

Abstract

Lead halide perovskite quantum dots (LHP QDs) CsPbX3 generate immense interest as narrow-band emitters for displays, lasers, and quantum light sources. All QD applications rely on suited engineering of surface capping ligands. The first generation of LHP QDs employed oleic acid/oleyl amine capping and have found only a limited use in photoredox catalysis. These catalysts have been reported to be unstable and decompose over the course of the reaction, thus reducing turnover numbers (TONs) and limiting their synthetic ability. Herein, the impact of eight distinct surface ligands on monodisperse CsPbBr3 QDs is reported, affording a thorough comprehension of their performance in photocatalytic C–H brominations. These efforts yielded QDs operating at extremely low catalyst loadings (<100 ppb) with TONs over 9,000,000 per LHP QD. We emphasize that the optimal catalytic performance requires increased QD surface accessibility without compromising the QD structural and colloidal integrity. Control experiments indicated that well-known photoredox catalysts such as Ir(ppy)3, Ru(bpy)3Cl2, or 4CzlPN are ineffective in the same reaction. Mechanistic studies reveal that the C–Br bond reduction in CH2Br2 is the rate-limiting step and is likely facilitated through interaction with the CsPbBr3 QD surface. This work outlines a holistic approach toward the design of practically useful photocatalysts out of QDs comprising structurally soft QD cores and dynamically bound capping ligands.
Introduction
Colloidal cesium lead halide perovskite quantum dots (LHP QDs) CsPbX3 (X = Cl, Br, I), first introduced in 2015, are of immense interest for applications in displays, photodetectors, solar cells, lasers, and quantum light sources.1 The physical properties of LHP QDs, in particular large extinction coefficients, near unity photoluminescence quantum yields, small exciton binding energies, and shallow trap states, make them compelling as photocatalysts in the service of organic synthesis.2 Furthermore, the LHP QD band gap can be tuned by varying the QD size and composition.1a Photoredox catalysis in organic synthetic methods has been dominated by Ir-, Ru-, and organo-photocatalysts, which typically perform with turnover numbers (TONs) of up to 10,000.3 Yet, for the generation of sustainable processes, even higher TONs while maintaining high turnover frequencies are desirable, resulting in lower catalyst loadings. Metals displaying low carbon footprints are particularly preferable (Fe < Pb < Cu < Ni < 20 kg CO2/kg metal vs Pt group metals ≈3000 kg CO2/kg metal).4 Herein, we report detailed studies of LHP QDs with eight distinct surface capping ligands to elucidate their photochemical performance as well as moisture and solvent stability and provide a blueprint for developing photoredox reactions with LHP QDs. The C–H brominations of alkylarenes, ketones, and β-ketoesters were chosen as model reactions. A salient feature of LHP QDs studied herein is their exceptionally high efficiency with TONs of >9,000,000 per QD at ppb catalyst loading, whereas traditional photocatalysts did not yield any products.5 We attribute this difference to the accessibility of the LHP QD surface, to allow for a photochemical reaction step to proceed efficiently (Figure 1).
Figure 1.

Investigation of LHP
QDs in organic synthesis.  = Cs,
= Cs,  = Pb,
= Pb,  = Br,
= Br,  = starting material, and
= starting material, and  = product. For structures of BRPE, DDAB, and ASC18, see Figure 2.
= product. For structures of BRPE, DDAB, and ASC18, see Figure 2.
Well-performing LHP QD photocatalysts were observed to provide surface accessibility by either of two scenarios: the use of dynamically binding ligands or static ligands, which cover the LHP QD partially. Taken together, this study highlights the unique features and advantages of LHP QDs, which are poised to be harnessed in organic synthesis.
Oleic acid/oleyl amine has been used as a surface ligand system for LHP QDs applied in photoredox catalysis. The reported transformations include the homocoupling of benzyl bromides, alkylation of aldehydes, annellation of benzylidene malononitriles, bromination of electron rich arenes and activated olefins, as well as the bromination of three benzylic substrates in low yield, among others.6 In their application as photocatalysts, these QDs undergo irreversible structural degradation both during storage and over the course of the reaction.2b,7 Furthermore, these LHP QDs have usually been prepared with large size distributions, resulting in a range of different redox potentials and poorly defined catalytic systems.6c,6d,6i We envision that future applications in organic synthesis would significantly benefit from the availability of LHP QDs with narrow size distributions that are stable over an extended time.
Since the discovery of LHP QDs, the prevailing notion has been to seek effective stabilization by high coverage of tightly binding ligands.7,8 Over the course of this study, we have noted that for catalysis to occur more efficiently, ligand-capped LHP QDs need to exhibit a proper balance between stability and surface accessibility.9 This necessary condition can be met in two different ways: LHP QDs with reversibly dissociating surface ligands or, alternatively, nondissociating ligands at a persistent surface coverage.
Results and Discussion
Selective bromination of activated C–H bonds has been a reoccurring challenge in industry and academia.10 We envisioned the use of LHP QDs as photoredox catalysts to provide new and efficient routes for the monobromination of organic substrates. We decided to investigate ethylbenzene (1a) as a model substrate. As a bromine source, CH2Br2 was chosen because it is an inexpensive and abundant commodity chemical produced in 500–9000 tons per year in the United States alone.11 It is less mass intensive than NBS, generally considered inert, and has found industrial applications, e.g., as solvent and water treatment products.12 As a Br transfer reagent, CH2Br2 remains underappreciated.13
Some of us have recently reported sulfobetaine-, phosphocholine-, and phosphoethanolamine-based zwitterionic ligands as well as quaternary ammonium ligands for improved LHP QD stability compared to oleic acid/oleyl amine-capped ones.7,8,14 However, no studies are available that examine the influence of these surface ligands on the photoredox performance of LHP QDs. At the onset of the study, we decided to probe the utility of commercially available ligands with the objective of covering a broad spectrum of different head groups. In five separate experiments, ethylbenzene (1a, 0.60 mmol) and CH2Br2 (1.8 mmol) were dissolved in 1.0 mL anhydrous cyclohexane and OA-QDs (oleic acid/oleyl amine capped), OGPE-QDs (phosphoethanolamine capped), ASC18-QDs (sulfobetaine capped), DDAB-QDs (ammonium capped), or OGPC-QDs (phosphocholine capped) were added as photocatalysts, respectively (Figure 2a). The use of LHP QDs as catalysts at very low loadings would constitute an important contribution to the synthetic methodology. Accordingly, we examined catalyst loadings of 6.4 to 14 × 10–6 mol % (64–140 ppb; for details, see the Supporting Information). The reactions were irradiated with blue light (λmax = 446 nm, 350 W blue-LED photoreactor; for technical details, see the Supporting Information) for 3 h.
Figure 2.
