

Abstract
The building block of hepatitis C virus (HCV) nucleocapsid, the core protein, together with viral RNA, is composed of different domains involved in RNA binding and homo-oligomerization. The HCV core protein 1-169 (CHCV169) and its N-terminal region from positions 1 to 117 (CHCV117) were expressed in Escherichia coli and purified to homogeneity suitable for biochemical and biophysical characterizations. The overall conformation and the oligomeric properties of the resulting proteins CHCV169 and CHCV117 were investigated by using analytical centrifugation, circular dichroism, intrinsic fluorescence measurements, and limited proteolysis. Altogether, our results show that core protein (CHCV169) behaves as a membranous protein and forms heterogeneous soluble micelle-like aggregates of high molecular weight in the absence of detergent. In contrast, it behaves, in the presence of mild detergent, as a soluble, well-folded, noncovalent dimer. Similar to findings observed for core proteins of HCV-related flaviviruses, the HCV core protein is essentially composed of α-helices (50%). In contrast, CHCV117 is soluble and monodispersed in the absence of detergent but is unfolded. It appears that the folding of the highly basic domain from positions 2 to 117 (2-117 domain) depends on the presence of the 117-169 hydrophobic domain, which contains the structural determinants ensuring the binding of core with cellular membranes. Finally, our findings provide valuable information for further investigations on isolated core protein, as well as for attempts to reconstitute nucleocapsid particles in vitro.
Hepatitis C virus (HCV) infection is a major cause of chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. It is estimated that 1% of the general population in Western Europe and North America and 380 million people globally are infected with HCV worldwide (19). A protective vaccine does not exist to date, and therapeutic options are still limited. HCV has been classified in the Hepacivirus genus within the Flaviviridae family of viruses. It contains a 9.6-kb plus-strand RNA genome (10, 59, 70) composed of a 5′ noncoding region (5′ NCR), a long open reading frame (ORF) encoding a polyprotein precursor of about 3,000 amino acids (aa), and a 3′ NCR. The HCV polyprotein precursor is co- and posttranslationally processed by cellular and viral proteases to yield the mature structural and nonstructural (NS) proteins. The structural proteins include the core protein, which forms the viral nucleocapsid, and the envelope glycoproteins E1 and E2. They are separated from the NS proteins by the viroporin p7. The NS proteins include the NS2-3 autoprotease and the NS3 serine protease, an NTPase/RNA helicase located in the C-terminal two-thirds of NS3, the NS4A cofactor of NS3, the NS4B and NS5A proteins, and the NS5B RNA-dependent RNA polymerase (for reviews, see references 55 and 58). Interestingly, alternative reading frames (ARF) were recently identified in the HCV core region that has the potential to encode proteins designated ARFP or F (frameshift) proteins (3, 77, 83).
The HCV core protein is a highly basic, RNA-binding protein that presumably forms the viral nucleocapsid. The biochemical features of this protein have been relatively poorly characterized, and its structure is unknown. It is 191 aa in length and consists of three distinct predicted domains: an N-terminal two-thirds hydrophilic domain of 120 aa or so (named domain D1), a C-terminal one-third hydrophobic domain of 50 aa or so (named domain D2), and approximately the last 20 aa serving as the signal peptide of the downstream protein E1 (20, 21, 44). The domain D1 includes numerous positively charged amino acids and is mainly involved in RNA binding. Its N terminus contains immunodominant antigenic sites, and nuclear magnetic resonance (NMR) three-dimensional structure analysis has revealed a helix-loop-helix motif at amino acids 17 to 37 (see PDB entry 1CWX [M. Jolivet, M. F. Penin, P. Dalbon, L. Ladaviere, and X. Lacoux, patent application EP 1015481, WO 98/39360, 1997]). The hydrophobic domain D2 is responsible for core association with lipid droplets in mammalian cells and with endoplasmic reticulum membranes (1, 24, 47). The initial polyprotein cleavage generates the immature core protein (P23) that undergoes additional processing by the intramembrane-cleaving protease SPP (for signal peptide peptidase) (31, 45, 53). This yields mature core P21 whose C terminus is not precisely known but probably lies between residues 170 and 179 (27, 37, 45, 60, 84).
As the building block of the nucleocapsid, the core protein would be anticipated to undergo homo-oligomerization, as well as specific association with the viral RNA. Regions for homotypic interaction of core have been localized in sequences between residues 36 and 91 (42) and residues 82 and 102 (51). In contrast, no clear specific binding of core to HCV RNA sequences has been demonstrated (60). We recently reported that HCV core possesses nucleic acid chaperone properties similar to that reported for retroviral NC proteins (13). Little is known about the mechanisms of HCV nucleocapsid assembly. In vitro nucleocapsid reconstitution experiments with core segments from positions 1 to 124 or 1 to 179 and structured RNA molecules (domains of the HCV IRES or tRNA but not with the full-length HCV genome) have thus far yielded irregular particles larger than those isolated from infected subjects (34). More recently, it has been proposed that the first 79 N-terminal amino acids of core were sufficient for nucleocapsid formation in vitro (40).
Intriguingly, the core protein has been reported to interact with a variety of cellular proteins and to influence numerous host cell functions such as gene transcription, lipid metabolism, apoptosis and cell signaling (for reviews, see references 19, 44, and 57). Moreover, the core protein has been implicated in HCV-related steatosis (49) and carcinogenesis (48) in transgenic mice. It has been recently suggested that core protein drives liver injury in transgenic mice by increasing Fas-mediated apoptosis and liver infiltration of peripheral T cells (67). However, the relevance of all of these observations, derived mainly from heterologous overexpression experiments, for the natural course and pathogenesis of hepatitis C is currently unknown. A better understanding of the structural properties of core protein is essential to provide a framework for elucidation of HCV nucleocapsid assembly and to further evaluate the core molecular interactions with these numerous host compounds.
We describe here the cloning and expression in Escherichia coli of the region from positions 1 to 117 (1-117 region) and the 1-169 region of HCV core protein (1b genotype) fused with a C-terminal six-histidine tag and the subsequent analysis of these proteins (named CHCV117 and CHCV169) by using various biophysical techniques, including analytical centrifugation, circular dichroism (CD), intrinsic tryptophan fluorescence, and limited proteolysis. Overall, the results show that the resulting CHCV169 protein behaves as a membrane protein and forms soluble aggregates in the absence of detergent. In contrast it is monodispersed in the presence of a mild detergent and is in equilibrium between monomer and dimer forms. CHCV169 is essentially composed of α-helices (50%), whereas the CHCV117 is mainly unfolded. It appears that the folding of the 2-117 region is dependent on the presence of the 117-169 region, which contains a putative membrane-interacting domain. We provide here novel insights into the biochemistry and structure of the HCV core protein and provide a basis for further investigations, as well as for attempts to reconstitute HCV nucleocapsid particles in vitro.
MATERIALS AND METHODS
Protein sequence analyses.
Sequence analyses were performed using the IBCP HCV database website facilities (HCVDB; http://hepatitis.ibcp.fr) (11). The core protein sequence of the HCV H strain consensus cDNA (GenBank TM accession number AF009606) was used to retrieve all reported isolates from the EMBL/GenBank databases using the FASTA homology search program (54). Multiple sequence alignment of the final set of 414 nonredundant sequences of various genotypes was carried out with the CLUSTAL W program (75). The repertoire of residues at each amino acid sequence position and their frequency of observation were computed by using a program developed in our laboratory (C. Combet, F. Dorkelt, F. Penin, and G. Deleage, unpublished). The secondary structure of proteins was predicted by using a large set of methods available at the HCVDB website, including DSC, HNNC, SIMPA96, SOPM, GOR4, PHD, and Predator. Various methods (TMHMM, PHDhtm, TopPred2, and DAS) were combined for the prediction of transmembrane sequences, as detailed previously (8). Hydrophobic cluster analysis (HCA) plots (18) were drawn by using the drawHCA program (http://www.lmcp.jussieu.fr/∼soyer/www-hca/hca-form.html).
Plasmid construction and protein purification.
Cloning, expression, and purification of HCV core protein 1 to 169 of (CHCV1-169) have been previously described (3). For the CHCV1-117 domain, a 351-bp fragment corresponding to aa 1 to 117 of HCV core protein, was amplified by PCR from sequence with EMBL accession number D89872 encoded by the plasmid PCMV-980 (a gift from K. Shimotohno) and two specific primers containing a NdeI site or a PstI site, respectively. The NdeI-PstI fragment was cloned into the expression vector pT7-7, including a C-terminal His6 tag (12). As for CHCV1-169 (3), the resulting plasmid encoded CHCV1-117 polypeptide in fusion with a C-terminal LQHHHHHH sequence. Proteins were expressed from the resulting plasmids after transformation in the E. coli strain BL21 SI (Life Technologies) or classical BL21(DE3). Both strains are producing the T7 RNA polymerase, which is respectively induced by salt or by IPTG (isopropyl-β-d-thiogalactopyranoside). Because of the processing of the N-terminal methionine (see Results), the expression products were named CHCV169* and CHCV117*, the asterisk indicating the presence of the C-terminal His6 tag.
Escherichia coli BL21 SI transformants were grown at 37°C in LB medium without NaCl until the culture reached an optical density at 600 nm of 0.7. Expression was induced by adding NaCl to a final concentration of 200 mM, and incubation was continued for 3 h. The same procedure was used for E. coli BL21(DE3) except that induction was done with 1 mM IPTG. Cells were harvested by centrifugation at 5,500 × g for 10 min at 4°C and resuspended in 25 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride, and 100 U of benzonase/ml. The cells were lysed by using a SLM-Aminco French pressure cell press at 1,200 lb/in2 and centrifuged at 30,000 × g for 30 min. In both cases the expressed protein was recovered in the pellet. The purification procedure used for CHCV117* was the same as previously described for CHCV169* (3) with few modifications as described in Results. After the last step of high-pressure liquid chromatography (HPLC) purification, proteins corresponding to the main peak were lyophilized and identified by mass spectrometry (MS) and N-terminal sequencing. Prior to lyophilization, the HPLC fractions were supplemented or not with a detergent (i.e., either n-dodecyl β-d-maltoside [DM], octaethyleneglycol dodecylether [C12E8], or l-α-lysophosphatidyl choline [LPC], as indicated) to yield a final detergent concentration of 0.1% for a 1-mg/ml protein solution after solubilization of the lyophilisate in the appropriate buffer.
