

Significance
Successful establishment of a profitable microalgae process is only possible by securing customized strains that are fit for purpose. Gene editing using the Cas9 system has provided a shortcut for genetically tailoring microalgae, but its application has been hindered by the low gene-editing frequency due to the poor nuclear import of the nuclease. Our results show that functional fusion of phytopathogen-derived nuclear localization signals, which are highly affine to the microalgal importin alpha, to Cas9 results in enhanced nuclear import, thereby doubling the gene-editing frequency (computed from a Sanger-based approach) in both a model and an industrial algal species compared to the conventional approach. This finding underscores the importance of nuclear accessibility for programmable gene editors to elevate their utility.
Keywords: CRISR/Cas9, microalgae, agrobacterium, nuclear localization signal (NLS), gene editing frequency
Abstract
Pathogen-derived nuclear localization signals (NLSs) enable vigorous nuclear invasion in the host by the virulence proteins harboring them. Herein, inspired by the robust nuclear import mechanism, we show that NLSs originating from the plant infection–associated Agrobacterium proteins VirD2 and VirE2 can be incorporated into the Cas9 system as efficient nuclear delivery enhancers, thereby improving the low gene-editing frequency in a model microalga, Chlamydomonas reinhardtii, caused by poor nuclear localization of the bulky nuclease. Prior to evaluation of the NLSs, IPA1 (Cre04.g215850) was first defined in the alga as the nuclear import-related importin alpha (Impα) that serves as a counterpart adaptor protein of the NLSs, based on extensive in silico analyses considering the protein’s sequence, tertiary folding behavior, and structural basis when interacting with a well-studied SV40TAg NLS. Through precursive affinity explorations, we reproducibly found that the NLSs mediated the binding between the Cas9 and Impα with nM affinities and visually confirmed that the fusion of the NLSs strictly localized the peptide-bearing cargoes in the microalgal nucleus without compensating for their cleavage ability. When employed in a real-world application, the VirD2 NLS increases the mutation frequency (~1.12 × 10−5) over 2.4-fold compared to an archetypal SV40TAg NLS (~0.46 × 10−5) when fused with Cas9. We demonstrate the cross-species versatility of the Impα-dependent strategy by successfully applying it to an industrial alga, Chlorella Sp. HS2. This work, focused on affinity augmentation, provides insights into increasing the frequency of gene editing, which can be advantageously used in programmable mutagenesis with broad applicability.
Microalgae are emerging as a promising carbon-neutral chassis (1, 2). Among various genetic toolkits for improving the technical readiness, the clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein 9 (Cas9) system has garnered substantial interest for its programmability and cross-species applicability (3, 4). However, the applications have been often frustrated by the low gene-editing frequency, which increases the workload and significantly reduces the likelihood of isolating transformants with desired functionality (5, 6). Since it relies on the enzymatic interaction between the bulky Cas9 protein (~160 kDa) and a target gene embedded in the genomic DNA (gDNA), endocytosis of the ribonucleoprotein (RNP) complex and its orientation to the gene — that requires overcoming several cellular obstacles—is considered one of the decisive frequency-determining factors (5, 7).
The nuclear envelope likely serves as an unavoidable last bastion, lowering the frequency. This is because: i) whereas the permeability of other barriers, including the cell wall and plasma membrane, can be simply increased, it is challenging to physicochemically increase the nuclear membrane permeability while maintaining the integrity of the living cell (5, 8, 9); and ii) the nuclear entry of the voluminous molecule is essential during the editing, regardless of the mutation strategy employed (e.g., expression vector-driven and extracellularly preassembled RNP-based methods) (SI Appendix, Fig. S1) (3, 4, 10). Thus, exploiting the innate, elaborately regulated nuclear pore complex (NPC)-mediated transportation mechanism (11) would be one of the remaining options that enables an efficient traverse of a macromolecule to the nucleus.
NPC-mediated nuclear internalization is initiated by the protein–protein interaction between nuclear localization signal (NLS)-bearing cargo and importin α (Impα) adaptor proteins (12–15). During this process, an NLS, a short peptide embedded in eukaryotic nuclear proteins or viral proteins, acts as a signal to direct the NLS-carrying protein into the nucleus (12). It is notable that cargo-adaptor binary complex formation determines the efficiency of nuclear transport based on the specific recognition of the NLS by Impα (14). Consistently, it is known that cargo, whose nuclear targeting signal shows higher specificity —and thus higher affinity—for Impα, is more likely to be destined for the nucleus in competition with other nuclear targeting proteins in vivo (16). Thus, it was hypothesized that the fusion of NLS with high affinity for microalgal Impα to the protein payload, Cas9, would help localize the RNP near gDNA, thereby increasing the gene-editing frequency. Without serious deliberation, so far, an NLS discovered in a viral protein, simian virus 40 large T antigen (SV40TAg)—which plays a critical role in the viral replication during primate infection—has been used as a delivery enhancer in most cases due to its multispecies applicability (3, 6, 10, 17, 18).
To significantly increase the gene-editing frequency, the sequence of the NLS used as a Cas9 delivery enhancer should be carefully selected based on the organism to be genetically manipulated. Two prospects can be devised for the selection of an NLS with high affinity for Impα in microalgae: utilization of i) an NLS derived from the endogenous nuclear protein; and ii) an NLS originating from infecting organisms. The former may ensure the proper function, but there is no guarantee that it would vigorously facilitate delivery of the cargo into the nucleus because its affinity would be essentially comparable to that of the existing nuclear targeted competitors. Instead, an NLS from the latter category could be expected to have a higher competitiveness in terms of affinity, likely achieved through putative coevolution in host–pathogen interactions during increasing its infectivity (19–22), albeit with the need to validate its fidelity to the host.
In this work, inspired by a well-designed plant-phytopathogen infection mechanism, we functionally attached two different NLSs found in virulence proteins VirD2 and VirE2, which are derived from a phytoinfecting bacterium, Agrobacterium tumefaciens (23–27), to Cas9 and compared their nuclear import performance and resulting gene-editing frequency with those induced by the typically used NLS (SV40TAg NLS), in a model alga, Chlamydomonas reinhardtii. Prior to practical use, since the identity of CrImpα itself and its affinities for the NLSs were not clearly determined, the nuclear adaptor protein was specified among several homolog candidates using state-of-the-art in silico tools and the true affinity was thoroughly evaluated qualitatively and quantitatively. The results showed that the NLSs from VirD2 reproducibly exhibit higher affinity for CrImpα than the archetypal NLS. By mimicking the infection-like nuclear import process in vivo, covalent fusion of the pathogen-derived NLSs to Cas9 significantly increased the targeted gene-editing frequency, with the VirD2 NLS increasing the frequency more than the SV40TAg NLS (>one-fold increase). When applied to the mutagenesis of an industrial microalga, Chlorella Sp. HS2, the VirD2 NLS also induced more frequent mutagenesis compared to the conventional NLS, demonstrating the cross-species versatility of the affinity enhancement strategy. On balance, this straightforward strategy enables precise, high-frequency alteration of genomic sequences in microalgae by facilitating the delivery of Cas9 into the nucleus.
Results
Selection of VirD2 and VirE2 NLS Subtypes.
A. tumefaciens, the origin of the phytopathogen-derived VirD2 and VirE2 NLSs, largely falls into two categories, nopaline- and octopine-type, depending on the opine compounds that the pathogen forces the plant host to produce for use as major nutrient sources (28–30). Although a plethora of putative NLSs from two types of A. tumefaciens have been proposed, only a few sequences have been empirically verified to show nuclear import abilities (28–30), which are listed in Table 1. As an initial estimate of the nuclear import capabilities of the infection-related NLSs, the cumulative charge of each sequence was calculated as a marker of NLS strength, assuming that a higher positive charge leads to a higher NLS strength (19). Intriguingly, the values were found to be identical for all the listed NLSs, despite the length and sequence difference, suggesting that both VirD2 and VirE2 NLSs from nopaline- and octopine-type A. tumefaciens may function as promising nuclear delivery enhancers similar to the universally used SV40TAg NLS. Moreover, NLSs can be structurally classified into monopartite and bipartite NLSs according to the number of positively charged amino acid clusters contained in the sequences (14). Monopartite NLSs such as SV40TAg NLS include one cluster, preferentially binding to the major NLS binding site (BS) located in the N-proximate of Impα, while bipartite NLSs like VirD2 and VirE2 NLSs include two clusters linked with a short peptide spacer sequence that bind to both major and minor (located in the C-proximate) NLS BSs in Impα in an antiparallel orientation (Table 1) (30). Given the structural and length dissimilarities, the same NLS strength computed from the different NLS sequences underscores the need for further investigation to measure the practical binding affinity for Impα before actual utilization. Since the structures and interaction modes of nopaline-type NLSs (i.e., VirD2N and VirE2N NLSs) have been intensively studied in plant systems (30–32), octopine-type NLSs (i.e., VirD2O and VirE2O NLSs) were selected for in-depth analysis in this study to provide broader insights into the interaction between the phytoinfection-associated bipartite NLSs and plant Impα proteins.
Table 1.