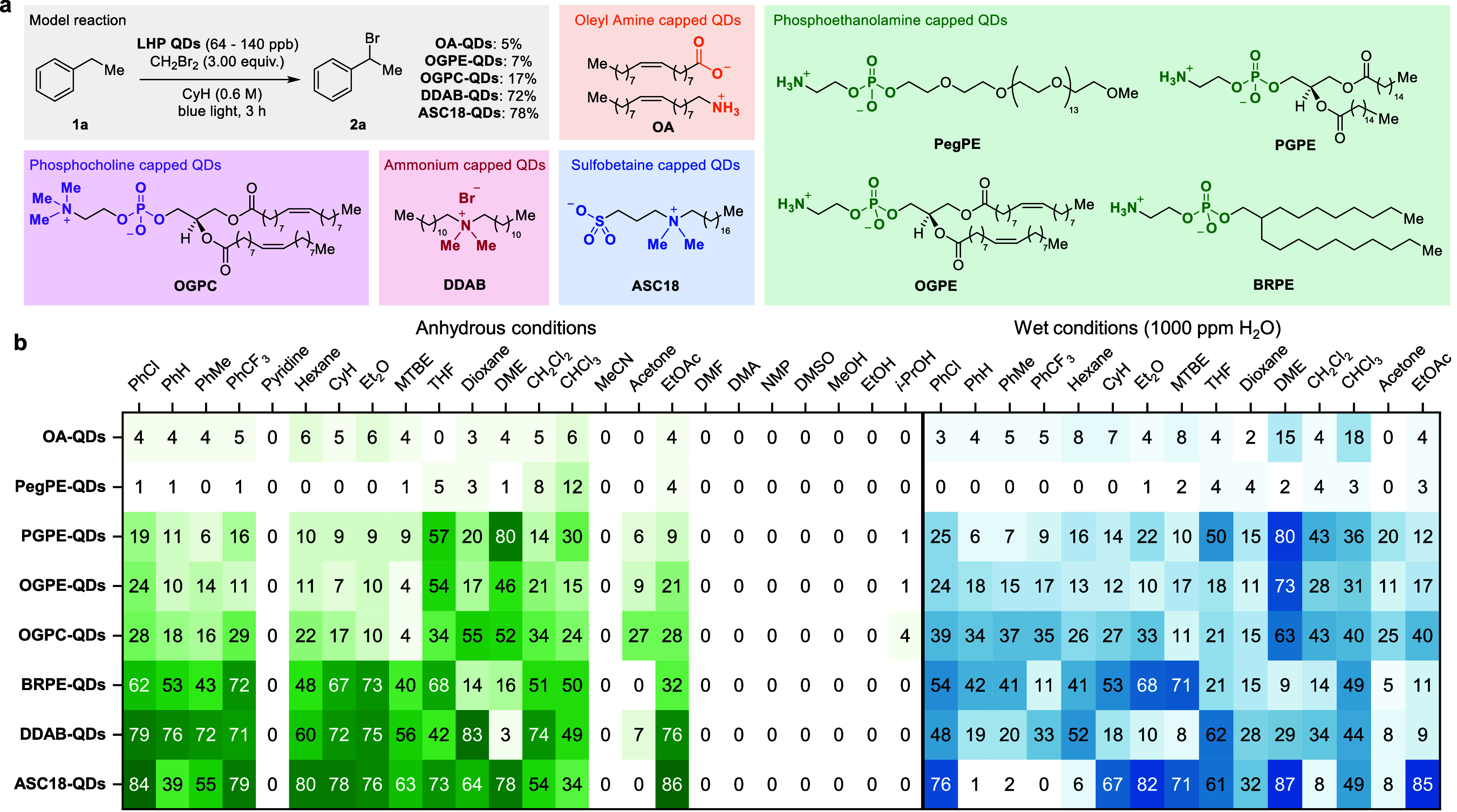
Photoredox performance of LHP QDs. (a) Benzylic bromination of ethylbenzene and structure of the five classes of ligands investigated in this study. (b) The green heat map displays 1H NMR yields with mesitylene as an internal standard for the benzylic bromination of PhEt (1a) under anhydrous conditions in 24 different solvents. The blue heat map displays 1H NMR yields with mesitylene as an internal standard for the benzylic bromination of PhEt (1a) in the presence of 1000 ppm of H2O in 15 different solvents as part of a moisture study.
Intriguingly, α-bromo ethylbenzene (2a) was afforded in a wide range of yields 5% (OA-QDs), 7% (OGPE-QDs), 17% (OGPC-QDs), 72% (DDAB-QDs), and 78% (ASC18-QDs). The fact that the reaction mixtures containing OA-QDs, OGPE-QDs, and OGPC-QDs were not photoluminescent after the reaction can be attributed to irreversible catalyst degradation. Only the reactions containing ASC18-QDs and DDAB-QDs remained photoluminescent. In the absence of either an LHP QD or light, no reaction was observed. Similarly, no conversion of PhEt was detected when only ASC18 or DDAB was employed instead of the corresponding QDs. To benchmark LHP QD performance against photocatalysts which cover a wide range of excited-state reduction potentials, bromination of 1a was attempted with 1.00 mol % of N,N,N,N-tetramethylbenzene-1,4-diamine, Ir(ppy)3, 12-phenyl-12H-benzo[5,6][1,4]thiazino [2,3-b]quinoxaline, Ru(bpy)3Cl2, 4CzlPN, and Eosin Y. None promoted the transformation as only traces (<1%) of 2a were observed in the 1H NMR spectra of the unpurified reaction mixtures with the mass balance being remaining starting material. We were intrigued by the fact that LHP QDs have unique properties that are unmatched by those of established catalysts. Furthermore, the stark differences observed in the initial experiments led us to investigate the effect of ligands on catalyst performance and stability of CsPbBr3 QDs.
Adding to the five different head groups examined in the preliminary experiment, we investigated 2-ammonioethyl (poly(ethylene)glycol) phosphate (PegPE), 2-ammonioethyl (dipalmitoyl-sn-glycero) phosphate (PGPE), and 2-ammonioethyl (2-octyldodecyl) phosphate (BRPE) to identify the influence of different tail groups (saturated, unsaturated, and heteroatom rich). With this selection, we aimed to gain insights into the effect of ligand structure on the integrity of the QD over the course of the reaction, along with its catalyst performance.
LHP QD structural integrity is susceptible to solvent effects.15 Consequently, we studied the influence of 24 organic solvents commonly employed in synthesis. In these experiments, ethylbenzene (0.60 mmol), 64–177 ppb LHP QD (see the Supporting Information for details about QD colloids), and 3.0 equiv of CH2Br2 were added to 1.0 mL of the solvent. After 3 h of blue-light irradiation, the NMR yields of 2a were determined (Figure 2b). The study revealed that sulfobetaine-capped ASC18-QDs performed best in the widest range of solvents (>50% yield in 12 solvents), followed by ammonium-capped DDAB-QDs (>50% yield in 11 solvents) and phosphoethanolamine-capped BRPE-QDs (>50% yield in 7 solvents). For PGPE-QDs (>50% yield in 2 solvents), OGPE-QDs (>50% yield in 1 solvent), OA-QDs (<10% yield in every solvent), and PegPE-QDs (<5% yield in every solvent), poor performance was observed. Very polar solvents like DMF can dissolve LHP QDs which was also observed in our investigations with the notable exceptions of EtOAc, THF, and DME.16
Moisture has generally been considered detrimental for LHP QD stability as water can dissolve CsPbBr3; however, studies investigating the influence of water in ligand-capped LHP QD photocatalysis remain elusive.17 Consequently, we set out to investigate the benzylic bromination of ethylbenzene in the presence of 1000 ppm of H2O (1 μL/1 mL solvent) (Figure 2b). In PhCl and ethers, the added water had minimal influence on the product formation catalyzed by ASC18-QDs and DDAB-QDs (for more details, see the Supporting Information). These examples highlight that water is not necessarily detrimental to QD photoredox catalysis, allowing their use under ambient conditions (vide infra). When considering new avenues for future photocatalyst design for organic synthesis, we conclude that the QD performance can be tuned with surface ligands. Especially, hydrocarbon-based side chains, as seen in BRPE, DDAB, and ASC18, appear to be advantageous.
The observation that PegPE-QDs performed worst among all investigated QDs came as a particular surprise, considering that PegPE-QDs are colloidally stable in the widest range of solvents (polar and apolar).8 Evidently, broad colloidal stability of QDs does not necessarily translate into well-performing photocatalysts. Colloidal instability may be caused by at least two pathways, namely, LHP QD degradation by solvents or antisolvent-induced precipitation of LHP QDs via interactions between surface ligands without altering their core integrity.16 For example, OA-QDs aggregated to bulk material in Et2O and MTBE, which shut down catalytic activity of the QD. By contrast, the use of antisolvents as reaction media for PGPE-QDs, OGPE-QDs, OGPC-QDs, BRPE-QDs, DDAB-QDs, and ASC18-QDs led to turbidity, but the product still formed in up to 86% yield (for details, see the Supporting Information). Surprisingly, this indicates that surface-ligand aggregates of LHP QDs retain catalytic activity as long as their core structure remains intact.
