

Abstract
Multiple myeloma (MM) is a malignancy of terminally differentiated plasma cells. MM cells localize to the bone marrow, where cell adhesion–mediated autocrine or paracrine activation of various cytokines, such as interleukin 6, insulin-like growth factor 1, and interferon α, results in their accumulation mainly because of loss of critical apoptotic controls. Resistance to apoptosis, a genetically regulated cell death process, may play a critical role in both pathogenesis and resistance to treatment of MM. Abnormalities in regulation and execution of apoptosis can contribute to tumor initiation, progression, as well as to tumor resistance to various therapeutic agents. Apoptosis is executed via 2 main pathways that lead to activation of caspases: the death receptor (extrinsic) pathway and the mitochondrial (intrinsic) pathway. Ionizing radiation and chemotherapeutic agents act primarily through the intrinsic pathway, in which mitochondria play the central role. Various therapeutic modalities that are effective in MM modulate levels of the proapoptotic and antiapoptotic Bcl-2 family of proteins and of inhibitors of apoptosis, expression of which is primarily regulated by p53, nuclear factor κB, and STAT (signal transducers and activators of transcription) factors. This review focuses on the key concepts and some of the most recent studies of signaling pathways regulated in MM and summarizes what is known about the clinical role of these pathways.
Keywords: Multiple myeloma, Apoptosis, Bcl-2 family, TNF ligand and receptor, Ionizing radiation
1. Introduction
Multiple myeloma (MM) is a malignancy of terminally differentiated B-lymphocytes, also known as plasma cells. MM is characterized by accumulation of a monotypic plasma cell population in the bone marrow (BM), serum and/or urine monoclonal immunoglobulin, and osteolytic lesions. Because MM cells are postgerminal, further mutation does not occur [1]. MM constitutes 10% of the hematopoietic malignancies and ranks just behind non-Hodgkin’s lymphoma as the second most common of these diseases in the United States [2]. MM is a malignancy characterized by very slow proliferation of malignant plasma cells, which leads to accumulation of these cells within the bone marrow. The existence of this phenomenon suggests that resistance to apoptosis may play a critical role in both pathogenesis and treatment resistance of MM. Moreover, inducers of apoptosis not only may have a lethal effect but also may support their immortalization in BM.
Apoptosis is a morphologically and biochemically distinct form of eukaryotic cell death that occurs under a variety of physiological and pathological conditions [3]. Apoptosis is executed via 2 main pathways that lead to the activation of caspases: the death receptor (DR) pathway and the mitochondrial pathway [4]. Caspases are members of a cysteine protease family that are synthesized as inactive zymogens. They are responsible for activation of various cellular proteases and endonucleases. The result of activation is cleavage of structural and regulatory cellular proteins and of nuclear DNA. This process leads to morphologic and biochemical cellular changes that are characteristic of apoptosis. The DR pathway is activated by ligation of members of the tumor necrosis factor (TNF) family, such as Fas ligand (FasL; also called Apo1), TNF-α, and Apo2 ligand (Apo2L; also called TNF-related apoptosis-inducing factor [TRAIL]), to DR on the plasma membrane. The receptors of these ligands—Fas, TNFR1 (TNF receptor 1), and DR4/DR5—are members of the TNF receptor superfamily, which is characterized by similar, cysteine-rich extracellular domains and homologous cytoplasmic death domains [5]. Binding of these DRs by their respective ligands recruits the adaptor proteins Fas-associated death domain (FADD) and/or TNFR1-associated death domain (TRADD) and caspase 8 to the death-inducing signaling complex, triggering the proteolytic activation of caspase 8. Caspase 8 in turn activates the effector caspases 3 and 7 [6] (Figure 1). The result is proteolytic targeting of key apoptotic or cell cycle regulatory proteins, such as Bcl-2 [7] and cyclin E [8].
Figure 1.
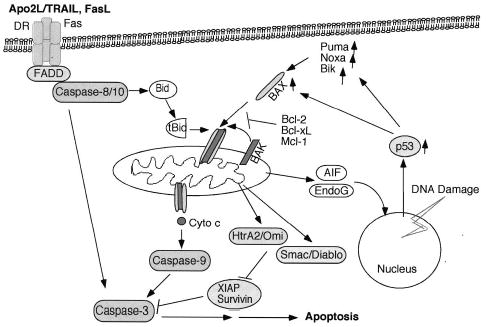
Apoptotic pathways. The death receptor (DR) pathway is activated by ligation of the death ligands (eg, Apo2L/TRAIL [tumor necrosis factor–related apoptosis-inducing factor ligand] and FasL) to their cognate receptors on the cell surface. This process results in sequential binding of Fas-associated death domain (FADD) and pro–caspase 8. Active caspase 8 cleaves and activates caspase 3. In addition, caspase 8 cleaves Bid, and truncated Bid translocates to the mitochondria to promote the release of cytochrome c (Cyto c). The mitochondrial pathway is activated by a number of stimuli, including chemotherapeutic drugs and ionizing radiation (IR). All these stimuli result in activation and oligomerization of Bax and Bak. These changes contribute to pore formation in the outer mitochondrial membrane and the release of cyto c and other apoptogenic factors. Cyto c promotes activation of caspase 9 and of the effector caspases. DNA-damaging agents such as IR induce activation of the p53 that promotes transcription of proapoptotic Bcl-2 family members such as the multidomain Bax and the Bcl-2 homology 3 (BH3)-only proteins Puma, Noxa, and Bik. Activation of the BH3-only molecules either directly or indirectly results in activation of Bax and Bak. AIF indicates apoptosis-inducing factor.
The mitochondrial pathway is regulated primarily by members of the Bcl-2 family, which comprises both proapoptotic and antiapoptotic proteins. The ratio between these subsets determines the susceptibility of cells to various death signals [4]. All contain at least 1 of the 4 conserved Bcl-2 homology (BH) domains [9]. The antiapoptotic members include Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and Bfl-1/A1, which are characterized by the presence of all 4 BH domains (BH1-BH4). Most members also contain a C-terminal hydrophobic tail, which can target these proteins predominantly to the mitochondria and/or endoplasmic reticulum. These proteins prevent cell death by binding and sequestering proapoptotic proteins. The death promoter members are subdivided according to their function and biochemical structure into multidomain proteins, such as Bax and Bak, that closely resemble Bcl-2 but lack the N-terminal BH4 domain. The other subgroup is represented by BH3-only members that possess the BH3 domain required for binding to other members and for death-promoting activity. The BH3-only proteins include Bik, Bid, Puma/BBC3, and Noxa/APR, each thought to be responsible for transducing a specific cell death signal [10].
