

Abstract
Propagation of the yeast prion [PSI+], a self-replicating aggregated form of Sup35p, requires Hsp104. One model to explain this phenomenon proposes that, in the absence of Hsp104, Sup35p aggregates enlarge but fail to replicate thus becoming diluted out as the yeast divide. To test this model, we used live imaging of Sup35p-GFP to follow the changes that occur in [PSI+] cells after the addition of guanidine to inactivate Hsp104. After guanidine addition there was initially an increase in aggregation of Sup35p-GFP; but then, before the yeast divided, the aggregates began to dissolve, and after ≈6 h the Sup35-GFP looked identical to the Sup35-GFP in [psi+] cells. Although plating studies showed that the yeast were still [PSI+], this reduction in aggregation suggested that curing of [PSI+] by inactivation of Hsp104 might be independent of cell division. This was tested by measuring the rate of curing of [PSI+] cells in both dividing and nondividing cells. Cell division was inhibited by adding either alpha factor or farnesol. Remarkably, with both of these methods, we found that the rate of curing was not significantly affected by cell division. Thus, cell division is not a determining factor for curing [PSI+] by inactivating Hsp104 with guanidine. Rather, curing apparently occurs because Sup35-GFP polymers slowly depolymerize in the absence of Hsp104 activity. Hsp104 then counteracts this curing possibly by catalyzing formation of new polymers.
Prions (infectious proteins) are proteins that are normally soluble but autocatalytically propagate as amyloid, which is a fibrous polymer-like aggregate rich in β-sheet secondary structure (1). Normally folded mammalian prion protein has a high α-helical content, but its aggregated form contains much more β-sheet and is highly resistant to proteolysis. When this autocatalytic conversion occurs on a large scale in the central nervous system, the amyloid form accumulates and is associated with the neurodegeneration in fatal diseases such as scrapie in sheep, bovine spongiform encephalopathy in cows, and Creutzfeld–Jacob disease in humans (2). Other proteins form amyloid in an autocatalytic manner, but only prion protein is infectious.
Prions also occur in yeast (3). The best-characterized yeast prion proteins are Sup35p, a translation termination factor, and Ure2p, which is involved in nitrogen metabolism (4). More recently, Rnq1p was identified as a prion protein, and New1p was shown to contain a domain that can substitute for the prion-determining region of Sup35p (5–7). Amyloidogenic yeast prion proteins have an Asn/Gln-rich prion-forming domain that, like mammalian prion protein, can be converted to a β-sheet-rich, protease-resistant conformation in an autocatalytic manner. When yeast cells containing soluble Sup35p, for example, are mated to cells containing the prion form of Sup35p, the soluble form is converted to the aggregated form, which then continues to propagate as the yeast divide. The term [PSI+] refers to the Sup35p prion as well as to yeast cells propagating the prion form of Sup35p. Normally, Sup35p is soluble, and the yeast are referred to as [psi–].
Although almost nothing is known about the requirement of accessory proteins for prion propagation in mammals, genetic experiments have shown that propagation of amyloidogenic yeast prions requires Hsp104, a member of the ClpB class of protein chaperones that form ring-shaped polymers and disassemble protein aggregates (5, 8–10). Paradoxically, high levels of Hsp104 also convert [PSI+]to[psi–], but this effect is unique for [PSI+] and does not occur with the other yeast prions. In vitro, Hsp104 has a dual function in that it both facilitates formation of Sup35p aggregates and breaks up Sup35p aggregates (11, 12).
The mechanism by which Hsp104 maintains [PSI+] is not yet understood. Lindquist and colleagues (11, 13) have suggested that the ability of Hsp104 both to nucleate Sup35p aggregation and to break up these aggregates plays a role in maintaining [PSI+]. On the other hand, others have suggested that Hsp104 is required only to break up aggregates to replicate them so that they are passed on to daughter cells when yeast divide (14–16). It was further proposed that Hsp104 breaks up only the small aggregates, whereas large aggregates form dead-end complexes unable to propagate the prion trait (15, 17–19).