CD measurements.
CD spectra were recorded on a Jobin-Yvon CD6 spectrometer calibrated with ammonium d-10-camphorsulfonate. Routinely, measurements were done at 25°C in a 0.02-cm-path-length quartz cuvettes (Hellma) with protein concentrations ranging from 10 to 30 μM in 20 mM sodium phosphate buffer (pH 7.4) containing 5 mM 2-mercaptoethanol. Spectra were recorded in the 180- to 250-nm wavelength range with 0.2-nm increments and 2-s integration times and averaged over four scans. The baseline-corrected spectra were smoothed by using a third-order least squared polynomial fit. Spectral units were changed in molar ellipticity per residue using protein concentrations determined by amino acid analysis. Protein secondary structure content was estimated by using mainly the Varselec method of spectral deconvolution with a basic set of 33 reference proteins of known secondary structure (28). Calculation was performed with Varselec program integrated in Jobin-Yvon (CDMax) or Antheprot (Dicroprot) processing softwares (15).
Analytical ultracentrifugation.
CHCV169* was purified as described previously (3) by reversed-phase HPLC (RP-HPLC), which also allows the removal of DM. Known amounts of detergent (DM or C12E8) were added to CHCV169* aliquot RP-HPLC fractions, and the detergent was colyophilized with protein. Freeze-dried powder was dissolved with 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 1 mM DTT. The buffer volume was adjusted to obtain a protein concentration of 30 to 40 μM and a detergent concentration of 0.1% (i.e., 1.92 and 1.82 mM for DM and C12E8, respectively). Lower protein concentrations were obtained by dilution with the buffer supplemented with 0.1% DM or C12E8. Analytical ultracentrifugation was carried out at 20°C by using a Beckman Optima XL-I analytical ultracentrifuge equipped with an An-60 Ti four-hole rotor. Sedimentation velocity experiments were performed at 42 000 rpm using 400 μl of samples in two-channel 12-mm-path-length centerpieces. Absorbance scans were taken at 5-min intervals at 277 nm for more than 12 h. Sedimentation equilibrium experiments were performed at three concentrations for CHCV169* in the presence of C12E8 in 3- or 12-mm-path-length centerpieces. Radial scans of the absorbance at 277 nm were obtained every 3 h for a total of 27 h at three rotor speeds (13,000, 16,000, and 22,000 rpm). The Sednterp software (developed by Haynes, Laue, and Philo; http://www.bbri.org/RASMB/rasmb.html) has been used to evaluate the solvent density and viscosity, ρ and η (1.005 g ml−1 and 1.02 cp, respectively, neglecting the presence of detergent), and the partial specific volume, molar mass, and molar extinction coefficient of CHCV169* from its amino-acid composition (0.726 ml/g, 19,421 g/mol, and 35,000 M−1 cm−1, respectively). Partial specific volumes of 0.824 and 0.973 ml/g were used for DM and C12E8, respectively (46). Sedimentation velocity and equilibrium data were analyzed with the Schuck's programs sedfit (14) and sedphat (76). The two programs are available at http://www.AnalyticalUltracentrifugartion.com. They use numerical solutions of the Lamm equation and allow the evaluation of time and radial systematic noise (62). Typically, a selection of 20 sedimentation profiles was analyzed with Sedfit, considering 200 discrete independent species, using a frictional ratio of 1.2 and partial specific volumes of 0.77 and 0.8 ml · g−1 for the proteins solubilized in the presence of DM and C12E8, respectively, with sedimentation coefficients of between 0.2 and 10 S. Their sedimentation profiles were generated on a grid of 200 radial points and combined with a regularization procedure (confidence level of 0.7) to represent the experimental data as a continuous size distribution. The sedimentation of detergent-solubilized protein depends on the amount of bound detergent (71). The experimental sedimentation coefficients were compared to that calculated for a complex of frictional ratio f/f° composed of Ni molecules of protein (P) and detergent (det) of molar masses Mi and partial specific volumes 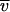 i (i being “P” or “det”):
i (i being “P” or “det”):
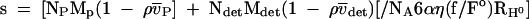 |
(1) |
NA is Avogadro's number and RH° is the minimum hydrodynamic radius for the nonhydrated complex: RH° = [3(NP Mp 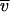 P + Ndet Mdet
P + Ndet Mdet 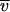 det)/4pNA]1/3. In equation 1, we used a f/f° value of 1.2, assuming a globular particle. More anisotropic shapes would correspond to larger values of f/f°.
det)/4pNA]1/3. In equation 1, we used a f/f° value of 1.2, assuming a globular particle. More anisotropic shapes would correspond to larger values of f/f°.
Data from equilibrium sedimentation were analyzed by using Sedphat and considering a reversible self-associating system between monomers and dimers or a mixture of monomers and dimers.
Fluorescence measurements.
Fluorescence measurements were performed at 25°C by using an SLM-Aminco 8000C spectrofluorometer equipped with a 450-W xenon lamp (excitation and emission slits set to 4 nm). The excitation wavelength was set to 295 nm to limit fluorescence to tryptophan only. Emission spectra were collected from 315 to 380 nm. Both emission and excitation spectra were corrected for buffer blank, and the variations of the lamp emission power were automatically corrected with a rhodamine solution as standard in the reference channel. The protein concentration used was 0.1 μM, and the measurements were performed in a buffer containing 20 mM Tris-HCl (pH 7.4), 1 mM DTT, 0.1% DM, and 10% glycerol in the presence or absence of 6 M guanidine hydrochloride (Gdn-HCl). Fluorescence quenching experiments were performed with acrylamide and iodide (I−, potassium salt) as external quenchers. Additions of quenchers were made from concentrated stock solutions, the potassium iodide solution containing 0.1 mM thiosulfate to prevent I3− formation. Fluorescence intensity values obtained in the presence of increasing concentrations of KI were corrected for the ionic-strength effect by reference to control performed with equal concentrations of KCl. The dilution and absorption effect of acrylamide on fluorescence were corrected according to the method of Calhoun et al. (6). The fluorescence quenching data were analyzed according to the Stern-Volmer relationship, where the static quenching is neglected (36):
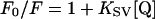 |
(2) |
where F0 and F are the fluorescence intensities in the absence and in the presence of quencher, respectively; KSV is the collisional Stern-Volmer constant; and [Q] is the quencher concentration. The plot of F0/F versus [Q] is linear for a homogenous population of emitting fluorophores but bends downward for multiple heterologous fluorophores. To estimate the accessible fluorophore fraction fa quenched with a constant KQ in such heterogeneous systems, modified Stern-Volmer plots were used, according to the relation of Lehrer (36):
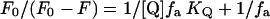 |
(3) |
The plot of F0/(F0 − F) versus 1/[Q] allows a graphical determination of fa and KQ.
MS and peptide sequencing.
All LC/MS analyses were carried out by using a Sciex API 165 quadrupole mass spectrometer coupled to an Applied Biosystems ABI 140D capillary LC system. The mass spectrometer was operated with two electrospray ionization sources (microspray and ionspray) in the positive ion mode. The microspray source was used for direct infusion of protein solutions with 0.2 μl/min flow rate in CH3OH-H2O (50:50 [vol/vol]) mixture containing 0.1% of HCOOH. LC/MS was carried out on a C18 HPLC microcolumn (Brownlee; 150- by-0.5-mm internal diameter, 5-mm particle size, 300-Å pore size) at a flow rate of 10 μl/min connected to a 785A Absorbance Detector (Applied Biosystem) and an ionspray source. The V8 digested peptides were separated using mobile phases A (0.05% trifluoro acetic acid [TFA]) and B (90% acetonitrile in 0.04% TFA) with a four-step linear gradient as follow: 0 to 5 min, 10% B; 65 min, 70% B; 75 min, 95% B; and hold at 95% B up to 85 min. Absorbance detection was set at 214 nm. The scan range was set at m/z 700 to 2,200. The peptides were sequenced by automatic Edman degradation using a Procise 492A liquid-phase sequencer (Applied Biosystems).
Endoproteinase Glu-C (V8) proteolysis.
Proteins were incubated with endoproteinase Glu-C (enzyme/substrate ratio of 5/100 [wt/wt]) at 37°C in a buffer containing 20 mM Tris-HCl (pH 7.4), 1 mM DTT, 0.1% DM, and 10% glycerol. Aliquots were withdrawn at different times of incubation and subjected to 15% polyacrylamide Tricine gel (61) or LC/MS analysis.
RESULTS
HCV core sequence analysis and design of expressed proteins.
