

Abstract
Background
Three-dimensional (3D) chromatin organization is emerging as a key factor in gene regulation in eukaryotes. Recent studies using high-resolution Hi-C analysis have explored fine-scale local chromatin contact domains in plants, as exemplified by the basic contact domains established at accessible gene border regions in Arabidopsis (Arabidopsis thaliana). However, we lack effective tools to identify these contact domains and examine their structural dynamics.
Results
We developed the Hi-C-based 3D Gene Domain analysis Tool (Hi-GDT) to identify fine-scale local chromatin contact domains in plants, with a particular focus on gene borders. Hi-GDT successfully identifies local contact domains, including single-gene and multigene domains, with high reproducibility. Hi-GDT can also be used to discover local contact domains that are differentially organized in association with differences in gene expression between tissue types, genotypes, or in response to environmental stimuli.
Conclusions
Hi-GDT is a valuable tool for identifying genes regulated by dynamic 3D conformational changes, expanding our understanding of the structural and functional relevance of local 3D chromatin organization in plants. Hi-GDT is publicly available at https://github.com/CDL-HongwooLee/Hi-GDT.
Keywords: Arabidopsis, Hi-C, fine-scale contact domain, gene domain, gene domain analysis tool
Key Points:
We developed the Hi-C-based 3D Gene Domain analysis Tool (Hi-GDT) to identify fine-scale local chromatin contact domains.
Hi-GDT successfully identifies local contact domains, including single-gene and multigene domains, with high reproducibility.
Hi-GDT also identifies 3D contact domains that are dynamically organized based on their transcriptional states.
Introduction
Three-dimensional (3D) chromatin organization is important for the proper structural arrangement of the genome and for controlling gene transcription [1–7]. Eukaryotic genomes are organized into structural units known as topologically associating domains (TADs), which exhibit strong self-interaction and thus appear as triangular shapes in 3D chromatin contact maps [8–10]. TADs can be classified into 2 types depending on the presence or absence of strong contacts at anchor sites (shown as corner dots in 3D Hi-C chromatin contact maps) [11–15]. The 3D contact domains with strong contacts at anchor sites (called loop domains) are established by cohesin and chromatin insulator proteins, including CCCTC-binding factor (CTCF), through a loop-extrusion mechanism [16–18]. By contrast, 3D contact domains without strong contacts at anchor sites (called compartment domains) form independently of CTCF and cohesin and are instead associated with local A/B compartments and epigenetic states [11–13, 15, 19].
The CTCF core architectural protein is not conserved across various plant species [20]. Consistent with this finding, most plant 3D chromatin domains lack strong interactions at anchor sites. Instead, their formation is likely to be dependent on epigenetic states, as observed in tomato (Solanum lycopersicum), rice (Oryza sativa), sorghum (Sorghum bicolor), and foxtail millet (Setaria italica) [21–24]. Plants with large genomes, such as maize (Zea mays), cotton (Gossypium hirsutum), pepper, and wheat (Triticum aestivum), also frequently contain 3D chromatin domains demarcated by gene-to-gene loops, which are enriched with active chromatin marks [25–29], suggesting that epigenetic states are critical for organizing the 3D conformation of chromatin in plants. Although the molecular mechanisms of 3D chromatin domain formation vary, the sizes of such domains in these plant species are comparable to those of TADs in mammals. Therefore, conventional TAD analysis tools have been reasonably applied to identify and analyze large 3D chromatin domains in plants.
The compartment domains in plant species with small genomes have recently been explored. Using ultra-deep Hi-C sequencing and advanced Hi-C-based techniques, fine-scale local chromatin domains have been identified in the Arabidopsis (Arabidopsis thaliana) genome, which are intimately associated with epigenetic states [30–33]. Notably, representative local chromatin domains (called gene domains) are organized at gene border regions with high chromatin accessibility in Arabidopsis [34]. These contact domains form at the single-gene scale through a process driven by self-interaction between transcriptional start sites (TSSs) and transcriptional end sites (TESs). They also form at chromatin regions spanning a series of multiple genes with high chromatin accessibility at their borders [34]. Although gene domains are conserved across diverse plant species, including tomato, maize and Marchantia polymorpha (Marchantia), the functional importance of these fine-scale local chromatin domains in plants has received little attention. One study examined the Arabidopsis biosynthetic gene clusters of the specialized metabolites thalianol and marneral, finding that each biosynthetic gene cluster is located in a 3D chromatin domain whose structure is likely reorganized in response to internal and/or external cues to ensure the transcriptional coordination of the associated genes [35]. Otherwise, this area of research has been limited, largely due to the lack of proper analysis tools for such contact domains. In particular, most existing contact domain callers were originally designed for analyzing animal TADs. However, given that gene domains are much smaller than conventional TADs or compartment domains and exhibit relatively low contact frequencies between anchor sites, new tools are needed that can analyze the fine-scale chromatin domains with high sensitivity and precision from high-resolution 3D contact maps.
In this study, we describe the Hi-C-based Gene Domain analysis Tool (Hi-GDT), which we designed to identify 3D contact domains with a focus on gene border regions, enabling more stringent fine-scale gene domain analysis. This tool successfully identified local contact domains, including single-gene and multigene domains, across various Hi-C datasets from Arabidopsis, tomato, maize and Marchantia. Hi-GDT also identified genes with differential 3D contact domains that are dynamically organized based on their transcriptional states in different tissue types, genotypes, and in response to environmental stimuli. Hi-GDT serves as a valuable tool for analyzing fine-scale local chromatin organization in plants.
Materials and Methods
Plant materials
Arabidopsis (A. thaliana) accession Columbia (Col-0) was grown at 23°C under long-day conditions (16 hours light/8 hours dark) using white fluorescent lamps (120 μmol photons m−2 s−1). For callus induction, the third and fourth leaves from 2-week-old seedlings were excised and incubated on callus-inducing medium (CIM; Murashige and Skoog medium supplemented with 0.5 μg/mL 2,4-D and 0.05 μg/mL kinetin) for 7 days. The callus tissues were selectively harvested for Hi-C analyses.
RNA sequencing experiments
Leaf explant samples were harvested immediately after excision (0 day), and leaf explant–derived callus samples were harvested at 7 days after incubation on CIM. RNA libraries were constructed with 1 μg total RNA using a TruSeq stranded mRNA library kit (Illumina) as previously described [36].
Hi-C experiments and processing
Leaf explant–derived callus samples were harvested at 7 days after incubation on CIM. Hi-C data for callus tissue were generated using an Arima-HiC kit (Arima Genomics) according to the manufacturer’s protocol, followed by next-generation sequencing performed by Macrogen. The publicly available Hi-C sequencing files were downloaded from the NCBI Sequence Read Archive (SRA; Supplementary Table S1) [24, 37–42]. Fastq files were extracted from the SRA files and processed using Juicer (v2.0) (RRID:SCR_017226) [43] with the corresponding reference genome (TAIR10 for Arabidopsis, M82v1.0 for tomato, B73v5 for maize, and MpTak1v5 for Marchantia). Mapped reads were filtered with a mapping quality cutoff value of 30 in Arabidopsis, tomato, and Marchantia, whereas no mapping quality cutoff was applied in maize. SCALE normalization was applied to the Hi-C contact matrices to correct for sequencing bias. The compressed binary files (.hic) generated by Juicer were used as inputs for Hi-GDT.
Single-gene domain analysis by Hi-GDT
Hi-GDT identifies single-gene domains by comparing intragenic contact frequencies with those in designated control regions. Given the relatively high contact frequencies at TSS–TES anchor sites, the intragenic target region near the anchor site is visualized as a triangle in Fig. 1B. The size of this region was defined as half the length of the corresponding gene. The control regions were set to be equidistant with a target region from the diagonal axis of a Hi-C contact map, including intragenic regions at one side of the anchor (Fig. 1B). The distance-normalized intragenic contact frequencies (OE values; observed contact frequency/expected contact frequency) within both intragenic and control regions were collected and tested using the Mann–Whitney U test. Genes with significantly higher contact frequencies within intragenic regions compared to the surrounding control regions were defined as single-gene domains. A Benjamini–Hochberg corrected P value (Q value) cutoff of 0.05 was applied for the identification of single-gene domains from the Hi-C datasets at a 250-bp resolution.
Figure 1:

Workflow and strategy of Hi-GDT for identifying local contact domains. (A) The Hi-GDT workflow. Hi-GDT analysis begins with a .hic file generated by Juicer software. Hi-GDT identifies single- and multigene domains (Hi-GDTsingle and Hi-GDTmulti, respectively). Hi-GDT also provides an analysis of the structural dynamics of single-gene domains, which are associated with changes in gene expression (Hi-GDTdiff). (B, C) Schematic diagrams illustrating the target and control regions in a Hi-C contact map, which are defined for gene domain identification by Hi-GDT. The regions used for single-gene domain identification (B) and multigene domain identification (C) are shown. Target regions are shown in red, and control regions are shown in green or yellow. The line at the bottom indicates the diagonal axis in a Hi-C contact map, and the black triangle indicates an individual gene. In (C), target and control regions used to identify quadruple-gene domains are shown as an example. Boundaries of a multigene domain are indicated by blue lines.
For differential single-gene domain analysis, the OE values within (i) the surrounding 2-kb regions and (ii) the gene body regions of individual genes from 2 different Hi-C datasets were collected. The OE values within the surrounding regions were subjected to a Wilcoxon signed-rank test to identify genes with differential (Q < 0.05) surrounding contact frequencies between 2 Hi-C datasets. In addition, a cutoff value of 0.1 for mean fold change of OE values was used to filter the differentially insulated genes that exhibited both increased contact frequencies within gene body regions and decreased contact frequencies within surrounding 2-kb regions. For example, fold change filters of OE values to identify single-gene domains with enhanced insulation under certain treatment condition are applied as follows.