After their activation, Bax and Bak undergo conformational changes that contribute to increased permeability of the outer mitochondrial membrane through formation of pores. Bax and Bak also facilitate the release of cytochrome c (cyto c) and sequential activation of the caspase cascade [11]. Cytosolic cyto c binds the adaptor molecule APAF-1 (apoptotic protease activating factor 1) and caspase 9, forming a macromolecular complex, the apoptosome. This process leads to activation of caspase 9 [12]. Activated caspase 9 cleaves caspase 3, which cleaves and activates the other effector caspases (caspases 6 and 7). Additional molecules released from the mitochondria include apoptosis-inducing factor [13] and endonuclease G [14], which exert their apoptotic activity on nuclei. Smac/Diablo [15–17] and Omi/HtrA2 [18,19] facilitate caspase activation by antagonizing molecules belonging to the inhibitor of apoptosis (IAP) family.
A number of recent reviews describe the biology of apoptosis and the clinical applications in MM [1,20–23]. We focus on the key concepts and some of the most recent studies that summarize the field of MM apoptosis.
2. Role of the Mitochondrial Pathway in Apoptosis
Ionizing radiation (IR) and chemotherapeutic agents act primarily through the intrinsic pathway, in which mitochondria play the central role. DNA-damaging agents signal cell death by altering the mitochondrial transmembrane potential (ΔΨm), activating Bcl-2 family members with subsequent cyto c release, and activating the caspase family of proteins [7,21]. Bcl-2, frequently expressed in follicular lymphomas bearing the t(14;18) chromosomal translocation, is also widely expressed in many other B- and T-cell lymphomas without involvement of a bcl-2 rearrangement [9,21].
The effect of genotoxic agents, such as IR and chemotherapeutic drugs, is directed through the mitochondrial pathway in a p53-dependent manner. BH3-only members of the Bcl-2 family—Bik, Puma, and Noxa—are believed to play a central role in p53-activated cell death [24,25]. We observed that after IR the BH3-only genes puma, noxa, and bik were up-regulated in MM and lymphoma tumor cells. Eight and 16 hours after IR, ribonuclease protection assays indicated dramatic transcriptional induction of Bik, and there were similar changes in protein levels. In contrast, an increase in Noxa messenger RNA (mRNA) levels was observed as early as 0.5 hours after IR, and Puma levels had increased by 4 hours after IR. The differences in kinetics of induction of these BH3-only proteins indicated their distinct role in apoptosis activation in MM cells (M.O., A.A., unpublished data). Because Bid is not activated by IR [7], the identity of the Bax- and/or Bak-activating BH3-only protein is of great interest.
3. Bcl-2 Proteins Are Key Targets of Therapeutics
Imbalances in expression levels of the Bcl-2 family members result in defects in programmed cell death associated with chemoresistance, malignancy, and aggressiveness of tumors. The expression pattern of the Bcl-2 family of proapoptotic and antiapoptotic genes in MM have been the subject of multiple studies in which the investigators found increased levels of expression of Bcl-2, Bcl-xL, and Mcl-1 are linked to MM cell survival and resistance to chemotherapeutic agents [21,26-28]. The expression pattern of the Bcl-2 family separates the malignant phenotype of MM from normal plasma cells. In MM there is higher expression of the antiapoptotic Bcl-2 and Mcl-1 but not of Bcl-xL, and there is a lower level of expression of Bax [29]. On the other hand, targeted overexpression of Bcl-xL and c-Myc in B-lymphoid cells in mice resulted in lymphoproliferative disease and plasma cell malignancies. These findings were evidence that Bcl-xL can contribute to plasmacytomagenesis [30]. Bcl-xL expression is also associated with drug resistance in MM patients [31].
Chemotherapeutic agents, such as doxorubicin (Dox) induce apoptosis by causing cyto c release from mitochondria and subsequent activation of caspases, which are blocked by overexpression of Bcl-2. Treatment of U266 cells with Dox increased activation of Bax and Bak as well as of the BH3-only proteins Bid and Bik [32]. Arsenic trioxide (ATO) has been shown to induce apoptosis in MM cells [33] by directly inducing cyto c release from mitochondria via the mitochondrial permeability transition pore. The voltage-dependent anion channel was identified as a biological target of ATO [34]. Recent studies showed 2 distinct pathways for ATO-induced death in MM, depending on their p53 status. ATO treatment of cells with mutated p53 resulted in G2/M cell-cycle phase block. In contrast, cells with wild-type p53 were blocked in G1. Moreover, apoptosis may be activated differentially by ATO, with cells having mutated p53 engaging the extrinsic pathway and those having functional p53 engaging the intrinsic pathway. Finally, ATO treatment led to up-regulation of Apo2L (TRAIL) receptors and down-regulation of decoy receptors, observations that help explain the synergistic effect of ATO with Apo2L [35]. Recent published data from a phase 2 study showed that ATO as monotherapy has therapeutic efficacy in relapsed or refractory MM and that this agent was well tolerated with manageable adverse effects [36].
Overexpression of the antiapoptotic members has been linked to resistance to various chemotherapeutic agents. Increased expression of these proteins after exposure to chemotherapeutic agents of MM cell lines suggested that these agents might contribute to acquired chemoresistance. Thus regulation of antiapoptotic proteins may represent an important strategy for sensitizing MM cells to various therapeutic agents. Using an antisense strategy, investigators found that Mcl-1, rather than Bcl-2 or Bcl-xL, is an essential survival factor, because Mcl-1 down-regulation induced rapid apoptosis of MM. Although it had no effect, Bcl-2 antisense treatment alone sensitized myeloma cell lines to dexamethasone (Dex), whereas Bcl-xL antisense in combination with Dex had no effect [37]. Pretreatment with Bcl-2–antisense (oblimersen [Genasense]) at clinically relevant doses potentiated Dex-, paclitaxel (Taxol)-, and Ad-p53–induced apoptosis in drug-resistant and freshly isolated myeloma cells [38]. The drug also was effective in patients treated with vincristine and adriamycin and those with Dex-refractory MM [39,40].