Information on the pathogen-derived NLSs considered in this study
| The number of positive-charged residues† | The number of negative-charged residues† | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NLS | Origin protein | Target host of the pathogen | Type | Peptide sequence of NLS* | Lys (K) | Arg (R) | Asp (D) | Glu (E) | Cumulative charge‡ | Ref. |
| SV40TAg NLS | Simian virus 40 large T antigen | Primates | Monopartite | PKKKRKV | 4 | 1 | 0 | 0 | 5 | (30) |
| VirD2O NLS | A. tumefaciens octopine-type VirD2 | Plants | Bipartite | KR(RNDEEAGPSGAN) RKGLK | 3 | 2 | 0 | 0 | 5 | (28) |
| VirE2O NLS | A. tumefaciens octopine-type VirE2 | Plants | Bipartite | KLR(PEDRYVQTE)RYGRR | 1 | 4 | 0 | 0 | 5 | (29) |
| VirD2N NLS | A. tumefaciens nopaline-type VirD2 | Plants | Bipartite | KR(PREDDDGEPSER)KRERR | 2 | 4 | 0 | 1 | 5 | (30) |
| VirE2N NLS | A. tumefaciens nopaline-type VirE2 | Plants | Bipartite | KLR(PEDRYIQTE)KYGRR | 2 | 3 | 0 | 0 | 5 | (30) |
*The sequences in the parentheses indicate the linker sequence between the BSs in the bipartite NLSs.
†The numbers were counted, except for the residues included in the linker sequence.
‡The charge, a measure of NLS strength, was calculated by the number of positive-charged amino acid residues (i.e., lysine and arginine) minus the number of negative-charged amino acid residues (i.e., aspartic acid and glutamic acid) in the sequence, excluding that of residues in the linker sequence (19).
Determination of the Nuclear Import-Related Impα in C. reinhardtii.
Development of in silico computational tools for functional and structural prediction of proteins is increasingly reducing our reliance on tedious, time-consuming, and capital-intensive legacy interpretation methods, such as X-ray crystallography (33). Taking advantage of recent advances, the Impα protein, which is essentially involved in nuclear transport in C. reinhardtii but has not been clearly described, was identified among three proteins annotated as Impα-like proteins (SI Appendix, Fig. S2 A–I). Based on the AlphaFold-assisted comparative structural analysis described in http://www.pnas.org/lookup/doi/10.1073/pnas.2415072122#supplementary-materialsSI Appendix, Supplementary Note 1, CrIPA1 (Cre04.g215850) was predicted to be the only Impα responsible for nuclear import in C. reinhardtii (34–36). We then observed the structural basis of binding of the conventional NLS (i.e., SV40TAg NLS) to CrIPA1 using another in silico tool, HDOCK algorithm—a template-based protein–protein docking predictor (37), to validate whether the predicted adaptor protein would form a stable dimer with the nuclear signal via explainable interresidual interactions. Using the cocrystallography data derived from the binding between OsISAΔIBB—OsISA lacking the autoinhibitory IBB domain—and SV40TAg NLS (PDB ID: 4B8O) as a template (14), the structural basis of CrIPA1ΔIBB:SV40TAg NLS was predicted. Since the monopartite NLS can interact with two sites in an Impα protein separately (i.e., the major and minor NLS BSs), the most plausible pose for each site was determined (Fig. 1 A and D and SI Appendix, Fig. S3 A to E and Supplementary Note 2). As a result of comparing the binding configuration of SV40TAg NLS with OsISA and CrIPA1 at their major NLS BS (Fig. 1A), a low RMSD of 1.779 Å was observed, implying that CrIPA1 interacts with SV40TAg NLS very similarly to the way OsISAΔIBB:SV40TAg NLS binds. Two models also share various interacting residues (14 out of 18 residues for CrIPA1ΔIBB) while forming antiparallel dimers (Fig. 1 B and C and Dataset S1). Moreover, a number of identical bondings were identified from both models, including electrostatic attraction between K (P2 position in the NLS) and D132 (in CrIPA1ΔIBB; corresponding to D188 in OsISA), hydrogen bonds between P (P−1 position) and R174 (R230), and between P (P0 position) and W167 (W223), which are the conserved fingerprint interactions in dimerization of SV40TAg NLS and various Impα proteins (SI Appendix, Fig. S4 A and B and Dataset S1) (14). Regarding the binding configurations of SV40TAg NLS at the minor NLS BSs in both IBB-truncated Impα proteins (Fig. 1D), the structural comparison resulted in a marginal RMSD (0.004 Å), suggesting that the two configurations are nearly identical and that the NLS interacts with the respect adaptor proteins in a very similar manner. Correspondingly, a large proportion of interacting residues (11 out of 12 residues for CrIPA1ΔIBB) were found to be common to both antiparallel binding complexes (Fig. 1 E and F and Dataset S1). Several conserved interresidual bonds—which are hallmark interactions of SV40TAg NLS and various Impα proteins, including OsISAΔIBB:SV40TAg NLS (14)—were also observed in the predicted model, representatively such as a hydrogen bond between R (P2′ position in the NLS) and E332 (in CrIPA1ΔIBB; corresponding to E388 in OsISA) (SI Appendix, Fig. S4A and Dataset S1). As to the docking scores that were copredicted from the CrIPA1ΔIBB:SV40TAg NLS simulation, the score related to the major BS (−278.31; Fig. 1A) was much lower than that related to the minor BS (−156.00; Fig. 1D), demonstrating that SV40TAg NLS forms a more stable dimer at the major NLS BS of CrIPA1ΔIBB, consistent with the fact that the NLS preferentially binds to the major NLS BS in Impα proteins. Throughout the comprehensive inspections using several in silico tools, it was confirmed that CrIPA1 faithfully follows the consensus features of a nuclear import-functioning Impα protein. On balance, IPA1 (termed CrImpα hereafter) was determined to be the protein with a pivotal role in nuclear import in C. reinhardtii and was thus used for further assessments.
Fig. 1.

Structural basis of the CrIPA1ΔIBB:SV40TAg NLS reveals that CrIPA1 (Cre04.g215850) functions as a nuclear import adaptor Impα protein in C. reinhardtii. (A) Comparison of the cocrystal structures of OsISAΔIBB:SV40TAg NLS and CrIPA1ΔIBB:SV40TAg NLS when the encounters occur at the major NLS BS of each adaptor protein. (B and C) Residual interactions at the major NLS BS of (B) OsISAΔIBB:SV40TAg NLS and (C) CrIPA1ΔIBB:SV40TAg NLS. (D) Comparison of the cocrystal structures of OsISAΔIBB:SV40TAg NLS and CrIPA1ΔIBB:SV40TAg NLS when the encounters occur at the minor NLS BS of each adaptor protein. (E and F) Residual interactions at the minor NLS BS of (E) OsISAΔIBB:SV40TAg NLS and (F) CrIPA1ΔIBB:SV40TAg NLS. (A and D) The RMSD values of the NLS between two structures are indicated. For CrIPA1ΔIBB:SV40TAg NLS, the copredicted docking scores from the simulation are present. N and C termini are shown. (B and C, and E and F) Bonds are indicated by dashed lines of the color specified in the legend. Residues commonly observed in two cocrystal structures are colored in blue.
Standardizing the Method to Negate the Outer Membrane Structure.
To test our hypothesis that instillation of the plant infection–motivated nuclear transport strategy would be beneficial for Cas9-based algal gene editing, we adopted the extracellularly preassembled RNP system (Fig. 2A), which has been recently preferred over the vector-driven method (SI Appendix, Fig. S1) due to its simplicity and low cellular toxicity without concern for genetically modified organism-related issues, such as proliferation of artificial genes in the environment (6). When using the system, lowering the threshold for cytoplasmic internalization through other cellular barricades (i.e., the outer membrane structure), including the cell wall and plasma membrane, should precede a reliable assay of the performance of the NLS candidates. As illustrated in Fig. 2B, of the two widely used reagents (3, 5, 18), gametolysin (GL)—also known as autolysin, which is produced during sexual reproduction of C. reinhardtii (SI Appendix, Fig. S5A), showed better performance than a commercial cell wall-degrading reagent (see Materials and Methods for details) in attenuating the cell wall, as indicated by the extent of cell integrity loss. As the cells were treated with a chemical plasma membrane-lysing surfactant (see Materials and Methods for details) prior to enervate the barrier effect, the combined use of detergent to weaken the plasma membrane and GL to weaken the cell wall appeared too lethal for the microalgal cells to efficiently undermine both barriers while maintaining viability (Fig. 2B). Thus, we changed the plasma membrane poration method to electropulse application because of its well-known effect on the plasma membrane (8) and visually examined the effect on the penetration of a proteinaceous material into the cytoplasm by initially using a fluorescent protein, mCherry (Fig. 2 C–E).
Fig. 2.

Standardization of the method for increasing the cellular permeability of proteinaceous materials across the cell wall and plasma membrane. (A) A simplified schematic depicting the nuclear localization of the Cas9 RNP. The internalization pathway is indicated by a thick arrow. The associated molecular interactions are shown with thin arrows. The NPC is highlighted with a dashed circle. The cartoon is not to scale. (B) Cell wall attenuation was tested by cotreatment with a plasma membrane-lysing detergent, Triton X-100. (Scale bar, 18.5 μm.) (C) Localization of mCherry reporter without NLS by confocal microscopy. (Scale bar, 5 μm.) (D) Box and whisker plots demonstrate the fluorescence ratio (as a measure of localization) for mCherry reporter. (E) Reagent-dependent permeability was reproducibly observed using flow cytometry. (F) Localization of Alexa 488-labeled Cas9 without NLS (Cas9ΔNLS) by confocal microscopy. (Scale bar, 5 μm.) (G) Box and whisker plots demonstrate the fluorescence ratio for Alexa 488-labeled Cas9ΔNLS. (H) Reagent-dependent permeability was reproducibly observed using flow cytometry for Alexa 488-labeled Cas9ΔNLS. (C and F) NM, CB, and GL stand for normal medium (TAP medium), commercial buffer for transformation (see Materials and Methods for details), and gametolysin, respectively. (D and G) Data were derived from four biological replicates and quantified using ImageJ software. The median (the center line) ± whiskers (1.5 × the interquartile range of the lower and upper quartiles) are shown. Student’s t test was performed. Statistical significances and fold-changes are presented. The region of interest for the quantification is visualized in the inset. (E and H) PE and FITC represent the phycoerythrin and fluorescein isothiocyanate channels, respectively, for detection of the fluorescence emitted by the proteins. Populations (%) above the threshold intensity are indicated.