We subsequently focused our study on ASC18-QDs for the photochemical bromination, as they provided the best compromise between stability and activity. We initially investigated the scope of the alkylarene substrates (Figure 3). Guided by the data in Figure 2, and a preliminary screening, we chose PhCl as the solvent for the substrate scope. When PhEt was subjected to the optimized conditions (3.0 equiv of CH2Br2 and 99 ppb ASC18-QDs in 0.6 M PhCl) and irradiated for 3 h, bromide 2a was observed in 84% yield. To highlight the synthetic utility of our transformation, bromination of 1a was conducted on a 10 mmol scale (6.0 M). Gratifyingly, 2a was formed in a 75% yield. The reaction conditions tolerated differently substituted arenes, affording 2b–2j in 49–99% yield.
Figure 3.

Substrate scope. Reaction conditions: substrate (0.60 mmol), CH2Br2 (1.8 mmol), ASC18-QDs (9.9 × 10–6 mol %, 99 ppb) in PhCl (1.0 mL), irradiation in a 350 W photoreactor for 1.5–5 h. aYield obtained by 1H NMR with mesitylene as an internal standard. bConducted on a 10 mmol scale. cConducted without exclusion of air and moisture. The CCDC deposition number for 4i is 2404075.
An ethylparaben-derived substrate yielded 2k in 81%, and bromides 2l and 2m, derived from the active pharmaceutical ingredients ketoprofen and probenecid, were isolated in 67 and 65% yield, respectively. The consistently high performance of 99 ppb ASC18-QDs throughout the reaction scope is remarkable, with TONs reaching as high as 9,650,000 per LHP QD. Additionally, we were able to effect the benzylic bromination reaction without the exclusion of air and moisture with a minimal decrease in yield for 2b and 2c (for details, see the Supporting Information).
The performance of ASC18-QDs prompted us to investigate the α-bromination of ketones under the same conditions next. Acetophenone was successfully converted to α-bromo acetophenone 4a in 73% yield (Figure 3). Other aryl alkyl ketones were also successfully brominated, giving rise to secondary or tertiary bromides 4b–4h in 50–95% yield. Dialkyl ketones were amenable to the reaction conditions, affording 4i–4m in 66 to 81% yield.
From our previous work on photocatalytic cyclopropanation of unactivated olefins with α-bromo-β-ketoesters, we were aware that monobromination of β-ketoesters often suffers from dibrominated side products.10b,18 Consequently, β-ketoesters were also subjected to the reaction conditions (Figure 3). Methyl 2-bromo-4,4-dimethyl-3-oxopentanoate (6a) was isolated in 93% yield. On a larger scale (10 mmol), 5a was brominated in 97% yield without formation of the dibrominated product. The reaction tolerated different ester groups, giving rise to 6b and 6c in 79% and 54% yield, respectively. Methyl 3-oxobutanoate (5d) was brominated in 65% yield. α-Alkyl β-ketoesters were also amenable to the reaction conditions, affording 6e and 6f in 66% and 57% yield, respectively.
When ethyl 3-cyclohexyl-3-oxopropanoate (5g) was subjected to 99 ppb ASC18-QDs and CH2Br2 (3.0 equiv) in PhCl and irradiated for 5 h with blue light, no α-bromo-β-ketoester was obtained. Instead, selective bromination at the γ-position was observed, and 6g was isolated in 75% yield. Bozzelli and co-workers have shown that α-alkyl substitution of a ketone diminishes the α-C–H bond dissociation energy by up to 8 kcal/mol.19 Since γ-brominated products are of interest as synthetic intermediates in drug and agrochemical research, further substrates were investigated.20 γ-Alkyl-β-ketoesters were subjected to the reaction conditions and only γ-bromo-β-ketoesters 6g–6j were isolated in 66–83% yield.
With the substrate scope in hand, we proceeded to investigate whether the LHP QDs change in size and shape over the course of the reaction. Because ASC18-QDs were the most general catalyst in our solvent study, we studied the QDs post-reaction with UV–vis and photoluminescence spectroscopy (Figure 4a). The spectroscopic analysis revealed only minute shifts in the absorbance and photoluminescence maxima (compared to native ASC18-QDs; for a full comparison, see the Supporting Information). This indicates that the average particle size does not significantly change during the reaction.7 When the benzylic bromination was conducted in the presence of 1000 ppm of water in PhCl, a significant red shift was observed in UV–vis (+9 nm) and PL (+8 nm), indicating particle growth.
Figure 4.

Characterization of ASC18-QDs post reaction. (a) UV–vis and photoluminescence (PL) spectra of ASC18-QDs before and after the reaction in PhCl. (b) Scanning transmission electron microscopy (STEM) images of ASC18-QDs in PhCl, dioxane, and EtOAc after the bromination reaction. The unpurified reaction mixture was used to prepare the TEM grids.
To further assess the quality of the LHP QD catalyst at the end of the reaction, we recorded transmission electron microscopy (TEM) images of the residuals from all anhydrous reactions which gave >10% yield catalyzed by ASC18-QDs (for full comparisons including DDAB-QDs and BRPE-QDs, see the Supporting Information). Interestingly, there was no direct correlation between the product yields and QD morphology after the reaction. As an example, ASC18-QDs catalyze the formation of 2a in PhCl, dioxane, and EtOAc in comparable yield (84%, 64%, and 86%, respectively). The STEM images after the reaction showed QDs of varying quality (Figure 4b). While in anhydrous PhCl the size of ASC18-QDs were retained, etching to smaller or sintering to larger particles was observed in dioxane and EtOAc. We conclude that a change in shape during the reaction is not necessarily detrimental to the catalyst’s performance.
A prerequisite for catalysis is the proximity between reactants and catalysts. However, the presence of surface-bound ligands on LHP QDs required for stabilization can have the undesired consequence of hampered accessibility to the surface by the reactants.9c These two opposing conditions (surface coverage for stability vs surface accessibility for reactivity) can be nonetheless satisfied in two scenarios: (1) QDs with nondissociating ligands at some optimal coverage to ensure stability and persistent open sites or (2) QDs with ligands that reversibly dissociate in a process that enables surface access. It was unclear which of these scenarios was operative for the various QDs in the solvent study (Figure 2). We envisioned that both scenarios could be distinguished by diffusion-ordered NMR spectroscopy (DOSY, Figure 5a–c).21
Figure 5.

Investigation of ligand dynamics. (a–c) DOSY traces for DDAB-, ASC18-, and BRPE-capped QDs before and after three washing cycles. The experiments were carried out using a double-stimulated echo sequence with a diffusion time (Δ) of 200 ms and a gradient time (δ) of 4 ms.24 (d) Ratio between the fast and slow components of the biexponential fit outlining the fraction of the bound ligand on the QD. (e) Graphical representation of the dynamic and static scenario.