Investigations into the mechanism of action of Hsp104 have been facilitated by experiments showing that Hsp104 in vivo can be inactivated by treating yeast with low levels of guanidine-hydrochloride (Gdn) (20–22). Whereas it was demonstrated many years ago that such treatment cures [PSI+], recently it was shown that Gdn acts by inhibiting Hsp104 ATPase activity (23, 24). Because Gdn inactivates Hsp104 immediately, kinetics of [PSI+] curing after this inactivation can be monitored straight-forwardly. When growing cells are exposed to Gdn there is a lag phase during which no curing occurs followed by a linear increase in the number of cured cells over time (25).
These kinetic studies have supported the model that Hsp104 acts by breaking up prion polymers, thereby generating new prion “seeds.” During the lag phase that occurs after Hsp104 activity is inhibited, the prion polymers or seeds are thought to grow in size but, failing to replicate, become diluted among the increasing number of dividing cells. Once the number of seeds per cell is sufficiently low, further cell division produces daughter cells lacking seeds, which are therefore [psi–]. Curing also appears to require cell division because it does not occur during stationary phase. In fact, if Gdn-treated cells enter stationary phase before becoming [psi–], curing of [PSI+] is interrupted.
In our previous studies of yeast expressing a full-length Sup35-GFP fusion protein (NGMC), we found that NGMC appeared diffuse in both [psi–] and [PSI+] cells (26). When we measured the rate of fluorescence recovery after photobleaching (FRAP), however, the rate of recovery of the NGMC fluorescence in [PSI+] cells was significantly slower than in [psi–] cells. This reduced rate reflects the presence of small NGMC aggregates in the [PSI+] cells. This ability to detect NGMC aggregates allows observation of the seeds thought to be responsible for [PSI+] propagation and, thereby, the change that occurs when Hsp104 activity is inhibited by Gdn.
In this study, we monitored Sup35p aggregation by using real-time imaging of yeast expressing NGMC and followed the changes after addition of Gdn. Our results showed that curing cells of [PSI+] by Hsp104 inactivation does not require cell division, which has important implications regarding current understanding of the dynamics of Sup35p aggregates in [PSI+] cells.
Materials and Methods
Strains, Plasmids, Media, and Growth Conditions. Yeast strains were [PSI+] and [psi–] derivatives of strain 780-1D (MATa kar1-1 SUQ5 ade2-1 his3Δ202 leu2Δ1 trp1Δ63 ura3-52 sup35::KanMX/pJ510 or pJ533) described in ref. 26. Plasmids pJ510 and pJ533, described in ref. 26, are single-copy plasmids with the LEU2 marker and NGMC or unmodified SUP35, respectively. Selection for plasmid maintenance is not necessary because Sup35p is essential. YPD medium contains 0.5% yeast extract, 2% peptone, and 2% dextrose. Cultures were maintained in log phase by periodic dilution with fresh medium. For monitoring NGMC in growing cells, yeast were grown at 30°C on synthetic medium (SD; 0.7% yeast nitrogen base, 2% glucose) with complete supplement mixture (CSM; 25 mg/liter adenine, histidine, leucine, tryptophan, and uracil). This medium has no autofluorescence and is suitable for imaging living cells by fluorescence microscopy. To arrest cell division, cells were treated with 50 μM α-factor (Zymo Research) or 100 μM farnesol (Sigma). This latter treatment arrests cell division by inhibiting DNA ligase and histone acetyltransferase (27).
[PSI+] Curing. Gdn was added to growing cultures to a concentration of 5 mM. The presence of [PSI+], detected by its ability to suppress the ade2-1 allele in our strains (28), was monitored by plating culture aliquots on 1/2YPD. On these plates, [PSI+] (suppressed) cells are white and [psi–] (nonsuppressed) cells are red. Growth was monitored by optical density (OD600). When assaying [PSI+] curing in the presence of α-factor (Zymo Research), the cells were plated and counted because OD600 was not an accurate measure of cell division.