Histone deacetylase (HDAC) inhibitors such as depsipeptide induce apoptosis in MM cell lines and primary MM cells by down-regulating Bcl-2, Bcl-xL, and Mcl-1 and increasing Bax levels [41]. Suberoylanilide hydroxamic acid (SAHA) treatment promoted cleavage of Bid in a calpain-dependent but caspase-independent manner. The cleavage was abrogated by overexpression of Bcl-2. In addition, SAHA sensitized MM cells to DR-induced apoptosis, this effect being mediated by down-regulation of the antiapoptotic proteins FLIP (Fas-associated death domain–like interleukin 1β–converting enzyme [FLICE]-like inhibitory protein), cIAP2, and XIAP [42].
Resistance of tumor cells to standard therapeutic agents has led to the continuous search for identification and characterization of new agents that augment the effects of older ones. Resveratrol treatment induces down-regulation of Bcl-xL and Mcl-1 and significant up-regulation of Bax and Apaf-1 and thus sensitizes MM cell lines to paclitaxel-induced apoptosis [43]. The selective mTOR (mammalian target of rapamycin) inhibitor rapamycin sensitized MM cell lines as well as primary myeloma cells to Dex-induced apoptosis. This effect is associated with concomitant down-regulation of cyclin D2 and the antiapoptotic protein survivin [44].
4. Reactive Oxygen Species as Apoptosis Mediators
An increase in generation of reactive oxygen species (ROS) within cells has been shown to induce loss of ΔΨm, release of cyto c and other apoptotic factors, and activation of caspases. IR treatment of MM cells results in ROS production associated with an increase in permeability transition pore opening and a decrease in ΔΨm and glutathione, effects associated with caspase activation and cyto c release [45]. IR, exogenous ROS, and caspase 3 all induce a decrease in ΔΨm and release of cyto c from mitochondria, effects that may be prevented by molecular (dominant-negative caspase 9) and pharmacologic (zVAD-fmk) caspase inhibitors and overexpression of Bcl-2. Exogenous ROS also induces mitochondrial permeability transition pore opening and cyto c release in isolated mitochondria that may be blocked by inhibition of the permeability transition pore with cyclosporin A. Results of studies conducted on MM cell lines sensitive or resistant to Dex indicated that treatment with 2-methoxyestradiol (2ME2) but not Dex induced generation of superoxide (O2−). 2ME2-induced apoptosis was associated with a decrease in ΔΨm, an increase in O2−, and release of cyto c and Smac [46]. Dex-triggered apoptosis involved a decrease in ΔΨm without a concurrent increase in O2−, a process associated with Smac release. The results of that study suggested that 2ME2 triggers O2− generation, which occurs upstream of caspase activation [46]. The synthetic oleanane triterpenoid 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO) and its derivative CDDO imidazolide ester (CDDO-Im) have been shown to induce apoptosis in MM and primary cells by decreasing the intracellular amount of reduced glutathione and increasing ROS levels, this effect being associated with FLIP down-regulation and caspase 8 activation [47]. Amplifying the levels of ROS within the tumor cells with inhibitors such as 2ME2 or CDDO may help increase the efficacy of standard treatments. It is interesting that 2ME2 and PS-341 activate c-jun NH2-terminal kinase (JNK), which translocates to mitochondria [48]. JNK recently was shown to activate a novel apoptotic pathway by releasing Smac rather than cyto c during TNF-induced apoptosis [49], a process similar to Dex-induced apoptosis in MM [23].
5. Role of the DR Pathway in Apoptosis
Activation of cell surface DRs, such as DR4 and DR5, by the Apo2L ligand induces a signaling cascade culminating in activation of caspases [5] (Figure 2). In addition to being tumor specific, Apo2L-based therapy is attractive because it is not dependent on p53 and therefore should be effective in p53-deficient tumors. Apo2L potently induces apoptosis of MM cells from patients and the majority of MM cell lines, including cells sensitive or resistant to Dex, Dox, melphalan, and mitoxantrone. Apo2L also has overcome the survival effect of interleukin 6 (IL-6). A recent report indicated that Apo2L is expressed in plasma cells and may be responsible for apoptosis of plasma cells in a caspase-independent manner after antibody secretion [50].
Figure 2.
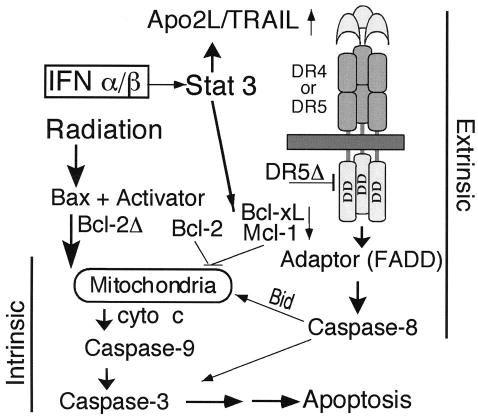
Model for activation of apoptosis in multiple myeloma by interferons (IFNs). After transcriptional induction by IFNs, Apo2 ligand (Apo2L) engages its death receptor 5 (DR5) or DR4 and through an adaptor Fas-associated death domain (FADD) recruits caspase 8 to the cell membrane, which can be blocked by a dominant-negative DR5Δ. After caspase 8 activation by proteolysis, Bid is cleaved and translocates to mitochondria, causing release of low levels of cytochrome c (cyto c) into the cytosol, a process that leads to caspase 9 and 3 activation. This process results in attack of the antiapoptotic protein Bcl-2 on the mitochondrial membranes, producing a truncated Bcl-2Δ protein, which causes release of more cyto c, caspase activation, and apoptosis. Bcl-xL or Mcl-1 transcriptional down-regulation mediated by signal transducers and activators of transcription 3 (Stat 3) is an additional mechanism by which IFNs may decrease levels of antiapoptotic proteins and shift the balance toward a proapoptotic state. (Modified from [6], with permission.) TRAIL indicates tumor necrosis factor–related apoptosis-inducing factor ligand; DD, death domain.
Interferons (IFNs) have been used as effective pharmacological agents in treating a variety of cancers and viral diseases in the last 30 years. Our studies have shown that type I but not type II IFNs induce apoptosis through activation of the Apo2L pathway and modulation of the Bcl-2 family of proteins in MM cell lines and patient-derived primary cells [6,21]. IFNs play an important role in the immune system and may influence expression of a number of genes associated with both apoptosis and cell cycle progression [51]. One mechanism by which IFNs may achieve their role in immune surveillance may be through regulation of other cytokines. Our data provided evidence that IFNs exert their profound effect by inducing apoptosis in MM. The data also suggested that Apo2L may be an important mediator of these effects. We have identified and isolated the promoter region of Apo2L and found that it contains IFN-stimulated regulatory elements that can be regulated in MM [52]. Cytokines suppress apoptosis by preventing Apo2L expression through modulation of the forkhead FOXO3a transcription factor [53]. These findings support a rationale for exploring Apo2L as well as the therapies that modulate its expression in the management of MM, especially in instances in which IFN-α and IFN-β may be effective, such as in advanced MM. Given the lack of cytotoxicity of Apo2L toward most other types of blood cells, as well as other cell types in mice and nonhuman primates, it would be of interest to examine whether Apo2L, which is expected to enter clinical trials, could be used for treating MM patients.