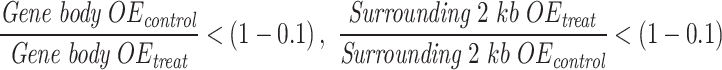 |
Multigene domain analysis by Hi-GDT
The identification of multigene domains involved 2 types of comparisons between intradomain regions and outside control regions: (i) comparisons of gene-to-gene contact frequencies at the domain boundaries with those in outside control regions and (ii) comparisons of intradomain contact frequencies at each domain boundary with those in respective control regions (Fig. 1C; Supplementary Fig. S1). The control and target regions were set to be equidistant from the diagonal axis of a Hi-C contact map. Mann–Whitney U tests were employed to compare OE values between pairs of target and control regions. A multigene domain was defined as a cluster of adjacent genes that exhibited significantly higher OE values within target regions compared to all their respective control regions.
To analyze multigene domain dynamics, multigene domains were identified at a 500-bp resolution. Multigene domains were identified based on Q value cutoffs of 0.05. Dual- and triple-gene domains identified in each specific tissue were compared to analyze the structural dynamics of multigene domains. Genes forming dual-gene domains with a TSS at one boundary, without forming single-gene domains, were used to analyze changes in gene expression.
Identification of 3D contact domains using existing domain callers
Domain calling with Arrowhead [43], HiCExplorer (RRID:SCR_022111) [44–46], and OnTAD [47] was conducted at a 250-, 500-, and 1,000-bp resolution for benchmarking with Hi-GDT. The parameter “-m 100 -k SCALE” was used for Arrowhead, and the parameter “-penalty 0.1 -minsz 3 -maxsz 200 -hic_norm SCALE” was used for OnTAD. For HiCExplorer, SCALE-normalized .hic files were converted to .h5 format using the hicConvertFormat command, and domain calling was conducted using hicFindTADs with the parameter “–correctForMultipleTesting fdr.”
Analysis of A and B compartments
To define the local A/B compartment, Pearson’s correlation matrix of the SCALE-normalized Hi-C contact map at a 750-bp resolution was obtained using Juicer Tools [43]. The first eigenvector of the correlation matrix (compartment eigenvector) was calculated using numpy.linalg.eig (RRID:SCR_008633) [48]. For single-gene domain analysis, compartment eigenvector values were measured at the TSS, the center region of the gene body, and the TES. Compartment eigenvector values >0 were assigned to the A compartment, and values <0 were assigned to the B compartment.
Metaplot analysis
Metaplot analysis for directionality index (DI) score, chromatin accessibility, and compartment eigenvector was performed using deepTools (RRID:SCR_016366) [49]. The DI score with a 5-kb window was calculated at a 250-bp resolution as described previously [8]. The processed BigWig file of Assay for Transposase-Accessible Chromatin (ATAC) sequencing data was downloaded from the Plant Chromatin State Database (PCSD) [50, 51]. The compartment eigenvector at a 750-bp resolution was converted to a BigWig file and used as input for metaplot analysis. For single-gene domain analysis, protein-coding genes without a single-gene domain structure were used for comparison.
Pile-up analysis
The SCALE-normalized OE values of Hi-C contact matrices at a 250-bp resolution were used for pile-up analysis. The OE values within identified contact domains and their surrounding regions (0.5× domain size) were obtained with a bin size of 10 bp and subsequently resized to an 80 × 80 matrix using bicubic interpolation. For single-gene domain analysis, the images of ( )-stranded genes were flipped to synchronize gene orientation. Pile-up analysis was conducted using PileUpDomain.py [52].
)-stranded genes were flipped to synchronize gene orientation. Pile-up analysis was conducted using PileUpDomain.py [52].
Analysis of chromatin state
Information about the 26 chromatin states was obtained from a previous report [53]. For single-gene domain analysis, chromatin states that overlapped with the TSS, the center of the gene body, and the TES were measured. For multigene domain analysis, chromatin states overlapping with the centers of genes located at multigene domain borders, at internal regions of multigene domains, and at outside regions of multigene domains were investigated. Genes forming single-gene domains were excluded from multigene domain analysis. To investigate the similarity of chromatin states between gene pairs in quadruple-gene domains, both genes in chromatin at the same states (constitutive heterochromatin [H], facultative heterochromatin [F], intergenic [I], and euchromatin [E]) were examined.
RNA sequencing analysis
The RNA sequencing reads used in this study were downloaded from the SRA database (Supplementary Table S1) [21, 24, 38, 42, 54]. The reads were mapped to the corresponding reference genome using STAR (v2.7.10a) (RRID:SCR_004463) [55] with the parameter “–peOverlapNbasesMin 12 –peOverlapMMp 0.1 –twopassMode Basic.” Transcript abundance was quantified using RSEM (v1.3.1) (RRID:SCR_000262) [56]. Differentially expressed genes (DEGs) and fold change values were identified by Deseq2 (v1.34.0) (RRID:SCR_015687) [57] with a threshold of log2(fold change) >1 and Q value <0.05. Log2(transcripts per million [TPM] + 1) values were used to quantify gene expression levels.
Calculation of Tau score
Tau score, a measure of tissue-specific gene expression, was calculated as previously described [58]. The expression levels of genes from various Arabidopsis tissues were downloaded from a previous report [59]. The corrected reads per kilobase of transcript per million mapped reads (cRPKM) values were used to calculate the Tau score.
Gene Ontology analysis
Gene Ontology (GO) analysis was performed using AmiGO2 powered by PANTHER (RRID:SCR_004869) [60–62]. The top 10 or 20 significantly enriched GO terms with P < 0.05 and number of gene hits >1 were visualized.
Colinear block analysis
Colinear blocks were identified using a pipeline that utilizes MCScanX (RRID:SCR_022067) [63]. Briefly, interspecies BLASTP analysis (RRID:SCR_001010) [64] was conducted between the Arabidopsis and tomato genomes, as well as between the Arabidopsis and maize genomes. A pairwise BLASTP was conducted by switching the reference genome, and the best 5 hits with an E-value cutoff of 1 × 10−10 were kept for the detection of colinear blocks. Colinear blocks between 2 different species were defined using MCScanX [65] with the default parameters. Among the genes within the colinear blocks in the tomato and maize genomes, those homologous to Arabidopsis single-gene domains and adjacent gene pairs homologous to Arabidopsis dual-gene domains were collected and visualized.
Data visualization
All heatmaps and 1-dimensional plots were generated with matplotlib (RRID:SCR_008624) [66] and seaborn (RRID:SCR_018132) [67]. Hi-C maps were visualized using JuiceBox (RRID:SCR_021172) [43].
Results
Development of Hi-GDT to analyze local contact domain structures
The discovery of gene domains in plants motivated us to develop Hi-GDT, a specialized tool for identifying fine-scale gene domain structures [34]. Since gene domains are prevalent in Arabidopsis, which have been analyzed with various high-resolution Hi-C datasets, we employed high-resolution Arabidopsis Hi-C data to validate the performance of our tool [38]. We designed a rule-based algorithm considering key features of gene domains, which include self-interaction patterns and strong insulation at gene borders in Hi-C contact maps, to identify such domains (Hi-GDTsingle for single-gene domains and Hi-GDTmulti for multigene domains) (Fig. 1A). For further analysis of the structural dynamics of single-gene domains, we also developed Hi-GDTdiff as a means to identify single-gene domains showing the 3D structural changes that are associated with differential gene expression in different tissue types, genotypes, or in response to environmental stimuli.
Given that single-gene domains display a self-interaction pattern with boundaries of TSSs and TESs that have high chromatin accessibility [34], we compared the distance-normalized intragenic contact frequencies (observed contact frequency/expected contact frequency, or OE) within a genic region defined by a TSS and TES with those of control regions outside genic regions to identify single-gene domains (Fig. 1B). For this comparison, we extracted OE values from the triangular region of a Hi-C contact map whose size was defined based on gene length (0.5× gene length). The target region (red triangle in Fig. 1B) was defined as the intragenic region near gene borders, whereas the control regions (green triangles) were determined based on 2 criteria: (i) the distance from the diagonal axis of a Hi-C contact map should be equal to that of the target region to minimize distance-dependent bias, and (ii) they should consist of interacting chromatin pairs, with one located outside and the other inside a gene body. We conducted a Mann–Whitney U test to compare OE values between target and control regions. Genes with significantly higher OE values in intragenic regions compared to those in both control regions were considered to form single-gene domains (Fig. 1B).
To identify multigene domains, we conducted 2 distinct comparisons of OE values between target (red squares) and control regions (green and yellow squares) that were equidistant from the diagonal axis of a Hi-C contact map (Fig. 1C; Supplementary Fig. S1): (i) OE values of gene-to-gene contact sites located at multigene domain boundaries were compared to those within the external control regions, and (ii) OE values at domain boundaries were compared to those in the 2 external control regions positioned in opposite directions (Fig. 1C; Supplementary Fig. S1). Adjacent gene sets that passed all of these statistical comparisons were annotated as multigene domains. In summary, Hi-GDT identified single-gene and multigene domains based on image analysis of Hi-C contact maps.
Hi-GDT is suitable for analyzing local contact domains in Arabidopsis
Since Hi-GDT was designed specifically for gene domain identification, we examined its possible advantages over conventional TAD callers, including Arrowhead [43], HiCExplorer [44–46], and OnTAD [47], in terms of the fine-scale identification of local contact domains in Arabidopsis. Hi-GDT identified a substantial number of local contact domains: 13,049 at a 250-bp resolution and 10,089 at a 500-bp resolution. These numbers were higher than the numbers of local contact domains identified by Arrowhead and HiCExplorer (Fig. 2A). The majority of contact domains identified by Hi-GDT were small in terms of size (<10 kb; Fig. 2B; Supplementary Fig. S2A). Notably, conventional domain callers generally predicted local contact domains at gene border regions (Supplementary Fig. S3), in agreement with the local contact domains formed mainly at gene boundaries in Arabidopsis [34]. We conducted pile-up analysis of the local contact domains identified by each domain caller to estimate their collective structural features. The contact domains identified by Hi-GDT and Arrowhead showed stronger contact domain structures that were more obvious than those identified by HiCExplorer and OnTAD across all tested resolutions, from 250 to 1,000 bp (Fig. 2C; Supplementary Fig. S2B).