Confocal Microscopy. To immobilize cells for imaging, two-well chamber 25-mm2 coverslips (Lab-Tek) were pretreated with 2 mg/ml Con A (Sigma-Aldrich) for 10 min. Cells were imaged on a Zeiss LSM 510 confocal microscope and photobleached as described in ref. 26. For each condition, 15–20 cells were photobleached. After normalizing each bleach experiment by setting the maximum fluorescence to 100% and the minimum to 0%, the data were compiled and used to calculate averages and standard deviations for each condition.
Results
Live cell imaging was used to monitor the changes that occurred in NGMC after addition of Gdn to [PSI+] cells that had been transferred directly from a YPD plate to a coverslip for imaging. We first noted that, before Gdn addition, in contrast to our previous study in which the NGMC appeared completely diffusive, the NGMC had a granular appearance. Then, as shown in Fig. 1A, in the first hour after Gdn addition, the NGMC granules clearly coalesced, which, in about one-third of the cells, led to the formation of a single granule. However, remarkably, with longer incubation times all of the granules began to dissolve so that at 2 h there was significantly less granularity. This dissolution of the NGMC granules continued with further incubation with Gdn so that at 3 h the NGMC appeared diffuse in most cells. FRAP studies on NGMC also showed a biphasic response after Gdn treatment (Fig. 1B). One hour after Gdn treatment, although the NGMC in the large granules was completely immobilized (data not shown), the cytosol surrounding the NGMC granules showed a slower rate of fluorescence recovery compared with the rate observed before Gdn addition. But after 5 h of incubation in Gdn, the rate of FRAP had again increased, becoming identical to the rate observed in [psi–] cells. Note that the extent of fluorescence recovery was only ≈50% in these experiments because photobleaching caused a decrease in the total fluorescence of the NGMC in the cells.
Fig. 1.

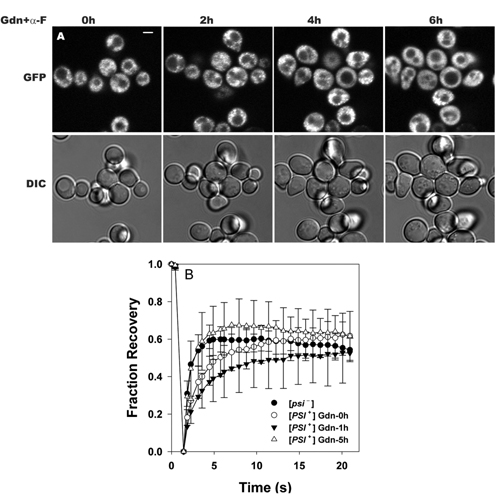
Dynamics of aggregate formation after Gdn treatment. (A) NGMC in [PSI+] cells was imaged by using confocal fluorescence microscopy before and after incubating with 5 mM Gdn for the indicated times. Fluorescence and differential interference contrast images are of the same fields. DAPI staining showed that the granules were not in the nucleus. (Scale bar, 2 μm.) (B) FRAP of NGMC in [PSI+] cells measured prior and after Gdn treatments. (C) Cells first treated with Gdn for 6 h (filled triangles) and then transferred to medium without Gdn for 2 h (open triangles). FRAP of NGMC in [psi–] cells is shown for comparison (filled circles).
Because the fluorescence of [PSI+] cells used in these studies was different from that observed in our previous studies in which the diffuse appearance of NGMC was similar in [PSI+] and [psi–] cells, we investigated the effect of growth conditions on the appearance of NGMC in [PSI+] cells. We found that NGMC was much more granular in [PSI+] cells in late log phase than in early log phase where the NGMC was completely diffuse. Therefore, for the remainder of our studies, cells were always kept in early log phase unless otherwise noted.
In examining the changes that occurred upon addition of Gdn to [PSI+] cells in early log phase, we found that Gdn treatment first caused the initially diffuse NGMC to become slightly aggregated with the appearance of small granules occurring over the first hour (see Fig. 6A, which is published as supporting information on the PNAS web site). Then, with further incubation with Gdn, the NGMC again became diffuse. Similarly, the FRAP studies showed that there initially was a slight decrease in the rate of fluorescence recovery after Gdn treatment, followed by an increase in this rate until it became identical to that of NGMC in [psi–] cells (see Fig. 6B). As expected, Gdn had no effect on the FRAP rate of NGMC in [psi–] cells (see Fig. 6C). Therefore, the addition of Gdn to [PSI+] cells in early log phase caused essentially the same biphasic effect on NGMC aggregation that occurred with [PSI+] cells in late log phase, although the results were much more dramatic with the latter cells than with the former.