Apo2L induction was at least partially necessary for proteolytic cleavage-dependent activation of apoptotic activity of Bcl-2 and Bid (Figure 2). In addition, we observed that IFN-α induces down-regulation of Bcl-xL, which is expressed at high levels in these cells (A.A., Q. Chen, unpublished data, 2002). Other reports have indicated that the effect of several therapeutic agents could be mediated by down-regulation of Bcl-2 family protein expression [21]. Expression of these proteins also may be regulated by the myeloma survival factors IL-6, IFNs, and insulin-like growth factor 1 (IGF-1) [54].
Recent studies have further demonstrated the role of the mitochondrial pathway in IFN-α–induced apoptosis in MM by showing that IFNs activate sequentially the proapoptotic Bcl-2 family members Bak and Bax [32]. IFN-α–induced apoptosis can be blocked through inhibition of the phosphatidylinositol 3-kinase (PI3K)/mTOR pathway. Because many tumor types are resistant to IFN treatment, these findings are an aid to understanding the variability of cellular response to IFN treatment and emphasize the importance of knowledge of the signaling pathways responsible for apoptosis induction [6,55].
6. Initiation and Amplification Stages in Apoptosis
The link between the extrinsic and intrinsic pathways of apoptosis through engagement of mitochondria by truncated Bid provides amplification of the apoptotic signal and further supports the critical role of mitochondria in apoptosis. In contrast, genotoxic agents such as IR and IFNs cause MM apoptosis by 2 distinct stages of cyto c release and a positive feedback loop linking caspase activation to cyto c release and mitochondrial dysfunction [6,7]. In addition, we found that in MM and all other hematopoietic tumor cells examined, a caspase 3–generated p18-kd proteolytic fragment of cyclin E produced during genotoxic stress-induced apoptosis [8] will further amplify the process by activating Bax. Bax is sequestered to cytoplasmic proteins, such as the DNA repair protein Ku70, and is released after p18 cyclin E binding to Ku70 (A.A., S. Mazumder, unpublished data, 2004) or acetylation [56]. Radiotherapeutics and chemotherapeutics also induce apoptosis through increased expression of cyclin E [57], perhaps by providing more substrate for genesis of p18 cyclin E.
7. Growth and Survival Factors Prevent Apoptosis
MM cells accumulate in the BM, where they acquire growth and survival properties conferred by the BM microenvironment [22]. BM stromal cells (BMSCs) secrete the chemokine stromal cell–derived factor 1α (SDF-1α). This chemokine plays a major role in homing, because MM cells express the SDF-1α receptor, CXCR4 (Figure 3). Once in the BM, integrin α4B1 (VLA-4) mediates attachment of MM cells to the stroma [58], and this process confers the cells a survival advantage. Adhesion of MM cells to fibronectin (FN) is known to protect the cells from drug-induced apoptosis [23]. Although nearly all myeloma cells respond to growth and survival factors, such as IL-6, only some require them, and only some produce them. Those that do not produce IL-6 and require it rely on its production by BMSCs [1, 59]. The pathophysiology of MM has been attributed to dysregulation of autocrine growth loops (including IL-6 and IL-1) and various survival pathways, including NF-κB/IκB (nuclear factor κB/inhibitory unit of NF-κB), JAK2/STAT3 (Janus kinase 2/signal transducers and activators of transcription 3), PI3K/Akt, and Ras/Raf/MAPK (Ras/Raf/mitogen-activated protein kinase) [23,60], which act through targets such as Bcl-2 or IAPs to prevent their apoptotic demise.
Figure 3.
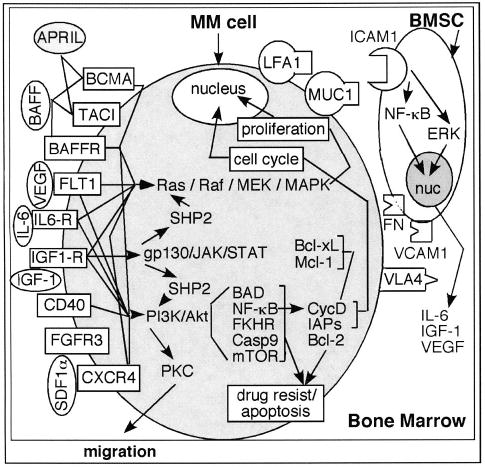
Growth and survival pathways. Receptors for a proliferation-inducing ligand (APRIL) and B-cell–activating factor (BAFF) include B-cell maturation antigen (BCMA), transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI), and BAFF receptor (BAFFR). FLT1 is a high-affinity vascular endothelial growth factor (VEGF) receptor. CXCR4 is a receptor for stromal cell–derived factor (SDF1α). These receptors, along with interleukin 6 receptor (IL6-R), insulin-like growth factor 1 receptor (IGF1-R), and CD40, mediate downstream activation of the various pathways. These pathways include nuclear factor κB (NF-κB), Janus kinase/signal transducers and activators of transcription (JAK/STAT), phosphatidylinositol 3-kinase (PI3K)/Akt, and Ras/Raf/mitogen-activated protein kinase (MAPK). These pathways ultimately lead to cytokine production (IL-6, IGF-1, VEGF), resistance to apoptosis (Bcl-2, inhibitors of apoptosis [IAPs]), cell proliferation, and migration of multiple myeloma (MM) cells. The bone marrow stromal cell (BMSC)/MM cell interaction is mediated by the surface receptors lymphocyte function–associated antigen 1 (LFA1), mucin, (MUC1) integrin α4β1 (VLA4), intracellular adhesion molecule 1 (ICAM1), vascular adhesion molecule 1 (VCAM1), and fibronectin (FN). Activation of ICAM by cell-cell interaction leads to secretion of various cytokines from the BMSCs. FGFR3 indicates fibroblast growth factor receptor 3; MEK, mitogen-activated protein/ERK kinase; SHP2, Src homology region 2 domain-containing phosphatase 2; gp130, glycoprotein 130; PKC, protein kinase C; BAD, Bcl-2 antagonist of cell death; FKHR, forkhead homolog of rhabdomyosarcoma; Casp9, caspase 9; mTOR, mammalian target of rapamycin; CycD, cyclin D; ERK, extracellular signal–regulated kinase; nuc, nucleus.