Figure 2:

Benchmarking of Hi-GDT against conventional domain callers. (A) Comparison of the number of contact domains identified by Arrowhead, HiCExplorer, OnTAD, and Hi-GDT. Local contact domains were identified at a 250-, 500-, and 1,000-bp resolution. (B) Density plots showing the size distribution of contact domains identified by each domain caller at a 250- and 500-bp resolution. (C) Pile-up images of Hi-C contact matrices for contact domains identified by each domain caller. The images of contact domains identified at a 250-bp (top) and 500-bp (bottom) resolution are shown. Black lines indicate the boundaries of the identified contact domains. (D) Comparison of the Jaccard index values of contact domains identified by each domain caller across different datasets. Domain calling was conducted with 2 technical replicates subsampled (80%) from a merged Hi-C dataset, 2 biological replicates, and 2 distinct datasets from different batches at a 250-, 500-, and 1,000-bp resolution. Jaccard index was calculated as the intersection over the union between 2 sets of identified contact domains.
We also assessed the reproducibility of local contact domain identification by each domain caller by comparing the contact domains identified from technical replicates with 80% subsampling of a merged Hi-C dataset, biological replicates, or 2 Hi-C datasets from different batches. The reproducibility of the conventional domain callers for domain identification was low, ranging from 29% to 61% between technical replicates, 4% to 29% between biological replicates, and 1% to 10% between datasets from different batches (Supplementary Fig. S4A), suggesting that conventional domain callers have a limited ability to identify fine-scale local contact domains. In comparison, Hi-GDT showed higher reproducibility (Supplementary Fig. S4A). Since, unlike the other domain callers, Hi-GDT primarily focuses on predefined boundaries (such as TSSs and TESs), we reanalyzed the reproducibility of the conventional domain callers for gene domain identification by allowing the callers to adjust domain boundaries to coincide with gene border regions for a fair comparison. Although this increased the reproducibility of the conventional domain callers to some extent, Hi-GDT still exhibited the highest reproducibility score as well as Jaccard index, which represents the similarity between 2 groups (Fig. 2D; Supplementary Fig. S4B). Taken together, these results indicate that Hi-GDT is highly optimized for fine-scale local contact domain identification in Arabidopsis with high reproducibility.
Validation of single-gene domains identified by Hi-GDTsingle
We investigated whether the single-gene domains identified by Hi-GDTsingle showed key characteristics of previously reported single-gene domains [34]. Pile-up analysis and metagene analysis of DI revealed that the Hi-GDTsingle-identified single-gene domains at a 250-bp resolution were insulated at the TSS and TES regions (Fig. 3A, B). Furthermore, the border regions of these Hi-GDTsingle-identified gene domains had highly accessible chromatin conformations (Fig. 3C), as observed in previously reported single-gene domains [34]. We then analyzed the compartment eigenvector at a high resolution (750 bp) to explore the distribution of local A/B compartments in these single-gene domains. The compartment eigenvector in Hi-GDTsingle-identified gene domains showed positive values (A compartments) at gene domain borders but negative values (B compartments) inside gene domains (Fig. 3D). Consistent with the local compartment distribution of single-gene domains in Arabidopsis [34], the Hi-GDTsingle-identified gene domains predominantly had an A-B-A compartment pattern: A compartments at TSS and TES regions and B compartments at gene bodies (Fig. 3D, E). This observation is also in line with the finding that active TSSs are primarily found within local A compartments in both plants and animals [34, 68].
Figure 3:

Validation of single-gene domains identified by Hi-GDTsingle. (A) Pile-up images of Hi-C matrices for single-gene domains identified by Hi-GDTsingle. Pile-up images of genes with single-gene domains (Hi-GDTsingle) and without single-gene domains (other genes) at a 250-bp resolution are shown. Black lines indicate gene borders. (B–D) Metagene plots of DI score (B), chromatin accessibility (C), and compartment eigenvector (D) for Hi-GDTsingle-identified (Hi-GDTsingle) or remaining genes (other genes). (E) The proportion of local A/B compartments within genic regions with or without single-gene domains. The local A/B compartments at TSS, gene body, and TES regions were analyzed. (F) Expression levels of genes with or without single-gene domains. Log2(transcripts per million [TPM] + 1) values were used to quantify gene expression levels. The P value was calculated by a 2-sided Mann–Whitney U -test. (G) Distribution of chromatin states at TSS, gene body, and TES regions for genes with or without single-gene domains. Twenty-six chromatin states were used for analysis, including 6 constitutive heterochromatin (H1–H6), 6 facultative heterochromatin (F1–F6), 3 accessible intergenic (I1–I3), and 11 euchromatin (E1–E11) states.
We further explored the association of Hi-GDTsingle-identified gene domains with transcriptional states. In general, genes with local contact domain structures had higher expression levels than control genes without local contact domains, implying that transcriptionally active genes are likely to form single-gene domains (Fig. 3F). We also analyzed the chromatin states of Hi-GDTsingle-identified gene domains based on the 26 previously reported chromatin states, including 6 constitutive heterochromatin states (H1–H6), 6 facultative heterochromatin states (F1–F6), 3 intergenic states with the highest chromatin accessibility (I1–I3), and 11 euchromatin states (E1–E11) [53]. Genes with local contact domain structures were frequently located in euchromatin, whereas genes without these structures were more frequently located in facultative heterochromatin (Fig. 3G), indicating that Hi-GDTsingle successfully identifies gene domains in transcriptionally active states.
Validation of multigene domains identified by Hi-GDTmulti
In addition to identifying single-gene domains, Hi-GDT also enabled us to identify multigene domains by recognizing their insulating gene borders (Fig. 1C). Using Hi-GDTmulti, we identified 6,420 multigene domains at a 500-bp resolution, with a median size of 11,550 bp comprising an average of 3.66 genes in Arabidopsis (Fig. 4A, B). Numerous multigene domains with distinct domain boundaries were observed in Hi-C contact maps (Fig. 4C; Supplementary Fig. S5A). Pile-up images of Hi-GDTmulti-identified contact domains also revealed strong domain boundaries, regardless of the number of constituent genes (Fig. 4D). Genes located at the borders of the multigene domain were predominantly oriented in a convergent direction, leading to highly accessible TSS–TSS interactions (Supplementary Fig. S5B–D), which is consistent with previous findings [34].
Figure 4:

Validation of multigene domains identified by Hi-GDTmulti. (A) The number of multigene domains identified by Hi-GDTmulti based on their number of constituent genes. (B) The size distribution of identified multigene domains. (C) An example region in a Hi-C contact map showing multigene domains identified by Hi-GDTmulti. The blue lines indicate the boundaries of multigene domains, and black boxes indicate gene borders. (D) Pile-up images of Hi-C matrices for identified multigene domains based on the number of constituent genes. Black lines indicate the boundaries of identified multigene domains. (E, F) Metaplots of chromatin accessibility (E) and compartment eigenvector (F) for the identified multigene domains. (G) The distribution of chromatin states of genes located at boundaries of multigene domains (border), internal regions of multigene domains (inner), and outside of multigene domains (other genes). Twenty-six chromatin states were used for analysis, including 6 constitutive heterochromatin (H1–H6), 6 facultative heterochromatin (F1–F6), 3 accessible intergenic (I1–I3), and 11 euchromatin (E1–E11) states. (H) Expression levels of genes located at boundaries of multigene domains, internal regions of multigene domains, and outside of multigene domains. Log2(transcripts per million [TPM] + 1) values were used to quantify gene expression levels. P -values were calculated by a Kruskal–Wallis with Dunn’s post hoc test. In (G) and (H), genes forming single-gene domains were excluded. (I) The proportion of gene pairs with similar chromatin states in quadruple-gene domains. Four nearby genes lacking quadruple-gene domains were used for the random control (control genes). Gene 1 (g1) to gene 4 (g4) indicate the sequential order of genes from the 5′ to 3′ direction within a quadruple-gene domain. The ratio between gene pairs from quadruple-gene domains and control nearby genes is indicated above the graph.
Similar to single-gene domains, the borders of Hi-GDTmulti-identified multigene domains exhibited high chromatin accessibility (Fig. 4E) and were usually located in the local A compartment, leading to strong A-B-A compartmentalization (Fig. 4F). Consistent with this finding, genes located at the borders of multigene domains resided in euchromatin regions with high transcriptional activity, whereas genes inside multigene domains were present in facultative heterochromatin, which usually has low transcriptional activity (Fig. 4G, H; Supplementary Fig. S6). Furthermore, pairs of genes located at the contact domain borders were coordinated to have active chromatin states in common, whereas series of internal genes within a multigene domain had similar transcriptionally repressive heterochromatic states (Fig. 4I). These results suggest that Hi-GDT can reliably identify multigene domains in Arabidopsis.
Identification of differentially insulated single-gene domains associated with transcriptional activity depending on tissue type
Taking advantage of Hi-GDTsingle, we analyzed independent Hi-C datasets and compared them to examine the potential dynamics of single-gene domain structures. Our goal was to determine whether Hi-GDTsingle is reliable across various resolutions and when using different Hi-C datasets. Despite the variation in the number of identified contact domains depending on the resolution (Supplementary Fig. S7A), we obtained relatively consistent results at all resolutions examined: Hi-GDTsingle-identified single-gene domains were characterized by strong anchors at the TSS–TES regions and transcriptionally active states (Fig. 3A, F; Supplementary Fig. S7B, C). We also confirmed the reliability of Hi-GDTsingle for processing independent Arabidopsis Hi-C datasets obtained from different tissues or environmental conditions (Supplementary Fig. S7D).
Given that TADs are closely linked with cellular identity in animals [69–73], we reasoned that local contact domains might differ in Arabidopsis depending on tissue identity. We employed Hi-C and RNA sequencing datasets obtained independently from shoot and root tissues and extracted “tissue-specific single-gene domains” [38, 39, 54]. While most single-gene domains were common between shoot and root tissues, ∼25% of single-gene domains from each tissue were defined as tissue-specific single-gene domains (Fig. 5A). Pile-up analysis of these gene domains revealed stronger contact strengths in their respective tissues (Fig. 5B).
