

Abstract
In addition to activating members of the STAT transcription factor family, IFN α/β activates the NF-κ B transcription factor. To determine the role of the JAK-STAT pathway in NF-κ B activation by IFN, we examined NF-κ B activation in JAK1-deficient mutant human fibrosarcoma cells. In wild-type fibrosarcoma cells (2fTGH) IFN activates STAT1, STAT2 and STAT3, as well as NF-κB complexes comprised of p50 and p65. In contrast, in JAK1-deficient cells IFN induces NF-κB activation and NF-κB dependent gene transcription, but does not activate these STAT proteins and has no effect on STAT-dependent gene transcription. Expression of a catalytically-inactive TYK2 tyrosine kinase in JAK1-deficient cells, as well as in the highly IFN-sensitive Daudi lymphoblastoid cell line, abrogates NF-κB activation by IFN. Moreover, IFN does not promote NF-κB activation in TYK2-deficient mutant fibrosarcoma cells. Our results demonstrate a dichotomy between the classical JAK-STAT pathway and the NF-κB signaling pathway. In the IFN signaling pathway leading to STAT activation both JAK1 and TYK2 are essential, while NF-κB activation requires only TYK2.
IFNs are a family of multifunctional cytokines that block viral infection, inhibit cell proliferation, and modulate cell differentiation. IFNs transduce signals from the cell surface, resulting in selective gene induction through the activation of Janus tyrosine kinases (JAKs) and Signal transducers and activators of transcription (STAT) proteins (1–3). Upon JAK-mediated tyrosine phosphorylation, the STATs (STAT1, STAT2, and STAT3) dimerize and translocate into the nucleus to activate transcription of IFN-stimulated genes (3).
Besides activating members of the STAT transcription factor family, IFNα/β also activates the transcription factor NF-κB. Viruses, cytokines, lipopolysaccharides, and other stimulating agents also promote NF-κB translocation to the nucleus and DNA binding. Dimers of the various Rel proteins (p50, p52, c-Rel, v-Rel, RelA/p65, and RelB) regulate the expression of genes involved in cell signaling, stress responses, growth, survival, and apoptosis. In most cell types, the predominant form of NF-κB is the p50:p65 heterodimer. NF-κB dimers are retained in the cytoplasm of unstimulated cells in an inactive state by binding to a family of inhibitory IκB proteins.
In human lymphoblastoid cells, NF-κB activation in response to IFN is associated with promoting cell survival (4). IFN mediated NF-κB activation involves the tyrosine phosphorylation-dependent association of STAT3 with the IFNAR1 chain of the IFN receptor, and results in the sequential activation of a serine kinase cascade involving phosphatidylinositol-3 kinase (PI-3K) and Akt (4,5). Recent studies with normal fibroblasts and NF-κB knockout (KO) fibroblasts demonstrate that a subset of IFN-stimulated genes is regulated through an NF-κB-dependent pathway, and that NF-κB regulates the sensitivity of cells to IFN-mediated antiviral activity (6).
To further characterize NF-κB activation by IFN, we examined the crosstalk between the JAK-STAT signaling pathway and NF-κB activation in human fibrosarcoma cell lines with defined defects in the IFNα/β signaling pathway. The parental 2fTGH cell line is sensitive to gene induction by IFNα/β, while JAK1-deficient (U4A) mutant cells are IFN-nonresponsive (7). IFN induces NF-κB activation in JAK1-deficient (U4A) cells and in the parental fibrosarcoma cells (2fTGH). In contrast, IFN induces STAT activation in 2fTGH cells but not in the JAK1-deficient cells. IFN induction of NF-κB activity in JAK1-deficient cells involves the TYK2 tyrosine kinase. These results demonstrate a clear distinction between the signaling components in the JAK-STAT pathway and the NF-κB signaling pathway.
EXPERIMENTAL PROCEDURES
Biological reagents, plasmids and cell culture.
The biological activity of recombinant human IFNα (IFNCon1), provided by InterMune, was assayed by protection against the cytopathic effect of vesicular stomatitis virus (VSV) on human fibroblasts, using the NIH human IFNα standard for reference. 2fTGH human fibrosarcoma, TYK2-deficient U1A mutant cells and JAK1-deficient U4A mutant cells (provided by Dr. George Stark, Cleveland Clinic Foundation, Cleveland, OH) have been previously described (8), and were maintained in monolayer cultures in Dulbecco’s modified essential medium (DMEM) supplemented with 10% defined calf serum (DCS) (HyClone Labs) and hygromycin (250 μg/ml). The K930R kinase-inactive mutant of TYK2, subcloned into the pRc/CMV-neo expression vector, was generously provided by Dr. S. Pellegrini (9). Transient transfection of cells (107) was accomplished by electroporation (capacitance 300 μF, 250 V) with 500 μg of salmon sperm DNA and 20 μg of plasmid DNA for each sample (4).