NF-κB refers to a group of dimeric transcription factors regulating various genes the functions of which include cell growth, angiogenesis, cell adhesion, and protection from apoptosis. NF-κB exists in the cytoplasm in an inactive form bound to IκBα, one of its interacting negative regulators, which has to be phosphorylated to mark it for degradation by the 26S proteasome. This process leaves NF-κB free to translocate to the nucleus [61], where it binds to its targets, such as cyclin D1 [61], Bcl-xL, and IAPs [62]. NF-κB, because of its striking importance in MM, continues to be a compelling target for drug development. Dex, thalidomide, PS-341, and curcumin all inhibit NF-κB [23,63]. Combined with the Chk1 abrogator UCN-01, Bay 11-7082, an irreversible inhibitor of IκBα phosphorylation, and the NF-κB inhibitor SN50 effectively induce apoptosis [60]. NF-κB is responsible for up-regulation of IAPs and Bcl-2 [62] to control cell survival and for expression of ICAM1 and VCAM1 (intercellular and vascular adhesion molecules 1) on MM cells and BMSCs [1]. Adhesion molecules such as these and LFA-1 (lymphocyte function–associated antigen 1) facilitate the BMSC-MM cell interaction [20, 64].
Posttranslational modifications involve ubiquitination and proteasome degradation. Proteasome inhibitors block accumulation of IκB in the cytoplasm, inhibiting the NF-κB cell survival pathway and leading to apoptosis. A transcriptional profile of MM.1S cells treated with the bortezomib/proteasome inhibitor PS-341 (Velcade) revealed distinct patterns of coordinated changes in a range of transcripts. The changes included down-regulation of growth and antiapoptotic transcripts and induction of apoptotic members, such as the Fas ligand and receptor. The effect of the agent was blocked by a dominant-negative Fas [65]. Moreover, coadministration of proteasome and HDAC inhibitors in MM cell lines sensitive or resistant to established cytotoxic agents resulted in a synergistic increase in mitochondrial injury, caspase activation, and apoptosis [66]. Furthermore, IL-6–induced growth was associated with high levels of CDC34 (ubiquitin-conjugating enzyme [UBC3]), IL-6–mediated protection from Dex-induced apoptosis being dampened by blocking of CDC34. CDC34 is responsible for ubiquitination of various proteins, including IκBα. MM cells express high levels of CDC34 at both the mRNA and the protein levels. Dex, 2ME2, and PS-341 treatments are associated with decreased CDC34 expression. Blocking CDC34 augments sensitivity to these therapies [67]. Targeting mitochondria may be an effective way to overcome conventional resistance and Velcade resistance in MM [68].
MM cell adhesion to BMSCs up-regulates secretion of vascular endothelial growth factor (VEGF) [64] and IL-6 by BMSCs. IL-6 secretion by BMSCs induces MM cells to produce their own IL-6. Secretion by MM cells in turn increases IL-6 secretion by BMSCs. VEGF is produced and secreted by both MM cells and BMSCs [64]. FLT1, a high-affinity VEGF receptor on MM cells, when phosphorylated by VEGF activates p42/p44/MAPK, leading to MM cell proliferation. VEGF is known to stimulate angiogenesis but also induces up-regulation of MCL-1 (myeloid cell leukemia 1), thus protecting MM cells against apoptosis [69]. In a PI3K/Akt/PKC (protein kinase C)-dependent manner, VEGF promotes migration of MM cells from BM [1]. IGF-1 mediates migration through PI3K as well [70]. CD40, a TNF family transmembrane protein expressed in most MM cells, may have a role in MM cell homing and migration by induction of VEGF [71].
IL-6, a major survival factor for MM tumor cells, induces signaling through the STAT proteins. STAT3 is constitutively activated in BM mononuclear cells from patients with MM and in the IL-6–dependent human MM cell line U266 [27]. IL-6–induced Dex resistance occurs in part through JAK/STAT3 signaling [1,31]. MM cells from patients have been shown to express constitutively active forms of NF-κB and STAT3, whereas cells from healthy subjects have not [31]. Up-regulation of Bcl-xL and Mcl-1 via retroviral insertion of activated STAT3 and STAT5A, targets of both IL-6 and IGF-1, causes cell growth, oncogenesis, and cytokine independence [59]. Fibroblast growth factor receptor 3 (FGFR3), frequently overexpressed owing to a t(4:14) translocation, also confers a growth and survival advantage, possibly by activating STAT3 and resulting in Bcl-xL expression [72]. Bcl-xL expression can be inhibited by blocking IL-6 receptor signaling from JAKs to the STAT3 protein, demonstrating that STAT3 signaling is essential for the survival of MM tumor cells [27]. IL-6, via Shp2/RAFTK (related adhesion focal tyrosine kinase) prevents a Dex-induced increase in Smac, thus preventing apoptosis. Dex, in addition to triggering apoptosis, has the effect of transiently increasing expression of IL-6 receptor and transforming growth factor β receptor II and increasing production and release of IL-6 [22].
IL-6 and IGF-1 afford protection against Dex-induced apoptosis by up-regulation of Bcl-xL, and Mcl-1 (through STATs) and activation of the MAPK and PI3K/Akt pathways, which contribute to inactivation of caspase 9 [61,64]. IGF-I has been reported to have multiple functions that lead to sustained activation of NF-κB and Akt, phosphorylation of FKHRL-1 (forkhead homolog of rhabdomyosarcoma receptor ligand 1), up-regulation of FLIP, survivin, cIAP-2, Bfl-1, and XIAP, and a decrease in Apo2L sensitivity [23,73]. More recently it has been shown that, via extracellular signal–regulated kinase (ERK), IGF-1 also induces VEGF secretion [70]. IL-6 and IGF-1 have separate receptors but have substantial downstream overlap of pathways. Simvastatin (a 3-hydroxy-3-methylglutaryl coenzyme A inhibitor) induces apoptosis in vitro despite protective influences of IL-6, IGF-1, and VLA-4 (which allows MM cell adhesion to FN). This finding suggests a common downstream pathway for IL-6, IGF-1, and VLA-4, possibly through PI3K [74].