Figure 5:

Identification of single-gene domains in shoot and root tissues. (A) Venn diagram illustrating the number of Hi-GDTsingle-identified single-gene domains from shoot and root tissues. (B) Pile-up images of Hi-C matrices for single-gene domains identified from shoot and root tissues based on their tissue specificity. Images of shoot (top) and root (bottom) Hi-C contact maps are shown. Black lines indicate the boundaries of the identified single-gene domains; +, the presence of a single-gene domain; −, the absence of a single-gene domain. (C, D) Changes in gene expression levels for single-gene domains grouped by their tissue specificity. Absolute value log2(fold change) of gene expression levels (C) and the proportions of overlap with DEGs between shoot and root tissues (D) in each group are shown. (E) Tau scores of single-gene domains grouped by their tissue specificity. In (C) and (E), different letters indicate statistically significant differences determined by a Kruskal–Wallis with Dunn’s post hoc test (P < 0.05).
Genes with single-gene domains in both tissues exhibited constitutive expression patterns, with little differential expression (Fig. 5C, D) as well as relatively low Tau scores, a measure of tissue-specific expression (Fig. 5E), suggesting that constitutively organized single-gene domains are associated with stable gene expression. Unexpectedly, however, despite the association of single-gene domains with transcriptional activity, there was little correlation between tissue-specific gene domain formation and tissue-specific gene expression (Fig. 5D; Supplementary Fig. S8). Given that gene domain formation is associated with transcriptional activity, our results imply that the Hi-GDT analysis for 2 independent, different Hi-C datasets should be more optimized.
To better define the linkage of single-gene domain structural dynamics with gene expression changes, we put our focus on the gene domain structure changes of DEGs between shoot and root tissues and found that differences in gene expression correlated positively with intragenic contact frequency changes but also negatively with contact frequency changes within surrounding chromatin regions outside gene bodies, consistent with previous findings (Supplementary Fig. S9) [12, 33, 34]. We therefore developed Hi-GDTdiff to compare the gene domain insulation strength, which is represented by chromatin contact frequencies within intragenic regions over those within the 2-kb surrounding regions of individual genes, across different Hi-C datasets. Hi-GDTdiff identified differentially insulated single-gene domains in one tissue compared to the other tissue (Fig. 6A; Supplementary Table S2). As expected, single-gene domains with enhanced insulation strength, which are marked by increased intragenic contact frequencies and decreased surrounding contact frequencies, exhibited higher transcription activity in the corresponding tissue (Fig. 6B–E). To further validate our findings, we performed GO analysis on differentially insulated single-gene domains between shoot and root tissues. Shoot-specific GO terms, including “Carbohydrate biosynthetic process” and “Photosynthesis, light harvesting in photosystem I,” were significantly enriched for single-gene domains having enhanced insulation strength in shoots (Fig. 6F; Supplementary Table S3), whereas GO terms related to transport systems, including “Phloem unloading” and “Vascular transport,” were enriched for those with enhanced insulation strength in roots (Fig. 6G; Supplementary Table S3). These results support the relevance of our approach for searching for differentially insulated single-gene domains that are associated with changes in gene expression.
Figure 6:

Identification of shoot- and root-specific active single-gene domains. (A) Pile-up images showing differences between shoot and root Hi-C matrices for differentially insulated single-gene domains identified by Hi-GDTdiff. The differences in pile-up images of single-gene domains with enhanced insulation strength in shoots (upper) and roots (lower) are shown. Black lines indicate the boundaries of the identified single-gene domains. (B, C) Changes in gene expression levels for single-gene domains exhibiting enhanced insulation strength in shoots. Log2(fold change) of expression levels (B) and the proportions of overlap with DEGs between shoot and root tissues (C) in each group are shown. (D, E) Changes in gene expression levels for single-gene domains exhibiting enhanced insulation strength in roots. Log2(fold change) of expression levels (D) and the proportions of overlap with DEGs (E) in each group are shown. In (B) and (D), P values were calculated by 2-sided Mann–Whitney U -tests. (F, G) Top 10 significantly enriched GO terms of single-gene domains exhibiting enhanced insulation strength in shoots (F) and roots (G). GO terms with P < 0.05 and number of genes in each category >1 are shown.
We further applied our tool to other Hi-C datasets generated using Arabidopsis shoot tissues exposed to different environmental conditions or using different genotypes. Hi-GDTdiff identified only a small number of single-gene domains (n = 11) with differential insulation strength between mock and heat shock conditions, even with a weaker Q value cutoff (Q < 0.1), leading to mild changes in gene expression (Supplementary Fig. S10A, B; Supplementary Table S2) (see Discussion). In contrast, when we applied Hi-GDTdiff to different genotypes, it identified a substantial number of differentially insulated single-gene domains between wild type (Col-0) and bmi1a bmi1b bmi1c (bmi1abc), which also exhibited differential gene expression, supporting the reliability and broad applicability of our tool (Supplementary Fig. S10C–F; Supplementary Table S2). Overall, these results suggest that Hi-GDTdiff can be used to compare single-gene domain structures across different, independent Hi-C datasets and extract differentially insulated single-gene domains associated with changes in transcriptional activity. They also suggest that gene domain dynamics are more closely associated with tissue identity than with plant responses to environmental stimuli.
Single-gene domains are dynamically regulated during cellular reprogramming in Arabidopsis
Considering the close association between local contact domains and tissue identity, we hypothesized that gene domains are dynamically regulated during the cellular reprogramming process, similar to TADs in animal systems [69, 71, 74–76]. To test this hypothesis, we generated high-resolution Hi-C data with 4 biological replicates from Arabidopsis leaf explant–derived calli, which undergo genome-wide reprogramming of cellular identity (Supplementary Table S4). Based on the fact that the 4 replicates of callus Hi-C data showed a high Spearman correlation coefficient with each other, we merged them into single-callus Hi-C data that exhibited a distance-decay model similar to that of shoot and root Hi-C data (Supplementary Fig. S11A, B). We also performed callus RNA sequencing in parallel and confirmed that the data were also highly correlated each other (Supplementary Fig. S11C). The high-resolution Hi-C contact maps of callus tissue showed numerous local contact domains, most with boundaries near gene borders (Fig. 7A), as observed in other Arabidopsis tissues (Fig. 4C), confirming that gene domains are prevalent 3D chromatin conformation units in Arabidopsis.
Figure 7:

Identification of callus-specific active single-gene domains. (A) Hi-C contact map of callus tissue with local contact domains identified by Hi-GDT. Black squares indicate gene borders, and blue lines indicate the boundaries of the identified contact domains. (B) Pile-up images showing differences between shoot and callus Hi-C matrices for differentially insulated single-gene domains identified by Hi-GDTdiff. The differences in pile-up images of single-gene domains with enhanced insulation strength in shoot (left) and callus tissues (right) are shown. Black lines indicate the boundaries of the identified single-gene domains. (C, D) Changes in gene expression levels for single-gene domains exhibiting enhanced insulation strength in calli. Log2(fold change) of expression levels (C) and the proportions of overlap with DEGs between leaf explant and callus tissues (D) in each group are shown. In (C), the P value was calculated by a 2-sided Mann–Whitney U test. (E) Top 20 significantly enriched GO terms of single-gene domains exhibiting enhanced insulation strength in calli. GO terms with P < 0.05 and number of genes in each category >1 are shown.
We then extracted genes showing dynamic changes in contact domain structures between shoots and calli. Although global Hi-C contact maps of calli showed a high degree of similarity to those of shoots, as evidenced by a Spearman correlation coefficient of 0.88 at a 25-kb resolution, we identified 961 differentially insulated single-gene domains, including 697 genes with enhanced insulation in calli (Fig. 7B; Supplementary Table S2), which were expressed at higher levels in calli compared to leaf explants (Fig. 7C, D). Moreover, GO terms related to callus development, such as “auxin biosynthetic process,” and “regulation of cell growth,” were significantly enriched for these single-gene domains (Fig. 7E; Supplementary Table S3). These results indicate that gene domain structures are tightly reorganized during the reprogramming of tissue identity, which may play a key role in this process.
Multigene domains are dynamically regulated by tissue type
We then analyzed dynamic changes in multigene domain structures in different tissues. We extracted tissue-specific multigene domains consisting of up to 10 genes: 2,109 shoot-specific, 4,305 root-specific, and 4,311 common multigene domains (Fig. 8A). Since diverse types of structural changes can occur in multigene domains, we focused on a specific case: a tissue-specific change in multigene domain structure from a dual-gene domain to a triple-gene domain with one side of the multigene domain border fixed. For instance, the shoot-specific dual-gene domain consisting of AT5G46260 and AT5G46270 changed to the triple-gene domains incorporating their 3′-downstream gene AT5G46280 in roots (Fig. 8B). Pile-up images demonstrated the expansion of multigene domain structures and tissue-dependent structural dynamics (Fig. 8C; Supplementary Fig. S12A).
Figure 8:

Identification of dynamically regulated multigene domains in different tissue types. (A) Venn diagram illustrating the number of multigene domains identified from shoot and root Hi-C datasets at a 500-bp resolution. (B) An example region showing the transition from a dual-gene domain to a triple-gene domain depending on tissue type. Upper-right triangles represent Hi-C maps in shoots, while lower-left triangles represent Hi-C maps in roots. Black boxes indicate the gene borders, and green and navy blue lines indicate the boundaries of identified multigene domains in shoot and root tissues, respectively. (C) Pile-up images of Hi-C matrices for differential multigene domains between shoot and root tissues. Dual-gene domains specifically identified in shoots and expanded to triple-gene domains in roots were collected. Pile-up images of dual-gene domains expanded to the 5′ direction (top) and the 3′ direction (bottom) in the formation of triple-gene domains are shown. Black lines indicate the boundaries of dual-gene domains. (D, E) Changes in gene expression levels for domain-internalized genes in roots (internalized). Log2(fold change) of expression levels (D) and the proportions of overlap with DEGs between shoot and root tissues (E) for domain-internalized genes from dual-gene domains compared to the control genes located in dual-gene domains without structural changes in both tissues (unchanged) are shown. (F, G) Changes in gene expression levels for domain-internalized genes in shoots (internalized). Log2(fold change) of expression levels (F) and the proportions of overlap with DEGs between shoot and root tissues (G) for domain-internalized genes from dual-gene domains compared to the control genes located in dual-gene domains without structural changes in both tissues (unchanged) are shown. (H, I) Changes in gene expression levels for domain-insulated genes in callus tissue, which are located within triple-gene domains in shoots but insulated at a border of a dual-gene domain in callus (insulated). Log2(fold change) of expression levels (H) and the proportions of overlap with DEGs between leaf explant and callus tissues (I) for domain-insulated genes from triple-gene domains compared to the control genes located in dual-gene domains without structural changes in both tissues (unchanged) are shown. In (D), (F), and (H), P values were calculated by 2-sided Mann–Whitney U -tests. In (B) to (I), genes that form dual-gene domains with their TSSs at the domain border were included in the analysis.