Even though NGMC in [PSI+] cells showed a [psi–] FRAP rate after 6 h of Gdn treatment, it is unlikely that these cells had, in fact, become [psi–] because by plating, we found that there was no significant curing of [PSI+] during the first 10 h of Gdn incubation (Fig. 2). Therefore, it is not surprising that, when Gdn was removed after 6 h of treatment, the rate of fluorescence recovery once again slowed, returning over a period of ≈2 h to the rate observed in the [PSI+] cells before Gdn treatment (Fig. 1C). Nevertheless, the observation that Gdn significantly reduced NGMC aggregation in mother cells as well as daughter cells raised the possibility that, over a longer period, Gdn could cure [PSI+] cells without cell division occurring.
Fig. 2.

Gdn curing of [PSI+] is independent of cell division when division is arrested by α-factor. (A) [PSI+] cells were cultured in SD+CSM with the indicated additions of Gdn and α-factor. Curing of [PSI+] was assessed by plating on 1/2YPD. Each time point represents an average of two independent experiments. (B) Growth rates of cells in A. (Inset) Example of colonies on 1/2YPD arising from cells of a 43-h sample. α-F, α-factor.
To test whether cell division was indeed necessary for curing of [PSI+], we followed the kinetics of [PSI+] curing in the presence and absence of α-factor by periodically removing aliquots of the Gdn-treated cultures and plating cells. As expected, without Gdn the yeast cells remained 100% [PSI+] over a period of 40 h both in the presence and absence of α-factor (Fig. 2 A and B). An example of this plating is shown in Fig. 2B Inset, which shows the curing at 43 h, as indicated by the red colonies. Therefore, neither the presence of α-factor nor the absence of cell division affected [PSI+] stability. Without α-factor, the time course of conversion of [PSI+] to [psi–] caused by Gdn was very similar to what we and others observed previously (25, 28). After a lag period of ≈12 h, there was a steady decrease in the fraction of [PSI+] cells until almost all of the cells were [psi–] at ≈43 h. Remarkably, however, when this same experiment was carried out in the presence of α-factor, where almost no cell division occurred, the time course of conversion from [PSI+] to [psi–] was very similar. In this case, there was a lag of ≈15 h, followed by the complete disappearance of [PSI+] after ≈43 h (Fig. 2, open triangles). Experiments were performed to test the viability of cells treated with α-factor and Gdn by determining cell density with a hemocytometer, followed by plating. The results showed that cells treated with α-factor and Gdn for 40 h were >80% viable. Furthermore, using fluorescence, we found that the level of NGMC was not significantly changed during the 40-h incubation period with α-factor and Gdn.
Essentially identical results were obtained when cell division was reversibly arrested by treatment with farnesol (27). As shown in Fig. 3A, farnesol-treated cells showed the same rate of conversion from [PSI+] to [psi–] as the control cells. However, the cell division of the farnesol-treated cells was much slower than the control cells, especially during the first 25 h, during which the farnesol-treated cells divided only three times while the control cells went through 13 generations (Fig. 3B). Finally, we found that the same changes in NGMC fluorescence occurred when Gdn was added to [PSI+] cells in the presence of α-factor as in its absence (see Fig. 7, which is published as supporting information on the PNAS web site). Therefore, it appears that the conversion of [PSI+]to[psi–] by inhibition of Hsp104 occurs not only without cell division but also, more remarkably, is not significantly affected by cell division.
Fig. 3.

Gdn curing of [PSI+] is independent of cell division when division is arrested by farnesol. (A) Curing of [PSI+] cells with the indicated additions of Gdn and farnesol. Each time point represents average of two independent experiments. (B) Growth rates of cells in A. (Inset) Example of colonies from cells in an aliquot of a 43-h sample. FOH, farnesol.