8. Future Directions
Novel therapeutic targets in MM emerge from proteins identified through microarray [1,75] and proteomics strategies [20]. Gene expression profiling provides essential information on expression and regulation of many genes in MM, including cell DRs (eg, DR4/5, BAFFR [B-cell–activating factor receptor], BCMA [B-cell maturation antigen], TACI [transmembrane activator and calcium modulator and cyclophilin ligand interactor]) and their ligands (Apo2L/TRAIL, APRIL [a proliferation-inducing ligand], BAFF, and CD40) [75] (M.O., A.A., unpublished data). Exogenous cytokine-dependent MM cells were reported to overexpress receptors for APRIL and BAFF. All 3 BAFF receptors—TACI, BCMA, and BAFFR—activate NF-κB. APRIL induces up-regulation of Mcl-1 and Bcl-2, and IL-6 up-regulates Mcl-1 only. Both BAFF and APRIL have been shown to be as effective as IL-6 in preventing Dex-induced apoptosis. IL-6 has induced phosphorylation of STAT3, MAPK, and Akt (activating the JAK/STAT, MAPK, and PI3K/Akt cascades, respectively) [2]. IL-6–producing MM cells, via activated STAT3, are resistant to Fas [59]. IGF-1 has been shown to activate MAPK and PI3K/Akt. BAFF and APRIL have induced phosphorylation of only MAPK and Akt (activating the ERK1/2 and PI3K/Akt cascades, respectively) [76]. Establishing the function of these molecules in MM and their validation as targets in clinical therapy will broaden our understanding of the molecular determinants of the disease and provide additional tools that may help to cure it.
Acknowledgments
Supported in part by research grants CA81504 and CA82858 to A.A. from the National Cancer Institute.
References
- 1.Hideshima T, Anderson KC. Molecular mechanisms of novel therapeutic approaches for multiple myeloma. Nat Rev Cancer. 2002;2:927–937. doi: 10.1038/nrc952. [DOI] [PubMed] [Google Scholar]
- 2.Hussein MA, Juturi JV, Lieberman I. Multiple myeloma: present and future. Curr Opin Oncol. 2002;14:31–35. doi: 10.1097/00001622-200201000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Arends MJ, Wyllie AH. Apoptosis: mechanisms and roles in pathology. Int Rev Exp Pathol. 1991;32:223–254. doi: 10.1016/b978-0-12-364932-4.50010-1. [DOI] [PubMed] [Google Scholar]
- 4.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 5.Almasan A, Ashkenazi A. Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev. 2003;14:337–348. doi: 10.1016/s1359-6101(03)00029-7. [DOI] [PubMed] [Google Scholar]
- 6.Chen Q, Gong B, Mahmoud-Ahmed A, et al. Apo2L/TRAIL and Bcl-2–related proteins regulate type I interferon-induced apoptosis in multiple myeloma. Blood. 2001;98:2183–2192. doi: 10.1182/blood.v98.7.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Q, Gong B, Almasan A. Distinct stages of cytochrome c release from mitochondria: evidence for a feedback amplification loop linking caspase activation to mitochondrial dysfunction in genotoxic stress induced apoptosis. Cell Death Differ. 2000;7:227–233. doi: 10.1038/sj.cdd.4400629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazumder S, Chen Q, Gong B, Drazba JA, Buchsbaum JC, Almasan A. Proteolytic cleavage of cyclin E leads to inactivation of associated kinase activity and amplification of apoptosis in hematopoietic cells. Mol Cell Biol. 2002;22:2398–2409. doi: 10.1128/MCB.22.7.2398-2409.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cory S, Adams JM. The bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 10.Huang DC, Strasser A. BH3-only proteins: essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 11.Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–377. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- 12.Strasser A, O’Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 13.Joza N, Susin SA, Daugas E, et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 14.Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 15.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 16.Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 17.Srinivasula SM, Hegde R, Saleh A, et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410:112–116. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 18.Hegde R, Srinivasula SM, Zhang Z, et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem. 2002;277:432–438. doi: 10.1074/jbc.M109721200. [DOI] [PubMed] [Google Scholar]
- 19.Martins LM, Iaccarino I, Tenev T, et al. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem. 2002;277:439–444. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- 20.Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood. 2004;104:607–618. doi: 10.1182/blood-2004-01-0037. [DOI] [PubMed] [Google Scholar]
- 21.Chen Q, Ray S, Hussein MA, Srkalovic G, Almasan A. Role of Apo2L/TRAIL and Bcl-2-family proteins in apoptosis of multiple myeloma. Leuk Lymphoma. 2003;44:1209–1214. doi: 10.1080/1042819031000068052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chauhan D, Anderson KC. Mechanisms of cell death and survival in multiple myeloma (MM): therapeutic implications. Apoptosis. 2003;8:337–343. doi: 10.1023/a:1024164700094. [DOI] [PubMed] [Google Scholar]
- 23.Chauhan D, Hideshima T, Anderson KC. Apoptotic signaling in multiple myeloma: therapeutic implications. Int J Hematol. 2003;78:114–120. doi: 10.1007/BF02983378. [DOI] [PubMed] [Google Scholar]
- 24.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 25.Oda E, Ohki R, Murasawa H, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 26.Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002;99:1885–1893. doi: 10.1182/blood.v99.6.1885. [DOI] [PubMed] [Google Scholar]
- 27.Catlett-Falcone R, Landowski TH, Oshiro MM, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 28.Tu Y, Renner S, Xu F, et al. BCL-X expression in multiple myeloma: possible indicator of chemoresistance. Cancer Res. 1998;58:256–262. [PubMed] [Google Scholar]