Among the electroporated cells, the highest amount of the fluorophore was observed in the cytoplasm of the gametolysin-treated group (Fig. 2C and SI Appendix, Fig. S5 B–E), suggesting that this combination is appropriate for delivering proteins to live cells. This result was quantitatively verified by image analysis (Fig. 2D). In the other groups, most of the fluorescent protein was trapped by the barriers, whereas in the GL-treated group, a large amount of the protein successfully entered the cells, leading to the lowest periphery/cytoplasm fluorescence ratio—3.65 and 4.34 times lower than those of the normal medium (NM)- and commercial buffer (CB)-treated groups, respectively (P values<10−4). However, in all groups, no significant concentration in any organelle was evident. This trend was reproducibly observed by flow cytometry analysis (Fig. 2E and SI Appendix, Fig. S6 A–D). Only the GL-treated group showed a distinguishable peak above the fluorescence intensity (FI) of 400 at the mCherry-detecting channel, and almost a quarter of the single-cell population was included in this range. In contrast, no noticeable peak was observed in the high-fluorescence region (i.e., FI>400) of the other groups, resulting in less than 10% of the parent population in the range.
The identical assays were performed with Alexa 488-labeled Cas9 to visually confirm the cellular internalization of the protein of interest in this study. Similar to the previous results, GL treatment allowed the Cas9 proteins to be internalized into the cells (Fig. 2F and SI Appendix, Fig. S7 A–D), resulting in the lowest periphery/cytoplasm fluorescence ratio—2.54 and 3.69 times lower than those of the NM- and CB-treated cells, respectively (P values < 3 × 10−4) (Fig. 2G). In flow cytometry, GL treatment also induced a dramatic peak shift at the FITC channel. As of FI=20, 85.2% of GL-treated single cells were above, whereas single cells in the other groups were below 10%, reproducibly indicating that GL is the most efficient cell wall attenuating reagent tested (Fig. 2H and SI Appendix, Fig. S8 A–D). In summary, the combination of gametolysin pretreatment and electroporation was found to be efficient for the translocation of protein molecules into the cells and was therefore adopted as the basic protocol for in vivo translocation of proteinaceous molecules (vide infra).
Phytoinfection-Related NLSs Strongly Bind to CrImpα.
To evaluate the true affinity between the NLS-bearing cargoes and the adaptor CrImpα, we synthesized the proteins of interest using the Escherichia coli BL21(DE3) expression system and performed three in vitro assays (13), coimmunoprecipitation (co-IP), enzyme-linked immunosorbent assay (ELISA), and microscale thermophoresis (MST). In these analyses, we employed Cas9 proteins harboring the NLS candidates as test molecules to investigate the practical affinities of the localizing peptides for the receptor (i.e., CrImpαΔIBB) while the bulky cargo is functionally attached to the sequence given the purpose of this study. Prior to the preparation of the proteins, Cas9 variants fused with three different NLSs (i.e., VirD2O, VirE2O, and SV40TAg NLSs) and CrImpαΔIBB were designed and tagged with poly-histidine (H × 6) and FLAG (DYKDDDDK) peptides on their far C and N termini, respectively, enabling the purification of the recombinant proteins (Fig. 3A and SI Appendix, Table S1). More specifically, to examine the positional effect of NLS labeling, each NLS was fused alternatively to the N and C termini of Cas9. The proteins produced were termed DN, DC, EN, EC, SN, and SC Cas9. In the abbreviations, the first letters, D, E, and S, correspond to VirD2O, VirE2O, and SV40TAg NLSs, respectively, while the last letters, N and C, indicate the N- and C-terminus, respectively, where the designated NLS is attached. The expression was confirmed using western blotting employing the corresponding tag-specific antibodies (Fig. 3A).
Fig. 3.

Binding of Cas9 nucleases fused independently with each NLS at the N- and C-terminus to CrImpαΔIBB. (A) Western blot analysis to confirm the proper expressions of the recombinant adaptor, CrImpαΔIBB, and the Cas9 variants in E. coli, including Cas9ΔNLS used for visual localization assays (Fig. 2 F–H), detected by anti-FLAG and anti-His antibodies, respectively. GroEL: an endogenous control. The diagram shows the secondary structure of each NLS-tagged Cas9 protein, its nomenclature, and the corresponding NLS sequence it contains. The antiparallel binding modes are presented. BS stands for the BS. (B) co-IP assay was conducted with anti-FLAG antibody on lysates from E. coli cells expressing FLAG-tagged CrImpαΔIBB (as bait) and poly-histidine-tagged Cas9 variants, which were alternatively fused to each NLS at both termini (as prey), as shown in the right panel. CL: cell lysate. (C) ELISA result. In the legend, the best-fit values from each 4-parameter logistic sigmoidal fit are given as binding affinities. Two biological and two technical replicates (a total of quadruplication) were tested for each combination. Inset: magnified graph around the inflection point. Dashed box briefly illustrates the microplate platform of the assay. (D) MST results reproducibly show comparable affinities to the ELISA for the same positionally NLS-fused Cas9 variants (i.e., DN and SN Cas9).
Based on the synthesized proteins, we first performed a co-IP test to qualitatively witness the binding of the adaptor protein against respective NLS-bearing cargo variants while using them as bait and prey (Fig. 3B). We found that all Cas9 variants were bound by the adaptor and formed cargo-adaptor heterodimers, as evidenced by the presence of bands around the expected molecular sizes, confirming the interactivity of the plant-infection-associated NLSs, as well as the archetypal SV40TAg NLS, with microalgal Impα, regardless of the position to which each NLS was bound (Fig. 3B and SI Appendix, Fig. S9).
Because it was difficult to extract quantitative information from the co-IP outcomes, we then conducted ELISA measurements to quantitatively determine the binding affinities of direct interaction between the respective Cas9 variants and CrImpαΔIBB, all of which were purified in advance from the E. coli cell lysates. From this assay, every Cas9 variant showed strong, nanomolar affinities as indicated by the dissociation constants (KD), DN: 4.785 (95% CI: 4.478 to 5.052) nM; DC: 5.013 (95% CI: 4.642 to 5.343) nM; EN: 7.811 (95% CI: 7.324 to 8.330) nM; EC: 6.901 (95% CI: 6.315 to 7.491) nM; SN: 8.728 (95% CI: 8.205 to 9.257) nM; and SC: 9.209 (95% CI: 8.051 to 10.51) nM for CrImpαΔIBB (Fig. 3C), where CI represents the CI. The R2 values of the fitting curves were greater than 0.97, indicating that the data fit well to the function. All binding affinities of Cas9 variants for CrImpαΔIBB were apparently high, although notable binding affinity trends depending upon the NLS species harbored were observed. Namely, VirD2O NLS was found to confer the strongest affinity for CrImpαΔIBB on the nuclear targeting cargo protein, followed by VirE2O and SV40TAg NLSs. For the same N-terminally fused variants, DN Cas9 showed approximately 1.63-fold (P < 0.0001) and 1.82-fold (P < 0.0001) higher affinity than EN and SN Cas9s, respectively.
Comparable affinity values to the previous microtiter-plate binding assay were obtained using MST analyses, with the most preferred N-terminal NLS-conjugated variant (i.e., DN Cas9) binding CrImpαΔIBB with high affinity (4.481 nM), whereas the least preferred variant (i.e., SN Cas9) showed lower affinity (11.42 nM) (Fig. 3D). In summary, in both quantitative and qualitative investigations, VirD2O, VirE2O, and SV40TAg NLSs showed comparatively high affinities for the recombinant CrImpα during their respective direct interactions, while the phytopathogen-derived NLSs showed superior affinity values to SV40TAg NLS without notable fusion position predilection in vitro.
Effects of NLS Fusion on Localization and Cleavage Performance.
Before applying the Cas9 variants for algal gene editing, it is necessary to validate the nuclear localization effect of a fusion NLS in vivo, as well as the DNA cleavage performance of the NLS-conjugated Cas9 proteins. To this end, we observed whether the respective NLSs promoted cargo localization in the target subcellular region (i.e., the nucleus) of C. reinhardtii using the mCherry reporter and Cas9 fused with the NLSs and also performed an in vitro cleavage appraisal to check whether the desired functionality of the target cargo, Cas9, was negatively affected by NLS fusion, potentially due to steric interference (38).
First, we synthesized the respective NLS-tagged mCherry fluorescent proteins using the E. coli BL21(DE3) expression system (Fig. 4A). As per the previous nomenclature (Fig. 3A), mCherry variants were prepared and confirmed using western blotting (Fig. 4B). After delivering the reporters intracellularly as described (Fig. 2C), the detailed subcellular localization of the mCherry reporters and Cas9 proteins (representatively for DN and SN Cas9 proteins) were traced by confocal microscopy. For both mCherry protein without NLS (the same mCherry used in Fig. 2C) and no NLS Cas9 (Cas9ΔNLS; the same Cas9 used in Fig. 2F), no specific localization was evident, which was consistent with the previous result (Fig. 4 C and D and SI Appendix, Figs. S10 A–G and S11 A–C). The fluorescence of the others tagged with any NLS was concentrated near the nucleus, whose location was indicated by the nuclear staining dye, DAPI. This was also quantitatively confirmed by image analysis (Fig. 4E). We defined the ratio of the areal fluorescence intensity of the nucleus to that of the total cytoplasm as an index of the extent of fluorescence localization—therefore, the ratio cannot exceed the value of 1. We found that the cells with the NLS-harboring mCherry reporters and Cas9 proteins showed at least 2.61-fold and 1.69-fold higher levels than those with the no NLS mCherry and Cas9ΔNLS proteins, respectively, and these differences were statistically significance (P values<0.05). We thus visually corroborated that all NLSs used effectively promote the delivery of the cargo to the nucleus of C. reinhardtiiin vivo.