One line of supporting evidence that inhibition of Hsp104, by itself, cannot convert yeast from [PSI+] to [psi–] without cell division is that Gdn does not induce this conversion in stationary-phase cells (25). We monitored stationary-phase [PSI+] cells by fluorescence microscopy and plating before and after adding Gdn. As we observed previously (26), when [PSI+] cells expressing NGMC were in stationary phase, the NGMC aggregates into numerous visible fluorescent foci. During a 72-h period of exposure to Gdn, the cells retained these large foci, and the overall fluorescence did not change (Fig. 4). Furthermore, throughout this time period the cells remained 100% [PSI+] as determined by periodic plating. Therefore, Gdn had no effect on NGMC fluorescence in stationary cells, and as was observed for unmodified Sup35p, Gdn alone could not convert cells expressing NGMC from [PSI+] to [psi–] when the cells were in stationary phase.
Fig. 4.

Gdn cannot cure stationary phase [PSI+] cells. After addition of Gdn to stationary phase cells, NGMC was examined periodically over 72 h by confocal fluorescence microscopy. Shown are cells before and 44 and 72 h after adding Gdn.
Another line of evidence suggesting that cell division is required for loss of [PSI+] is that the addition of ethanol to Gdn-treated [PSI+] cells slows both growth rate and rate of conversion from [PSI+] to [psi–] to the same extent (25). Thus, it appears that there is a quantitative relationship between how rapidly cells divide and how rapidly [PSI+] is lost. In contrast, if our observation that cell division is not required for [PSI+] loss is correct, it would imply that the effect of ethanol is unrelated to its effect on growth rate. We therefore tested the effect of ethanol on both cell division and the time course of conversion of [PSI+] to [psi–], both in the presence and absence of α-factor (Fig. 5). To make a more direct comparison with earlier experiments, we used yeast cells expressing unmodified Sup35p.
Fig. 5.

Delayed Gdn curing of [PSI+] by ethanol (3%) is unrelated to the reduced rate of cell division caused by ethanol. (A) [PSI+] cells expressing native Sup35p were grown under the indicated conditions, and [PSI+] curing was monitored by plating. (B) Growth rates of the cultures shown in A. α-F,α-factor; EtOH, ethanol.
In agreement with the results of Eaglestone et al. (25), we indeed observed that ethanol doubled the yeast cell division time (Fig. 5B, open squares). Similarly, we found that the presence of ethanol caused a doubling in the lag time that occurred before [PSI+] began to linearly decrease after Gdn treatment. However, ethanol also doubled this lag time in the presence of α-factor (Fig. 5A, filled squares), even though essentially no cell division occurred under this condition (Fig. 5B). Therefore, the delay in the Gdn-induced conversion of [PSI+] to [psi–] caused by ethanol is apparently unrelated to its effect on cell division.
Discussion
We used our previously described full-length GFP construct of Sup35p to investigate the mechanism of curing [PSI+] by inhibiting Hsp104 activity with Gdn. After adding Gdn to growing yeast, we saw a biphasic response first with an increase in the aggregation of NGMC, and then with a dissolution of the aggregates resulting in their disappearance over a period of ≈5–6 h. These changes in the aggregation state of NGMC were independent of cell division. Therefore, our data contradict the possibility that Gdn curing of [PSI+] occurs because mother cells retain large NGMC aggregates that are not passed on to daughter cells during cell division thus causing daughters to become [psi–].
The dissolution of NGMC aggregates that began after 1 h in both mother and daughter cells showed that, even when Hsp104 activity is inhibited, NGMC aggregates undergo dissolution. Furthermore, this dissolution was not limited to large NGMC granules. Even after the large aggregates dissolved, our FRAP analysis showed that the level of NGMC aggregation continued to decrease over the next 3–4 h until the FRAP became identical to that observed for [psi–] cells. However, despite having [psi–] FRAP kinetics, these cells were still [PSI+], reverting to the slower [PSI+] FRAP after Gdn was removed. It should be noted that preliminary studies using both FRAP and fluorescence correlation spectroscopy suggest that NGMC in [psi–] cells is much larger than GFP perhaps because even monomeric Sup35p is associated with ribosomes or other proteins in the cytosol. Therefore, even when the FRAP becomes identical to that observed in the [psi–] cells, the Sup35p may still be aggregated, which could explain why, despite having [psi–] FRAP, cells can still be [PSI+].
