite Targeting the Ribosome. J. Am. Chem. Soc 2020, 142 (43), 18369–18377. 10.1021/jacs.0c04675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Edwards MJ; Flatman RH; Mitchenall LA; Stevenson CEM; Le TBK; Clarke TA; McKay AR; Fiedler H-P; Buttner MJ; Lawson DM; Maxwell A A Crystal Structure of the Bifunctional Antibiotic Simocyclinone D8, Bound to DNA Gyrase. Science 2009, 326 (5958), 1415–1418. 10.1126/science.1179123. [DOI] [PubMed] [Google Scholar]
- (6).Zhanel GG; Schweizer F; Karlowsky JA Oritavancin: Mechanism of Action. Clin. Infect. Dis 2012, 54 (suppl_3), S214–S219. 10.1093/cid/cir920. [DOI] [PubMed] [Google Scholar]
- (7).Corey GR; Kabler H; Mehra P; Gupta S; Overcash JS; Porwal A; Giordano P; Lucasti C; Perez A; Good S; Jiang H; Moeck G; O’Riordan W Single-Dose Oritavancin in the Treatment of Acute Bacterial Skin Infections. N. Engl. J. Med 2014, 370 (23), 2180–2190. 10.1056/NEJMoa1310422. [DOI] [PubMed] [Google Scholar]
- (8).Martin JK; Sheehan JP; Bratton BP; Moore GM; Mateus A; Li SHJ; Kim H; Rabinowitz JD; Typas A; Savitski MM; Wilson MZ; Gitai Z A Dual-Mechanism Antibiotic Kills Gram-Negative Bacteria and Avoids Drug Resistance. Cell 2020, 181 (7), 1518–1532.e14. 10.1016/j.cell.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yuan D; Liu S; Li S; Liu R; Zhu X Design, Synthesis and Biological Evaluation of 7-Substituted-1,3-diaminopyrrol[3,2- f ]Quinazolines as Potential Antibacterial Agents. ChemMedChem 2023, 18 (12), e202300078. 10.1002/cmdc.202300078. [DOI] [PubMed] [Google Scholar]
- (10).World Health Organization Model List of Essential Medicines – 23rd List, 2023. https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2023.02. [Google Scholar]
- (11).Nzila A The Past, Present and Future of Antifolates in the Treatment of Plasmodium falciparum Infection. J. Antimicrob. Chemother 2006, 57 (6), 1043–1054. 10.1093/jac/dkl104. [DOI] [PubMed] [Google Scholar]
- (12).Sharma M; Chauhan PMS Dihydrofolate Reductase as a Therapeutic Target for Infectious Diseases: Opportunities and Challenges. Future Med. Chem 2012, 4 (10), 1335–1365. 10.4155/fmc.12.68. [DOI] [PubMed] [Google Scholar]
- (13).Modest EJ Chemical and Biological Studies on 1,2-Dihydro-s-Triazines. II. Three-Component Synthesis. J. Org. Chem 1956, 21 (1), 1–13. 10.1021/jo01107a001. [DOI] [Google Scholar]
- (14).Modest EJ; Levine P Chemical and Biological Studies on 1,2-Dihydro-s-Triazines. III. Two-Component Synthesis. J. Org. Chem 1956, 21 (1), 14–20. 10.1021/jo01107a002. [DOI] [Google Scholar]
- (15).Lowe G; Carr C; Quarrell R Racemisation and Rearrangement of 1,2-Dihydro-1,3,5-Triazines: A Novel Reversible Thermal Electrocyclic Reaction. Chem. Commun 2001, No. 8, 737–738. 10.1039/b101245m. [DOI] [Google Scholar]
- (16).Afonin S; Glaser RW; Berditchevskaia M; Wadhwani P; Gührs K; Möllmann U; Perner A; Ulrich AS 4-Fluorophenylglycine as a Label for 19F NMR Structure Analysis of Membrane-Associated Peptides. ChemBioChem 2003, 4 (11), 1151–1163. 10.1002/cbic.200300568. [DOI] [PubMed] [Google Scholar]
- (17).Chain C; Sheehan JP; Xu X; Ghaffari S; Godbole A; Kim H; Freundlich JS; Rabinowitz JD; Gitai Z A Folate Inhibitor Exploits Metabolic Differences in Pseudomonas aeruginosa for Narrow-Spectrum Targeting. Nat. Microbiol 2024, 9 (5), 1207–1219. 10.1038/s41564-024-01665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Krucinska J; Lombardo MN; Erlandsen H; Estrada A; Si D; Viswanathan K; Wright DL Structure-Guided Functional Studies of Plasmid-Encoded Dihydrofolate Reductases Reveal a Common Mechanism of Trimethoprim Resistance in Gram-Negative Pathogens. Commun. Biol 2022, 5 (1), 459. 10.1038/s42003-022-03384-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kim KH; Dietrich SW; Hansch C; Dolnick BJ; Bertino JR Inhibition of Dihydrofolate Reductase. 3. 4,6-Diamino-1,2-Dihydro-2,2-Dimethyl-1-(2-Substituted-Phenyl)-s-Triazine Inhibition of Bovine Liver and Mouse Tumor Enzymes. J. Med. Chem 1980, 23 (11), 1248–1251. 10.1021/jm00185a022. [DOI] [PubMed] [Google Scholar]