Fig. 4.

Visual confirmation of localization using NLS-bearing mCherry reporters and Cas9 variants, and in vitro cleavage assay of NLS-fused Cas9 variants. (A) Design and preparation of mCherry reporters with two different NLS fusion positions, as indicated. mCherry without NLS was identically used in the previous assay (Fig. 2 C–E). (B) Proper expression of the reporters was confirmed by western blot. GroEL: an endogenous control. Circled numbers denote the reporter variants according to the assignments in Fig. 4A. (C) Confocal microscopy-based localization assay using various mCherry variants. (Scale bar, 5 μm.) (D) Confocal microscopy-based localization assay using various Alexa 488-labeled Cas9 variants. (Scale bar, 5 μm.) (C and D) Cells were simultaneously stained with DAPI to detect the nucleus. (E) Box and whisker plots show the fluorescence ratio (as a localization measure) derived from four biological replicates and quantified using ImageJ software. The median (the center line) ± whiskers (1.5 × the interquartile range of the lower and upper quartiles) are shown. Student’s t test was performed. The fold-changes are presented with the statistical significance. The region of interest for the quantification is indicated in the inset. (F) Expected cut site and lengths of the resulting cleavage fragments when PCR-amplified MAA7 (Cre03.g161400) gene of C. reinhardtii is subjected to in vitro cleavage. cCas9 indicates a commercial Cas9. cPAM: complementary protospacer adjacent motif; U.D.: uncleaved DNA (i.e., intact PCR product); and C.F.: cleavage fragment. (G) Representative cleavage result using different molecular combinations at the end of the assay (after 90 min). No cleavage was evident when one of the essential components was missing (for cases I, II, and III). M: marker; N.A.: not applicable. (H) Kinetic analysis of in vitro cleavage performance. Two biological replicates were tested. Uncleaved fraction (%) was analyzed from the gel images in SI Appendix, Fig. S12 using ImageJ software. The data were fitted based on one phase exponential decay model. The Arabic numbers indicate the Cas9 variants according to the numbering in Fig. 4G.
Second, we performed an in vitro cleavage assay of the previously described Cas9 variants (Fig. 3A), targeting a PCR-amplified MAA7 (tryptophan synthase beta subunit; Cre03.g161400) gene in C. reinhardtii, an obligate tryptophan auxotrophic marker gene (3)—given the subsequent in vivo application (vide infra). To enable the Cas9 effectors to recognize the target, a chimeric single guide RNA (sgRNA) was transcribed in vitro (39), and combined with each Cas9 variant, resulting in the formation of various RNP complexes (Fig. 4F). Within 90 min, the target gene was completely cleaved into two DNA fragments of the expected lengths by regular recognition of the protospacer adjacent motif (PAM) and dissection of the proximity to the site in the target molecule (Fig. 4 F and G), demonstrating the normal function of the NLS-fused programmable nucleases, including a commercial Cas9 protein (as a control; see Materials and Methods for details). We also conducted a time-lapse assay of the DNA cleavage performance to observe the kinetics of the nuclease variants (Fig. 4H and SI Appendix, Fig. S12). We found that fusion of any NLS did not make a notable difference to the cleavage kinetics of our Cas9 proteins. Even though the commercial Cas9 showed the fastest cleavage activity, the reaction was completed in no more than 60 min for all variants. Considering the general incubation time of gene editing for algae (i.e., over 12 h) (3, 18), the kinetic difference within 60 min would be negligible and may not significantly influence the practical gene-editing frequency and efficiency in vivo. In summary, the use of the NLS candidates enabled the delivery of the cargoes (e.g., mCherry and Cas9) to the nucleus in vivo (confirmed by confocal analysis; Fig. 4 C and D) without compromising the essential recognition- and cleavage-related functions (confirmed by in vitro cleavage assay; Fig. 4 G and H).
Phytoinfection-Inspired Strategy Doubles the Mutagenesis Frequency in C. reinhardtii.
Based on the preceding multilateral characterization of the NLS candidates, we introduced the Cas9 variants into the real-world application of gene editing in C. reinhardtii, targeting the MAA7 gene (Fig. 5A and SI Appendix, Fig. S13A). As no DNA template was applied, libraries of nonhomologous end joining (NHEJ)-mediated null mutant strains were constructed, allowing the frequency of gene editing by the respective Cas9 variants to be assessed as a direct result of NLS-influenced nuclear accessibility, without confounding factors such as homology-directed repair efficiency (5, 40). The MAA7 knockout mutant needs L-Trp feeding for survival and simultaneously confers tolerance to a toxic compound, 5-fluoroindole (FI), on the transgenic strains, which enabled antibiotic-independent, auxotrophic selection (Fig. 5B and SI Appendix, Fig. S13B) (3). For simplicity, the groups of strains generated by treatment with the respective Cas9 variants (Fig. 3A) were named DN-, DC-, EN-, EC-, SN-, and SC-ΔMAA7.
Fig. 5.

In vivo application of Cas9 variants for algal mutagenesis and investigation of the genetic variance as a result of NHEJ. (A) Representative plate images obtained from the mutagenesis of C. reinhardtii by each Cas9 variant. (B) Simplified description of a strain selection strategy based on tryptophan auxotrophy, when the MAA7 gene is disrupted. The synthesis of the proteinogenic amino acid, L-Trp, from intracellularly synthesized indole is impaired by ΔMAA7. It also deprives cells of the ability to synthesize the cytotoxic, nonproteinogenic amino acid, 5-Fluoro-L-Trp, which is produced when 5-fluoroindole is supplied extracellularly. (C) Mutagenesis frequencies from two scoring methods, namely the number of colony formations and short-read deep sequencing (SR deep seq.), are compared. The absence and presence of L-Trp and 5-FI in each assay are indicated below two vertical axes. The mutation frequencies from the SR deep seq. were corrected with the error rate from the WT sample (SI Appendix, Table S2) and are presented as bars. The individual colony count data are shown as dots with biological triplicates. Floating bars represent the mean. Student’s t test was performed on the frequencies from the colony counts. The fold-changes are presented together with the statistical significance. The mutagenesis frequency is further calibrated with the mutagenesis efficiency calculated from the Sanger sequencing results (Table 2). (D) Exemplified Sanger sequencing data processing is shown (Dataset S2). In: insertion; Del: deletion; Sub: substitution; M: marker; PAM: PAM; and N.A.: not applicable. The nomenclature of the mutant library follows the previous convention, while the numbering of each strain follows the order of random selection for Sanger sequencing. (E) Statistics of genetic variation resulting from NHEJ. CI: complex indel; TN: true negative, genetically identical to WT; NS: not sequenced, including sequencing errors and unamplified cases; and ME: mutagenesis efficiency (%). (F) Length variation is shown with normalized counts (%). The numbers of Sanger sequenced strains are given.
First, the gene-editing frequency was intuitively estimated by the number of colonies formed on auxotrophic solid media supplemented with L-Trp and 5-FI (Fig. 5 A and C, and SI Appendix, Fig. S13A). In good agreement with the previous in vitro assay results, mutagenesis using the Cas9 variants associated with VirD2O generated the first- and second-largest numbers of individual colonies — on average, 230.3 and 176.3 colonies from DN and DC Cas9 treatment, respectively (n = 3) (Fig. 5C). In particular, DN Cas9-based gene editing spawned 2.39-fold more colonies than SN Cas9-based mutagenesis (96.3 colonies on average; n = 3)—the lowest colony-forming Cas9 variant—with statistical significance (P < 0.0016). We noticed a similar quantitative difference pattern to that in the previous in vitro ELISA and MST analyses on the NLS species and the recombinant CrImpα (Fig. 3 C and D).
In parallel with colony counting, a short-range PCR followed by SR deep seq. (41) was performed to recheck the NLS-dependent gene-editing frequency (Fig. 5C and SI Appendix, Table S2). In this case, no 5-FI was provided to keep the wild-type (WT) alive to obtain unbiased outcomes. The results reproducibly showed that ΔMAA7 mutants were most frequently produced by mutagenesis with the VirD2O-associated Cas9 variants, while DC and DN Cas9 treatments were the first (6.24 × 10−4) and second (3.84 × 10−4) most frequent, respectively. Although there was a ranking discrepancy between the frequency outcomes obtained by two different approaches according to the fusion position on the cargo, we found that the VirD2O NLS significantly increased the frequency of gene editing compared to the other NLSs. Moreover, VirE2O was observed by both quantification methods to show some advantage in increasing the gene-editing frequency over the typically used SV40TAg NLS, as expected from the previous in vitro analysis (Fig. 3C). However, the difference in the colony counting assay was not found to be statistically significant, and, even in deep sequencing, a change in ranking was evident depending on the tag position of the NLSs for EC Cas9- and SN Cas9-treated groups (Fig. 5C), all of which made it difficult to assess the general superiority of the VirE2O NLS over the SV40TAg NLS in terms of promoting nuclear localization in vivo.
To derive a mutagenesis efficiency (ME) considering gene-editing frequency based on the number of colonies, from which the potential false-positive frequency is excluded—termed the targeted mutagenesis frequency—we further genotyped the colonies on the auxotrophic agarized media using PCR followed by Sanger sequencing that enabled comparably long reads (Fig. 5D and SI Appendix, Fig. S14 A–F) and compared the outcomes with those obtained from SR deep seq. (Table 2). For this purpose, approximately a quarter of the total algal colonies from the respective ΔMAA7 libraries were randomly selected and Sanger sequenced (Fig. 5D), and the sequence modifications were categorized (SI Appendix, Data S2) into insertions (In), deletions (Del), substitutions (Sub), and complex indels (CI; also known as complex local rearrangements) (41) (Fig. 5E).