The steady decrease in aggregation during the 6 h after inactivating Hsp104 occurred in both mother and daughter cells, suggesting that cell division is not involved in this phenomenon. However, as Tuite and his colleagues proposed (15), it still seemed possible that the decreased level of aggregation was caused by cell division gradually diluting out NGMC aggregates too small to be detected by fluorescence methods. Remarkably, however, nondividing α-factor- or farnesol-treated cells began to become [psi–] after a lag of ≈10 h, the same time course seen by us and others for Gdn curing of [PSI+] in dividing cells. These data imply that the decrease in NGMC aggregation, which we observed by FRAP during the first 5 h, continued as long as Gdn was present, whether or not cell division occurred, until all [PSI+] seeds were eliminated.
The same phenomenon occurred in the presence of ethanol, which caused the same decrease in the rates of cell division and of [PSI+] curing (25). However, this correlation seems to be fortuitous because we showed that ethanol caused the same decrease in the rate of [PSI+] curing when cell division was arrested by α-factor. The reduced rate of curing when ethanol was present might have been due to ethanol causing a stress response, thus increasing the abundance of Hsp104 and correspondingly decreasing the efficacy of Gdn. Because we did this experiment with cells expressing native Sup35p, our observation that cell division was not the determining factor in curing [PSI+] was not due to our use of the NGMC fusion.
Although the experiments with α-factor and farnesol may provide the best evidence that cell division is not required for curing [PSI+], it is interesting to note that, before it was even known that [PSI+] was a prion, it was found that subjecting yeast to osmotic shock converted the cells from [PSI+] to [psi–] without cell division occurring (23, 29). In addition, Ness et al. (15) observed that when Gdn curing of [PSI+] was done under conditions of limiting glucose, which greatly reduces cell division, it caused a marked decrease in sedimentable Sup35p. They proposed that this might be due to the small amount of cell division that still occurred, but in light of our results with α-factor and farnesol it seems possible that it was due to dissolution of Sup35p aggregates independently of cell division.
It has been proposed that the number of seeds in [PSI+] cells can be estimated by determining the number of cell divisions required to convert the cells to [psi–] (25). However, our observation that cell division does not affect the time course of [PSI+] curing strongly suggests that until a [PSI+] cell actually becomes [psi–] the number of seeds in the cell is very large. Otherwise, it might be expected that cell division would cause enough dilution of the seeds to speed up the curing process, an effect that we did not observe. Apparently, dissolution of individual seeds within a cell has a much greater effect on the time course of curing than the reduction in seed number caused by cell division.
A question that arises is why Tuite and his collaborators (30) observed a relatively small number of cells that were not cured in colonies grown on plates containing Gdn. They proposed that the seeds in the cell that began the colony remained in some of the progeny after cell division diluted them, but another explanation could be that cells at the center of colonies are in stationary state, which would prevent them from being cured by Gdn. In any case, because curing occurs without cell division, it is not possible to estimate the number of seeds in the cells by determining the number of cell divisions required to cure [PSI+].
From our data we conclude first that Hsp104 is involved in preventing formation of large Sup35p aggregates, because we initially observed an increase in aggregation after Hsp104 was inactivated. Second, as originally suggested by Ness et al. (15), the seeds that maintain [PSI+] do not have to be relatively large aggregates, because the FRAP in [PSI+] cells can be as fast as the rate observed in [psi–] cells. Of course, the seeds could be relatively large oligomers of Sup35p because, as we noted above, FRAP studies suggest that even Sup35p in [psi–] cells is much larger than GFP. Finally, inactivation of Hsp104 does not cure [PSI+] by preventing either large aggregates or small seeds from being broken up and passed on to daughter cells because cell division is not required to cure [PSI+]. Rather, in the absence of both cell division and Hsp104 activity, an as yet unknown mechanism completely rids the cells first of the large aggregates and then the smaller seeds.