This approach allowed us to identify genes with multigene domain dynamics in association with gene expression levels by simply comparing the Hi-GDTmulti results from different Hi-C datasets. Genes located at the borders of multigene domains are transcriptionally active, whereas genes within multigene domains are usually silenced (Fig. 4G, H) [34]. Thus, structural changes from a dual-gene domain to a triple-gene domain may induce changes in the expression of genes comprising the domain: border genes of dual-gene domains that become internalized in triple-gene domains during the structural transition are expected to be silenced. It should also be noted that since a TSS has higher chromatin accessibility than a TES [77, 78], changes in gene expression were more obvious for genes whose TSSs, located at domain borders, were moved to the internal region during structural changes (Supplementary Fig. S13A–C).
Genes of shoot-specific dual-gene domains with their TSSs at domain borders that were internalized in roots (domain-internalized genes in roots) exhibited high expression in shoots but low expression in roots (Fig. 8D, E; Supplementary Table S5). We also identified domain-internalized genes in shoots, meaning those that formed dual-gene domains in roots but were internalized into triple-gene domains in shoots. These genes strongly overlapped with DEGs that were more highly expressed in roots than in shoots (Fig. 8F, G; Supplementary Table S5).
Finally, we identified dynamic multigene domain structures by comparing dual- and triple-gene domains between callus and shoot tissues. In particular, we focused on genes in the middle of triple-gene domains in shoots that moved to the borders of dual-gene domains in calli (domain-insulated genes in calli) (Supplementary Fig. S12B, C). These genes, including CINNAMOYL COA REDUCTASE (CCR2) and NITRATE TRANSPORTER (NRT1.8), were indeed transcriptionally activated in callus tissues (Fig. 8H, I; Supplementary Table S5). Overall, our results demonstrate that Hi-GDT is an effective tool for identifying local chromatin structures, including single-gene and multigene domains. Furthermore, this tool can also be used to identify gene domains with dynamic structural changes in association with transcriptional activity.
Application of Hi-GDT in other plant species
Given that gene domains have been identified in other plant species, including tomato, maize and Marchantia (34), we examined the applicability of Hi-GDT to various plant species. As expected, both Hi-GDTsingle and Hi-GDTmulti successfully identified single-gene and multigene domains characterized by strong internal contact frequencies (Fig. 9A, B; Supplementary Fig. S14). Furthermore, consistent with the findings in Arabidopsis, genes with single-gene domain structures or genes located at the borders of multigene domains were transcriptionally active, whereas genes located in the middle of multigene domains were transcriptionally repressed (Fig. 9C, D). However, given a weak association between changes in insulation strength and gene expression levels in single-gene domains from tomato and Marchantia, Hi-GDTdiff is most effective in high-depth Arabidopsis Hi-C analysis (Supplementary Fig. S15).
Figure 9:

Application of Hi-GDT on other plant species. (A) Pile-up images of Hi-C matrices for single-gene domains identified by Hi-GDTsingle in tomato, maize, and Marchantia. Pile-up images of genes with single-gene domains (Hi-GDTsingle) and without single-gene domains (other genes) at a 250-bp resolution are shown. Black lines indicate gene borders in each species. (B) Pile-up images of Hi-C matrices for identified multigene domains in tomato, maize, and Marchantia based on the number of constituent genes. Black lines indicate the boundaries of identified multigene domains. (C) Expression levels of genes with or without single-gene domains. P values were calculated by 2-sided Mann–Whitney U tests. (D) Expression levels of genes located at boundaries of multigene domains, internal regions of multigene domains, and outside of multigene domains. P values were calculated by Kruskal–Wallis with Dunn’s post hoc tests. Genes forming single-gene domains were excluded from all categories. In (C) and (D), log2(transcripts per million [TPM] + 1) values were used to quantify gene expression levels. (E) Pile-up image of Hi-C matrices for tomato genes homologous to Arabidopsis single-gene domains. All other genes in the tomato genome were used as control genes. Black lines indicate gene borders. (F) Pile-up image of Hi-C matrices for tomato-adjacent gene pairs homologous to Arabidopsis dual-gene domains. All other adjacent gene pairs in the tomato genome were used as control gene pairs. Black lines indicate the boundaries of identified adjacent gene pairs. In (E) and (F), homologous genes or gene pairs within the colinear block of tomato and Arabidopsis were used.
Taking advantage of the broad applicability of Hi-GDT, we examined whether the gene domain structure is evolutionarily conserved across plant species. To this end, we identified colinear blocks between Arabidopsis and tomato genomes, as well as between Arabidopsis and maize genomes. Notably, tomato and maize genes, which are homologous to Arabidopsis single-gene and dual-gene domains in colinear blocks, exhibited single- or dual-gene domain structures (Fig. 9E, F; Supplementary Fig. S16). Overall, these results indicate that gene domain structures and their formation mechanisms are widely conserved across various plant species.
Discussion
Numerous bioinformatics tools, including those using deep learning models, have been developed to identify hierarchical chromatin structures in animal genomes [43–47, 79, 80]. Although some of them have been applied to analyze TAD-like structures and compartment domains in large genome plants [28, 43–46], they usually showed low reliability and reproducibility in identifying fine-scale chromatin contact domains (Supplementary Fig. S4A). This limitation is mainly due to the combined effects of weak interaction strength within contact domains and high noise of the Hi-C matrix at a high resolution. To overcome this limitation, we developed a novel tool designed to analyze fine-scale local chromatin contact domains, with a particular focus on single-gene and multigene domains that are established at gene boundaries (Supplementary Fig. S3) [34]. This semi-targeted approach allows for high sensitivity and reproducibility, outperforming existing tools (Fig. 2; Supplementary Fig. S4B). For example, although OnTAD, which is optimized to identify hierarchical (nested) contact domain structures [47], identified a large number of potential fine-scale contact domains (Fig. 2A, B), its low reproducibility may limit the broad application (Fig. 2; Supplementary Fig. S4B). Indeed, our tool successfully identified local contact domains in various plant species, including Arabidopsis, tomato, soybean, rice, maize, Jatropha curcas, and Marchantia, and found that gene domains are conserved across plant species. Hi-GDT showed higher reproducibility than conventional domain callers, enabling effective analysis at a high resolution. This advantage makes it feasible to analyze 3D gene domain dynamics in different tissues and environmental conditions in association with their transcriptional activity.
Our findings confirm that changes in local chromatin structure, especially gene domains, are associated with transcriptional changes. In general, local contact domains identified by Hi-GDT showed high chromatin accessibility at their boundaries (Figs. 3C and 4E). Moreover, RNA polymerase II (RNA Pol II) is highly enriched at the gene domain boundaries (Supplementary Fig. S17), and the binding of multiple transcription factors is crucial for the formation of local 3D chromatin structure [7, 34, 81]. Hence, we suggest that the concerted action of transcription factors and RNA Pol II at gene borders plays a crucial role in shaping local chromatin structure in Arabidopsis. Indeed, transcriptional activity and RNA Pol II binding influence the single-gene domain structures of both animals and plants, as revealed by the finding that inhibiting RNA Pol II activity alters local chromatin structure at individual genes [12, 33].
Despite the strong association between local contact domain formation and transcriptional activity, the biochemical relevance of local contact domain formation to the regulation of 3D-conformation-dependent gene expression remains unclear. Considering the extremely short intergenic sequences in the small genome of Arabidopsis, local contact domain formation might be particularly important in that proximal cis-regulatory elements near gene borders play an additional role in regulating adjacent genes. Indeed, deletion of the promoter region of a gene affects expression of its adjacent gene connected by a promoter–promoter loop [82]. Alternatively, given that these local contact domains frequently form at gene borders, efficient transcription might be facilitated by the anchoring of border regions, similar to the transcription factory model that has been suggested for plants with large genomes [26, 28]. Integrative analysis of Hi-GDT data with intergenic genomic components, such as super enhancers, could provide a comprehensive understanding of the intricate principles governing the relationship between 3D chromatin structure and gene regulation.
Our understanding of the biological relevance of local contact domains in plants also remains limited. Using Hi-GDT, we determined that stable local contact domain structures are associated with constitutive gene expression. In addition, genes with the structural dynamics of local contact domains are frequently related to tissue identity. Notably, the structural plasticity of gene domains in response to environmental stimuli (such as heat) is relatively low (Supplementary Fig. S10), suggesting that local chromatin structures are more tightly linked with cell or tissue identity compared to environmental conditions. In this context, the low structural dynamics of gene domains in response to environmental stimuli might also be due to the dilution effect of heterogeneous cell types. Hi-GDT should facilitate functional studies of local contact domains in Arabidopsis and provide broader insight into the biological impact of local chromatin organization in plants.
We placed a particular emphasis on the relevance of our tool for analyzing dynamic structural changes associated with transcriptional activity at the gene scale, as this issue has not previously been analyzed due to the lack of optimal analytical tools. Although the analysis of local chromatin domains requires high sequencing depth for precise analysis, this limitation can be overcome using micrococcal nuclease–based techniques such as highly sensitive transposase-mediated analysis of chromatin (Hi-TrAC), Micro-C, or chemical-crosslinking assisted proximity capture (CAP-C) [81–84]. Notably, the newer cutting-edge techniques known as CAP-C and Micro-C-XL can be used to generate contact maps at an extremely high resolution of 200 bp to support analysis of fine-scale chromatin structure in plants [33, 82]. Hi-GDT is compatible with high-resolution contact maps, and the combinations of these techniques with Hi-GDT should be a powerful tool for analyzing fine-scale local contact domains.