Table 2.
Performance on the targeted mutagenesis frequency of each Cas9 variant in C. reinhardtii gene editing
| Total number of apparent mutants from three replicates (Rep.) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Initial pool size | Cas9 variants | Rep. 1 | Rep. 2 | Rep. 3 | Total | Mean | The number of genotyped mutants (genotyping method used) | Apparent mutagenesis frequency* | Mutagenesis efficiency (%)† | Targeted mutagenesis frequency (TMF)‡ | Average of TMF from the same NLS affiliation |
| 2.0 × 107 | cCas9§ | 96 | - | - | 96 | 96¶ |
24 (Sanger sequencing)# |
0.48 × 10–5 | 95.83 | 0.46 × 10–5 | 0.46 × 10–5 |
| 2.0 × 107 | DN Cas9 | 251 | 242 | 198 | 691 | 230.3 |
166 (Sanger sequencing)# |
1.15 × 10–5 | 96.99 | 1.12 × 10–5 | 0.99 × 10–5 |
| 2.0 × 107 | DC Cas9 | 170 | 197 | 162 | 529 | 176.3 |
127 (Sanger sequencing)# |
0.88 × 10–5 | 96.85 | 0.85 × 10–5 | |
| 2.0 × 107 | EN Cas9 | 113 | 101 | 97 | 311 | 103.7 |
75 (Sanger sequencing)# |
0.52 × 10–5 | 93.33 | 0.49 × 10–5 | 0.53 × 10–5 |
| 2.0 × 107 | EC Cas9 | 115 | 129 | 95 | 339 | 113.0 |
81 (Sanger sequencing)# |
0.57 × 10–5 | 98.77 | 0.56 × 10–5 | |
| 2.0 × 107 | SN Cas9 | 109 | 90 | 90 | 289 | 96.33 |
68 (Sanger sequencing)# |
0.48 × 10–5 | 97.06 | 0.46 × 10–5 | 0.48 × 10–5 |
| 2.0 × 107 | SC Cas9 | 122 | 99 | 77 | 298 | 99.33 |
72 (Sanger sequencing)# |
0.50 × 10–5 | 98.61 | 0.49 × 10–5 | |
*Apparent mutagenesis frequency was calculated as the mean number of apparent mutants from three replicates over the size of the initial pool (3).
†Mutagenesis efficiency (%) was estimated as the percentage of the number of null mutants confirmed by genotyping out of the total number of genotyped (Sanger sequenced) mutants, including potential true negative (TN; genetically identical to WT) and unsequenced (NS) cases.
‡Targeted mutagenesis efficiency was determined by the mean number of null mutants confirmed by genotyping over the size of the initial pool. Therefore, the targeted mutagenesis efficiency can be also derived by multiplying the apparent mutagenesis frequency by the mutagenesis efficiency (%), and it can be interpreted as an index of the comprehensive mutagenesis performance of each Cas9 variant.
§A commercial Cas9 (see Materials and Methods for details) was used as a control. No replicate was performed.
¶As there was no replicate, this value has no statistical meaning.
#Approximately a quarter of the colonies from three biological replicates were randomly selected and genotyped (Sanger sequenced).
Of the variations, deletions were the most common, consistent with the previous SR deep seq. outcomes (SI Appendix, Table S2). Based on the classification, the ME of each treatment was calculated by excluding true negative (TN; genetically identical to WT) and not sequenced (NS; albeit encompassed in the total statistical population for rigorous analysis) cases (Fig. 5E). As all groups generally showed high efficiencies — even the lowest case of EN Cas9 was over 93%, there was no dramatic discrepancy between apparent (i.e., derived only based on the number of colony appearances) and targeted mutagenesis frequencies (i.e., derived by further considering the mutagenesis efficiency), with the previous ranking of NLSs by increasing colony formation largely maintained (Table 2). In summary, although the targeted gene-editing frequencies estimated from the two genotyping methods differed by ca. 1.7 orders of magnitude in quantitative terms (i.e., VirD2O NLS: 5.04 × 10−4 and 0.99 × 10−5; VirE2O NLS: 3.30 × 10−4 and 0.53 × 10−5; SV40TAg NLS: 2.42 × 10−4 and 0.48 × 10−5 on average; the former from SR deep seq. and the latter from Sanger sequencing), the frequency induction ranking was almost the same (Fig. 5C, Table 2, and SI Appendix, Table S2), with VirD2O reproducibly yielding the most frequent mutagenesis with statistical significance, followed by the VirE2O and SV40TAg NLS peptides. Notably, we found that the VirD2O NLS showed an even higher in vivo gene-editing frequency than commercial Cas9 (Table 2), whose C-terminus is known to be fused to the SV40TAg NLS, which showed the best kinetic performance in vitro (Fig. 4H), strongly suggesting not only that NLS fusion does not disturb the function of the nuclease in vivo but also that the high-affinity VirD2O NLS is indeed effective in inducing high-frequency gene editing despite the possible kinetic disadvantage.
Sanger sequencing also allowed the length change of each mutant gene to be observed (Fig. 5F), which merits further investigation (41). We found that the respective Cas9 variants induced a wide variety of length alterations, from unexpectedly large insertions (e.g., ~700 bp) to deletions of tens of bp via NHEJ. Intriguingly, −3 and −6 bp variations were the first- and second-most frequent for all mutagenesis groups, in good agreement with the result of a previous study on the NHEJ pattern of C. reinhardtii (3).
Cross-Species Versatility.
Chlorella Sp., the industrial microalgal genera, are promising organisms for various future valorization applications (42). Like other species, however, the programmable gene editing suffers from the limited efficiency of the conventional Cas9 system (43). Thus, using Chlorella Sp. HS2 (44), we further extended our scope to validate whether the phytopathogenic NLS-fused Cas9 could contribute to increasing the frequency of programmable gene editing in an industrial alga with its potential cross-species versatility. First, to forecast the applicability of the NLS-mediated strategy to this species, we predicted the structure of the potential Impα protein (Gene ID: Hscell_00006266) using AlphaFold (Fig. 6A) (33). Based on the sequence and structural homologies (SI Appendix, Fig. S15 A–C), we evaluated the performance of Cas9 variants of DN Cas9, SN Cas9, and cCas9 in gene editing while also targeting the putative MAA7 gene in Chlorella Sp. HS2 (Gene ID: HScell_00004316) in vivo. In the colony formation assay, DN Cas9 treatment led to producing the highest number of survived cells on the selective auxotrophic media apparently (Fig. 6B and SI Appendix, Fig. S16D and Table S3). Sanger sequencing was then conducted to estimate the mutagenesis efficiency (SI Appendix, Fig. S15 E–G), which was 46.88%, 59.38%, and 50.00% for the DN Cas9-, SN Cas9-, and cCas9-treated groups, respectively (SI Appendix, Table S3). Taking colony number and mutagenesis efficiency together, DN Cas9 treatment showed significantly improved targeted mutagenesis frequency (3.60 × 10−4 on average), which is 2.07- and 22.3-fold higher than SN Cas9 and cCas9 treatment, respectively (Fig. 6C). In parallel, SR deep seq. was also performed (SI Appendix, Table S4). The result reproducibly demonstrated that the DN Cas9-treated group triggered the most frequent targeted mutagenesis compared to the other groups, although the fold differences between the experimental groups differ from those of the Sanger sequencing-based assay (Fig. 6C).
Fig. 6.
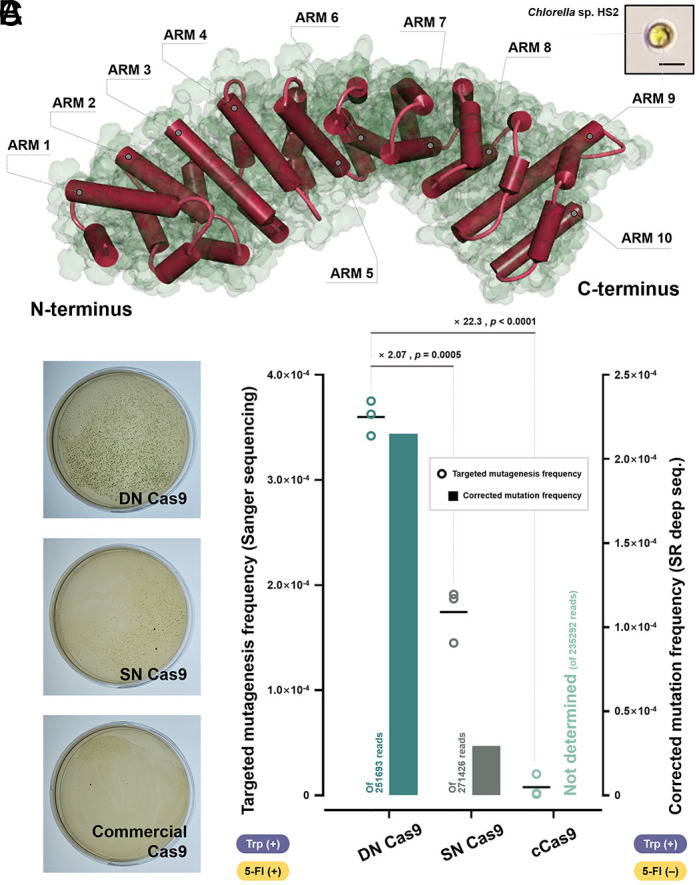
Validation of cross-species versatility using Chlorella Sp. HS2. (A) The three-dimensional structure of the putative Impα (Hscell_00006266) in Chlorella Sp. HS2 was predicted using AlphaFold. The structural homology provided a rationale for applying the NLS-mediated delivery enhancement strategy. Armadillo repeat motifs (Arms) in the predicted structure are presented. N and C termini are indicated. A microscopic image of Chlorella Sp. HS2 is shown at the Upper Right. (Scale bar, 3 μm.) (B) Representative plate images (from replicate 1 in SI Appendix, Table S3) obtained from the mutagenesis of Chlorella Sp. HS2 targeting the MAA7 gene by each Cas9 variant. (C) Mutagenesis frequencies from the targeted mutagenesis frequency derived from colony formation followed by Sanger sequencing and SR deep seq. are compared. The absence and presence of L-Trp and 5-FI in each assay are indicated below two vertical axes. The mutation frequencies from the SR deep seq. were corrected with the error rate from the WT sample (SI Appendix, Table S4) and are presented as bars. The individual targeted mutagenesis frequency data are shown as dots with technical triplicates. Floating bars represent the mean. Student’s t test was performed on the targeted mutagenesis frequencies. The fold-changes are presented together with the statistical significance.