By using crude sedimentation analysis, Ness et al. (15) showed that, after addition of Gdn, Sup35p aggregates did not appear to depolymerize or to be proteolyzed during the first four cell divisions. They also showed that newly synthesized Sup35p was added to existing aggregates in the presence of Gdn. They concluded that Gdn blocks seed replication but not aggregate assembly and that cell division diluted out the Sup35p aggregates, resulting in curing of [PSI+]. However, because we find that cell division does not affect the time course of [PSI+] curing, our data show, in fact, that proteolysis or net depolymerization of the Sup35p aggregates must occur faster than formation of new aggregates over the 40-h period in Gdn.
A clue to a possible curing mechanism comes from the observation that, after ≈6 h of Gdn treatment, when we observe complete dissolution of the Sup35p aggregates, which are presumably bundles of Sup35p fibers, individual fibers have actually undergone an ≈7-fold increase in their length (ref. 16 and our unpublished data). This finding suggests that it is the ability of Hsp104 to sever the individual Sup35p fibers and keep them relatively short that maintains the normal aggregates of Sup35p observed in [PSI+] cells. On this basis, inhibition of Hsp104 activity first causes the individual Sup35p fibers to grow longer, increasing the size of the fiber bundles. However, as the individual fibers become longer and possibly change their structure, the Sup35p aggregates actually become unstable and, perhaps because they become more susceptible to the action of chaperones other than Hsp104, fall apart into individual long fibers. Our FRAP studies would suggest that these long fibers are much smaller than the Sup35p aggregates in [PSI+] cells but about the same size as Sup35p in [psi–] cells. Finally, these long fibers themselves either depolymerize or are degraded resulting in the cells becoming [psi–].
It is possible that Hsp104 maintains the Sup35p aggregates or fiber bundles in the [PSI+] cells not only by severing existing fibers but also by simultaneously catalyzing the formation of new fibers in the aggregates using the original fibers as templates. Such catalysis by Hsp104 could explain why in stationary phase, when more Hsp104 is present, the aggregates become even larger. In any event, this model suggests first that it is Hsp104 that maintains the aggregates at an intermediate size in [PSI+] cells, and second that if the fibers become too long, the aggregates fall apart and the individual fibers themselves become unstable.
Of course, this is not the only model of Hsp104 activity that can explain our data; other models involving capping of old filaments or nucleation of new ones by Hsp104 might also work. Independent of any model, however, our data clearly show that dissolution of aggregated Sup35p can occur in the cell when both Hsp104 activity and cell division are blocked. Understanding how this dissolution takes place is of great importance because it may lead to understanding how cells rid themselves of toxic aggregates such as prions or polyglutamine fragments that pose a danger to cell viability.
Supplementary Material
Acknowledgments
We thank Dr. Xufeng Wu for technical assistance on the confocal microscope.
Author contributions: L.E.G. and E.E. designed research; Y.-X.W. performed research; D.C.M. contributed new reagents/analytic tools; D.C.M. analyzed data; and L.E.G. and E.E. wrote the paper.
Abbreviations: FRAP, fluorescence recovery after photobleaching; Gdn, guanidine-hydrochloride; YPD, yeast extract/peptone/dextrose.