- (20).Dale GE; Broger C; Hartman Ronald DeHoogt PG; Á se Jolidon S; Kompis I; Labhardt Hanno Langen AM; Locher H; P Page MG; Stu È ber Rudolf Then DL; Wipf B; Oefner C A Single Amino Acid Substitution in Staphylococcus aureus Dihydrofolate Reductase Determines Trimethoprim Resistance The Problem of Bacterial Resistance to Many of Todays Antimicrobial Agents Is Well Known. J Mol Biol 1992, 266, 23–30. [DOI] [PubMed] [Google Scholar]
- (21).Selassie CD; Guo ZR; Hansch C; Khwaja TA; Pentecost S A Comparison of the Inhibition of Growth of Methotrexate-Resistant and -Sensitive Leukemia Cells in Culture by Triazines. Evidence for a New Mechanism of Cell Resistance to Methotrexate. J. Med. Chem 1982, 25 (2), 157–161. 10.1021/jm00344a013. [DOI] [PubMed] [Google Scholar]
- (22).Sawaya MR; Kraut J Loop and Subdomain Movements in the Mechanism of Escherichia Coli Dihydrofolate Reductase: Crystallographic Evidence. Biochemistry 1997, 36 (3), 586–603. 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- (23).Holm L; Laiho A; Törönen P; Salgado M DALI Shines a Light on Remote Homologs: One Hundred Discoveries. Protein Sci 2023, 32 (1), e4519. 10.1002/pro.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Stokes A; Lacey RW Effect of Thymidine on Activity of Trimethoprim and Sulphamethoxazole. J. Clin. Pathol 1978, 31 (2), 165–171. 10.1136/jcp.31.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wray R; Wang J; Blount P; Iscla I Activation of a Bacterial Mechanosensitive Channel, MscL, Underlies the Membrane Permeabilization of Dual-Targeting Antibacterial Compounds. Antibiotics 2022, 11 (7), 970. 10.3390/antibiotics11070970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Baba T; Ara T; Hasegawa M; Takai Y; Okumura Y; Baba M; Datsenko KA; Tomita M; Wanner BL; Mori H Construction of Escherichia Coli K-12 In-frame, Single-gene Knockout Mutants: The Keio Collection. Mol. Syst. Biol 2006, 2 (1), 2006.008. 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yamamoto N; Nakahigashi K; Nakamichi T; Yoshino M; Takai Y; Touda Y; Furubayashi A; Kinjyo S; Dose H; Hasegawa M; Datsenko KA; Nakayashiki T; Tomita M; Wanner BL; Mori H Update on the Keio Collection of Escherichia coli Single-gene Deletion Mutants. Mol. Syst. Biol 2009, 5 (1), 335. 10.1038/msb.2009.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zarou MM; Vazquez A; Vignir Helgason G Folate Metabolism: A Re-Emerging Therapeutic Target in Haematological Cancers. Leukemia 2021, 35 (6), 1539–1551. 10.1038/s41375-021-01189-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cho S; Lee HS; Franzblau S Microplate Alamar Blue Assay (MABA) and Low Oxygen Recovery Assay (LORA) for Mycobacterium tuberculosis. In Mycobacteria Protocols; Parish T, Roberts DM, Eds.; Methods in Molecular Biology; Springer New York: New York, NY, 2015; Vol. 1285, pp 281–292. 10.1007/978-1-4939-2450-9_17. [DOI] [PubMed] [Google Scholar]
- (30).Pethe K; Sequeira PC; Agarwalla S; Rhee K; Kuhen K; Phong WY; Patel V; Beer D; Walker JR; Duraiswamy J; Jiricek J; Keller TH; Chatterjee A; Tan MP; Ujjini M; Rao SPS; Camacho L; Bifani P; Mak PA; Ma I; Barnes SW; Chen Z; Plouffe D; Thayalan P; Ng SH; Au M; Lee BH; Tan BH; Ravindran S; Nanjundappa M; Lin X; Goh A; Lakshminarayana SB; Shoen C; Cynamon M; Kreiswirth B; Dartois V; Peters EC; Glynne R; Brenner S; Dick T A Chemical Genetic Screen in Mycobacterium tuberculosis Identifies Carbon-Source-Dependent Growth Inhibitors Devoid of in Vivo Efficacy. Nat. Commun 2010, 1 (1), 57. 10.1038/ncomms1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hunt-Serracin AC; Parks BJ; Boll J; Boutte CC Mycobacterium abscessus Cells Have Altered Antibiotic Tolerance and Surface Glycolipids in Artificial Cystic Fibrosis Sputum Medium. Antimicrob. Agents Chemother 2019, 63 (7), e02488–18. 