DOSY allows for the separation of 1H NMR signals of different species based on their diffusion coefficients. The measurement consists of a series of spin echo experiments paired with pulsed field gradients of increasing strength. The application of the gradient pulses results in diffusion-dependent 1H NMR signal decay which can be analyzed.22 For ligand-capped QDs, two distinct species could be differentiated: free ligands in solution displaying fast diffusion (10–9 m2/s) and ligands bound to the LHP QD surface showing slow diffusion (10–11 m2/s).7
DOSY experiments were carried out for the three well-performing ligands with characteristically different headgroups (BRPE, DDAB, and ASC18) to elucidate whether the ligands are dissociating (dynamic vs static). In the experiment, the LHP QDs were freshly prepared with excess ligand, precipitated, centrifuged, and redispersed in deuterated cyclohexane. Acquisition of the first set of NMR data (labeled non-washed) was followed by QD precipitation, centrifugation, and washing (3 times; for details, see the Supporting Information). A second set of NMR data was acquired for comparison (labeled as triple-washed). The acquired data of the non-washed LHP QDs was fitted first by using a two-component version of the Stejskal–Tanner equation (eq 1). All three samples displayed a two-component decay (biexponential fit) of the 1H NMR signal corresponding to the terminal methyl groups of the ligand tails (Figure 5a–c).23
| 1 |
The fast component, which dominates
the signal decay up until a
gradient of 20 G/cm, has a diffusion coefficient on the order of 10–9 m2/s, consistent with the free ligand
in solution below its critical micelle concentration. The slow component
fits a diffusion coefficient of 5 × 10–11 m2/s consistent with the solvo-dynamic radius of an LHP QD.
We define the ratio between the fast and slow components as the ratio
of the free to bound ligand (bound ligand fraction, Figure 5d). The bound ligand fraction
of the non-washed DDAB, ASC18, and BRPE samples was determined to be 52% ( ), 23% (
), 23% ( ), and 54% (
), and 54% ( ), respectively. We concluded that for
the nonwashed case, not all ligands are bound to the QD surface, which
can be attributed to excess ligands used in the LHP QD synthesis.
), respectively. We concluded that for
the nonwashed case, not all ligands are bound to the QD surface, which
can be attributed to excess ligands used in the LHP QD synthesis.
After three washing cycles, the DOSY data of DDAB-QDs and ASC18-QDs still displayed biexponential behavior
with fast and slow diffusing components (triple-washed, Figure 5a,b). As shown in Figure 5d, this corresponds
to bound ligand fractions of 70% for DDAB ( ) and 78% for ASC18 (
) and 78% for ASC18 ( ). As the washing steps removed excess DDAB and ASC18 from the LHP QD solution, the
presence of the free ligand (29% and 22%, respectively) is the result
of the re-establishment of equilibria between bound and free ligands.
This unveils the dynamic nature of DDAB and ASC18 which can generate catalytically active surface sites (Figure 5e). Triple-washed
LHP QDs capped with BRPE show slow monoexponential signal
decay in the DOSY experiment (Figure 5c). With a bound ligand fraction of 100% (
). As the washing steps removed excess DDAB and ASC18 from the LHP QD solution, the
presence of the free ligand (29% and 22%, respectively) is the result
of the re-establishment of equilibria between bound and free ligands.
This unveils the dynamic nature of DDAB and ASC18 which can generate catalytically active surface sites (Figure 5e). Triple-washed
LHP QDs capped with BRPE show slow monoexponential signal
decay in the DOSY experiment (Figure 5c). With a bound ligand fraction of 100% ( ), no ligands are dissociating from the
surface (Figure 5d,e),
which corresponds to scenario one.
), no ligands are dissociating from the
surface (Figure 5d,e),
which corresponds to scenario one.
With these observations, we conclude that high-yielding QDs fall into one of two categories: Triple-washed ASC18-QDs (78% yield) and DDAB-QDs (72% yield) are covered with ligands that are able to generate open surface sites via reversible, dynamic dissociation, which allows for substrates to approach the QD surface. BRPE (67% yield) has persistent ligand coverage that retains active surface sites. In all three cases, the ligand sufficiently stabilizes the LHP QD for catalysis to occur. In contrast, the low-yielding QDs (from Figure 2), OA-QDs, OGPE-QDs, and OGPC-QDs, do not fulfill the stability criteria under the reaction conditions (for details, see the Supporting Information). For PegPE-QDs, we hypothesize that the surface is inaccessible because the oxygen-rich ligand tail interacts with the QD surface, further covering it in the process.
At the onset of our mechanistic investigation, we were intrigued that a range of photoredox catalysts (vide supra) did not promote C–H bromination in our hands. While for the related oleic acid/oleyl amine-capped quantum dots an excited-state reduction potential in the range of −1.15 V vs SHE has been measured (Pt electrode, 50 mM TPAB in 4:1 MeCN/PhMe), numerous reduction potentials for CH2Br2 can be found in the literature.25 Fedurco and co-workers attributed the differences in the reduction potential to solvent effects, which influence the overpotential during the electron transfer from the electrode to CH2Br2. For instance, the authors report a reduction potential of −2.60 V in DMF/0.1 M TBAPF6 vs Fc/Fc+ and −1.05 V in H2O/1 M NaClO4 vs NHE.26 The reduction potential of oleic acid/oleyl amine-capped QDs would lie within the range of investigated photocatalysts. We concluded that LHP QDs may have a unique property for the reaction to proceed. Given the heterogeneous nature of LHP QDs, we hypothesize that CH2Br2 may adsorb onto the CsPbBr3 QD surface to facilitate subsequent C–Br bond cleavage (Figure 6a, Step A and B).6k,27 Conventional homogeneous photocatalysts, however, lack an analogous surface, hampering reductive C–Br bond cleavage.25,26 When Stern–Volmer quenching studies were conducted with ASC18-QDs, the observed quenching of the excited-state QD photocatalyst, albeit weak in intensity, suggests energy or charge transfer from ASC18-QDs to CH2Br2 (Figure 6b).
Figure 6.

Mechanistic studies. (a) Proposed catalytic cycle. (b) Stern–Volmer quenching studies. (c) Reaction order for each reactant. (d) Kinetic isotope effect (KIE) for CD2Br2 vs CH2Br2 and 1a vs 1a–d10.
We determined the reaction order of ASC18-QDs, CH2Br2, and PhEt by measuring the initial rate at varying reactant concentrations (Figure 6c). We found that the reaction is first order in ASC18-QDs, while CH2Br2 has a calculated order of 0.71. To explain the subfirst-order value of the latter, a number of effects can be hypothesized: Surface saturation of ASC18-QDs by CH2Br2 and thermodynamic equilibria in the adsorption of CH2Br2 could alter the reaction rate at varying CH2Br2 concentrations. The rate could further be limited by the diffusion rate of CH2Br2 to the surface of ASC18-QDs.28 Alternatively, with increasing CH2Br2 concentration, the physicochemical properties of the reaction media change, which could expedite partial catalyst decomposition. The reaction order in ethylbenzene is zero.
KIE experiments (Figure 6d) from parallel reactions (initial rate; for details, see the Supporting Information) furthermore revealed a secondary KIE for CD2Br2 (KIE = 1.8), while 1a–d10 displayed a negligible KIE of only 1.05. From these experiments, we concluded that the C–Br bond reduction of CH2Br2 is the rate-determining step.29
Notably, in the course of the KIE study, we also observed the formation of CD2HBr and 1a–d9. This is consistent with a bromomethyl radical abstracting a benzylic H atom in Step C to generate benzyl radical 9. The latter can subsequently proceed through two productive pathways: Br-atom abstraction from CH2Br2 (Step D), which leads to product formation consistent with a radical chain mechanism, or oxidation to a benzyl cation (Step E), which is subsequently trapped by bromide (Step F) and corresponds to a closed catalytic cycle.
Radical quenching experiments were conducted next. TsCN (1.00 equiv) was employed as a radical scavenger, as TEMPO (1.00 equiv) was not tolerated by ASC18-QDs.30 In the experiment, α-cyanoethylbenzene and bromoacetonitrile were observed in the 1H NMR spectrum of the unpurified reaction mixture (for details, see the Supporting Information). These products substantiate the formation of intermediates 7 and 9 in the catalytic cycle. Lastly, we determined the reaction quantum yield for the benzylic bromination of 1a as Φ = 0.011 (for details, see the Supporting Information). This value is consistent with a closed catalytic cycle but does not rule out a radical chain mechanism.