On balance, we found that the VirD2O NLS confers a high affinity to the nucleus-localized protein payload for Impα of microalgae, C. reinhardtii and Chlorella Sp. HS2, thereby increasing the frequency of the resulting NHEJ-mediated genome modification by more than twofold compared with the conventional SV40TAg NLS (based on the data from the colony formation followed by Sanger sequencing) without compensating for the essential role of the programmable nuclease. We thus confirmed that the plant–pathogen interaction-inspired gene-editing strategy would be effective and versatile in algal mutagenesis by addressing the key issue of low gene-editing frequency due to low transport of programmable nucleases into the nucleus.
Discussion
Cas9 is expected to provide a breakthrough in realizing the potential of microalgae as CO2 scavengers by enabling targeted gene manipulation. To overcome the low applicability in microalgae, we proposed that the use of A. tumefaciens–derived NLSs as delivery enhancers of Cas9 would provide a promising answer. For all the in vitro and in vivo experiments, VirD2O exhibited the highest fidelity to the algal nuclear adaptor protein among the NLSs tested, helped the protein payload enter the nucleus most efficiently in the competitive nuclear-peripheral environment, and ultimately produced the most frequent mutation, agreeing in principle with previously reported results related to Impα in other plants (14, 30). The high affinity of the VirD2O NLS for CrImpα can be explained by the simultaneous exertion of the high NLS strength (Table 1) and the tight dimer formation in an antiparallel bipartite joining manner, all of which confers high interactivity with plant-derived Impα proteins (30). In contrast, although the VirE2O NLS is also a plant-specific bipartite NLS with the same high NLS strength score (14, 30), the in vivo performance was inferior to that of the VirD2O NLS. This is likely because the VirE2O NLS has a comparatively short linker peptide (Table 1 and SI Appendix, Table S2) between the major and minor BSs, making it difficult to induce a stable, two-point binding to the adaptor protein (30). Although the typical SV40TAg NLS is a monopartite NLS, it also showed a passable affinity for the algal Impα, consistent with the compatibility of SV40TAg NLS-fused Cas9 in C. reinhardtii (3, 5, 17, 18). The cross-domain versatility of SV40TAg NLS may be attributed to its highly concentrated positive charge within a single binding cluster (Table 1), contributing to the strong unilateral binding of NLS to the major NLS binding pocket of Impα, which is highly conserved in different organisms (SI Appendix, Figs. S2 B and S4 B). Even so, given that the bipartite NLSs bound more strongly with the Impα protein than the monopartite NLS in this study, it is likely that the stable configuration formed by properly distributed positive charge throughout the major and minor BSs, resulting in the formation of two-point binding, would increase the binding affinity more effectively than a single-point dimerization generated by the concentrated positive charge within a sole cluster, provided that the NLS strengths are equal. To further investigate the contribution of each cluster in the bipartite NLS to CrImpα binding, we prepared additional N-terminally NLS-fused Cas9 variants (SI Appendix, Fig. S16A) and tested their binding affinities using ELISA (SI Appendix, Fig. S16 B–D). Prior to the quantitative analysis, their binding to CrImpα was explored using co-IP (SI Appendix, Fig. S17). As a result, DK1AR2AN Cas9, in which two positively charged residues in the minor BS were each substituted with alanine, showed an impaired KD value (13.29 nM). On the other hand, DR15AN Cas9, in which the first single positively charged residue in the major BS was replaced by alanine, showed a slightly more impaired KD value (15.20 nM) compared to the DN Cas9 (4.785 nM; Fig. 3). These results imply that the major BS residue in the NLS has a higher binding contribution than the minor BS residue. As a combined case, DK1AR2AR15AN Cas9 showed a synergistically reduced affinity (42.60 nM) compared to the previous sets. Although three positively charged residues were incapacitated, the binding affinity was not completely abolished. These results suggest that the three residues contribute somewhat to binding but may not be the irreplaceable residues for the function of the NLS.
As an extension of the NLS assays, a version with increased cumulative charge in the major BS (DR15KG17KL18KN Cas9; SI Appendix, Fig. S16E and http://www.pnas.org/lookup/doi/10.1073/pnas.2415072122#supplementary-materialsTable S5) was tested in an attempt to improve the binding affinity of the VirD2O NLS, but was not quite effective (KD = 6.416 nM). This implies that multiple factors should be carefully considered in addition to cumulative charge when designing an artificial NLS. The binding affinity of a putative endogenous NLS in C. reinhardtii, the CrPAP1 NLS (SI Appendix, Supplementary Note 3 for details) (45, 46), was also evaluated as a potential alternative to the VirD2O NLS as a delivery enhancer. However, the PN Cas9 showed an inferior binding (KD = 17.07 nM; SI Appendix, Fig. S16F) compared to the DN Cas9. Albeit based on limited information, this result supports the idea that pathogenic NLSs may be useful in engineering applications compared to endogenous NLSs. Finally, the nopaline-type NLSs, DNopN NLS and ENopN NLS, were also wet-tested in parallel as part of the thorough inspection, which resulted in the KD values of 7.875 nM and 18.00 nM, respectively (SI Appendix, Fig. S16 G and H). Namely, NLSs derived from nopaline-type A. tumefaciens showed weaker binding to CrImpα than those from octopine-type. Given the reasonable concordance of the quantitative order derived from previous binding affinity in vitro (Fig. 3C) and mutagenesis frequency in vivo (Fig. 5C and Table 2) assays, the VirD2O NLS may have an advantage over the other NLSs tested in increasing nuclear accessibility.
When examining the mutagenesis frequency as a result of gene editing, we found that both the error-corrected frequency derived from SR deep seq. (Figs. 5 C and 6 C and SI Appendix, Tables S2 and S4) — a method relied upon by many researchers (41) — and the targeted mutagenesis frequency derived from colony PCR followed by Sanger sequencing (Fig. 6C, Table 2 and SI Appendix, Fig. S14 A–F and Table S3) — the method used as the gold standard for validation (47) — broadly followed the same trend, although some perturbations were observed depending on the position of the NLS attachment (Fig. 5C). Meanwhile, for the C. reinhardtii application, the results of the two approaches differed by orders of magnitude (Table 2 and SI Appendix, Table S2) — while maintaining the hierarchy of the binding affinity according to the NLS species. However, the values are likely to be sufficiently reconciled by the survival rate in plating and 5-FI toxicity in the colony formation assay (Fig. 5 B and C). On the other hand, for the Chlorella application, the mutagenesis frequencies from both assays were similar by and large with no order of magnitude difference (Fig. 6C). Rather, the frequency results from colony formation-based estimation showed slightly higher values for all treatment groups compared to those from the SR deep seq. This may be due to the high tolerance of the Chlorella to abiotic stress factors (44), such as selection markers, which interfered with a sophisticated selection and caused a large gap between the number of apparent colonies and true null mutants (SI Appendix, Table S3). It is noteworthy that while SN Cas9, which is expected to have the same NLS as cCas9, was functional in Chlorella Sp. HS2, cCas9 was barely functional. This will require future investigation, which may provide a reason for the often frustrated applications of Cas9 in Chlorella spp. so far and a clue to improve the efficiency of Cas9 in this genus (43).
The advantages of this phytoinfection-inspired strategy in facilitating nuclear localization of Cas9 and thus increasing mutation frequency are i) the applicability to both vector-driven and RNP-based systems (SI Appendix, Fig. S1); and ii) the possibility of the precise targeted delivery of the nuclease by hijacking the in situ sophisticated nuclear import mechanism. This approach can be functionally incorporated into various state-of-the-art nucleases, which, despite the trend toward miniaturization, still suffer from low nuclear delivery (48). In addition, as its practicality was confirmed in the representative model (i.e., C. reinhardtii) and industrial (i.e., Chlorella Sp. HS2) alga, it is likely to be compatible with several commercially viable algae, including Euglena (23), Schizochytrium (24), and Haematococcus (25), all of which are known to be susceptible to Agrobacterium infection. Based on the sequence homology of the receptor of the phytovirulence-associated NLS (i.e., Impα in organisms), it seems to be further exertable to other highlighted algae, such as Nannochloropsis and Phaeodactylum, and possibly to other plants and fungi based on the high conservation of Impα across different phylogenetic groups (SI Appendix, Figs. S2 C and S4 B). Overall, we believe that this phytoinfection-inspired, host-specificity-focused nuclear localization augmentation strategy would open a possibility for realizing diverse applications of microalgae (49), based on emerging genetic engineering technologies, such as synthetic biology (2), by providing a powerful platform tool that helps address the fundamental challenge of programmable mutagenesis.
Materials and methods
Strain Availability and Culture Conditions.
C. reinhardtii WT strains were obtained from the Chlamydomonas Resource Center (University of Minnesota, USA). All mutant strains of C. reinhardtii analyzed in this study share the same parental background with C. reinhardtii CC-124. Chlorella Sp. HS2 was obtained from the algae collection of Korea Research Institute of Bioscience and Biotechnology. E. coli WT strains were purchased from Real Biotech Corporation (Taiwan). Details of culture conditions are described in SI Appendix, Method S1.