The biochemical and biophysical characterizations of HCV core protein require the mature core protein. However, the precise position of the SPP cleavage site remain unknown but has been proposed to lay close to aa 172 or 179 (27, 37, 60, 84). To assess the putative SPP cleavage site and to identify significant structural features of core protein, sequence comparisons were performed to analyze the degree of amino acid conservation among different HCV isolates. In addition, the HCV core sequence has been compared to that of GB virus B (GBV-B), which is the most closely related of all animal viruses to HCV (66) and constitutes an attractive surrogate model for HCV study (4). Figure 1A shows the comparison of GBV-B core sequence with the amino acid repertoire of HCV core sequence deduced from the analysis of 414 HCV isolates of various genotypes. Five sequence stretches showing substantial similarities between both viruses were identified within rather well conserved segments of HCV sequences (boxed sequences). For the N-terminal region (positions 1 to 105 of HCV core), sequence similarities appear to be limited. In contrast, the region 106-167 of HCV including the hydrophobic domain D2 appears to be well conserved. Finally, apart the fact that they are mainly hydrophobic, the primary sequences of the signal peptide regions exhibited limited similarities. Sequence examination revealed that HCV core shares some characteristics of natively unfolded proteins such as long stretches of hydrophilic residues. In addition, sequence analysis using BLAST program revealed the presence of two low-complexity regions including residues 32 to 47 and 138 to 158. Furthermore, secondary structure predictions using a set of various methods showed obvious discrepancies in most of the few regions predicted to be structured (data not shown). Taken together, these features suggest that secondary structure prediction of core is globally poorly reliable and must be used with caution. To refine the comparison of HCV and GBV-B core sequences, we thus preferred to use HCA, which allows an overall appreciation of many structural features of proteins at a glance (7, 18). This two-dimensional representation of protein sequences (Fig. 1B) allows the detection of structural relationships between protein sequences sharing low levels of sequence identity (typically <20% identity) by adding information on secondary structures to the comparison process. Indeed, hydrophobic clusters mainly correspond to the inner faces of regular secondary structures involved in the hydrophobic core of folded proteins (81). The corresponding clusters are much better conserved than sequences themselves and constitute signatures helping the comparison of remote sequences. The visualization of the hydrophobic clusters edges in HCA plots presented in Fig. 1B clearly highlight the structural limit between domains D1 and D2 of HCV core, as well as its counterpart in GBV-B (positions 118/119 and 86/87 for the HCV and GBV-B core proteins, respectively). Examination of HCV domain D1 reveals two hydrophobic clusters (clusters 2 and 4 in Fig. 1B) and three highly basic clusters (clusters 1, 3, and 5 in Fig. 1B) bearing potential nuclear localization signals. A similar of hydrophobic and basic clusters is observed in GBV-B domain D1, but both the hydrophobic clusters (clusters c and e) and the basic clusters (clusters b, d, and f) are shorter than in HCV. However, sequence similarities between HCV and GBV-B basic clusters 1 and b, 3 and d, and 5 and f point to a similar organization and features. In contrast, the equivalences between GBV-B and HCV hydrophobic clusters are less obvious. However, the various similarities between the hydrophobic/hydrophilic patterns and amino acid composition suggest that cluster 2 of HCV might correspond to the combination of clusters a and c of GBV-B, assuming that the basic cluster “b” corresponds to an insertion between these clusters, whereas its HCV counterpart (basic cluster 1) is located at the N terminus of HCV core. In summary, despite their differences in length and hydrophobic clusters location, the folding patterns of domains D1 of HCV and GBV-B core proteins are probably similar.
FIG. 1.

Sequence analysis of HCV core protein and comparison with GBV-B core protein. (A) alignment of the amino acid sequences of the HCV and GBV-B core proteins. For HCV core, amino acids repertoire at each sequence position was deduced from the CLUSTAL W multiple alignments of 414 nonredundant core sequences of various genotypes (see Materials and Methods). Amino acids are listed in decreasing order of observed frequencies, from bottom to top. Amino acids observed at a given position in <1% of the sequences were not included. For GBV-B core (EMBL accession number U22304), no amino acid variability has been reported to date. Alignment of GBV-B core sequence with those of HCV core was done with the T-Coffee program (52; http://www.ch.embnet.org/software/TCoffee.html), which is more accurate than CLUSTAL W for sequences with low identity. This alignment is consistent with the results obtained using the Local Similarity Program SIM (26; http://www.expasy.org/tools/sim-prot.html). Identical, strongly conservative, and weakly conservative amino acid are indicated by asterisks, colons, and dots, respectively, accordingly to CLUSTAL W convention (75). Note that the highest degree of conservation is indicated when the corresponding amino acid is present within HCV repertoire. Sequence stretches showing substantial similarities are boxed, and positions exhibiting identical or highly conserved residues are color shaded. Amino acid positions are indicated above the repertoire for HCV core and below the sequence for GBV-B core, respectively. (B) HCA plots of the aligned domains of HCV core of genotype 1b (EMBL accession number D89872) and GBV-B core protein (EMBL accession number U22304). In these plots, the amino acid sequences are written vertically (with an angle of nearly 25° to the right) on a duplicated α-helical net, and the clusters formed by contiguous hydrophobic residues are boxed (7, 18). The standard one-letter code for amino acids is used except for proline, glycine, threonine, and serine, which are represented by stars, diamonds, open squares, and dotted squares, respectively, to highlight the particular structural features of these residues. Basic and acidic amino acids are in blue and red, respectively. The HCA multisequence alignments involve the visual analysis of hydrophobic cluster shapes, lengths, and distribution, along with an analysis of the strictly and strongly conserved residues. Color-shaded amino acid positions highlight the conserved sequence stretches corresponding to that indicated in panel A. Hydrophobic clusters and nonhydrophobic spacing sequences observed in the basic domains of HCV and GBV-B core proteins are denoted 1 to 5 and a to f, respectively.
Concerning the domains D2 of HCV and GBV-B, HCA plots show that the hydrophobic clusters are comparable, which is in agreement with the sequence similarities detected in Fig. 1A. Interestingly, the high sequence homology observed between segment 154-167 in HCV and its counterpart 124-136 in GBV-B stops abruptly, indicating the end of this common structural element. Since the following 168-172 sequence Asn-Leu/Ile-Pro-Gly in HCV is almost fully conserved among the various genotypes, this short segment likely play an important role in HCV core. The Pro-Gly sequence 170-171 suggests the presence of a turn or a flexible junction, as expected for a limit between two secondary structure elements. There is no obvious counterpart in GBV-B, but the segment 139-141, including a glycine and a histidine, might play the same role. In contrast to these segment sequences that are specific for each virus, the sequence Ala-Thr-Gly is identical in HCV (aa 165 to 167) and GBV-B (aa 134 to 136), suggesting the same role in the capsid structure and/or maturation. Since this segment is also quite flexible and located at the end of a hydrophobic cluster, it is possible that it constitutes the structural end of domain D2. Overall, the limit of D2 domain seems to be within the segment 170-173, but it cannot be ruled out that the structural limit lay backward, i.e., within the fully conserved Ala-Thr-Gly sequence. In order to produce a HCV core protein that is structurally close to the mature form, we thus decided to overexpress the sequence 1-169, assuming that SPP cleavage site lay within segment 170-173. Although sequence 1-169 might not reflect the true mature core protein, it contains the essential structural elements that are necessary for proper core folding, including the hydrophobic residue leucine-169 that could participate to the domain D2 structure.
Efficient purification of core required the presence of a mild detergent.
The recombinant CHCV117* was easily produced in the classical BL21(DE3) strain after induction by IPTG. In contrast, no production of CHCV169* was detected in this strain after induction by IPTG. This is likely due to a cloning selection of defective mutants as previously described for HCV core protein (17), which is known to be toxic for bacteria (23, 64, 68). Indeed, the T7 promoter is well known to allow the production of a low quantity of protein even without IPTG induction. For a toxic protein, this T7 promoter leakage favors the selection of mutated plasmids unable to express the protein. To overcome this difficulty, BL21(DE3)/pLysS strain, which efficiently represses the T7 promoter in the absence of IPTG, was used. However, the highest level of CHCV169* production was obtained by using BL21(DE3)/SI strain, allowing the tight control of T7 promoter in the absence of salt but the subsequent production of the protein upon induction with 0.2 M NaCl. It should be noted that bacterial growth was strongly reduced after salt induction, indicating the toxic nature of CHCV169* for E. coli.
Both recombinant CHCV117* and CHCV169* proteins were essentially found associated to the insoluble fraction after cell lysis. Solubilization of CHCV117* was easily reached with limited amount of urea (2 M) or Gdn-HCl (1.2 M), and its purification to homogeneity was reached after Ni-nitrilotriacetic acid (NTA) agarose chromatography and RP-HPLC steps, as illustrated in Fig. 2 (lanes 2 to 4). This requirement of chaotropic agent for solubilization of CHCV117* highly basic domain while it is soluble after purification suggests that in E. coli extract this domain was likely associated with nucleic acid aggregates via electrostatic interactions. CHCV169* solubilization from the cell insoluble fraction was only partially efficient with a high amount of either urea (6 to 8 M) or Gdn-HCl (6 M) alone, even under reducing conditions, and subsequent purification by Ni-NTA agarose chromatography was poorly efficient (data not shown). By exploring the subcellular localization of CHCV169* to understand the origin of this poor solubilization efficiency with strong denaturating agents, we found that a significant amount of this protein was associated with the bacterium membrane fraction (data not shown). Interestingly, in this context, CHCV169* was efficiently solubilized by mild, nonionic detergent such as DM without addition of chaotropic compounds. This behavior is typical of membrane protein and indicates that HCV core must be handled as a membrane protein. As a result, the addition of detergent (1% DM), together with 6 M urea, allowed the efficient solubilization of overproduced CHCV169* from the cell insoluble fraction. Moreover, the presence of DM (0.1%) during the Ni-NTA agarose chromatography step was essential for the recovery of CHCV169* with a high yield. The subsequent step of RP-HPLC permitted the high purification of CHCV169* (Fig. 2, lane 8) and its separation from alternative forms of core protein due to frameshifting events described previously (3). After the RP-HPLC step, which also ensure the removal of the DM detergent, as well as any buffer compounds, the purified proteins were recovered by lyophilization. MS analysis of purified proteins gave a mass of 14,296 Da for CHCV117* and 19,421 Da for CHCV169*. These masses showed the removal of the N-terminal methionine that was also confirmed by amino-terminal sequencing (result not shown).
FIG. 2.

Expression and purification of CHCV117* and CHCV169* in E. coli. Coomassie blue staining after SDS-15% PAGE. Lanes 1 to 4 and lanes 5 to 8 correspond to expression and purification of CHCV117* and CHCV169*, respectively. Whole-cell extracts of noninduced (lanes 1 and 5) and induced (lanes 2 and 6) BL21(DE3)/SI cells expressing CHCV117* or CHCV169* are shown. Lanes 3 and 7 correspond to proteins purified by Ni-NTA affinity chromatography. CHCV117* (lane 4) and CHCV169* (lane 8) were purified by RP-HPLC.