Availability of Source Code and Requirements
Project name: Hi-GDT
Project homepage: https://github.com/CDL-HongwooLee/Hi-GDT
Operating system(s): Linux
Programming language: Python
Other requirements: Python 3.8, subprocess, numpy, pickle, multiprocessing, PIL, functools, itertools, scipy
License: MIT license
Software Heritage PID: swh:1:snp:a4d7864a7adfdf451f1058edde2c0014d7b51fe6
Supplementary Material
Hao Wu -- 11/9/2024
Hao Wu -- 1/12/2025
Yusen Ye -- 11/20/2024
Yusen Ye -- 1/20/2025
Author Contributions
P.J.S. and H.L. conceived and designed the study and wrote the manuscript. H.L. performed all analyses and experiments.
Contributor Information
Hongwoo Lee, Department of Chemistry, Seoul National University, Seoul 08826, Korea.
Pil Joon Seo, Department of Chemistry, Seoul National University, Seoul 08826, Korea; Plant Genomics and Breeding Institute, Seoul National University, Seoul 08826, Korea.
Additional Files
Supplementary Fig. S1. Strategy of Hi-GDTmulti for multigene domain identification. (A, B) Schematic diagrams illustrating the target and control regions in a Hi-C contact map, which were defined for multigene domain identification by Hi-GDTmulti. The regions compared for multigene domains composed of 2 (A) and 3 (B) adjacent genes are shown. Target regions are shown in red and control regions in green or yellow. Boundaries of a multigene domain are indicated by blue lines. The lines at the bottom indicate the diagonal axis in a Hi-C contact map, and each black triangle indicates an individual gene.
Supplementary Fig. S2. Benchmarking of Hi-GDT against conventional domain callers. (A) Density plot illustrating the size distribution of contact domains identified by each domain caller at 1,000 bp resolution. (B) Pile-up images of Hi-C contact matrices for contact domains identified by each domain caller at 1,000 bp resolution. Black lines indicate the boundaries of the identified contact domains.
Supplementary Fig. S3. Genomic contexts at boundary regions of local contact domains identified by conventional domain callers. (A , B) Proportions of domain boundaries overlapping with gene border, gene body, and intergenic regions at various resolutions. The overlap between identified contact domains and intra/intergenic regions of annotated protein-coding genes (A) or their 5-kb-shifted control regions (B) was investigated. The size of the domain boundaries was set to match the given resolution, and the size of gene borders was defined as 200 bp from both TSSs or TESs in order to measure the overlap.
Supplementary Fig. S4. Comparison of the reproducibility of local contact domains identified by Hi-GDT and conventional domain callers. (A) Reproducibility rates of local contact domain identification by conventional domain callers between technical replicates subsampled (80%) from a merged Hi-C dataset, biological replicates, and datasets from different batches. The identified contact domains, which exactly match their boundaries with another contact domain, were considered to represent a reproducible prediction. (B) Comparison of the reproducibility rates of local contact domain identification, with domain boundaries adjusted to coincide with gene border regions. In (A) and (B), reproducibility rates were calculated as the ratio of the number of overlapping domains to the number of domains in a single dataset.
Supplementary Fig. S5. Characteristics of Hi-GDTmulti-identified multigene domains. (A) An example region in a Hi-C OE map annotated with multigene domains identified by Hi-GDTmulti. The blue lines indicate the boundaries of multigene domains, and black boxes indicate gene borders. (B-D) The number of identified multigene domains categorized based on the orientations of their constituent genes. The distribution of gene orientations of constituent genes within dual-gene domains (B), triple-gene domains (C), and quadruple-gene domains (D) are shown. The colors indicate the gene orientations of gene pairs at the boundaries of multigene domains: convergent (pink), parallel (blue), and divergent (brown).
Supplementary Fig. S6. Expression levels of constituent genes of Hi-GDTmulti-identified multigene domains. (A-C) Expression levels of constituent genes of multigene domains. The expression levels of dual-gene domains (A), triple-gene domains (B), and quadruple-gene domains (C) identified by Hi-GDTmulti are shown. Gene 1 to Gene 4 indicate the sequential order of genes from the 5’ to 3’ direction within a multigene domain. Genes outside multigene domains were used as a control (other genes). Log2(transcripts per million [TPM] + 1) values were used to quantify gene expression levels. Different letters indicate statistically significant differences determined by a Kruskal–Wallis with Dunn’s post-hoc test (P-value < 0.05).
Supplementary Fig. S7. Identification of single-gene domains by Hi-GDTsingle at various resolutions using different datasets. (A-C) Profiles of single-gene domains identified by Hi-GDTsingle at 1-restriction fragment (1 f) and 2 f, 250 bp, and 500 bp resolution. In (A), the number of single-gene domains identified by Hi-GDTsingle at each resolution are shown. Pile-up images (B) and expression levels (C) for Hi-GDTsingle-identified (Hi- GDTsingle) or remaining (other genes) genes at each resolution are shown. In (C), P-values were calculated by two-sided Mann–Whitney U-tests. (D) Pile-up images of Hi-C matrices for genes with or without single-gene domains identified by Hi-GDTsingle from various Hi-C datasets at 250 bp resolution. Hi-C datasets obtained from roots or heat shock-exposed shoots were used for pile-up analysis. In (B) and (D), black lines indicate gene borders.
Supplementary Fig. S8. Transcript levels of genes with shoot- and root-specific single-gene domains. (A-C) Transcript levels of genes with single-gene domains grouped by their tissue specificity. Expression levels in shoot and root tissues (A), log2(fold change) of expression levels (B), and the number of single-gene domains overlapping with DEGs between shoot and root tissues (C) in each group are shown. +, the presence of a single-gene domain; −, the absence of a single-gene domain. In (A), log2(transcripts per million [TPM] + 1) values were used to quantify expression levels. In (A) and (B), different letters indicate statistically significant differences determined by a Kruskal-Wallis with Dunn’s post-hoc test (P-value < 0.05).
Supplementary Fig. S9. Structural alterations of single-gene domains in association with changes in gene expression. (A and B) Pile-up images showing differences between shoot and root Hi-C matrices for differentially expressed genes. Up-regulated genes (A) and down-regulated genes (B) in roots compared to shoots were included in the pile-up analysis. Genes with no significant changes in expression levels were used as control genes. Differences in pile-up images between up-regulated and control genes (A), and between down-regulated and control genes (B) are shown. Black lines indicate the boundaries of the identified single-gene domains.
Supplementary Fig. S10. Identification of differentially-insulated single-gene domains between different Hi-C datasets. (A and B) Changes in gene expression levels for single-gene domains with enhanced insulation after heat shock treatment. Log2(fold change) of expression levels (A) and the proportions of overlap with DEGs between mock and heat conditions (B) in each group are shown. (C and D) Changes in gene expression levels for single-gene domains with enhanced insulation in WT compared to bmi1abc mutant. Log2(fold change) of expression levels (C) and the proportions of overlap with DEGs between WT and bmi1abc mutant (D) in each group are shown. (E and F) Changes in gene expression levels for single-gene domains with enhanced insulation in bmi1abc mutant compared to WT. Log2(fold change) of expression levels (E) and the proportions of overlap with DEGs between WT and bmi1abc mutant (F) in each group are shown. In (A), (C) and (E), P-values were calculated by two-sided Mann–Whitney U-tests.
Supplementary Fig. S11. Validation of callus Hi-C and RNA-seq data. (A) Correlation plot for four callus Hi-C replicates. Spearman’s correlation coefficient values between each Hi-C replicate at 25-kb resolution are shown. (B) A distance-decay plot of callus Hi-C data with other public Hi-C data from shoot and root tissues. Four replicates were merged to estimate distance-decay model of callus Hi-C data. SCALE-normalized Hi-C counts at 25-kb resolution were estimated. (C) A correlation plot for RNA-seq data produced from leaf explant and callus tissues. Spearman’s correlation coefficient values between all RNA-seq replicates are shown. Hierarchical clustering was performed based on the Spearman’s correlation coefficient values.
Supplementary Fig. S12. Pile-up analysis of multigene domains with dynamic structural changes between different tissue types. (A) Pile-up images of Hi-C matrices for differential multigene domains between shoot and root tissues. Dual-gene domains specifically identified in shoots and expanded to triple-gene domains in roots were collected. Pile-up images of triple-gene domains expanded in the 5’ direction (top) or the 3’ direction (bottom) from dual-gene domains are shown. (B and C) Pile-up images of Hi-C matrices for differential multigene domains between shoot and callus tissues. Triple-gene domains specifically identified in shoots and insulated to dual-gene domains in callus were collected. In (B), pile-up images of dual-gene domains expanded to the 5’ direction (top) or the 3’ direction (bottom) in the formation of triple-gene domains are shown. In (C), pile-up images of triple-gene domains expanded in the 5’ direction (top) or the 3’ direction (bottom) from dual-gene domains are shown. In (A) and (C), black lines indicate the boundaries of triplegene domains. In (B), black lines indicate the boundaries of dual-gene domains.
Supplementary Fig. S13. Changes in the expression levels of genes with structural changes in multigene domains. (A and B) Changes in gene expression levels for domain-internalized genes in roots (internalized). Log2(fold change) of expression levels (A) and the proportions of overlap of DEGs between shoot and root tissues (B) for domain-internalized genes from dual-gene domains as compared to the control genes located in dual-gene domains without structural changes in both tissues (unchanged) are shown. In (A), the P-value was calculated by a two-sided Mann–Whitney U-test. (C) Log2(fold change) of gene expression levels between shoot and root tissues based on the orientations of constituent genes within dual-gene domains. Expression changes of dual-gene domains expanded to the 5’ direction (left) or the 3’ direction (right) in the formation of triple-gene domains are shown. Dual-gene domains expanded to triple-gene domains with internalization of TSSs at dual-gene domain borders are shown in orange.