In silico Docking Simulation and Pose Screening.
Gametolysin Preparation and Cell Wall Attenuation Test.
Gametolysin (GL) was prepared according to the “Autolysin Prep & Assay protocol” from the Chlamydomonas Resource Center (University of Minnesota, USA). See SI Appendix, Method S2 for details. The experimental procedure to test the effect of different reagents on cell wall weakening coupled with plasma membrane-lysing Triton X-100 (Sigma-Aldrich, USA) treatment is described in SI Appendix, Method S3.
Construction of Expression Vectors.
For the preparation of the recombinant adopter protein, CrImpαΔIBB, the coding sequence of CrIPA1 (Cre04.g215850) was obtained from the Phytozome database. A coding sequence of CrImpαΔIBB (498 amino acids; 1,494 bp) was amplified from a C. reinhardtii CC-124 cDNA library constructed according to the methods, as described in SI Appendix, Methods S4 and S5. All plasmids encoding Cas9 variants designed in this study were constructed using pET-NLS-Cas9-6 × His (Addgene #62934) (50) and pET-Cas9-NLS-6 × His (Addgene #62933) (50) as backbones with the primer sets listed (SI Appendix, Table S1) and appropriate enzymes, as described in SI Appendix, Method S6. To construct mCherry reporters, the mCherry coding sequence from the pA-RFP-rG2 vector (51) (Addgene #188969; kindly provided by Dr. Seung-Goo Lee) was used as a template. The subsequent procedure was conducted using appropriate primer sets (SI Appendix, Table S1) and enzymes as described in SI Appendix, Method S6.
Preparation and Purification of the Recombinant Proteins.
All plasmids were transformed into E. coli DH5α competent cells for plasmid amplification and E. coli BL21(DE3) competent cells for protein expression. The expression of the respective proteins was induced with 0.4 mM of isopropyl-β-D-thiogalactoside (IPTG) (LPS Solution, South Korea) as described in SI Appendix, Method S7. For protein extraction and purification, a VCX 750 sonicator (Sonics, USA) and appropriate affinity-based methods were used, respectively. Details are given in SI Appendix, Method S8.
Acquirement of Confocal Images and Quantification of the Localization.
The protocol for Cas9 labeling with Alexa Fluor® 488 NHS Ester (Succinimidyl Ester) (Invitrogen, USA) was described in SI Appendix, Method S9. Fluorescent protein delivery to algal cells was performed as described in SI Appendix, Method S9. Visual confirmation of subcellular localization of mCherry reporters was performed using a Zeiss LSM 800 confocal microscope (Carl Zeiss, Germany). The fluorescence of mCherry, Alexa 488, chlorophyll a, and DAPI was excited and detected at Ex587/Em610 nm, Ex493/Em517 nm, Ex655/Em667 nm, and Ex359/Em457 nm, respectively. Fluorescence localization was quantified using ImageJ software with respect to the regions of interest, as indicated (Figs. 2 C and F and 4 C and D).
Flow Cytometry.
mCherry reporters and Alexa 488-labeled Cas9 were delivered to algal cells by electroporation as described in SI Appendix, Method S9. To remove excessive, uninternalized mCherry and Alexa 488-labeled Cas9, each sample was vigorously washed thrice with PBS buffer. The fluorescence was measured using a BD FACSAria III flow cytometer (BD Bioscience, USA) and analyzed using FlowJo™ V10 software (BD Bioscience, USA). The samples were excited with a common 488 nm laser and the emission was detected with a 585/42 nm (PE channel) and 530/30 nm (FITC channel) bandpass filter for mCherry and Alexa 488 detection, respectively. The gating strategies are described in SI Appendix, Figs. S6 and S8.
Immunoblotting.
For western blot, SDS-PAGE was initially performed as described (SI Appendix, Method S2). Conventional immunoblotting with appropriate antibodies was then conducted. Details are given in SI Appendix, Method S11.
co-IP.
Anti-FLAG bead-mediated co-IP using total protein samples from E. coli BL21(DE3) was conducted based on the interaction between CrImpαΔIBB and NLS-fused Cas9 variants as described in SI Appendix, Method S12.
ELISA.
ELISA to quantify the KD values between CrImpαΔIBB and the corresponding Cas9 variants in this study was performed as described in SI Appendix, Method S13.
MST.
MST was used to perform affinity measurements using a Monolith NT.115 Pico apparatus (NanoTemper Technologies GmbH, Germany) and purified protein samples (13). The detailed procedure is given in SI Appendix, Method S14.
Design and Preparation of Cas9 RNP Complexes.
Single guide RNA (sgRNA) targeting the C. reinhardtii MAA7 gene (Cre03.g161400) (3) and the Chlorella Sp. HS2 gene (HScell_00004316) were synthesized using the Guide-it™ sgRNA In Vitro Transcription Kit according to the manufacturer’s protocol (Takara, Japan). The respective RNP complexes were prepared with 10 µg of sgRNA and 7.5 µg of the corresponding Cas9 protein by incubation at 37 °C for 30 min and used for in vitro test and in vivo application.
In Vitro Cleavage Assay.
In vitro cleavage assay targeting the PCR-amplified CrMAA7 gene to estimate the kinetic performance of the NLS-fused Cas9 variants was performed as described in SI Appendix, Method S15.
Colony Formation Assay.
RNP-based genome editing of C. reinhardtii and Chlorella Sp. HS2 were performed by electroporation according to a previous study (5) with some modifications as described in detail in SI Appendix, Method S16.
Sanger Sequencing.
PCR-based Sanger sequencing of C. reinhardtii and Chlorella Sp. HS2 strains was performed to estimate mutagenesis efficiency using the conventional method as described in SI Appendix, Method S17.
SR deep seq.
Mutagenesis was also conducted as described in SI Appendix, Method S16. Detailed sample preparation steps are described in SI Appendix, Method S18. The algorithm proposed in a previous study was used for the statistical process of genetic variances (52). The raw frequency elicited by each Cas9 variant was corrected by the error rate derived from the WT sample.
AlphaFold Prediction for Putative Impα Proteins.
Data Plot and Statistical Analysis.
Data visualization and statistical processing were mainly performed using Prism 9 software (GraphPad, USA). The number of replicates for all experiments is indicated in the figure legends. Where necessary, a two-tailed Student’s t test was performed to determine statistical significance. P < 0.05 was considered statistically different. The statistical differences between the affinity values in the ELISA experiment were calculated using the extra-sum-of-squares F test.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Acknowledgments
We would like to thank Dr. Seung-Goo Lee at Korea Research Institute of Bioscience and Biotechnology for providing the pA-RFP-rG2 plasmid for the preparation of the mCherry reporters. This work was supported by the Basic Science Research Program (2021R1C1C100342; Y.J.L.) and the STEAM Project (RS-2024-00459155; H.-S.K.) of the National Research Foundation funded by the Korean government.
Author contributions
Y.J.L. designed research; Y.J.L. and H.-S.K. supervised research; T.T.L., H.I.C., J.W.K., J.-H.Y., Y.H.L., E.J.J., K.K.K., D.-H.C., D.-Y.C., S.-B.P., H.R.Y., J.L., and E.J.S. performed research; T.T.L., H.I.C., J.W.K., J.L., Y.J.L., and H.-S.K. analyzed data; and T.T.L., H.I.C., J.W.K., Y.J.L., and H.-S.K. wrote the paper.
Competing interests
H.S.K., Y.J.L., J.W.K., T.T.L., and H.I.C. are inventors on a patent application related to this work filed by Korea Research Institute of Bioscience and Biotechnology [KR 10-2023-0061795 (filed on 12 May 2023) and PCT/KR2023/006493 (filed on 12 May 2023)].
Footnotes
This article is a PNAS Direct Submission. H.P. is a guest editor invited by the Editorial Board.
Contributor Information
Yong Jae Lee, Email: leeyj@kribb.re.kr.
Hee-Sik Kim, Email: hkim@kribb.re.kr.
Data, Materials, and Software Availability
The flow cytometry data were deposited at the FlowRepository under accession number FR-FCM-Z6Q3 (53). The short-read deep sequencing data were deposited at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under Accession No: PRJNA1013174 (54) and PRJNA1188325 (55). Information on the gene and protein sequence of Chlorella Sp. HS2 is available in the Chlorella sp. HS2 genome browser (http://web.seeders.co.kr/hs2/index.php/chl/browse) (56). The plasmid encoding the DN Cas9 is available for academic use upon proper request to Addgene (Addgene plasmid #232296) (57). All other data are included in the manuscript and SI Appendix.