Core protein behaves as a dimer in the presence of mild detergent.
The oligomerization state of CHCV169* was investigated by analytical centrifugation. The sedimentation velocity experiments were analyzed in terms of distribution of sedimentation coefficients, c(s), which allowed a qualitative evaluation of the protein homogeneity and auto-association capability. For CHCV169* solubilized in aqueous buffer without detergent, the sedimentation profiles describe a heterogeneous population with sedimentation coefficient ranging from 20 to 100 S (Table 1 and data not shown). The major population is at 20 to 25 S, which would correspond to a molecular mass of 600 to 700 kDa, as long as a globular hydrated molecule is considered (the molecular mass would be larger if the oligomer exhibit an asymmetric form). This molecular mass indicates the association of a minimum of 30 to 35 monomers of CHCV169*.
TABLE 1.
Sedimentation coefficients (s) for different concentrations of CHCV169* in the presence of DM or C12E8
| Concn (mM)
|
CHCV169* concn (μM) | s (S) | |
|---|---|---|---|
| DM | C12E8 | ||
| 0 | 0 | 9 | 20-100 |
| 0 | 0 | 20 | 20-100 |
| 0 | 0 | 34 | 20-100 |
| 1.92 | 0 | 9.2 | 3.1 |
| 1.92 | 0 | 19.3 | 3.1 |
| 1.92 | 0 | 34 | 3.1 |
| 0 | 1.82 | 3.1 | 1.7 |
| 0 | 1.82 | 9.6 | 1.7 |
| 0 | 1.82 | 31 | 1.7 ± 2.4 (mean, 2.1) |
We found that the addition of DM or C12E8 detergents to the CHCV169* solution was poorly efficient to reduce the heterogeneity of the preparation. In contrast, the addition of either DM or C12E8 in the RP-HPLC fractions of purified CHCV169* before the lyophilization step totally avoided the formation of high-molecular-mass species, as illustrated by the sedimentation profiles shown in Fig. 3A and B, respectively. The low dispersity of residuals (Fig. 3A2 and B2) reflects the good quality of the fit versus the experimental values. It should be mentioned that the analysis of analytical centrifugation data recorded in the presence of detergent is complicated by the fact that the amount of detergent bound to the studied protein is generally unknown, and thus several hypotheses must be considered (30, 80).
FIG. 3.

Sedimentation velocity of CHCV169*: Experiments were done in 20 mM Tris pH 8.0, 150 mM NaCl, 1 mM DTT, in the presence of 0.1% of DM (A panels) or C12E8 (B panels). Panels A1 and B1 show a selection of experimental (dots) and fitted (line) profiles. In panels A1 and B1, the last profile corresponds, respectively, to 8 h 30 min and 11 h of sedimentation at 42,000 rpm and 20°C. Panels A2 and B2 show the superposition of differences between the experimental and fitted profiles. Panels A3 and B3 show the results of the analysis made in terms of a continuous distribution of species (see the text).
In the presence of 0.1% DM, CHCV169* migrates as a homogeneous species with a sedimentation coefficient of 3.1 ± 0.1 S (Fig. 3A3 and Table 1) whatever its concentration (9.2 to 34 μM). This s value is not compatible with a detergent-free monomer for which the s value would be below 2.13 S, whatever the shape of the protein. This 3.1 ± 0.1 S value is also not explained by a monomer interacting with DM since the binding of at least 70 molecules of DM per monomer would be required, a stoichiometry that is higher than the disposable DM in the sample of highest protein concentration (protein/DM molar ratio of 1:58). In contrast, the data are compatible with a compact globular dimer solvated by a minimum number of five molecules of DM.
In the presence of 0.1% C12E8, the sedimentation profiles show a symmetrical boundary with s values of 1.7 ± 0.1 S for 3.1 and 9.6 μM CHCV169*. At 31 μM protein, the c(s) plot shows an additional boundary at 2.4 S, yielding a mean s value of 2.1 S (Fig. 3B and Table 1). This indicates an auto-association equilibrium. The shape of the distribution profiles on Fig. 3B3 depends on different factors: the sedimentation coefficients of the species, the equilibrium constant, and the kinetics of the equilibrium. It cannot be said, for example, that the boundary at 2.4 S corresponds to a species. Since the buoyancy of C12E8 is close to null, equilibrium sedimentation experiments were performed in order to determine the stoichiometry of CHCV169* (Fig. 4). We analyzed globally the sedimentation equilibrium profiles obtained for CHCV169* concentrations of 6, 16, and 36 μM at the three angular velocities. The comparison between the experimental and fitted profiles using a model of a monomer-dimer equilibrium leads to a dissociation constant of 40 ± 10 μM, estimated from different fitting procedures. The quality of the fit, based on the residuals is, however, not excessively good, except when systematic time-independent noise is considered (Fig. 4). This could be related to some aging of DTT during the long equilibrium experiments. However, a mixture of monomer and dimer for each of the samples (allowing only a floating baseline) gave essentially the same description (Kd values of 30 and 50 μM for the two samples with the largest concentrations, the precision being very low for the 6 μM sample). The value of the dissociation constant corresponds to 90 and 50% of monomer for 3.1 and 31 μM CHCV169*, the minimum and maximum concentrations investigated in sedimentation velocity experiments. These percentages agree well with the results of the sedimentation velocity experiments (Table 1 and Fig. 3): at 3.1 μM the experimental s value of 1.7 S corresponds to that calculated for a monomer (<2 S) and at 31 μM, the mean value of 2.1 S is intermediate between that calculated for the monomer and the dimer (less than 3 S, whatever the shape and amounts of bound detergent). Because analytical centrifugation experiments required long time recording and could not be done in the absence of oxygen, all experiments were performed in the presence of DTT to prevent any unexpected oxidation of the single cysteine residue present in CHCV169* at position 128. In fact, the time-dependent disulfide bond formation experiments that we performed with CHCV169* under various conditions revealed that, in the presence of DM, Cys-128 remained reduced, even in the absence of DTT and for incubation times as long as 40 h (results not shown). This indicates that the dimer observed in analytical centrifugation experiments performed in the presence of DM and DTT is not a covalent dimer.
FIG. 4.

Equilibrium sedimentation of CHCV169*. CHCV169* was in C12E8 0.1%-150 mM NaCl-1 mM DTT-20 mM Tris-HCl (pH 8.0). Experiments were done for three protein concentrations c (36, 16, or 6 μM) using either 3- or 12-mm-path-length cells (I) as indicated in the corresponding panels. The equilibrium sedimentation profiles were recorded for three rotor velocities (13,000, 16,000, and 22,000 rpm from top to bottom at the origin). The data analysis was made considering an monomer-dimer equilibrium and mass conservation for each channel; the protein concentration at each position in the cell was calculated from the optical density. The top panels show the experimental data (•), the fit (continuous lines), and the calculated invariant time independent noise (★); the differences between the experimental and modeled profiles (residuals) are reported in the corresponding bottom panels. The resulting value for the buoyancy molar mass is close to that expected for the monomer polypeptide (120%), and that for the dissociation constant is 50 μM.
Core protein is an α-helical protein.
The content of secondary structures of CHCV117* and CHCV169* protein was investigated by CD. The CD spectrum of CHCV169* solubilized in the presence of LPC (Fig. 5) is typical of folded proteins, and its shape exhibiting a maximum around 192 nm and two minima around 208 and 222 nm indicates a dominance of α-helix conformation (82). Very similar spectra were obtained in the presence of mild detergents such as DM (Fig. 5) or C12E8 (not shown), although the intensity of spectra were slightly smaller, suggesting a better stabilization of CHCV169* folding in the presence of natural phospholipids. In the absence of detergent, CHCV169* yielded noisy CD spectra (especially below 205 nm), likely due to the presence of large oligomers, as revealed above by analytical centrifugation. However, the shape and intensity of this spectrum (not shown) were similar to those recorded for CHCV169* solubilized in the presence of LPC or detergents. This indicates that the detergent added to prevent the formation of aggregates had little effect on CHCV169* secondary structure. Because of the poor quality of CHCV169* spectra recorded in absence of detergent, its secondary structure content analysis was only performed on spectra recorded in the presence of detergents. About 50% of the α-helix content was estimated with any of the various CD deconvolution methods used. In addition, analysis by Varselec method (28) indicated the presence of a quite large amount of turn (30%), a finding in agreement with the high number of Pro and Gly residues. In contrast, the β-sheet content was very low (<3%), and other structures represent ca. 20%. The absence of β-sheet structure is a good indication that no aggregated protein was present in the corresponding CD samples.
FIG. 5.

Far UV CD spectra of CHCV169* and CHCV117*. CHCV169* was dissolved in 20 mM sodium phosphate buffer (pH 7.4), 10 mM β-mercaptoethanol, and 0.1% LPC (solid line) or 0.1% DM (dashed line). CHCV117* was dissolved in 20 mM sodium phosphate buffer (pH 7.4)-20 mM β-mercaptoethanol in the absence (dotted line) or in the presence of DM (dashed line). Note that the presence of LPC did not significantly modify CHCV117* spectrum recorded in the presence of DM (data not shown for clarity).
Conversely to that observed for CHCV169*, the CD spectrum of CHCV117* (Fig. 5) shows a single negative band at 198 nm typical of polypeptide in random-coil conformation, while the spectral shape in the 210- to 240-nm region presents similarities to CD spectra of peptides exhibiting turn structures (82). The presence of DM or C12E8 (not shown) had almost no effect on CHCV117*. Fittingly, Varselec analysis indicate a very low amount of α-helix and ca. 40 and 55% of turns and other structures, respectively. Even if we assume that the residue molar ellipticity at 222 nm is exclusively due to the α-helix, the α-helical content estimated according to the method of Chen et al. (9) does not exceed 10%. This large deficit in α-helix contrasts with the 50% helix content observed for CHCV169* and suggests that the C-terminal part of CHCV169* is mandatory to ensure and/or stabilize some α-helical folding in the 2-117 region. Indeed, the 50% helix content of CHCV169* indicates that about 84 residues are actually folded in α-helix. This amount is much higher than the 52 residues of the 118-169 region present in CHCV169*. These data also indicate that the 2-117 region does not constitute an independent structural domain since its folding appears to be dependent from the presence of the 118-169 region.