References
- 1.Sipe, J. D. & Cohen, A. S. (2000) J. Struct. Biol. 130, 88–98. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner, S. B. (1998) Proc. Natl. Acad. Sci. USA 95, 13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wickner, R. B. (1994) Science 264, 566–569. [DOI] [PubMed] [Google Scholar]
- 4.Taylor, K. L. & Wickner, R. B. (2001) Contrib. Microbiol. 7, 21–31. [DOI] [PubMed] [Google Scholar]
- 5.Derkatch, I. L., Bradley, M. E., Zhou, P., Chernoff, Y. O. & Liebman, S. W. (1997) Genetics 147, 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santoso, A., Chien, P., Osherovich, L. Z. & Weissman, J. S. (2000) Cell 100, 277–288. [DOI] [PubMed] [Google Scholar]
- 7.Sondheimer, N. & Lindquist, S. (2000) Mol. Cell 5, 163–172. [DOI] [PubMed] [Google Scholar]
- 8.Chernoff, Y. O., Lindquist, S. L., Ono, B., Inge-Vechtomov, S. G. & Liebman, S. W. (1995) Science 268, 880–884. [DOI] [PubMed] [Google Scholar]
- 9.Moriyama, H., Edskes, H. K. & Wickner, R. B. (2000) Mol. Cell. Biol. 20, 8916–8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schirmer, E. C., Glover, J. R., Singer, M. A. & Lindquist, S. (1996) Trends Biochem. Sci. 21, 289–296. [PubMed] [Google Scholar]
- 11.Shorter, J. & Lindquist, S. (2004) Science 304, 1793–1797. [DOI] [PubMed] [Google Scholar]
- 12.Inoue, Y., Taguchi, H., Kishimoto, A. & Yoshida, M. (2004) J. Biol. Chem. 279, 52319–52323. [DOI] [PubMed] [Google Scholar]
- 13.Krobitsch, S. & Lindquist, S. (2000) Proc. Natl. Acad. Sci. USA 97, 1589–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paushkin, S. V., Kushnirov, V. V., Smirnov, V. N. & Ter-Avanesyan, M. D. (1996) EMBO J. 15, 3127–3134. [PMC free article] [PubMed] [Google Scholar]
- 15.Ness, F., Ferreira, P., Cox, B. S. & Tuite, M. F. (2002) Mol. Cell. Biol. 22, 5593–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kryndushkin, D. S., Alexandrov, I. M., Ter-Avanesyan, M. D. & Kushnirov, V. V. (2003) J. Biol. Chem. 278, 49636–49643. [DOI] [PubMed] [Google Scholar]
- 17.Borchsenius, A. S., Wegrzyn, R. D., Newnam, G. P., Inge-Vechtomov, S. G. & Chernoff, Y. O. (2001) EMBO J. 20, 6683–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wegrzyn, R. D., Bapat, K., Newnam, G. P., Zink, A. D. & Chernoff, Y. O. (2001) Mol. Cell. Biol. 21, 4656–4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ripaud, L., Maillet, L. & Cullin, C. (2003) EMBO J. 22, 5251–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung, G. & Masison, D. C. (2001) Curr. Microbiol. 43, 7–10. [DOI] [PubMed] [Google Scholar]
- 21.Jung, G., Jones, G. & Masison, D. C. (2002) Proc. Natl. Acad. Sci. USA 99, 9936–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferreira, P. C., Ness, F., Edwards, S. R., Cox, B. S. & Tuite, M. F. (2001) Mol. Microbiol. 40, 1357–1369. [DOI] [PubMed] [Google Scholar]
- 23.Tuite, M. F., Mundy, C. R. & Cox, B. S. (1981) Genetics 98, 691–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grimminger, V., Richter, K., Imhof, A., Buchner, J. & Walter, S. (2004) J. Biol. Chem. 279, 7378–7383. [DOI] [PubMed] [Google Scholar]
- 25.Eaglestone, S. S., Ruddock, L. W., Cox, B. S. & Tuite, M. F. (2000) Proc. Natl. Acad. Sci. USA 97, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song, Y., Wu, Y. X., Jung, G., Tutar, Y., Eisenberg, E., Greene, L. E. & Masison, D. C. (2005) Eukaryot. Cell 4, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Machida, K., Tanaka, T., Yano, Y., Otani, S. & Taniguchi, M. (1999) Microbiology 145, 293–299. [DOI] [PubMed] [Google Scholar]
- 28.Jung, G., Jones, G., Wegrzyn, R. D. & Masison, D. C. (2000) Genetics 156, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh, A., Helms, C. & Sherman, F. (1979) Proc. Natl. Acad. Sci. USA 76, 1952–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cox, B., Ness, F. & Tuite, M. (2003) Genetics 165, 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}