10.1128/AAC.02488-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hajian B; Scocchera E; Shoen C; Krucinska J; Viswanathan K; G-Dayanandan N; Erlandsen H; Estrada A; Mikušová K; Korduláková J; Cynamon M; Wright D Drugging the Folate Pathway in Mycobacterium tuberculosis: The Role of Multi-Targeting Agents. Cell Chem. Biol 2019, 26 (6), 781–791. 10.1016/j.chembiol.2019.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Foley GE Chemical and Biological Studies on 1,2-Dihydro-s-Triazines. IV. Activity in Pteroylglutamic Acid-Streptococcus faecaelis No. 8043 Systems. Exp. Biol. Med 1953, 83 (4), 733–739. 10.3181/00379727-83-20477. [DOI] [PubMed] [Google Scholar]
- (34).Foley GE; Modest EJ; Catalog JR; Riley HD Chemical and Biological Studies on 1:2-Dihydro-s-Trtazines—XIII: Inhibition of Response to p-Aminobenzoic Acid and Mechanism of Action in Lactobacillus arabinosus Bioassay Systems. Biochem. Pharmacol 1959, 3 (1), 18–30. 10.1016/0006-2952(59)90004-8. [DOI] [PubMed] [Google Scholar]
- (35).Hansch C; Hathaway BA; Guo Z; Selassie CD; Dietrich SW; Blaney JM; Langridge R; Volz KW; Kaufman BT Crystallography, Quantitative Structure-Activity Relationships, (QSAR) and Molecular Graphics in a Comparative Analysis of the Inhibition of Dihydrofolate Reductase from Chicken Liver and Lactobacillus casei by 4,6-Diamino-1,2-Dihydro-2,2-Dimethyl-1-(Substituted-Phenyl)-s-Triazines. J. Med. Chem 1984, 27 (2), 129–143. 10.1021/jm00368a006. [DOI] [PubMed] [Google Scholar]
- (36).Hathaway BA; Guo ZR; Hansch C; Delcamp TJ; Susten SS; Freisheim JH Inhibition of Human Dihydrofolate Reductase by 4,6-Diamino-1,2-Dihydro-2,2-Dimethyl-1-(Substituted-Phenyl)-s-Triazines. A Quantitative Structure-Activity Relationship Analysis. J. Med. Chem 1984, 27 (2), 144–149. 10.1021/jm00368a007. [DOI] [PubMed] [Google Scholar]
- (37).Wróbel A; Arciszewska K; Maliszewski D; Drozdowska D Trimethoprim and Other Nonclassical Antifolates an Excellent Template for Searching Modifications of Dihydrofolate Reductase Enzyme Inhibitors. J. Antibiot. (Tokyo) 2020, 73 (1), 5–27. 10.1038/s41429-019-0240-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhang K; Rathod PK Divergent Regulation of Dihydrofolate Reductase Between Malaria Parasite and Human Host. Science 2002, 296 (5567), 545–547. 10.1126/science.1068274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Cao H; Gao M; Zhou H; Skolnick J The Crystal Structure of a Tetrahydrofolate-Bound Dihydrofolate Reductase Reveals the Origin of Slow Product Release. Commun. Biol 2018, 1 (1), 226. 10.1038/s42003-018-0236-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ribeiro JA; Chavez-Pacheco SM; De Oliveira GS; Dos Santos Silva C; Giudice JHP; Libreros-Zúñiga GA; Dias MVB Crystal Structures of the Closed Form of Mycobacterium tuberculosis Dihydrofolate Reductase in Complex with Dihydrofolate and Antifolates. Acta Crystallogr. Sect. Struct. Biol 2019, 75, 682–693. 10.1107/S205979831900901X. [DOI] [PubMed] [Google Scholar]
- (41).Aragaw WW; Negatu DA; Bungard CJ; Dartois VA; Marrouni AE; Nickbarg EB; Olsen DB; Warrass R; Dick T Pharmacological Validation of Dihydrofolate Reductase as a Drug Target in Mycobacterium abscessus. Antimicrob. Agents Chemother 2024, 68 (1), e00717–23. 10.1128/aac.00717-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Aragaw WW; Lee BM; Yang X; Zimmerman MD; Gengenbacher M; Dartois V; Chui W-K; Jackson CJ; Dick T Potency Boost of a Mycobacterium tuberculosis Dihydrofolate Reductase Inhibitor by Multienzyme F420H 2-Dependent Reduction. Proc. Natl. Acad. Sci 2021, 118 (25), e2025172118. 10.1073/pnas.2025172118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zhang Y-M; Rock CO Membrane Lipid Homeostasis in Bacteria. Nat. Rev. Microbiol 2008, 6 (3), 222–233. 10.1038/nrmicro1839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.