The central tryptophan-rich region (region 76-107) is buried in core protein dimer.
The intrinsic tryptophan fluorescence is known to be very sensitive to a wide variety of environmental conditions and especially to a hydrophobic or hydrophilic environment. It is then currently used to probe the fold and the native conformation of a protein, as well as the accessibility of fluorophores. The intrinsic fluorescence of CHCV169* and CHCV117* is due to the five tryptophan residues clustered in the 76-107 region at positions 76, 83, 93, 96, and 107. The fluorescence excitation spectrum of CHCV169* and CHCV117* recorded in the presence of 0.1% DM exhibited a maximum near 280 nm (λmexc), which is consistent with tryptophan fluorescence (not shown). Excitation at either 280 or 295 nm resulted in emission spectra with maxima at wavelengths of 336 nm (λmem) for CHCV169* and 346 nm (λmem) for CHCV117* (Fig. 6). Under denaturing conditions (6 M Gdn-HCl), the λm exc of both proteins were unchanged, but their λmem were shifted to 350 nm (Fig. 6), indicating that all tryptophan residues become accessible to the solvent in these conditions. Indeed, this λmem value is close to that of the solute tryptophan model compound NATA (λmem of 355 nm) in the same conditions, whether 6 M Gdn-HCl was present or not (not shown). According to the Burnstein's classification (5), the rather low λmem of 336 observed for CHCV169* under native conditions indicates that most of the Trp residues are in a hydrophobic environment, whereas the relatively high λmem of 346 nm for CHCV117* indicates a hydrophilic environment around the Trp. In other words, Trp residues seem to be buried in CHCV169*, whereas they appear to be exposed in CHCV117*. To better estimate the degree of accessibility of these tryptophan residues, fluorescence quenching experiments were carried out with iodide and acrylamide as quenching agents. Iodide is a large, polar anion that is considered to have access only to surface tryptophan residues. Polar, nonionic acrylamide has good access to all tryptophan residues but those buried into the hydrophobic core of proteins. As expected, a progressive decrease of fluorescence intensity was observed for increasing concentrations of the quenchers, with a greater efficiency of quenching with acrylamide than with iodide (result not shown). Modified Stern-Volmer plots (36) were used to determine the tryptophan-accessible fraction fa and the quenching constant KQ for CHCV117* and CHCV169*, in the presence of either iodide or acrylamide. The corresponding data reported in Table 2 show that all of the tryptophan residues of CHCV117* are accessible both to iodide and acrylamide (fa = 1.0), as is the case with NATA used as a control. These results are consistent with the CD results presented above showing that CHCV117* is mainly unfolded under native conditions. In contrast, only a limited fraction of Trp residues of CHCV169* were found accessible since the total fluorescence emission was partially quenched by iodide and acrylamide (fa values = 0.27 and 0.56, respectively). These values are consistent with an overall hydrophobic environment for most of the Trp residues, as previously deduced from the value of λmem of 336 nm. As expected, denaturation of both CHCV117* and CHCV169* in 6 M Gdn-HCl rendered all Trp residues accessible to both iodide and acrylamide (fa value of 1.0 [results not shown]). To check whether the addition of detergent could modify the accessibility of Trp residues to the quenchers, similar fluorescence studies have been done in the absence of 0.1% DM for CHCV117*. We found that the accessible fraction of tryptophan residues to acrylamide was very close to that observed in the presence of 0.1% DM (fa values of 0.93 and 1.0 in the absence and in the presence of DM, respectively), showing that the detergent is not able to mask Trp residue accessibility. Interestingly, in the case of CHCV169*, the tryptophan residues were found to be clearly much more accessible to the quencher acrylamide than in the presence of detergent (fa values of 0.88 instead of 0.56). This indicates that the environment and/or folding of the tryptophan-rich region is obviously different in the aggregated form of CHCV169* compared to the monodispersed, dimeric form of CHCV169* obtained in the presence of detergent.
FIG. 6.

Fluorescence emission spectra of native and denaturated CHCV169* and CHCV117*. Emission spectra of CHCV169* (thick line) and CHCV117* (dashed line) were recorded in the presence of 0.1% DM as described in Materials and Methods. Emission spectra of unfolded CHCV169* (thin line) and CHCV117* (dotted line) were recorded after overnight incubation of the samples in 6 M Gdn-HCl.
TABLE 2.
Accessibility of tryptophan residues of CHCV117* and CHCV169* to quenchers
| Quencher | CHCV117*
|
CHCV169*
|
NATA
|
|||
|---|---|---|---|---|---|---|
| KQ | fa | KQ | fa | KQ | fa | |
| Iodide | 43.4 | 1.0 | 35.4 | 0.27 | 7.5 | 1.0 |
| Acrylamide | 6.0 | 1.0 | 10.6 | 0.56 | 14.7 | 1.0 |
CHCV169* folding and topology was further investigated by using limited proteolysis with endoproteinase Glu-C (V8), which cleaves after glutamate in the experimental conditions used. The resulting peptide mixtures obtained at various incubation time have been analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and LC/MS. The time course hydrolysis of CHCV169* is illustrated in Fig. 7 by the SDS-PAGE analysis showing that numerous fragments appeared completely digested after 5 min of incubation, whereas one fragment was completely digested only after 2 h of incubation. Analysis by LC/MS allowed the identification of each fragments (data not shown). The cleavage after glutamate 54 and 72 was achieved very rapidly (<5 min), whereas the cleavage after glutamate 159 appeared much slower and complete cleavage after glutamate 89 was only achieved after 2 h of incubation. The degree of accessibility of glutamate residues can thus be ranged as follows: 54, 72 > 159 ≫ 89. These results indicate a compact fold in the tryptophan-rich region containing Glu 89, as well as in the C-Terminal region containing Glu 159 (i.e., domain 2). The same experiments made in the absence of detergent yielded similar results, but the kinetics of cleavage were about twofold slower (data not shown). This relative resistance of CHCV169* to proteolysis in the absence versus presence of detergent was also observed with various proteases such as trypsin and chymotrypsin (data not shown). It is likely related to the aggregated nature of CHCV169* observed in the absence of detergent.
FIG. 7.

Cleavage pattern of CHCV169* with endoproteinase Glu-C. CHCV169* was treated with endoproteinase Glu-C as described in Materials and Methods and subjected to a 15% polyacrylamide Tricine gel electrophoresis (61). Lane 1, control CHCV169*. Lanes 2 to 7 show CHCV169*, digested during increasing lengths of time: lane 2, 5 min; lane 3, 15 min; lane 4, 30 min; lane 5, 1 h; lane 6, 2 h; and lane 7, 3 h.
DISCUSSION
Structural analysis of core protein is essential to provide a framework for elucidation of HCV nucleocapsid assembly and for further understanding of the core molecular interactions with numerous host compounds involved in crucial metabolic pathways (19, 33, 44, 65, 74). Current knowledge of HCV core structural features is still limited. This is mainly due to the absence of an efficient system for HCV particles production and the difficulties in overproducing, purifying, and handling this protein, as well as to the uncertainties concerning the C-terminal end of its mature form. Analysis of core amino acid sequence and comparison with GBV-B core protein allowed us to design CHCV169* that is thought to contain the essential structural elements necessary for proper core folding. We show here that this protein exhibits membrane protein features and essentially assembles as dimers in the presence of mild detergents used to mimic the membrane environment, whereas it yields soluble aggregates in the absence of detergent. In addition, core protein exhibits an overall α-helical fold and the folding of its N-terminal domain D1 (region 2-117) is dependent on the presence of its C-terminal domain D2 (region 118-169).
HCV core behaves as a membrane protein.
Several lines of evidence indicate that core protein behaves as a membrane protein. First, a large part of overexpressed CHCV169* was found to be associated with the bacterial membrane fraction and was easily solubilized after the addition of mild detergents, even in the absence of chaotropic agents (urea or Gdn-HCl). The presence of detergent also appeared to be essential during the Ni-NTA affinity chromatography step to avoid the aggregation of CHCV169*. Second, as shown by analytical centrifugation, the addition of mild detergent to core protein was mandatory to avoid the formation of soluble aggregates. It is worth mentioning that detergent addition to aggregated core was poorly efficient for solubilization. To be efficient, the detergent must be added to CHCV169* when solubilized in an organic solvent-water mixture (e.g., 65% acetonitrile in RP-HPLC fractions containing CHCV169*) and then lyophilized to remove the solvents. Third, several groups observed the presence of mature core protein attached to cellular membranes, such as endoplasmic reticulum (69); at the surface of lipid droplets in mammalian cells (1, 24, 47); and at the surface of the outer mitochondrial membrane (63). Moreover, McLauchlan and coworkers showed that the core domain D2 is responsible for core association with lipid droplets and bears a resemblance to plant oleosin, a lipid globule-binding protein (25). The fact that CHCV117* was highly soluble and did not require any detergent to yield monodispersed solution strengthens the hypothesis that the detergent-binding properties of core are only borne by the D2 domain. This is also in keeping with the mainly hydrophobic nature of this domain and our recent finding that the domain D2 fused with green fluorescent protein colocalized with lipid droplets when expressed in mammalian cells (S. Boulant, G. Hope, P. Targett-Adams, J. P. Lavergne, F. Penin, and J. McLauchlan, unpublished data). Taken together, these results clearly indicate that the domain D2 of core protein is a membrane-binding domain. The prediction of two amphipathic α-helices in this domain by us and others (69) suggests that core membrane association could be mediated by in-plane amphipathic helices as we recently described for the HCV NS5A membrane anchor domain (56). Nevertheless, HCV core protein must be handled as a membrane protein, i.e., in the presence of detergent or lipids. The absence of membrane mimetic compounds leads to the formation of high molecular soluble aggregates as observed here and by others (35). The formation of these aggregates can be explained by intermolecular hydrophobic interactions of domains D2 yielding an overall hydrophobic core surrounded by a layer of highly charged, hydrophilic domains D1.