Supplementary Fig. S14. Gene domains identified by Hi-GDT in various plant species. (A) Pile-up images of Hi-C matrices for single-gene domains identified by Hi-GDTsingle in rice, soybean, and Jatropha curcas (J. curcas). Pile-up images of genes with single-gene domains (Hi-GDTsingle) and without single-gene domains (other genes) at 250-bp resolution are shown. Black lines indicate gene borders in each species. (B) Pile-up images of Hi-C matrices for identified multigene domains in rice, soybean, and J. curcas based on the number of constituent genes. Black lines indicate the boundaries of identified multigene domains.
Supplementary Fig. S15. Identification of differentially-insulated single-gene domains between different Hi-C datasets from tomato and Marchantia. (A and B) Changes in gene expression levels for single-gene domains with reduced insulation after heat shock treatment in tomato. Log2(fold change) of expression levels (A) and the proportions of overlap with DEGs between mock and heat conditions (B) in each group are shown. (C and D) Changes in gene expression levels for single-gene domains with enhanced insulation after heat shock treatment in tomato. Log2(fold change) of expression levels (C) and the proportions of overlap with DEGs between mock and heat conditions (D) in each group are shown. (E and F) Changes in gene expression levels for singlegene domains with enhanced insulation in WT compared to tcp1 mutant in Marchantia. Log2(fold change) of expression levels (E) and the proportions of overlap with DEGs between WT and tcp1 mutant (F) in each group are shown. (G and H) Changes in gene expression levels for single-gene domains with enhanced insulation in tcp1 compared to WT in Marchantia. Log2(fold change) of expression levels (G) and the proportions of overlap with DEGs between WT and tcp1 mutant (H) in each group are shown. In (A), (C), (E) and (G), P-values were calculated by two-sided Mann–Whitney U-tests.
Supplementary Fig. S16. Local contact domain structures of maize genes homologous to Arabidopsis single-gene domains and dual-gene domains (A) Pile-up image of Hi-C matrices for maize genes homologous to Arabidopsis single-gene domains. All other genes in maize genome were used as control genes. Black lines indicate gene borders. (B) Pileup image of Hi-C matrices for maize adjacent gene pairs homologous to Arabidopsis dual-gene domains. All other gene pairs in maize genome were used as control gene pairs. Black lines indicate the boundaries of identified adjacent gene pairs. Homologous genes or gene pairs within the colinear block of maize and Arabidopsis were used.
Supplementary Fig. S17. Metaplots of RNA polymerase II (RNA Pol II) enrichment at the identified local contact domains. (A, B) RNA Pol II enrichment level at the single-gene domains (A) and multigene domains (B) identified by Hi-GDT. In (A), single-gene domains identified by Hi-GDTsingle and remaining genes (other genes) are shown. (C) RNA Pol II enrichment level at the local contact domains identified by HiCExplorer, OnTAD, or Hi-GDT at 250 bp resolution. The metaplot of those identified by Arrowhead is not displayed due to the low number of identified domains.
Supplementary Table S1. NGS datasets used in this study.
Supplementary Table S2. Differentially insulated single-gene domains identified by Hi-GDTdiff.
Supplementary Table S3. GO enrichment analysis for differentially insulated single-gene domains.
Supplementary Table S4. Reads alignment statistics of callus Hi-C and RNA-seq data.
Supplementary Table S5. Domain-internalized and domain-boundary genes of multigene domains in different tissue types.
Abbreviations
3D: three-dimensional; DEG: differentially expressed gene; DI: directionality index; GO: Gene Ontology; OE: observed contact frequency/expected contact frequency; TAD: topologically associating domain; TES: transcriptional end site; TPM: transcripts per million; TSS: transcriptional start site.
Funding
This work was supported by the Basic Science Research (RS-2025-00517108) and Basic Research Laboratory (NRF-2022R1A4A3024451) programs provided by the National Research Foundation of Korea.
Data Availability
All publicly available datasets used in this study are listed in Supplementary Table S1. The source codes and step-by-step instructions of Hi-GDT are available from the GitHub repository [85]. Snapshots of the code and data are available in Software Heritage [86]. Raw data and all implemented codes used in this study have been deposited at Zenodo [52]. Hi-C and RNA-seq data produced in this study were deposited in NCBI under accession number PRJNA1112149.
Competing Interests
The authors declare no competing interests.
References
- 1. Dixon JR, Jung I, Selvaraj S, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–36. 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fukaya T, Lim B, Levine M. Enhancer control of transcriptional bursting. Cell. 2016;166:358–68. 10.1016/j.cell.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gonzalez-Sandoval A, Gasser SM. On TADs and LADs: spatial control over gene expression. Trends Genet. 2016;32:485–95. 10.1016/j.tig.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 4. Gu B, Swigut T, Spencley A, et al. Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science. 2018;359:1050–55. 10.1126/science.aao3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaushal A, Mohana G, Dorier J, et al. CTCF loss has limited effects on global genome architecture in Drosophila despite critical regulatory functions. Nat Commun. 2021;12:1011. 10.1038/s41467-021-21366-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schoenfelder S, Fraser P. Long-range enhancer–promoter contacts in gene expression control. Nat Rev Genet. 2019;20:437–55. 10.1038/s41576-019-0128-0. [DOI] [PubMed] [Google Scholar]
- 7. Stadhouders R, Vidal E, Serra F, et al. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet. 2018;50:238–49. 10.1038/s41588-017-0030-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–80. 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nora EP, Lajoie BR, Schulz EG, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–85. 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rao SSP, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–80. 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rao SSP, Huang S-C, Glenn SHB, et al. Cohesin loss eliminates all loop domains. Cell. 2017;171:305–20. e24. 10.1016/j.cell.2017.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rowley MJ, Nichols MH, Lyu X, et al. Evolutionarily conserved principles predict 3D chromatin organization. Mol Cell. 2017;67:837–52. e7. 10.1016/j.molcel.2017.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwarzer W, Abdennur N, Goloborodko A, et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature. 2017;551:51–56. 10.1038/nature24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rowley MJ, Corces VG. Organizational principles of 3D genome architecture. Nat Rev Genet. 2018;19:789–800. 10.1038/s41576-018-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beagan JA, Phillips-Cremins JE. On the existence and functionality of topologically associating domains. Nat Genet. 2020;52:8–16. 10.1038/s41588-019-0561-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alipour E, Marko JF. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res. 2012;40:11202–12. 10.1093/nar/gks925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sanborn AL, Rao SSP, Huang S-C, et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A. 2015;112:E6456–65. 10.1073/pnas.1518552112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fudenberg G, Imakaev M, Lu C, et al. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016;15:2038–49. 10.1016/j.celrep.2016.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nichols MH, Corces VG. Principles of 3D compartmentalization of the human genome. Cell Rep. 2021;35:109330. 10.1016/j.celrep.2021.109330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heger P, Marin B, Bartkuhn M, et al. The chromatin insulator CTCF and the emergence of metazoan diversity. Proc Natl Acad Sci U S A. 2012;109:17507–12. 10.1073/pnas.1111941109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dong P, Tu X, Chu P-Y, et al. 3D Chromatin architecture of large plant genomes determined by local A/B compartments. Mol Plant. 2017;10:1497–509. 10.1016/j.molp.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 22. Dong Q, Li N, Li X, et al. Genome-wide Hi-C analysis reveals extensive hierarchical chromatin interactions in rice. Plant J. 2018;94:1141–56. 10.1111/tpj.13925. [DOI] [PubMed] [Google Scholar]
- 23. Dong P, Tu X, Li H, et al. Tissue-specific Hi-C analyses of rice, foxtail millet and maize suggest non-canonical function of plant chromatin domains. J Integr Plant Biol. 2020;62:201–17. 10.1111/jipb.12809. [DOI] [PubMed] [Google Scholar]
- 24. Huang Y, An J, Sircar S, et al. HSFA1a modulates plant heat stress responses and alters the 3D chromatin organization of enhancer-promoter interactions. Nat Commun. 2023;14(1):469. 10.1038/s41467-023-36227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang M, Wang P, Lin M, et al. Evolutionary dynamics of 3D genome architecture following polyploidization in cotton. Nat Plants. 2018;4:90–97. 10.1038/s41477-017-0096-3. [DOI] [PubMed] [Google Scholar]
- 26. Concia L, Veluchamy A, Ramirez-Prado JS, et al. Wheat chromatin architecture is organized in genome territories and transcription factories. Genome Biol. 2020;21:104. 10.1186/s13059-020-01998-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun Y, Dong L, Zhang Y, et al. 3D genome architecture coordinates trans and cis regulation of differentially expressed ear and tassel genes in maize. Genome Biol. 2020;21:143. 10.1186/s13059-020-02063-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liao Y, Wang J, Zhu Z, et al. The 3D architecture of the pepper genome and its relationship to function and evolution. Nat Commun. 2022;13:3479. 10.1038/s41467-022-31112-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pei L, Huang X, Liu Z, et al. Dynamic 3D genome architecture of cotton fiber reveals subgenome-coordinated chromatin topology for 4-staged single-cell differentiation. Genome Biol. 2022;23:45. 10.1186/s13059-022-02616-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang Y, Sicar S, Ramirez-Prado JS, et al. Polycomb-dependent differential chromatin compartmentalization determines gene coregulation in Arabidopsis. Genome Res. 2021;31:1230–44. 10.1101/gr.273771.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deng L, Zhou Q, Zhou J, et al. 3D organization of regulatory elements for transcriptional regulation in Arabidopsis. Genome Biol. 2023;24:181. 10.1186/s13059-023-03018-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yin X, Romero-Campero FJ, Yang M, et al. Binding by the Polycomb complex component BMI1 and H2A monoubiquitination shape local and long-range interactions in the Arabidopsis genome. Plant Cell. 2023;35:2484–503. 10.1093/plcell/koad112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun L, Zhou J, Xu X, et al. Mapping nucleosome-resolution chromatin organization and enhancer-promoter loops in plants using Micro-C-XL. Nat Commun. 2024;15:35. 10.1038/s41467-023-44347-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee H, Seo Pil J. Accessible gene borders establish a core structural unit for chromatin architecture in Arabidopsis. Nucleic Acids Res. 2023;51:10261–77. 10.1093/nar/gkad710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nützmann H-W, Doerr D, Ramírez-Colmenero A, et al. Active and repressed biosynthetic gene clusters have spatially distinct chromosome states. Proc Natl Acad Sci U S A. 2020;117:13800–9. 10.1073/pnas.1920474117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koo D, Lee HG, Bae SH, et al. Callus proliferation-induced hypoxic microenvironment decreases shoot regeneration competence in Arabidopsis. Mol Plant. 2024;17:395–408. 10.1016/j.molp.2024.01.009. [DOI] [PubMed] [Google Scholar]