Supporting Information
References
- 1.Choi H. I., et al. , Augmented CO2 tolerance by expressing a single H+-pump enables microalgal valorization of industrial flue gas. Nat. Commun. 12, 6049 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Long B., et al. , Machine learning-informed and synthetic biology-enabled semi-continuous algal cultivation to unleash renewable fuel productivity. Nat. Commun. 13, 541 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shin S. E., et al. , CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Sci. Rep. 6, 27810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeon S., et al. , Current status and perspectives of genome editing technology for microalgae. Biotechnol. Biofuels 10, 267 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Picariello T., et al. , TIM, a targeted insertional mutagenesis method utilizing CRISPR/Cas9 in Chlamydomonas reinhardtii. PLoS ONE 15, e0232594 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang W., Brueggeman A. J., Horken K. M., Plucinak T. M., Weeks D. P., Successful transient expression of Cas9 and single guide RNA genes in Chlamydomonas reinhardtii. Eukaryot. Cell 13, 1465–1469 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glass Z., Lee M., Li Y., Xu Q., Engineering the delivery system for CRISPR-based genome editing. Trends Biotechnol. 36, 173–185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park R. V., Asbury H., Miller S. M., Modification of a Chlamydomonas reinhardtii CRISPR/Cas9 transformation protocol for use with widely available electroporation equipment. MethodsX 7, 100855 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X., et al. , Rock the nucleus: Significantly enhanced nuclear membrane permeability and gene transfection by plasmonic nanobubble induced nanomechanical transduction. Chem. Commun. (Camb.) 54, 2479–2482 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Ajjawi I., et al. , Lipid production in Nannochloropsis gaditana is doubled by decreasing expression of a single transcriptional regulator. Nat. Biotechnol. 35, 647–652 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Wente S. R., Gatekeepers of the nucleus. Science 288, 1374–1377 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Lu J., et al. , Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Commun. Signal. 19, 60 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith K. M., et al. , Structural basis for importin alpha 3 specificity of W proteins in Hendra and Nipah viruses. Nat. Commun. 9, 3703 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang C. W., Counago R. L., Williams S. J., Boden M., Kobe B., Crystal structure of rice importin-α and structural basis of its interaction with plant-specific nuclear localization signals. Plant Cell 24, 5074–5088 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King F. W., Shtivelman E., Inhibition of nuclear import by the proapoptotic protein CC3. Mol. Cell. Biol. 24, 7091–7101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodel A. E., et al. , Nuclear localization signal receptor affinity correlates with in vivo localization in Saccharomyces cerevisiae. J. Biol. Chem. 281, 23545–23556 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Shin Y. S., et al. , Targeted knockout of phospholipase A2 to increase lipid productivity in Chlamydomonas reinhardtii for biodiesel production. Bioresour. Technol. 271, 368–374 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Baek K., et al. , DNA-free two-gene knockout in Chlamydomonas reinhardtii via CRISPR-Cas9 ribonucleoproteins. Sci. Rep. 6, 30620 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gussow A. B., et al. , Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. Proc. Natl. Acad. Sci. U.S.A. 117, 15193–15199 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rivas S., Genin S., A plethora of virulence strategies hidden behind nuclear targeting of microbial effectors. Front. Plant Sci. 2, 104 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Komorizono R., Sassa Y., Horie M., Makino A., Tomonaga K., Evolutionary selection of the nuclear localization signal in the viral nucleoprotein leads to host adaptation of the genus Orthobornavirus. Viruses 12, 1291 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long J. S., Mistry B., Haslam S. M., Barclay W. S., Host and viral determinants of influenza A virus species specificity. Nat. Rev. Microbiol. 17, 67–81 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Becker I., et al. , Agrobacterium tumefaciens-mediated nuclear transformation of a biotechnologically important microalga-Euglena gracilis. Int. J. Mol. Sci. 22, 6299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng R., et al. , Agrobacterium tumefaciens mediated transformation of marine microalgae Schizochytrium. Microbiol. Res. 167, 179–186 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Kathiresan S., Chandrashekar A., Ravishankar G. A., Sarada R., Agrobacterium-mediated transformation in the green alga Haematococcus pluvialis (Chlorophyceae, Volvocales). J. Phycol. 45, 642–649 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Pratheesh P. T., Vineetha M., Kurup G. M., An efficient protocol for the Agrobacterium-mediated genetic transformation of microalga Chlamydomonas reinhardtii. Mol. Biotechnol. 56, 507–515 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Sharma P. K., Goud V. V., Yamamoto Y., Sahoo L., Efficient Agrobacterium tumefaciens-mediated stable genetic transformation of green microalgae, Chlorella sorokiniana. 3 Biotech 11, 196 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shurvinton C. E., Hodges L., Ream W., A nuclear localization signal and the C-terminal omega sequence in the Agrobacterium tumefaciens VirD2 endonuclease are important for tumor formation. Proc. Natl. Acad. Sci. U.S.A. 89, 11837–11841 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Citovsky V., Zupan J., Warnick D., Zambryski P., Nuclear localization of Agrobacterium VirE2 protein in plant cells. Science 256, 1802–1805 (1992). [DOI] [PubMed] [Google Scholar]
- 30.Chang C. W., Williams S. J., Counago R. M., Kobe B., Structural basis of interaction of bipartite nuclear localization signal from Agrobacterium VirD2 with rice importin-α. Mol. Plant 7, 1061–1064 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Bhattacharjee S., et al. , IMPa-4, an Arabidopsis importin α isoform, is preferentially involved in Agrobacterium-mediated plant transformation. Plant Cell 20, 2661–2680 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dym O., et al. , Crystal structure of the Agrobacterium virulence complex VirE1-VirE2 reveals a flexible protein that can accommodate different partners. Proc. Natl. Acad. Sci. U.S.A. 105, 11170–11175 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldfarb D. S., Corbett A. H., Mason D. A., Harreman M. T., Adam S. A., Importin α: A multipurpose nuclear-transport receptor. Trends Cell. Biol. 14, 505–514 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Miyamoto Y., Yamada K., Yoneda Y., Importin α: A key molecule in nuclear transport and non-transport functions. J. Biochem. 160, 69–75 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Huang Y., Jiang Z., Gao X., Luo P., Jiang X., ARMC subfamily: Structures, functions, evolutions, interactions, and diseases. Front Mol. Biosci. 8, 791597 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan Y., Tao H., He J., Huang S. Y., The HDOCK server for integrated protein-protein docking. Nat. Protoc. 15, 1829–1852 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Zhou J., et al. , Unexploited performance of NLS in the dCas9-VPR-mediated transcriptional activation. ACS Chem. Biol. 18, 1246–1253 (2023). [DOI] [PubMed] [Google Scholar]
- 39.Greiner A., et al. , Targeting of photoreceptor genes in Chlamydomonas reinhardtii via zinc-finger nucleases and CRISPR/Cas9. Plant Cell 29, 2498–2518 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferenczi A., Pyott D. E., Xipnitou A., Molnar A., Efficient targeted DNA editing and replacement in Chlamydomonas reinhardtii using Cpf1 ribonucleoproteins and single-stranded DNA. Proc. Natl. Acad. Sci. U.S.A. 114, 13567–13572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park S. H., et al. , Comprehensive analysis and accurate quantification of unintended large gene modifications induced by CRISPR-Cas9 gene editing. Sci. Adv. 8, eabo7676 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yun J.-H., et al. , Toward a zero-waste microalgal biorefinery: Complete utilization of defatted Chlorella biomass as a sole heterotrophic substrate for Chlorella Sp. HS2 and an improved composite filler. Chem. Eng. J. 480, 147998 (2024). [Google Scholar]
- 43.Kim J. S., Lee S., Cho S., Jung Y., Inducing heritable genomic deletions in APT gene of Chlorella sorokiniana using CRISPR/Cas9. Algal Research 79, 103435 (2024). [Google Scholar]
- 44.Yun J.-H., et al. , Evaluation of the potential of Chlorella Sp. HS2, an algal isolate from a tidal rock pool, as an industrial algal crop under a wide range of abiotic conditions. J. Appl. Phycol. 31, 2245–2258 (2019). [Google Scholar]
- 45.Zimmer S. L., Fei Z., Stern D. B., Genome-based analysis of Chlamydomonas reinhardtii exoribonucleases and poly(A) polymerases predicts unexpected organellar and exosomal features. Genetics 179, 125–136 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kosugi S., Hasebe M., Tomita M., Yanagawa H., Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. U.S.A. 106, 10171–10176 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng C., Fei Z., Xiao P., Methods to improve the accuracy of next-generation sequencing. Front. Bioeng. Biotechnol. 11, 982111 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koch L., CRISPR systems go mini. Nat. Rev. Genet. 22, 690 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Choi H. I., et al. , Reconsidering the potential of direct microalgal biomass utilization as end-products: A review. Renew. Sustainable Ener. Rev. 155, 111930 (2022). [Google Scholar]
- 50.Zuris J. A., et al. , Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 33, 73–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim S. K., et al. , CRISPRi-based programmable logic inverter cascade for antibiotic-free selection and maintenance of multiple plasmids. Nucleic Acids Res. 50, 13155–13171 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hwang G. H., et al. , Web-based design and analysis tools for CRISPR base editing. BMC Bioinform. 19, 542 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi H. I., Lee Y. J., Cell wall permeability of microalgae. FlowRepository. http://flowrepository.org/id/FR-FCM-Z6Q3. Deposited 7 September 2023.
- 54.Lee Y. J., Choi H. I., Genetic variance of C. reinhardtii according to Cas9-mediated NHEJ. NCBI Sequence Read Archive (SRA). http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=PRJNA1013174. Deposited 6 September 2023.
- 55.Lee Y. J., Choi H. I., Genetic variance of Chlorella sp. HS2 according to Cas9-mediated NHEJ. NCBI Sequence Read Archive (SRA). http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=PRJNA1188325. Deposited 20 November 2024.
- 56.Kim H.-S., Lee Y. J., Chlorella HS2 Browser v2. http://web.seeders.co.kr/hs2/index.php/chl/browse. Accessed 24 May 2018.
- 57.Kim H.-S., pET-Octo_VirD2_NLS-Cas9-6xHis. Addgene. https://www.addgene.org/232296. Deposited 6 January 2025.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Data Availability Statement
The flow cytometry data were deposited at the FlowRepository under accession number FR-FCM-Z6Q3 (53). The short-read deep sequencing data were deposited at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under Accession No: PRJNA1013174 (54) and PRJNA1188325 (55). Information on the gene and protein sequence of Chlorella Sp. HS2 is available in the Chlorella sp. HS2 genome browser (http://web.seeders.co.kr/hs2/index.php/chl/browse) (56). The plasmid encoding the DN Cas9 is available for academic use upon proper request to Addgene (Addgene plasmid #232296) (57). All other data are included in the manuscript and SI Appendix.