HCV core is a dimeric α-helical protein.
Analytical centrifugation experiments in the presence of mild detergents avoid the formation of heterogeneous soluble aggregates and revealed that CHCV169* behaves as a dimer that can dissociate depending on the detergent. In DM, CHCV169* at 9 to 34 μM exhibits only one value of sedimentation coefficient corresponding to a dimer, whereas in C12E8 this value increases with the protein concentration. The sedimentation equilibrium experiments confirm the equilibrium between two forms, assigned to monomer and dimer, with a dissociation constant of ca. 40 μM. Since both detergents exhibit a C12 hydrophobic tail, the monomer-dimer equilibrium differences should be due to the hydrophilic part of the detergent used. Compared to DM, it is possible that the long polyoxyethylene chain for C12E8 interacts at the dimer interface and destabilizes the dimer formation. Whatever the explanation of this different behavior in DM and C12E8, CHCV169* dimer results from a noncovalent association. Indeed, the presence of detergent prevents intermolecular disulfide-bond formation, even in the absence of reducing compound.
CD analyses showed that CHCV117* is mainly unfolded, whereas the CD spectra of CHCV169* is typical of folded proteins and its deconvolution revealed a high content of α-helices (ca. 50%). These results first indicate that the D1 domain of the core protein is unstructured when isolated and that it is folded only in the presence of the D2 domain. This is in agreement with the findings of Kunkel and Watowitch (35) and strengthens the hypothesis of a heterotypic interaction between the N- and C-terminal domains of core (51). Regarding the relative content in secondary structures of CHCV169*, our data are not in agreement with those reported by Kunkel and Watowitch for HCVC179 (35). These authors found 16% α-helix, 29% β-sheet and turns, and 55% random coil, whereas we found ca. 50% α-helix, <3% β-sheet, 30% turn, and ca. 20% of other structures. The reason for this discrepancy might be the length of the protein studied (CHCV169 in the present study and CHCV179 in the case of Kunkel an Watowitch). Indeed, the additional 170-179 sequence, which is very hydrophobic, could induce and propagate aggregation along core protein and prevents its proper folding. In contrast, we showed by CD that the addition of detergent which prevents CHCV169* aggregation had no detectable effect on its folding. Moreover, the high α-helix content of CHCV169* is close to the 50% helix reported for core proteins of HCV-related yellow fever, dengue, and West Nile viruses from CD and NMR experiments (16, 29, 39). Altogether, these data indicate that the content in secondary structures we measured for CHCV169* should be close to the native structure of the core protein.
Our fluorescence experiments revealed that the five Trp residues clustered in the center of the core sequence appeared buried in CHCV169*, as indicated by the rather low λem (336 nm), the decrease of the KQ value in the presence of acrylamide compared to the value determined with NATA, and the only partial accessibility (fa) of Trp residues to Iodide and acrylamide. These findings are not in agreement with that reported for HCVC179, where the Trp residues were mainly observed exposed to the solvent (35) as we observed here for the unfolded CHCV117* domain. This again suggests that HCVC179 is likely not properly folded due to the absence of a membrane mimetic environment. The high KQ value observed with CHCV169* in the presence of iodide is probably due to a local environment of the fluorophores, rich in positively charged residues, as illustrated by Lehrer using synthetic polypeptides (36). That would mean that in the native core protein the highly charged D1 domain might surround the buried tryptophan-rich region upon its D2-dependent folding. The burying of this Trp-rich region into the molecule is further strengthened by the low accessibility of Glu 89 to V8 proteolysis compared to the high accessibility of Glu residues in the D1 domain (Glu 54 and 72). This result is in keeping with other studies suggesting that the Trp-rich region could constitute a homotypic interaction domain (42, 51).
Hepaciviruses core proteins are unique among the Flaviviridae.
The results reported here clearly show that HCV core protein behaves as a dimeric α-helical protein as was previously shown for other members of the Flaviviridae family such as yellow fever, dengue, and West Nile viruses (16, 29, 39). However, no detergent was required for the latter core proteins to keep them monodispersed in solution. Comparative amino acid sequence analysis from the three families of Flaviviridae (40) (i.e., Hepacivirus, Flavivirus, and Pestivirus) showed that core proteins from HCV and GBV-B are longer and contain a C-terminal hydrophobic domain. No such specialized domain required for membrane association is observed in the core proteins of flaviviruses, for which membrane association is mediated by an internal hydrophobic sequence (41). The three-dimensional structures of dengue and West Nile core proteins have been recently determined by NMR (16, 39). They are α-helixcal and dimeric proteins in solution, with the basic residues lying along one face of the dimer, while conserved hydrophobic residues form a concave hydrophobic cleft on the opposite side. This asymmetry led the authors of those studies to propose that the base-rich side interacts with RNA, whereas the apolar opposite region interacts with the viral membrane. By analogy to dengue core protein three-dimensional structure and taking into account our findings, a similar topology could be assumed for HCV and GBV-B core proteins, with the highly basic domain D1 interacting with viral RNA, whereas the hydrophobic domain D2 interacted with the membrane. In addition, the interactions between domains D1 and D2 should allow the stable folding and the compactness of the core dimer. In summary, although the presence of the hydrophobic membrane-binding domain D2 in the hepacivirus core proteins makes them unique among Flaviviridae, their topology in nucleocapsids might be similar to that of flaviviruses and pestiviruses. However, domain D2 and its interaction with the membrane are expected to play a crucial role in the mechanism of nucleocapsid assembly.
Implications for viral particle assembly.
Previous reports of nucleocapsid-like particles assembly from recombinant HCV core protein in vitro have shown that, in the presence of RNA with secondary structures such as the HCV internal ribosome entry site or tRNA, core protein assembles into particles that are heterogeneous in size and larger than the virus particles isolated from infected patients (34, 38). Moreover, no particle formation was observed in the presence of the full-length HCV RNA genome. However, neither lipids nor detergents were included in these reconstitution studies. Although the presence of detergent tends to reduce the heterogeneity of the reconstituted particles, we obtained similar data with our CHCV169 preparation, indicating that the addition of detergent is not sufficient to allow an efficient reconstitution of HCV nucleocapsid (data not shown). More recently, it has been reported that the presence of the C-terminal transmembrane domain serving as a peptide signal for E1 was needed to generate particles in yeast cells (40). Furthermore, full-length core protein (region 1-191) was shown to assemble into nucleocapsid-like particles upon de novo synthesis in cell-free systems made of rabbit reticulocyte lysate or wheat germ extracts (32). These data support our findings showing the membrane binding properties of core protein and suggest that nucleocapsid reconstitution requires the attachment of core protein to a phospholipid layer as a template for proper nucleocapsid reconstitution. In addition, one can wonder whether at least a part of the hydrophobic sequence downstream position 169 of the core protein could play a role in core-lipid interaction and subsequent core multimerization. Further studies centered on in vitro reconstitution of HCV nucleocapsid should involve the mature core protein with the proper C terminus, which, however, remains to be identified. In addition, the presence of membrane-mimetic environment such as mild detergents or lipids appears to be a critical parameter that must be included in in vitro protocols of HCV nucleocapsid-like particles assembly.
The basic building block of the HCV nucleocapsid is likely a dimer of core, which further oligomerize upon interaction with viral RNA. In the context of viral assembly into the cell, this process might take place in the vicinity of a membrane ensuring the interaction of core protein with the lipid bilayer by its putative amphipathic C-terminal binding domain D2 (possibly including a part of the downstream signal peptide sequence) and with RNA by its basic N-terminal domain D1. Consequently, the assembly pathway of HCV virions might not include a step of core preassembly prior to membrane budding, as reported for alphaviruses (50, 72, 73, 78, 79), but might proceed via coassembly at endoplasmic reticulum-derived membrane. Interestingly, such a mechanism of coassembly on membranes has been proposed for various retroviruses (see, for instance, references 2, 22, 43, and 85).
Acknowledgments
We thank Michel Becchi for excellent technical assistance in the LC/MS analysis and Dominique Mazzocut for protein sequencing and amino acid analyses. We also thank Jean Dubuisson, Darius Moradpour, and John McLauchlan for critical readings of the manuscript.
This study was supported by grants from the CNRS (PCV Program), the Université Claude Bernard Lyon I, the Association pour la Recherche contre le Cancer (grant 4301 to J.-P.L.), the ATC “hepatites” from INSERM (F.P.), and ANRS (J.-P.L. and F.P.).