Conclusions
We have developed a novel photocatalytic bromination reaction that allowed us to study the photocatalytic performance of CsPbBr3 QDs. To demonstrate the synthetic utility, a wide variety of functionalized substrates (alkyl benzenes, ketones, and β-ketoesters) were brominated. Interestingly, for certain β-ketoesters, high selectivity for γ-bromination over α is observed. The salient features of the transformation are its air and moisture tolerance and functional group compatibility. Additionally, ligand-capped CsPbBr3 QDs exhibit high activity as photocatalysts with loadings in the <100 ppb range, exceeding the activity of conventional photocatalysts substantively. We hypothesize that CsPbBr3 QDs exhibit surface effects that separate them from established small-molecule photocatalysts. Our comparative study featuring eight different ligand-capped LHP QDs revealed that colloidal stability and quality of the QDs after the reaction are only limited indicators for QD photocatalyst performance. DOSY experiments revealed two possible scenarios for generating catalytically active LHP QDs while maintaining stability: dynamic ligand coverage or use of static ligands at an optimal persistent surface coverage. Within these scenarios, ligand and LHP QD design offers opportunities for the next generation of photocatalysts in synthesis. We expect that the insights we provided in this work will lead to new applications with lead halide perovskite QDs in photoredox catalysis.
Acknowledgments
This publication was created as part of NCCR Catalysis (grant nos. 180544 and 225147), a National Centre of Competence in Research funded by the Swiss National Science Foundation. This work was in part financially supported by the European Union through the Horizon 2020 research and innovation program (grant agreement No. 819740, project SCALE-HALO). The authors are thankful for the access to the Scientific Center for Optical and Electron Microscopy (ScopeM) at ETH Zurich for the use of their facilities and Dr. F. Krumeich for STEM measurements. The authors furthermore thank Dr. M.-O. Ebert, R. Frankenstein, S. Burkhardt, and R. Arnold for support with NMR experiments, Dr. N. Trapp and M. Solar for X-ray crystallographic analysis, as well as C. A. Bärtschi, C. Marro, and H. Benz for the maintenance and construction of the photoreactor (all ETH Zurich). W. M. Amberg is grateful for partial support with funding from the SSCI (Scholarship Fund of the Swiss Chemical Industry).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c17013.
Experimental procedures, quantum dot preparation and characterization, TEM images, UV–vis and photoluminescence spectroscopy, 1H NMR spectra, 13C NMR spectra, IR, and HRMS (PDF)
Author Contributions
§ W.M.A., H.L., and Y.S. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- a Protesescu L.; Yakunin S.; Bodnarchuk M. I.; Krieg F.; Caputo R.; Hendon C. H.; Yang R. X.; Walsh A.; Kovalenko M. V. Nanocrystals of Cesium Lead Halide Perovskites (CsPbX3, X = Cl, Br, and I): Novel Optoelectronic Materials Showing Bright Emission with Wide Color Gamut. Nano Lett. 2015, 15, 3692–3696. 10.1021/nl5048779. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhu H.; Fu Y.; Meng F.; Wu X.; Gong Z.; Ding Q.; Gustafsson M. V.; Trinh M. T.; Jin S.; Zhu X. Y. Lead halide perovskite nanowire lasers with low lasing thresholds and high quality factors. Nat. Mater. 2015, 14, 636–642. 10.1038/nmat4271. [DOI] [PubMed] [Google Scholar]; c Fang Y.; Dong Q.; Shao Y.; Yuan Y.; Huang J. Highly narrowband perovskite single-crystal photodetectors enabled by surface-charge recombination. Nat. Photonics 2015, 9, 679–686. 10.1038/nphoton.2015.156. [DOI] [Google Scholar]; d Gao Y.; Huang C.; Hao C.; Sun S.; Zhang L.; Zhang C.; Duan Z.; Wang K.; Jin Z.; Zhang N.; Kildishev A. V.; Qiu C.-W.; Song Q.; Xiao S. Lead Halide Perovskite Nanostructures for Dynamic Color Display. ACS Nano 2018, 12, 8847–8854. 10.1021/acsnano.8b02425. [DOI] [PubMed] [Google Scholar]; e Rainò G.; Becker M. A.; Bodnarchuk M. I.; Mahrt R. F.; Kovalenko M. V.; Stöferle T. Superfluorescence from lead halide perovskite quantum dot superlattices. Nature 2018, 563, 671–675. 10.1038/s41586-018-0683-0. [DOI] [PubMed] [Google Scholar]; f Becker M. A.; Vaxenburg R.; Nedelcu G.; Sercel P. C.; Shabaev A.; Mehl M. J.; Michopoulos J. G.; Lambrakos S. G.; Bernstein N.; Lyons J. L.; Stöferle T.; Mahrt R. F.; Kovalenko M. V.; Norris D. J.; Rainò G.; Efros A. L. Bright triplet excitons in caesium lead halide perovskites. Nature 2018, 553, 189–193. 10.1038/nature25147. [DOI] [PubMed] [Google Scholar]; g Utzat H.; Sun W.; Kaplan A. E. K.; Krieg F.; Ginterseder M.; Spokoyny B.; Klein N. D.; Shulenberger K. E.; Perkinson C. F.; Kovalenko M. V.; Bawendi M. G. Coherent single-photon emission from colloidal lead halide perovskite quantum dots. Science 2019, 363, 1068–1072. 10.1126/science.aau7392. [DOI] [PubMed] [Google Scholar]; h Dey A.; Ye J.; De A.; Debroye E.; Ha S. K.; Bladt E.; Kshirsagar A. S.; Wang Z.; Yin J.; Wang Y.; Quan L. N.; Yan F.; Gao M.; Li X.; Shamsi J.; Debnath T.; Cao M.; Scheel M. A.; Kumar S.; Steele J. A.; Gerhard M.; Chouhan L.; Xu K.; Wu X.-g.; Li Y.; Zhang Y.; Dutta A.; Han C.; Vincon I.; Rogach A. L.; Nag A.; Samanta A.; Korgel B. A.; Shih C.-J.; Gamelin D. R.; Son D. H.; Zeng H.; Zhong H.; Sun H.; Demir H. V.; Scheblykin I. G.; Mora-Seró I.; Stolarczyk J. K.; Zhang J. Z.; Feldmann J.; Hofkens J.; Luther J. M.; Pérez-Prieto J.; Li L.; Manna L.; Bodnarchuk M. I.; Kovalenko M. V.; Roeffaers M. B. J.; Pradhan N.; Mohammed O. F.; Bakr O. M.; Yang P.; Müller-Buschbaum P.; Kamat P. V.; Bao Q.; Zhang Q.; Krahne R.; Galian R. E.; Stranks S. D.; Bals S.; Biju V.; Tisdale W. A.; Yan Y.; Hoye R. L. Z.; Polavarapu L. State of the Art and Prospects for Halide Perovskite Nanocrystals. ACS Nano 2021, 15, 10775–10981. 10.1021/acsnano.0c08903. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Kim J. S.; Heo J.-M.; Park G.-S.; Woo S.-J.; Cho C.; Yun H. J.; Kim D.-H.; Park J.; Lee S.-C.; Park S.-H.; Yoon E.; Greenham N. C.; Lee T.-W. Ultra-bright, efficient and stable perovskite light-emitting diodes. Nature 2022, 611, 688–694. 10.1038/s41586-022-05304-w. [DOI] [PubMed] [Google Scholar]; j Kaplan A. E. K.; Krajewska C. J.; Proppe A. H.; Sun W.; Sverko T.; Berkinsky D. B.; Utzat H.; Bawendi M. G. Hong–Ou–Mandel interference in colloidal CsPbBr3 perovskite nanocrystals. Nat. Photonics 2023, 17, 775–780. 10.1038/s41566-023-01225-w. [DOI] [Google Scholar]