- 29.Spets H, Stromberg T, Georgii-Hemming P, Siljason J, Nilsson K, Jernberg-Wiklund H. Expression of the bcl-2 family of pro- and anti-apoptotic genes in multiple myeloma and normal plasma cells: regulation during interleukin-6 (IL-6)-induced growth and survival. Eur J Haematol. 2002;69:76–89. doi: 10.1034/j.1600-0609.2002.01549.x. [DOI] [PubMed] [Google Scholar]
- 30.Linden M, Kirchhof N, Carlson C, Van Ness B. Targeted overexpression of Bcl-XL in B-lymphoid cells results in lymphoproliferative disease and plasma cell malignancies. Blood. 2004;103:2779–2786. doi: 10.1182/blood-2003-10-3399. [DOI] [PubMed] [Google Scholar]
- 31.Bharti AC, Shishodia S, Reuben JM, et al. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood. 2004;103:3175–3184. doi: 10.1182/blood-2003-06-2151. [DOI] [PubMed] [Google Scholar]
- 32.Panaretakis T, Pokrovskaja K, Shoshan MC, Grander D. Interferon-alpha–induced apoptosis in U266 cells is associated with activation of the proapoptotic Bcl-2 family members Bak and Bax. Oncogene. 2003;22:4543–4556. doi: 10.1038/sj.onc.1206503. [DOI] [PubMed] [Google Scholar]
- 33.Park WH, Seol JG, Kim ES, et al. Arsenic trioxide-mediated growth inhibition in MC/CAR myeloma cells via cell cycle arrest in association with induction of cyclin-dependent kinase inhibitor, p21, and apoptosis. Cancer Res. 2000;60:3065–3071. [PubMed] [Google Scholar]
- 34.Zheng Y, Shi Y, Tian C, et al. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene. 2004;23:1239–1247. doi: 10.1038/sj.onc.1207205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Q, Hilsenbeck S, Gazitt Y. Arsenic trioxide-induced apoptosis in myeloma cells: p53-dependent G1 or G2/M cell cycle arrest, activation of caspase-8 or caspase-9, and synergy with APO2/TRAIL. Blood. 2003;101:4078–4087. doi: 10.1182/blood-2002-10-3231. [DOI] [PubMed] [Google Scholar]
- 36.Hussein MA, Saleh M, Ravandi F, Mason J, Rifkin RM, Ellison R. Phase 2 study of arsenic trioxide in patients with relapsed or refractory multiple myeloma. Br J Haematol. 2004;125:470–476. doi: 10.1111/j.1365-2141.2004.04941.x. [DOI] [PubMed] [Google Scholar]
- 37.Derenne S, Monia B, Dean NM, et al. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002;100:194–199. doi: 10.1182/blood.v100.1.194. [DOI] [PubMed] [Google Scholar]
- 38.Liu Q, Gazitt Y. Potentiation of dexamethasone-, paclitaxel-, and Ad-p53–induced apoptosis by Bcl-2 antisense oligodeoxynucleotides in drug-resistant multiple myeloma cells. Blood. 2003;101:4105–4114. doi: 10.1182/blood-2002-10-3067. [DOI] [PubMed] [Google Scholar]
- 39.van de Donk NW, Kamphuis MM, van Dijk M, Borst HP, Bloem AC, Lokhorst HM. Chemosensitization of myeloma plasma cells by an antisense-mediated downregulation of Bcl-2 protein. Leukemia. 2003;17:211–219. doi: 10.1038/sj.leu.2402768. [DOI] [PubMed] [Google Scholar]
- 40.van de Donk NW, de Weerdt O, Veth G, et al. G3139, a Bcl-2 antisense oligodeoxynucleotide, induces clinical responses in VAD refractory myeloma. Leukemia. 2004;18:1078–1084. doi: 10.1038/sj.leu.2403363. [DOI] [PubMed] [Google Scholar]
- 41.Khan SB, Maududi T, Barton K, Ayers J, Alkan S. Analysis of histone deacetylase inhibitor, depsipeptide ( FR901228), effect on multiple myeloma. Br J Haematol. 2004;125:156–161. doi: 10.1111/j.1365-2141.2004.04882.x. [DOI] [PubMed] [Google Scholar]
- 42.Mitsiades N, Mitsiades CS, Richardson PG, et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood. 2003;101:4055–4062. doi: 10.1182/blood-2002-11-3514. [DOI] [PubMed] [Google Scholar]
- 43.Jazirehi AR, Bonavida B. Resveratrol modifies the expression of apoptotic regulatory proteins and sensitizes non-Hodgkin’s lymphoma and multiple myeloma cell lines to paclitaxel-induced apoptosis. Mol Cancer Ther. 2004;3:71–84. [PubMed] [Google Scholar]
- 44.Stromberg T, Dimberg A, Hammarberg A, et al. Rapamycin sensitizes multiple myeloma cells to apoptosis induced by dexamethasone. Blood. 2004;103:3138–3147. doi: 10.1182/blood-2003-05-1543. [DOI] [PubMed] [Google Scholar]
- 45.Chen Q, Chai Y-C, Mazumder S, et al. The late increase in intracellular free radical oxygen species during apoptosis is associated with cytochrome c release, caspase activation, and mitochondrial dysfunction. Cell Death Differ. 2003;10:323–334. doi: 10.1038/sj.cdd.4401148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chauhan D, Li G, Sattler M, et al. Superoxide-dependent and - independent mitochondrial signaling during apoptosis in multiple myeloma cells. Oncogene. 2003;22:6296–6300. doi: 10.1038/sj.onc.1206734. [DOI] [PubMed] [Google Scholar]
- 47.Ikeda T, Nakata Y, Kimura F, et al. Induction of redox imbalance and apoptosis in multiple myeloma cells by the novel triterpenoid 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid. Mol Cancer Ther. 2004;3:39–45. [PubMed] [Google Scholar]
- 48.Chauhan D, Li G, Hideshima T, et al. JNK-dependent release of mitochondrial protein, Smac, during apoptosis in multiple myeloma (MM) cells. J Biol Chem. 2003;278:17593–17596. doi: 10.1074/jbc.C300076200. [DOI] [PubMed] [Google Scholar]
- 49.Deng Y, Ren X, Yang L, Lin Y, Wu XA. JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 50.Ursini-Siegel J, Zhang W, Altmeyer A, et al. TRAIL/Apo-2 ligand induces primary plasma cell apoptosis. J Immunol. 2002;169:5505–5513. doi: 10.4049/jimmunol.169.10.5505. [DOI] [PubMed] [Google Scholar]
- 51.Sangfelt O, Erickson S, Castro J, Heiden T, Einhorn S, Grander D. Induction of apoptosis and inhibition of cell growth are independent responses to interferon-alpha in hematopoietic cell lines. Cell Growth Differ. 1997;8:343–352. [PubMed] [Google Scholar]
- 52.Gong B, Almasan A. Genomic organization and transcriptional regulation of the human Apo2L/TRAIL gene. Biochem Biophys Res Commun. 2000;278:747–752. doi: 10.1006/bbrc.2000.3872. [DOI] [PubMed] [Google Scholar]