REFERENCES
- 1.Barba, G., F. Harper, T. Harada, M. Kohara, S. Goulinet, Y. Matsuura, G. Eder, Z. Schaff, M. J. Chapman, T. Miyamura, and C. Brechot. 1997. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl. Acad. Sci. USA 94:1200-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barklis, E., J. McDermott, S. Wilkens, E. Schabtach, M. F. Schmid, S. Fuller, S. Karanjia, Z. Love, R. Jones, Y. Rui, X. Zhao, and D. Thompson. 1997. Structural analysis of membrane-bound retrovirus capsid proteins. EMBO J. 16:1199-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boulant, S., M. Becchi, F. Penin, and J. P. Lavergne. 2003. Unusual multiple recoding events leading to alternative forms of hepatitis C virus core protein from genotype 1b. J. Biol. Chem. 278:45785-45792. [DOI] [PubMed] [Google Scholar]
- 4.Bukh, J., C. L. Apgar, and M. Yanagi. 1999. Toward a surrogate model for hepatitis C virus: an infectious molecular clone of the GB virus-B hepatitis agent. Virology 262:470-478. [DOI] [PubMed] [Google Scholar]
- 5.Burstein, E. A., N. S. Vedenkina, and M. N. Ivkova. 1973. Fluorescence and the location of tryptophan residues in protein molecules. Photochem. Photobiol. 18:263-279. [DOI] [PubMed] [Google Scholar]
- 6.Calhoun, D. B., J. M. Vanderkooi, and S. W. Englander. 1983. Penetration of small molecules into proteins studied by quenching of phosphorescence and fluorescence. Biochemistry 22:1533-1539. [DOI] [PubMed] [Google Scholar]
- 7.Callebaut, I., G. Labesse, P. Durand, A. Poupon, L. Canard, J. Chomilier, B. Henrissat, and J. P. Mornon. 1997. Deciphering protein sequence information through hydrophobic cluster analysis (HCA): current status and perspectives. Cell Mol. Life Sci. 53:621-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrere-Kremer, S., C. Montpellier-Pala, L. Cocquerel, C. Wychowski, F. Penin, and J. Dubuisson. 2002. Subcellular localization and topology of the p7 polypeptide of hepatitis C virus. J. Virol. 76:3720-3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, Y. H., J. T. Yang, and K. H. Chau. 1974. Determination of the helix and beta form of proteins in aqueous solution by circular dichroism. Biochemistry 13:3350-3359. [DOI] [PubMed] [Google Scholar]
- 10.Choo, Q. L., K. H. Richman, J. H. Han, K. Berger, C. Lee, C. Dong, C. Gallegos, D. Coit, R. Medina-Selby, P. J. Barr, et al. 1991. Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. USA 88:2451-2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Combet, C., F. Penin, C. Geourjon, and G. Deleage. 2004. Hepatitis C virus sequences database. Appl. Bioinformatics 3:237-240. [DOI] [PubMed] [Google Scholar]
- 12.Cortay, J. C., D. Negre, M. Scarabel, T. M. Ramseier, N. B. Vartak, J. Reizer, M. H. Saier, Jr., and A. J. Cozzone. 1994. In vitro asymmetric binding of the pleiotropic regulatory protein, FruR, to the ace operator controlling glyoxylate shunt enzyme synthesis. J. Biol. Chem. 269:14885-14891. [PubMed] [Google Scholar]
- 13.Cristofari, G., R. Ivanyi-Nagy, C. Gabus, S. Boulant, J. P. Lavergne, F. Penin, and J. L. Darlix. 2004. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral positive-strand RNA in vitro. Nucleic Acids Res. 32:2623-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dam, J., and P. Schuck. 2004. Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles. Methods Enzymol. 384:185-212. [DOI] [PubMed] [Google Scholar]
- 15.Deleage, G., and C. Geourjon. 1993. An interactive graphic program for calculating the secondary structure content of proteins from circular dichroism spectrum. Comput. Appl. Biosci. 9:197-199. [DOI] [PubMed] [Google Scholar]
- 16.Dokland, T., M. Walsh, J. M. Mackenzie, A. A. Khromykh, K. H. Ee, and S. Wang. 2004. West Nile virus core protein; tetramer structure and ribbon formation. Structure 12:1157-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forns, X., J. Bukh, R. H. Purcell, and S. U. Emerson. 1997. How Escherichia coli can bias the results of molecular cloning: preferential selection of defective genomes of hepatitis C virus during the cloning procedure. Proc. Natl. Acad. Sci. USA 94:13909-13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaboriaud, C., V. Bissery, T. Benchetrit, and J. P. Mornon. 1987. Hydrophobic cluster analysis: an efficient new way to compare and analyze amino acid sequences. FEBS Lett. 224:149-155. [DOI] [PubMed] [Google Scholar]
- 19.Giannini, C., and C. Brechot. 2003. Hepatitis C virus biology. Cell Death Differ. 10(Suppl. 1):S27-S38. [DOI] [PubMed] [Google Scholar]
- 20.Grakoui, A., C. Wychowski, C. Lin, S. M. Feinstone, and C. M. Rice. 1993. Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 67:1385-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harada, S., Y. Watanabe, K. Takeuchi, T. Suzuki, T. Katayama, Y. Takebe, I. Saito, and T. Miyamura. 1991. Expression of processed core protein of hepatitis C virus in mammalian cells. J. Virol. 65:3015-3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill, C. P., D. Worthylake, D. P. Bancroft, A. M. Christensen, and W. I. Sundquist. 1996. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl. Acad. Sci. USA 93:3099-3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hitomi, Y., W. M. McDonnell, J. R. Baker, Jr., and F. K. Askari. 1995. High efficiency prokaryotic expression and purification of a portion of the hepatitis C core protein and analysis of the immune response to recombinant protein in BALB/c mice. Viral Immunol. 8:109-119. [DOI] [PubMed] [Google Scholar]
- 24.Hope, R. G., and J. McLauchlan. 2000. Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis C virus core protein. J. Gen. Virol. 81(Pt. 8):1913-1925. [DOI] [PubMed] [Google Scholar]
- 25.Hope, R. G., D. J. Murphy, and J. McLauchlan. 2002. The domains required to direct core proteins of hepatitis C virus and GB virus-B to lipid droplets share common features with plant oleosin proteins. J. Biol. Chem. 277:4261-4270. [DOI] [PubMed] [Google Scholar]
- 26.Huang, X. Q., R. C. Hardison, and W. Miller. 1990. A space-efficient algorithm for local similarities. Comput. Appl. Biosci. 6:373-381. [DOI] [PubMed] [Google Scholar]
- 27.Hussy, P., H. Langen, J. Mous, and H. Jacobsen. 1996. Hepatitis C virus core protein: carboxy-terminal boundaries of two processed species suggest cleavage by a signal peptide peptidase. Virology 224:93-104. [DOI] [PubMed] [Google Scholar]
- 28.Johnson, W. C., Jr. 1990. Protein secondary structure and circular dichroism: a practical guide. Proteins 7:205-214. [DOI] [PubMed] [Google Scholar]
- 29.Jones, C. T., L. Ma, J. W. Burgner, T. D. Groesch, C. B. Post, and R. J. Kuhn. 2003. Flavivirus capsid is a dimeric alpha-helical protein. J. Virol. 77:7143-7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Josse, D., C. Ebel, D. Stroebel, A. Fontaine, F. Borges, A. Echalier, D. Baud, F. Renault, M. Le Maire, E. Chabrieres, and P. Masson. 2002. Oligomeric states of the detergent-solubilized human serum paraoxonase (PON1). J. Biol. Chem. 277:33386-33397. [DOI] [PubMed] [Google Scholar]
- 31.Kato, T., M. Miyamoto, A. Furusaka, T. Date, K. Yasui, J. Kato, S. Matsushima, T. Komatsu, and T. Wakita. 2003. Processing of hepatitis C virus core protein is regulated by its C-terminal sequence. J. Med. Virol. 69:357-366. [DOI] [PubMed] [Google Scholar]
- 32.Klein, K. C., S. J. Polyak, and J. R. Lingappa. 2004. Unique features of hepatitis C virus capsid formation revealed by de novo cell-free assembly. J. Virol. 78:9257-9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kountouras, J., C. Zavos, and D. Chatzopoulos. 2003. Apoptosis in hepatitis C. J. Viral Hepat. 10:335-342. [DOI] [PubMed] [Google Scholar]
- 34.Kunkel, M., M. Lorinczi, R. Rijnbrand, S. M. Lemon, and S. J. Watowich. 2001. Self-assembly of nucleocapsid-like particles from recombinant hepatitis C virus core protein. J. Virol. 75:2119-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kunkel, M., and S. J. Watowich. 2004. Biophysical characterization of hepatitis C virus core protein: implications for interactions within the virus and host. FEBS Lett. 557:174-180. [DOI] [PubMed] [Google Scholar]
- 36.Lehrer, S. S. 1971. Solute perturbation of protein fluorescence. The quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry 10:3254-3263. [DOI] [PubMed] [Google Scholar]
- 37.Lo, S. Y., F. Masiarz, S. B. Hwang, M. M. Lai, and J. H. Ou. 1995. Differential subcellular localization of hepatitis C virus core gene products. Virology 213:455-461. [DOI] [PubMed] [Google Scholar]
- 38.Lorenzo, L. J., S. Duenas-Carrera, V. Falcon, N. Acosta-Rivero, E. Gonzalez, M. C. de la Rosa, I. Menendez, and J. Morales. 2001. Assembly of truncated HCV core antigen into virus-like particles in Escherichia coli. Biochem. Biophys. Res. Commun. 281:962-965. [DOI] [PubMed] [Google Scholar]
- 39.Ma, L., C. T. Jones, T. D. Groesch, R. J. Kuhn, and C. B. Post. 2004. Solution structure of dengue virus capsid protein reveals another fold. Proc. Natl. Acad. Sci. USA 101:3414-3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Majeau, N., V. Gagne, A. Boivin, M. Bolduc, J. A. Majeau, D. Ouellet, and D. Leclerc. 2004. The N-terminal half of the core protein of hepatitis C virus is sufficient for nucleocapsid formation. J. Gen. Virol. 85:971-981. [DOI] [PubMed] [Google Scholar]
- 41.Markoff, L., B. Falgout, and A. Chang. 1997. A conserved internal hydrophobic domain mediates the stable membrane integration of the dengue virus capsid protein. Virology 233:105-117. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto, M., S. B. Hwang, K. S. Jeng, N. Zhu, and M. M. Lai. 1996. Homotypic interaction and multimerization of hepatitis C virus core protein. Virology 218:43-51. [DOI] [PubMed] [Google Scholar]