- a Wu K.; Liang G.; Shang Q.; Ren Y.; Kong D.; Lian T. Ultrafast Interfacial Electron and Hole Transfer from CsPbBr3 Perovskite Quantum Dots. J. Am. Chem. Soc. 2015, 137, 12792–12795. 10.1021/jacs.5b08520. [DOI] [PubMed] [Google Scholar]; b De Roo J.; Ibáñez M.; Geiregat P.; Nedelcu G.; Walravens W.; Maes J.; Martins J. C.; Van Driessche I.; Kovalenko M. V.; Hens Z. Highly Dynamic Ligand Binding and Light Absorption Coefficient of Cesium Lead Bromide Perovskite Nanocrystals. ACS Nano 2016, 10, 2071–2081. 10.1021/acsnano.5b06295. [DOI] [PubMed] [Google Scholar]; c Wang H.; Wang X.; Chen R.; Zhang H.; Wang X.; Wang J.; Zhang J.; Mu L.; Wu K.; Fan F.; Zong X.; Li C. Promoting Photocatalytic H2 Evolution on Organic–Inorganic Hybrid Perovskite Nanocrystals by Simultaneous Dual-Charge Transportation Modulation. ACS Energy Lett. 2019, 4, 40–47. 10.1021/acsenergylett.8b01830. [DOI] [Google Scholar]; d Saris S.; Loiudice A.; Mensi M.; Buonsanti R. Exploring Energy Transfer in a Metal/Perovskite Nanocrystal Antenna to Drive Photocatalysis. J. Phys. Chem. Lett. 2019, 10, 7797–7803. 10.1021/acs.jpclett.9b03164. [DOI] [PubMed] [Google Scholar]; e Kobosko S. M.; DuBose J. T.; Kamat P. V. Perovskite Photocatalysis. Methyl Viologen Induces Unusually Long-Lived Charge Carrier Separation in CsPbBr3 Nanocrystals. ACS Energy Lett. 2020, 5, 221–223. 10.1021/acsenergylett.9b02573. [DOI] [Google Scholar]; f Loiudice A.; Saris S.; Buonsanti R. Tunable Metal Oxide Shell as a Spacer to Study Energy Transfer in Semiconductor Nanocrystals. J. Phys. Chem. Lett. 2020, 11, 3430–3435. 10.1021/acs.jpclett.0c00820. [DOI] [PubMed] [Google Scholar]; g Luo X.; Liang G.; Han Y.; Li Y.; Ding T.; He S.; Liu X.; Wu K. Triplet Energy Transfer from Perovskite Nanocrystals Mediated by Electron Transfer. J. Am. Chem. Soc. 2020, 142, 11270–11278. 10.1021/jacs.0c04583. [DOI] [PubMed] [Google Scholar]; h Liu M.; Xia P.; Zhao G.; Nie C.; Gao K.; He S.; Wang L.; Wu K. Energy-Transfer Photocatalysis Using Lead Halide Perovskite Nanocrystals: Sensitizing Molecular Isomerization and Cycloaddition. Angew. Chem., Int. Ed. 2022, 61, e202208241 10.1002/anie.202208241. [DOI] [PubMed] [Google Scholar]; i DuBose J. T.; Kamat P. V. Energy Versus Electron Transfer: Managing Excited-State Interactions in Perovskite Nanocrystal–Molecular Hybrids. Chem. Rev. 2022, 122, 12475–12494. 10.1021/acs.chemrev.2c00172. [DOI] [PubMed] [Google Scholar]; j DuBose J. T.; Kamat P. V. How Pendant Groups Dictate Energy and Electron Transfer in Perovskite–Rhodamine Light Harvesting Assemblies. J. Am. Chem. Soc. 2023, 145, 4601–4612. 10.1021/jacs.2c12248. [DOI] [PubMed] [Google Scholar]
- a Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Romero N. A.; Nicewicz D. A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]; c Yoon T. P. Photochemical Stereocontrol Using Tandem Photoredox–Chiral Lewis Acid Catalysis. Acc. Chem. Res. 2016, 49, 2307–2315. 10.1021/acs.accounts.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Staveness D.; Bosque I.; Stephenson C. R. J. Free Radical Chemistry Enabled by Visible Light-Induced Electron Transfer. Acc. Chem. Res. 2016, 49, 2295–2306. 10.1021/acs.accounts.6b00270. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Strieth-Kalthoff F.; James M. J.; Teders M.; Pitzer L.; Glorius F. Energy transfer catalysis mediated by visible light: principles, applications, directions. Chem. Soc. Rev. 2018, 47, 7190–7202. 10.1039/C8CS00054A. [DOI] [PubMed] [Google Scholar]; f Kron K. J.; Rodriguez-Katakura A.; Elhessen R.; Mallikarjun Sharada S. Photoredox Chemistry with Organic Catalysts: Role of Computational Methods. ACS Omega 2021, 6, 33253–33264. 10.1021/acsomega.1c05787. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Fischer D. M.; Freis M.; Amberg W. M.; Lindner H.; Carreira E. M. Organophotocatalytic carbo-heterofunctionalization of unactivated olefins with pendant nucleophiles. Chem. Sci. 2023, 14, 7256–7261. 10.1039/D3SC02250A. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Lindner H.; Amberg W. M.; Martini T.; Fischer D. M.; Moore E.; Carreira E. M. Photo- and Cobalt-Catalyzed Synthesis of Heterocycles via Cycloisomerization of Unactivated Olefins. Angew. Chem., Int. Ed. 2024, 63, e202319515 10.1002/anie.202319515. [DOI] [PubMed] [Google Scholar]; i Lindner H.; Carreira E. M. Cobalt-Catalyzed Photo-Semipinacol Rearrangement of Unactivated Allylic Alcohols. Angew. Chem., Int. Ed. 2024, 63, e202407827 10.1002/anie.202407827. [DOI] [PubMed] [Google Scholar]
- a Anastas P.; Eghbali N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. 10.1039/B918763B. [DOI] [PubMed] [Google Scholar]; b Fürstner A.; Leitner A.; Méndez M.; Krause H. Iron-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc. 2002, 124, 13856–13863. 10.1021/ja027190t. [DOI] [PubMed] [Google Scholar]; c Kettler P. B. Platinum Group Metals in Catalysis: Fabrication of Catalysts and Catalyst Precursors. Org. Process Res. Dev. 2003, 7, 342–354. 10.1021/op034017o. [DOI] [Google Scholar]; d Hierso J.-C.; Beaupérin M.; Meunier P. Ultra-Low Catalyst Loading as a Concept in Economical and Sustainable Modern Chemistry: The Contribution of Ferrocenylpolyphosphane Ligands. Eur. J. Inorg. Chem. 2007, 2007, 3767–3780. 10.1002/ejic.200700405. [DOI] [Google Scholar]; e Hoover J. M.; Stahl S. S. Highly Practical Copper(I)/TEMPO Catalyst System for Chemoselective Aerobic Oxidation of Primary Alcohols. J. Am. Chem. Soc. 2011, 133, 16901–16910. 10.1021/ja206230h. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Ashby M. F.Materials and the Environment: Eco-Informed Material Choice; Elsevier, 2012. [Google Scholar]; g Fürstner A. Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes To Make This Base Metal a Multitasking Champion. ACS Cent. Sci. 2016, 2, 778–789. 10.1021/acscentsci.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Shang R.; Ilies L.; Nakamura E. Iron-Catalyzed C–H Bond Activation. Chem. Rev. 2017, 117, 9086–9139. 10.1021/acs.chemrev.6b00772. [DOI] [PubMed] [Google Scholar]; i Kennedy C. R.; Zhong H.; Macaulay R. L.; Chirik P. J. Regio- and Diastereoselective Iron-Catalyzed [4 + 4]-Cycloaddition of 1,3-Dienes. J. Am. Chem. Soc. 2019, 141, 8557–8573. 10.1021/jacs.9b02443. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Ganesh K. N.; Zhang D.; Miller S. J.; Rossen K.; Chirik P. J.; Kozlowski M. C.; Zimmerman J. B.; Brooks B. W.; Savage P. E.; Allen D. T.; Voutchkova-Kostal A. M. Green Chemistry: A Framework for a Sustainable Future. Environ. Sci. Technol. 2021, 55, 8459–8463. 10.1021/acs.est.1c03762. [DOI] [PubMed] [Google Scholar]; k Lindner H.; Amberg W. M.; Carreira E. M. Iron-Mediated Photochemical Anti-Markovnikov Hydroazidation of Unactivated Olefins. J. Am. Chem. Soc. 2023, 145, 22347–22353. 10.1021/jacs.3c09122. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Zhang Z.-J.; Jacob N.; Bhatia S.; Boos P.; Chen X.; DeMuth J. C.; Messinis A. M.; Jei B. B.; Oliveira J. C. A.; Radović A.; Neidig M. L.; Wencel-Delord J.; Ackermann L. Iron-catalyzed stereoselective C–H alkylation for simultaneous construction of C–N axial and C-central chirality. Nat. Commun. 2024, 15, 3503. 10.1038/s41467-024-47589-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Sharpless K. B.; Amberg W.; Bennani Y. L.; Crispino G. A.; Hartung J.; Jeong K. S.; Kwong H. L.; Morikawa K.; Wang Z. M. The osmium-catalyzed asymmetric dihydroxylation: a new ligand class and a process improvement. J. Org. Chem. 1992, 57, 2768–2771. 10.1021/jo00036a003. [DOI] [Google Scholar]
- The reported turnover number is only valid under the assumption that the reaction proceeds with a closed catalytic cycle.