- 53.Ghaffari S, Jagani Z, Kitidis C, Lodish HF, Khosravi-Far R. Cytokines and BCR-ABL mediate suppression of TRAIL-induced apoptosis through inhibition of forkhead FOXO3a transcription factor. Proc Natl Acad Sci U S A. 2003;100:6523–6528. doi: 10.1073/pnas.0731871100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jourdan M, Veyrune JL, Vos JD, Redal N, Couderc G, Klein B. A major role for Mcl-1 antiapoptotic protein in the IL-6–induced survival of human myeloma cells. Oncogene. 2003;22:2950–2959. doi: 10.1038/sj.onc.1206423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thyrell L, Hjortsberg L, Arulampalam V, et al. Interferon alpha-induced apoptosis in tumor cells is mediated through the phosphoinositide 3-kinase/mammalian target of rapamycin signaling pathway. J Biol Chem. 2004;279:24152–24162. doi: 10.1074/jbc.M312219200. [DOI] [PubMed] [Google Scholar]
- 56.Cohen H, Lavu S, Bitterman K, et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell. 2004;13:627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 57.Mazumder S, Gong B, Almasan A. Cyclin E induction by genotoxic stress leads to apoptosis of hematopoietic cells. Oncogene. 2000;19:2828–2835. doi: 10.1038/sj.onc.1203623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parmo-Cabanas M, Bartolome RA, Wright N, Hidalgo A, Drager AM, Teixido J. Integrin alpha4beta1 involvement in stromal cell-derived factor-1alpha-promoted myeloma cell transendothelial migration and adhesion: role of cAMP and the actin cytoskeleton in adhesion. Exp Cell Res. 2004;294:571–580. doi: 10.1016/j.yexcr.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 59.Hodge DR, Xiao W, Wang LH, Li D, Farrar WL. Activating mutations in STAT3 and STAT5 differentially affect cellular proliferation and apoptotic resistance in multiple myeloma cells. Cancer Biol Ther. 2004;3:188–194. doi: 10.4161/cbt.3.2.621. [DOI] [PubMed] [Google Scholar]
- 60.Dai Y, Pei XY, Rahmani M, Conrad DH, Dent P, Grant S. Interruption of the NF-kappaB pathway by Bay 11-7082 promotes UCN-01-mediated mitochondrial dysfunction and apoptosis in human multiple myeloma cells. Blood. 2004;103:2761–2770. doi: 10.1182/blood-2003-09-3037. [DOI] [PubMed] [Google Scholar]
- 61.Panwalkar A, Verstovsek S, Giles F. Nuclear factor-kappaB modulation as a therapeutic approach in hematologic malignancies. Cancer. 2004;100:1578–1589. doi: 10.1002/cncr.20182. [DOI] [PubMed] [Google Scholar]
- 62.Mitsiades N, Mitsiades CS, Poulaki V, et al. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: therapeutic applications. Blood. 2002;99:4079–4086. doi: 10.1182/blood.v99.11.4079. [DOI] [PubMed] [Google Scholar]
- 63.Bharti AC, Donato N, Singh S, Aggarwal BB. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-kappa B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood. 2003;101:1053–1062. doi: 10.1182/blood-2002-05-1320. [DOI] [PubMed] [Google Scholar]
- 64.Greenstein S, Krett NL, Kurosawa Y, et al. Characterization of the MM. 1 human multiple myeloma (MM) cell lines: a model system to elucidate the characteristics, behavior, and signaling of steroid-sensitive and -resistant MM cells. Exp Hematol. 2003;31:271–282. doi: 10.1016/s0301-472x(03)00023-7. [DOI] [PubMed] [Google Scholar]
- 65.Mitsiades N, Mitsiades CS, Poulaki V, et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci U S A. 2002;99:14374–14379. doi: 10.1073/pnas.202445099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004;10:3839–3852. doi: 10.1158/1078-0432.CCR-03-0561. [DOI] [PubMed] [Google Scholar]
- 67.Chauhan D, Li G, Hideshima T, et al. Blockade of ubiquitin-conjugating enzyme CDC34 enhances anti-myeloma activity of bortezomib/proteasome inhibitor PS-341. Oncogene. 2004;23:3597–3602. doi: 10.1038/sj.onc.1207458. [DOI] [PubMed] [Google Scholar]
- 68.Chauhan D, Li G, Podar K, et al. Targeting mitochondria to overcome conventional and bortezomib/proteasome inhibitor PS-341 resistance in multiple myeloma (MM) cells. Blood [Epub ahead of print]. June 24, 2004. [DOI] [PubMed]
- 69.Le Gouill S, Podar K, Amiot M, et al. VEGF induces MCL-1 upregulation and protects multiple myeloma cells against apoptosis. Blood [Epub ahead of print]. June 24, 2004. [DOI] [PubMed]
- 70.Menu E, Kooijman R, Van Valckenborgh E, et al. Specific roles for the PI3K and the MEK-ERK pathway in IGF-1-stimulated chemotaxis, VEGF secretion and proliferation of multiple myeloma cells: study in the 5T33MM model. Br J Cancer. 2004;90:1076–1083. doi: 10.1038/sj.bjc.6601613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tai YT, Catley LP, Mitsiades CS, et al. Mechanisms by which SGN-40, a humanized anti-CD40 antibody, induces cytotoxicity in human multiple myeloma cells: clinical implications. Cancer Res. 2004;64:2846–2852. doi: 10.1158/0008-5472.can-03-3630. [DOI] [PubMed] [Google Scholar]
- 72.Paterson JL, Li Z, Wen XY, et al. Preclinical studies of fibroblast growth factor receptor 3 as a therapeutic target in multiple myeloma. Br J Haematol. 2004;124:595–603. doi: 10.1111/j.1365-2141.2004.04814.x. [DOI] [PubMed] [Google Scholar]
- 73.Tarte K, Jourdan M, Veyrune JL, et al. The Bcl-2 family member Bfl-1/A1 is strongly repressed in normal and malignant plasma cells but is a potent anti-apoptotic factor for myeloma cells. Br J Haematol. 2004;125:373–382. doi: 10.1111/j.1365-2141.2004.04908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Osadchy A, Drucker L, Radnay J, Shapira H, Lishner M. Microenvironment factors do not afford myeloma cell lines protection from simvastatin. Eur J Haematol. 2004;73:1–8. doi: 10.1111/j.1600-0609.2004.00284.x. [DOI] [PubMed] [Google Scholar]
- 75.Ray S, Hissong JG, Oancea M, Almasan A. Expression and regulation of death receptors in multiple myeloma and prostate carcinoma. In: El-Deiry WS, ed. Death Receptors in Cancer Therapy. Totowa, NJ: Humana Press; 2004:281–296.
- 76.Moreaux J, Legouffe E, Jourdan E, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood. 2004;103:3148–3157. doi: 10.1182/blood-2003-06-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]