- 37. Sayers EW, Bolton EE, Brister JR, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022;50:D20–26. 10.1093/nar/gkab1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun L, Jing Y, Liu X, et al. Heat stress-induced transposon activation correlates with 3D chromatin organization rearrangement in Arabidopsis. Nat Commun. 2020;11:1886. 10.1038/s41467-020-15809-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun L, Cao Y, Li Z, et al. Conserved H3K27me3-associated chromatin looping mediates physical interactions of gene clusters in plants. J Integr Plant Biol. 2023;65:1966–82. 10.1111/jipb.13502. [DOI] [PubMed] [Google Scholar]
- 40. Wang C, Liu C, Roqueiro D, et al. Genome-wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Res. 2015;25:246–56. 10.1101/gr.170332.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu C, Wang C, Wang G, et al. Genome-wide analysis of chromatin packing in Arabidopsis thaliana at single-gene resolution. Genome Res. 2016;26:1057–68. 10.1101/gr.204032.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Karaaslan ES, Wang N, Faiß N, et al. Marchantia TCP transcription factor activity correlates with three-dimensional chromatin structure. Nat Plants. 2020;6:1250–61. 10.1038/s41477-020-00766-0. [DOI] [PubMed] [Google Scholar]
- 43. Durand NC, Shamim MS, Machol I, et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016;3:95–98. 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ramírez F, Bhardwaj V, Arrigoni L, et al. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun. 2018;9:189. 10.1038/s41467-017-02525-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wolff J, Bhardwaj V, Nothjunge S, et al. Galaxy HiCExplorer: a web server for reproducible Hi-C data analysis, quality control and visualization. Nucleic Acids Res. 2018;46:W11–16. 10.1093/nar/gky504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wolff J, Rabbani L, Gilsbach R, et al. Galaxy HiCExplorer 3: a web server for reproducible Hi-C, capture Hi-C and single-cell Hi-C data analysis, quality control and visualization. Nucleic Acids Res. 2020;48:W177–84. 10.1093/nar/gkaa220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. An L, Yang T, Yang J, et al. OnTAD: hierarchical domain structure reveals the divergence of activity among TADs and boundaries. Genome Biol. 2019;20:282. 10.1186/s13059-019-1893-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harris CR, Millman KJ, van der Walt SJ, et al. Array programming with NumPy. Nature. 2020;585:357–62. 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ramírez F, Ryan DP, Grüning B, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–65. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lu Z, Hofmeister BT, Vollmers C, et al. Combining ATAC-seq with nuclei sorting for discovery of cis-regulatory regions in plant genomes. Nucleic Acids Res. 2017;45:e41. 10.1093/nar/gkw1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu Y, Tian T, Zhang K, et al. PCSD: a plant chromatin state database. Nucleic Acids Res. 2018;46:D1157–67. 10.1093/nar/gkx919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee H, Seo PJ. Hi-GDT: a Hi-C-based 3D gene domain analysis tool for analyzing local chromatin contacts in plants. Zenodo. 2025; 10.5281/zenodo.14728732. Accessed 25 January 2025. [DOI]
- 53. Jamge B, Lorković ZJ, Axelsson E, et al. Histone variants shape chromatin states in Arabidopsis. eLife. 2023;12:RP87714. 10.7554/eLife.87714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Potter KC, Wang J, Schaller GE, et al. Cytokinin modulates context-dependent chromatin accessibility through the type-B response regulators. Nat Plants. 2018;4:1102–11. 10.1038/s41477-018-0290-y. [DOI] [PubMed] [Google Scholar]
- 55. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011;12:323. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yanai I, Benjamin H, Shmoish M, et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–59. 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- 59. Martín G, Márquez Y, Mantica F, et al. Alternative splicing landscapes in Arabidopsis thaliana across tissues and stress conditions highlight major functional differences with animals. Genome Biol. 2021;22:35. 10.1186/s13059-020-02258-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Carbon S, Ireland A, Mungall CJ, et al. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;25:288–89. 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. The G, Ontology C, Aleksander SA, et al. The gene ontology knowledgebase in 2023. Genetics. 2023;224:iyad031. 10.1093/genetics/iyad031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang Y, Tang H, Wang X, et al. Detection of colinear blocks and synteny and evolutionary analyses based on utilization of MCScanX. Nat Prot. 2024;19:2206–29. 10.1038/s41596-024-00968-2. [DOI] [PubMed] [Google Scholar]
- 64. Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang Y, Tang H, DeBarry JD, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9:90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 67. Waskom ML. Seaborn: statistical data visualization. J Open Source Softw. 2021;60:3021. 10.21105/joss.03021. [DOI] [Google Scholar]
- 68. Harris HL, Gu H, Olshansky M, et al. Chromatin alternates between A and B compartments at kilobase scale for subgenic organization. Nat Commun. 2023;14:3303. 10.1038/s41467-023-38429-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bonev B, Mendelson CN, Szabo Q, et al. Multiscale 3D genome rewiring during mouse neural development. Cell. 2017;171:557–72. e24. 10.1016/j.cell.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ke Y, Xu Y, Chen X, et al. 3D Chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell. 2017;170:367–81. e20. 10.1016/j.cell.2017.06.029. [DOI] [PubMed] [Google Scholar]
- 71. Winick-Ng W, Kukalev A, Harabula I, et al. Cell-type specialization is encoded by specific chromatin topologies. Nature. 2021;599:684–91. 10.1038/s41586-021-04081-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rahman S, Dong P, Apontes P, et al. Lineage specific 3D genome structure in the adult human brain and neurodevelopmental changes in the chromatin interactome. Nucleic Acids Res. 2023;51:11142–61. 10.1093/nar/gkad798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rajderkar S, Barozzi I, Zhu Y, et al. Topologically associating domain boundaries are required for normal genome function. Commun Biol. 2023;6:435. 10.1038/s42003-023-04819-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fraser J, Ferrai C, Chiariello AM, et al. Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol Syst Biol. 2015;11:852. 10.15252/msb.20156492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang Y, Li T, Preissl S, et al. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat Genet. 2019;51:1380–88. 10.1038/s41588-019-0479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang J, Yu H, Ma Q, et al. Phase separation of OCT4 controls TAD reorganization to promote cell fate transitions. Cell Stem Cell. 2021;28:1868–83. e11. 10.1016/j.stem.2021.04.023. [DOI] [PubMed] [Google Scholar]
- 77. Starks RR, Biswas A, Jain A, et al. Combined analysis of dissimilar promoter accessibility and gene expression profiles identifies tissue-specific genes and actively repressed networks. Epigenet Chromatin. 2019;12:16. 10.1186/s13072-019-0260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sanghi A, Gruber JJ, Metwally A, et al. Chromatin accessibility associates with protein-RNA correlation in human cancer. Nat Commun. 2021;12:5732. 10.1038/s41467-021-25872-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang P, Wu Y, Zhou H, et al. CLNN-loop: a deep learning model to predict CTCF-mediated chromatin loops in the different cell lines and CTCF-binding sites (CBS) pair types. Bioinformatics. 2022;38:4497–504. 10.1093/bioinformatics/btac575. [DOI] [PubMed] [Google Scholar]
- 80. Zhang P, Wu H. An attention-based deeplearning model for identifying chromatin interactions. IEEE J Biomed Health Inform. 2023;27:4559–68. 10.1109/JBHI.2023.3292299. [DOI] [PubMed] [Google Scholar]
- 81. Liu S, Cao Y, Cui K, et al. Hi-TrAC reveals division of labor of transcription factors in organizing chromatin loops. Nat Commun. 2022;13:6679. 10.1038/s41467-022-34276-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang Y, Dong Q, Wang Z, et al. A fine-scale Arabidopsis chromatin landscape reveals chromatin conformation-associated transcriptional dynamics. Nat Commun. 2024;15:3253. 10.1038/s41467-024-47678-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hsieh T-HS, Weiner A, Lajoie B, et al. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell. 2015;162:108–19. 10.1016/j.cell.2015.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hsieh T-HS, Fudenberg G, Goloborodko A, et al. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat Methods. 2016;13:1009–11. 10.1038/nmeth.4025. [DOI] [PubMed] [Google Scholar]
- 85. Lee H, Seo PJ. Hi-GDT: a Hi-C-based 3D gene domain analysis tool for analyzing local chromatin contacts in plants. [Computer software]. Software Heritage. 2025. https://archive.softwareheritage.org/browse/snapshot/a4d7864a7adfdf451f1058edde2c0014d7b51fe6/directory/. Accessed 24 January 2025.
- 86. Lee H. Hi-GDT [Computer software]. GitHub. 2024; https://github.com/CDL-HongwooLee/Hi-GDT. Accessed 19 May 2024.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Lee H, Seo PJ. Hi-GDT: a Hi-C-based 3D gene domain analysis tool for analyzing local chromatin contacts in plants. Zenodo. 2025; 10.5281/zenodo.14728732. Accessed 25 January 2025. [DOI]
Supplementary Materials
Hao Wu -- 11/9/2024
Hao Wu -- 1/12/2025
Yusen Ye -- 11/20/2024
Yusen Ye -- 1/20/2025
Data Availability Statement
All publicly available datasets used in this study are listed in Supplementary Table S1. The source codes and step-by-step instructions of Hi-GDT are available from the GitHub repository [85]. Snapshots of the code and data are available in Software Heritage [86]. Raw data and all implemented codes used in this study have been deposited at Zenodo [52]. Hi-C and RNA-seq data produced in this study were deposited in NCBI under accession number PRJNA1112149.