- a Cao Q.; Feng J.; Chang K. T.; Liang W.; Lu H. Emerging Opportunities of Colloidal Quantum Dots for Photocatalytic Organic Transformations. Adv. Mater. 2024, 2409096. 10.1002/adma.202409096. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rosa-Pardo I.; Casadevall C.; Schmidt L.; Claros M.; Galian R. E.; Lloret-Fillol J.; Pérez-Prieto J. The synergy between the CsPbBr3 nanoparticle surface and the organic ligand becomes manifest in a demanding carbon–carbon coupling reaction. Chem. Commun. 2020, 56, 5026–5029. 10.1039/D0CC01339K. [DOI] [PubMed] [Google Scholar]; c Zhu X.; Lin Y.; Sun Y.; Beard M. C.; Yan Y. Lead-Halide Perovskites for Photocatalytic α-Alkylation of Aldehydes. J. Am. Chem. Soc. 2019, 141, 733–738. 10.1021/jacs.8b08720. [DOI] [PubMed] [Google Scholar]; d Zhu X.; Lin Y.; San Martin J.; Sun Y.; Zhu D.; Yan Y. Lead halide perovskites for photocatalytic organic synthesis. Nat. Commun. 2019, 10, 2843. 10.1038/s41467-019-10634-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Yuan Y.; Zhu H.; Hills-Kimball K.; Cai T.; Shi W.; Wei Z.; Yang H.; Candler Y.; Wang P.; He J.; Chen O. Stereoselective C–C Oxidative Coupling Reactions Photocatalyzed by Zwitterionic Ligand Capped CsPbBr3 Perovskite Quantum Dots. Angew. Chem., Int. Ed. 2020, 59, 22563–22569. 10.1002/anie.202007520. [DOI] [PubMed] [Google Scholar]; f Lin Y.; Avvacumova M.; Zhao R.; Chen X.; Beard M. C.; Yan Y. Triplet Energy Transfer from Lead Halide Perovskite for Highly Selective Photocatalytic 2 + 2 Cycloaddition. ACS Appl. Mater. Interfaces 2022, 14, 25357–25365. 10.1021/acsami.2c03411. [DOI] [PubMed] [Google Scholar]; g Ren K.; Yue S.; Li C.; Fang Z.; Gasem K. A. M.; Leszczynski J.; Qu S.; Wang Z.; Fan M. Metal halide perovskites for photocatalysis applications. J. Mater. Chem. A 2022, 10, 407–429. 10.1039/D1TA09148D. [DOI] [Google Scholar]; h Zhu Y.; Liu Y.; Miller K. A.; Zhu H.; Egap E. Lead Halide Perovskite Nanocrystals as Photocatalysts for PET-RAFT Polymerization under Visible and Near-Infrared Irradiation. ACS Macro Lett. 2020, 9, 725–730. 10.1021/acsmacrolett.0c00232. [DOI] [PubMed] [Google Scholar]; i Lin Y.; Yan Y. CsPbBr3 Perovskite Nanocrystals for Photocatalytic [3 + 2] Cycloaddition. ChemSusChem 2024, 17, e202301060 10.1002/cssc.202301060. [DOI] [PubMed] [Google Scholar]; j Li Y.; Gao Y.; Deng Z.; Cao Y.; Wang T.; Wang Y.; Zhang C.; Yuan M.; Xie W. Visible-light-driven reversible shuttle vicinal dihalogenation using lead halide perovskite quantum dot catalysts. Nat. Commun. 2023, 14, 4673. 10.1038/s41467-023-40359-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Li Y.; Wang T.; Wang Y.; Deng Z.; Zhang L.; Zhu A.; Huang Y.; Zhang C.; Yuan M.; Xie W. Tunable Photocatalytic Two-Electron Shuttle between Paired Redox Sites on Halide Perovskite Nanocrystals. ACS Catal. 2022, 12, 5903–5910. 10.1021/acscatal.2c01044. [DOI] [Google Scholar]
- Krieg F.; Ochsenbein S. T.; Yakunin S.; ten Brinck S.; Aellen P.; Süess A.; Clerc B.; Guggisberg D.; Nazarenko O.; Shynkarenko Y.; Kumar S.; Shih C.-J.; Infante I.; Kovalenko M. V. Colloidal CsPbX3 (X = Cl, Br, I) Nanocrystals 2.0: Zwitterionic Capping Ligands for Improved Durability and Stability. ACS Energy Lett. 2018, 3, 641–646. 10.1021/acsenergylett.8b00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morad V.; Stelmakh A.; Svyrydenko M.; Feld L. G.; Boehme S. C.; Aebli M.; Affolter J.; Kaul C. J.; Schrenker N. J.; Bals S.; Sahin Y.; Dirin D. N.; Cherniukh I.; Raino G.; Baumketner A.; Kovalenko M. V. Designer phospholipid capping ligands for soft metal halide nanocrystals. Nature 2024, 626, 542–548. 10.1038/s41586-023-06932-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zaera F. Designing Sites in Heterogeneous Catalysis: Are We Reaching Selectivities Competitive With Those of Homogeneous Catalysts?. Chem. Rev. 2022, 122, 8594–8757. 10.1021/acs.chemrev.1c00905. [DOI] [PubMed] [Google Scholar]; b Zhang Z.; Edme K.; Lian S.; Weiss E. A. Enhancing the Rate of Quantum-Dot-Photocatalyzed Carbon–Carbon Coupling by Tuning the Composition of the Dot’s Ligand Shell. J. Am. Chem. Soc. 2017, 139, 4246–4249. 10.1021/jacs.6b13220. [DOI] [PubMed] [Google Scholar]; c Kodaimati M. S.; McClelland K. P.; He C.; Lian S.; Jiang Y.; Zhang Z.; Weiss E. A. Viewpoint: Challenges in Colloidal Photocatalysis and Some Strategies for Addressing Them. Inorg. Chem. 2018, 57, 3659–3670. 10.1021/acs.inorgchem.7b03182. [DOI] [PubMed] [Google Scholar]; d Temerov F.; Baghdadi Y.; Rattner E.; Eslava S. A Review on Halide Perovskite-Based Photocatalysts: Key Factors and Challenges. ACS Appl. Energy Mater. 2022, 5, 14605–14637. 10.1021/acsaem.2c02680. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Zhong F.; Sheng J.; Du C.; He Y.; Sun Y.; Dong F. Ligand-mediated exciton dissociation and interparticle energy transfer on CsPbBr3 perovskite quantum dots for efficient CO2-to-CO photoreduction. Sci. Bull. 2024, 69, 901–912. 10.1016/j.scib.2024.01.027. [DOI] [PubMed] [Google Scholar]
- a Murray J. I.; Silva Elipe M. V.; Cosbie A.; Baucom K.; Quasdorf K.; Caille S. Kinetic Investigations To Enable Development of a Robust Radical Benzylic Bromination for Commercial Manufacturing of AMG 423 Dihydrochloride Hydrate. Org. Process Res. Dev. 2020, 24, 1523–1530. 10.1021/acs.oprd.0c00256. [DOI] [Google Scholar]; b Fischer D. M.; Lindner H.; Amberg W. M.; Carreira E. M. Intermolecular Organophotocatalytic Cyclopropanation of Unactivated Olefins. J. Am. Chem. Soc. 2023, 145, 774–780. 10.1021/jacs.2c11680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Center for Biotechnology Information . PubChem Compound Summary for CID 3024, Dibromomethane, 2024. https://pubchem.ncbi.nlm.nih.gov/compound/Dibromomethane (accessed August 27, 2024).