- 43.Mayo, K., M. L. Vana, J. McDermott, D. Huseby, J. Leis, and E. Barklis. 2002. Analysis of Rous sarcoma virus capsid protein variants assembled on lipid monolayers. J. Mol. Biol. 316:667-678. [DOI] [PubMed] [Google Scholar]
- 44.McLauchlan, J. 2000. Properties of the hepatitis C virus core protein: a structural protein that modulates cellular processes. J. Viral Hepat. 7:2-14. [DOI] [PubMed] [Google Scholar]
- 45.McLauchlan, J., M. K. Lemberg, G. Hope, and B. Martoglio. 2002. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 21:3980-3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moller, J. V., and M. le Maire. 1993. Detergent binding as a measure of hydrophobic surface area of integral membrane proteins. J. Biol. Chem. 268:18659-18672. [PubMed] [Google Scholar]
- 47.Moradpour, D., C. Englert, T. Wakita, and J. R. Wands. 1996. Characterization of cell lines allowing tightly regulated expression of hepatitis C virus core protein. Virology 222:51-63. [DOI] [PubMed] [Google Scholar]
- 48.Moriya, K., H. Fujie, Y. Shintani, H. Yotsuyanagi, T. Tsutsumi, K. Ishibashi, Y. Matsuura, S. Kimura, T. Miyamura, and K. Koike. 1998. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 4:1065-1067. [DOI] [PubMed] [Google Scholar]
- 49.Moriya, K., H. Yotsuyanagi, Y. Shintani, H. Fujie, K. Ishibashi, Y. Matsuura, T. Miyamura, and K. Koike. 1997. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 78:1527-1531. [DOI] [PubMed] [Google Scholar]
- 50.Mukhopadhyay, S., P. R. Chipman, E. M. Hong, R. J. Kuhn, and M. G. Rossmann. 2002. In vitro-assembled alphavirus core-like particles maintain a structure similar to that of nucleocapsid cores in mature virus. J. Virol. 76:11128-11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nolandt, O., V. Kern, H. Muller, E. Pfaff, L. Theilmann, R. Welker, and H. G. Krausslich. 1997. Analysis of hepatitis C virus core protein interaction domains. J. Gen. Virol. 78:1331-1340. [DOI] [PubMed] [Google Scholar]
- 52.Notredame, C., D. G. Higgins, and J. Heringa. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205-217. [DOI] [PubMed] [Google Scholar]
- 53.Okamoto, K., K. Moriishi, T. Miyamura, and Y. Matsuura. 2004. Intramembrane proteolysis and endoplasmic reticulum retention of hepatitis C virus core protein. J. Virol. 78:6370-6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pearson, W. R. 1998. Empirical statistical estimates for sequence similarity searches. J. Mol. Biol. 276:71-84. [DOI] [PubMed] [Google Scholar]
- 55.Penin, F. 2003. Structural biology of hepatitis C virus. Clin. Liver Dis. 7:1-21, vii. [DOI] [PubMed] [Google Scholar]
- 56.Penin, F., V. Brass, N. Appel, S. Ramboarina, R. Montserret, D. Ficheux, H. E. Blum, R. Bartenschlager, and D. Moradpour. 2004. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 279:40835-40843. [DOI] [PubMed] [Google Scholar]
- 57.Ray, R. B., and R. Ray. 2001. Hepatitis C virus core protein: intriguing properties and functional relevance. FEMS Microbiol. Lett. 202:149-156. [DOI] [PubMed] [Google Scholar]
- 58.Reed, K. E., and C. M. Rice. 2000. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr. Top. Microbiol. Immunol. 242:55-84. [DOI] [PubMed] [Google Scholar]
- 59.Robertson, B., G. Myers, C. Howard, T. Brettin, J. Bukh, B. Gaschen, T. Gojobori, G. Maertens, M. Mizokami, O. Nainan, S. Netesov, K. Nishioka, T. Shin i, P. Simmonds, D. Smith, L. Stuyver, A. Weiner, et al. 1998. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. Arch. Virol. 143:2493-2503. [DOI] [PubMed] [Google Scholar]
- 60.Santolini, E., G. Migliaccio, and N. La Monica. 1994. Biosynthesis and biochemical properties of the hepatitis C virus core protein. J. Virol. 68:3631-3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schagger, H., and G. von Jagow. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166:368-379. [DOI] [PubMed] [Google Scholar]
- 62.Schuck, P., and B. Demeler. 1999. Direct sedimentation analysis of interference optical data in analytical ultracentrifugation. Biophys. J. 76:2288-2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schwer, B., S. Ren, T. Pietschmann, J. Kartenbeck, K. Kaehlcke, R. Bartenschlager, T. S. Yen, and M. Ott. 2004. Targeting of hepatitis C virus core protein to mitochondria through a novel C-terminal localization motif. J. Virol. 78:7958-7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seong, Y. R., E. K. Lee, S. Choi, S. K. Chon, and D. S. Im. 1996. Overexpression and simple purification of a truncated, immunologically reactive GST-HCV core (1-123) fusion protein. J. Virol. Methods 59:13-21. [DOI] [PubMed] [Google Scholar]
- 65.Shimotohno, K. 2000. Hepatitis C virus and its pathogenesis. Semin. Cancer Biol. 10:233-240. [DOI] [PubMed] [Google Scholar]
- 66.Simons, J. N., T. J. Pilot-Matias, T. P. Leary, G. J. Dawson, S. M. Desai, G. G. Schlauder, A. S. Muerhoff, J. C. Erker, S. L. Buijk, M. L. Chalmers, et al. 1995. Identification of two flavivirus-like genomes in the GB hepatitis agent. Proc. Natl. Acad. Sci. USA 92:3401-3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Soguero, C., M. Joo, K. A. Chianese-Bullock, D. T. Nguyen, K. Tung, and Y. S. Hahn. 2002. Hepatitis C virus core protein leads to immune suppression and liver damage in a transgenic murine model. J. Virol. 76:9345-9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Songsivilai, S., T. Dharakul, R. Kunkitti, and C. Thepthai. 1996. Molecular cloning and expression of hepatitis C virus core protein and production of monoclonal antibodies to the recombinant protein. Asian Pacific J. Allergy Immunol. 14:31-41. [PubMed] [Google Scholar]
- 69.Suzuki, R., S. Sakamoto, T. Tsutsumi, A. Rikimaru, K. Tanaka, T. Shimoike, K. Moriishi, T. Iwasaki, K. Mizumoto, Y. Matsuura, T. Miyamura, and T. Suzuki. 2005. Molecular determinants for subcellular localization of hepatitis C virus core protein. J. Virol. 79:1271-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takamizawa, A., C. Mori, I. Fuke, S. Manabe, S. Murakami, J. Fujita, E. Onishi, T. Andoh, I. Yoshida, and H. Okayama. 1991. Structure and organization of the hepatitis C virus genome isolated from human carriers. J. Virol. 65:1105-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tanford, C., Y. Nozaki, J. A. Reynolds, and S. Makino. 1974. Molecular characterization of proteins in detergent solutions. Biochemistry 13:2369-2376. [DOI] [PubMed] [Google Scholar]
- 72.Tellinghuisen, T. L., A. E. Hamburger, B. R. Fisher, R. Ostendorp, and R. J. Kuhn. 1999. In vitro assembly of alphavirus cores by using nucleocapsid protein expressed in Escherichia coli. J. Virol. 73:5309-5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tellinghuisen, T. L., R. Perera, and R. J. Kuhn. 2001. In vitro assembly of Sindbis virus core-like particles from cross-linked dimers of truncated and mutant capsid proteins. J. Virol. 75:2810-2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tellinghuisen, T. L., and C. M. Rice. 2002. Interaction between hepatitis C virus proteins and host cell factors. Curr. Opin. Microbiol. 5:419-427. [DOI] [PubMed] [Google Scholar]
- 75.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vistica, J., J. Dam, A. Balbo, E. Yikilmaz, R. A. Mariuzza, T. A. Rouault, and P. Schuck. 2004. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 326:234-256. [DOI] [PubMed] [Google Scholar]
- 77.Walewski, J. L., T. R. Keller, D. D. Stump, and A. D. Branch. 2001. Evidence for a new hepatitis C virus antigen encoded in an overlapping reading frame. RNA 7:710-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wengler, G., U. Boege, H. Bischoff, and K. Wahn. 1982. The core protein of the alphavirus Sindbis virus assembles into core-like nucleoproteins with the viral genome RNA and with other single-stranded nucleic acids in vitro. Virology 118:401-410. [DOI] [PubMed] [Google Scholar]
- 79.Wengler, G., U. Boege, and K. Wahn. 1984. Establishment and analysis of a system which allows assembly and disassembly of alphavirus core-like particles under physiological conditions in vitro. Virology 132:401-412. [DOI] [PubMed] [Google Scholar]
- 80.Winstone, T. L., M. Jidenko, M. le Maire, C. Ebel, K. A. Duncalf, and R. J. Turner. 2005. Organic solvent extracted EmrE solubilized in dodecyl maltoside is monomeric and binds drug ligand. Biochem. Biophys. Res. Commun. 327:437-445. [DOI] [PubMed] [Google Scholar]
- 81.Woodcock, S., J. P. Mornon, and B. Henrissat. 1992. Detection of secondary structure elements in proteins by hydrophobic cluster analysis. Protein Eng. 5:629-635. [DOI] [PubMed] [Google Scholar]
- 82.Woody, R. W. 1985. Circular dichroism of peptides, vol. 7. Academic Press, Inc., New York, N.Y.
- 83.Xu, Z., J. Choi, T. S. Yen, W. Lu, A. Strohecker, S. Govindarajan, D. Chien, M. J. Selby, and J. Ou. 2001. Synthesis of a novel hepatitis C virus protein by ribosomal frameshift. EMBO J. 20:3840-3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yasui, K., T. Wakita, K. Tsukiyama-Kohara, S. I. Funahashi, M. Ichikawa, T. Kajita, D. Moradpour, J. R. Wands, and M. Kohara. 1998. The native form and maturation process of hepatitis C virus core protein. J. Virol. 72:6048-6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zuber, G., J. McDermott, S. Karanjia, W. Zhao, M. F. Schmid, and E. Barklis. 2000. Assembly of retrovirus capsid-nucleocapsid proteins in the presence of membranes or RNA. J. Virol. 74:7431-7441. [DOI] [PMC free article] [PubMed] [Google Scholar]