- REACH . Substance Infocard Dibromomethane. https://echa.europa.eu/de/substance-information/-/substanceinfo/100.000.750.
- For a recently reported example with low yield seeLin Y.; Yan Y. CsPbBr3 Perovskite Nanocrystals for Photocatalytic [3 + 2] Cycloaddition. ChemSusChem 2024, 17, e202301060 10.1002/cssc.202301060. [DOI] [PubMed] [Google Scholar]
- Bodnarchuk M. I.; Boehme S. C.; ten Brinck S.; Bernasconi C.; Shynkarenko Y.; Krieg F.; Widmer R.; Aeschlimann B.; Günther D.; Kovalenko M. V.; Infante I. Rationalizing and Controlling the Surface Structure and Electronic Passivation of Cesium Lead Halide Nanocrystals. ACS Energy Lett. 2019, 4, 63–74. 10.1021/acsenergylett.8b01669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J.; Zhu Q. Stability strategies of perovskite quantum dots and their extended applications in extreme environment: A review. Mater. Res. Bull. 2022, 156, 111987. 10.1016/j.materresbull.2022.111987. [DOI] [Google Scholar]
- Sun Y.; Zhang H.; Zhu K.; Ye W.; She L.; Gao X.; Ji W.; Zeng Q. Research on the influence of polar solvents on CsPbBr3 perovskite QDs. RSC Adv. 2021, 11, 27333–27337. 10.1039/D1RA04485K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Li Y.; Zheng C.; Gao D.; Huang W. Advancements in the stability of perovskite solar cells: degradation mechanisms and improvement approaches. RSC Adv. 2016, 6, 38079–38091. 10.1039/C5RA27424A. [DOI] [Google Scholar]
- Saikia I.; Borah A. J.; Phukan P. Use of Bromine and Bromo-Organic Compounds in Organic Synthesis. Chem. Rev. 2016, 116, 6837–7042. 10.1021/acs.chemrev.5b00400. [DOI] [PubMed] [Google Scholar]
- Hudzik J. M.; Bozzelli J. W. Thermochemistry and Bond Dissociation Energies of Ketones. J. Phys. Chem. A 2012, 116, 5707–5722. 10.1021/jp302830c. [DOI] [PubMed] [Google Scholar]
- Takaaki H.; Katsumasa H.. PRODUCTION OF GAMMA-HALOGENO-BETA-KETOESTERS. JP 2000256262 A, 2000.
- Li D.; Keresztes I.; Hopson R.; Williard P. G. Characterization of Reactive Intermediates by Multinuclear Diffusion-Ordered NMR Spectroscopy (DOSY). Acc. Chem. Res. 2009, 42, 270–280. 10.1021/ar800127e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Claridge T. D.High-resolution NMR Techniques in Organic Chemistry; Elsevier, 2016; Vol. 27. [Google Scholar]; b Stilbs P.Diffusion and Electrophoretic NMR; Walter de Gruyter GmbH & Co KG, 2019. [Google Scholar]
- Stejskal E. O.; Tanner J. E. Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient. J. Chem. Phys. 1965, 42, 288–292. 10.1063/1.1695690. [DOI] [Google Scholar]
- Connell M. A.; Bowyer P. J.; Adam Bone P.; Davis A. L.; Swanson A. G.; Nilsson M.; Morris G. A. Improving the accuracy of pulsed field gradient NMR diffusion experiments: Correction for gradient non-uniformity. J. Magn. Reson. 2009, 198, 121–131. 10.1016/j.jmr.2009.01.025. [DOI] [PubMed] [Google Scholar]
- Ravi V. K.; Markad G. B.; Nag A. Band Edge Energies and Excitonic Transition Probabilities of Colloidal CsPbX3 (X = Cl, Br, I) Perovskite Nanocrystals. ACS Energy Lett. 2016, 1, 665–671. 10.1021/acsenergylett.6b00337. [DOI] [Google Scholar]
- Fedurco M.; Sartoretti C. J.; Augustynski J. Medium Effects on the Reductive Cleavage of the Carbon–Halogen Bond in Methyl and Methylene Halides. J. Phys. Chem. B 2001, 105, 2003–2009. 10.1021/jp003646m. [DOI] [Google Scholar]
- a Liang J.; Chen D.; Yao X.; Zhang K.; Qu F.; Qin L.; Huang Y.; Li J. Recent Progress and Development in Inorganic Halide Perovskite Quantum Dots for Photoelectrochemical Applications. Small 2020, 16, 1903398. 10.1002/smll.201903398. [DOI] [PubMed] [Google Scholar]; b Qiao G.-Y.; Guan D.; Yuan S.; Rao H.; Chen X.; Wang J.-A.; Qin J.-S.; Xu J.-J.; Yu J. Perovskite Quantum Dots Encapsulated in a Mesoporous Metal–Organic Framework as Synergistic Photocathode Materials. J. Am. Chem. Soc. 2021, 143, 14253–14260. 10.1021/jacs.1c05907. [DOI] [PubMed] [Google Scholar]
- Klaewkla R.; Arend M.; Hoelderich W. F.. A Review of Mass Transfer Controlling the Reaction Rate in Heterogeneous Catalytic Systems; INTECH Open Access Publisher: Rijeka, 2011; Vol. 5. [Google Scholar]
- a Simmons E. M.; Hartwig J. F. On the Interpretation of Deuterium Kinetic Isotope Effects in C-H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem., Int. Ed. 2012, 51, 3066–3072. 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]; b Atkins P. W.; De Paula J.; Keeler J.. Atkins’ Physical Chemistry; Oxford University Press, 2023. [Google Scholar]; c Connors K. A.Chemical Kinetics: The Study of Reaction Rates in Solution; Wiley-VCH Verlag GmbH, 1990. [Google Scholar]
- Gaspar B.; Carreira E. M. Mild Cobalt-Catalyzed Hydrocyanation of Olefins with Tosyl Cyanide. Angew. Chem., Int. Ed. 2007, 46, 4519–4522. 10.1002/anie.200700575. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.