Nuclear extracts and DNA binding activity assays.
Nuclei isolated from control and IFN-treated cells were extracted with buffer (20 mM Tris-HCl pH 7.85, 250 mM sucrose, 0.4 M KCl, 1.1 mM MgCl2, 5 mM β-mercaptoethanol, 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 5 μg/ml soybean trypsin inhibitor, 5 μg/ml leupeptin, and 1.75 μg/ml benzamidine) (10). The nuclear extracts were incubated with a [32P]-labeled probe for the IFN-stimulus response element (ISRE) in ISG15 gene (5′-GATCCATGCCTCGGGAAGGGAAACCGAAACTGAAGCC-3′), the high affinity c-sis inducible element (SIE) in the c-fos gene (5′-AGCTTCATTTCCCGTAATCCCTAAAGCT-3′), or the κB site (5′-AGTTGAGGGGACTTTCCCAGG-3′) in the immunoglobulin gene promoter and subjected to electrophoretic mobility shift assay (EMSA) (11). To define the presence of specific STAT or Rel proteins in DNA-protein complexes, nuclear extracts were incubated with anti-STAT or anti-Rel antibodies at 25°C for 20 min prior to EMSA. Gels were quantitated by PhosphorImage autoradiography.
Immunoblot analysis.
For immunoblotting, cells were treated with IFNα (1,000 IU/ml) at 37°C for the indicated periods of time and lysed directly in sample buffer (4). Samples were run on SDS-7.5% PAGE and transferred to PVDF membranes (Millipore). To define the effect of IFN on STAT tyrosine phosphorylation membranes were probed with immunoaffinity-purified anti-STAT1, anti-STAT2, anti-STAT3, anti-phospho-STAT1 (Tyr701), anti-phospho-STAT2 (Tyr689) or anti-phospho-STAT3 (Tyr705) (dilution 1:1000, Upstate Biotechnology). For TYK2 analysis, total cell lysates were analyzed by immunoblotting for phospho-TYK2 (Cell Signaling) , TYK2 (Upstate Biotechnology) and actin (Santa Cruz). Blots were incubated with anti-IgG coupled to horseradish peroxidase (Santa Cruz), and developed using enhanced chemiluminiscence (ECL) with the Super Signal West Pico Chemiluminescent Substrate (Pierce).
Transcriptional assays.
For gene reporter assays, cells were transiently transfected by electroporation with either the IRF-1 3X GAS-chloramphenicol acetyltransferase (CAT) reporter plasmid containing three copies of the SIE (equivalent to GAS) site from the IRF-1 gene (provided by Drs. V. Viggiano and H. Young, National Cancer Institute-Fredrick Research Cancer and Development Center), the pUX-CAT 3XHLAκB CAT reporter construct containing three tandemly repeated copies of the NF-κB site from the HLA-B7 gene (12), or the corresponding promoter-less CAT construct. After 48 hrs the cells were treated with IFNα (1000 U/ml) for 4 hr and assayed for CAT activity. Acetylated and unacetylated [14C] chloramphenicol were separated by thin layer chromatography and radioactivity was measured by PhosphorImage autoradiography.
RESULTS
IFN induces STAT activation in parental 2fTGH cells but not in JAK1− mutant fibrosarcoma cells.
The JAK1 and TYK2 tyrosine kinases mediate the tyrosine phosphorylation of cytoplasmic STAT proteins, which is essential for their nuclear translocation and DNA-binding activity. For example, upon IFNα-induced phosphorylation STAT1 and STAT2 dimerize, complex with the DNA binding protein IRF-9, and translocate into the nucleus to bind to the ISRE of IFN-stimulated genes and activate ISG transcription. The tyrosine phosphorylation of STAT3 is essential for its DNA-binding activity to SIE-dependent responsive genes. Lysates prepared from control or IFNα-treated 2fTGH and JAK1-deficient (JAK1−) cells were immunoblotted with immunoaffinity-purified anti-STAT Abs and anti-STAT phosphospecific Abs. As shown in Fig. 1, IFN induces the tyrosine phosphorylation of STAT1, STAT2 and STAT3 in 2fTGH cells, but not in the JAK1− cells (Fig. 1). IFN treatment also induced DNA-binding activity attributable to STAT1, STAT2 and STAT3 in 2fTGH cells, but not in the JAK1− mutant cells (data not shown). These results are not unexpected since both JAK1 and TYK2 are required to transduce the signal that leads to STAT phosphorylation, STAT dimerization, STAT translocation and subsequent STAT-mediated gene induction (3,7).
FIGURE 1. IFN induces the tyrosine phosphorylation of STAT proteins in parental 2fTGH cells but not in JAK1− fibrosarcoma cells.
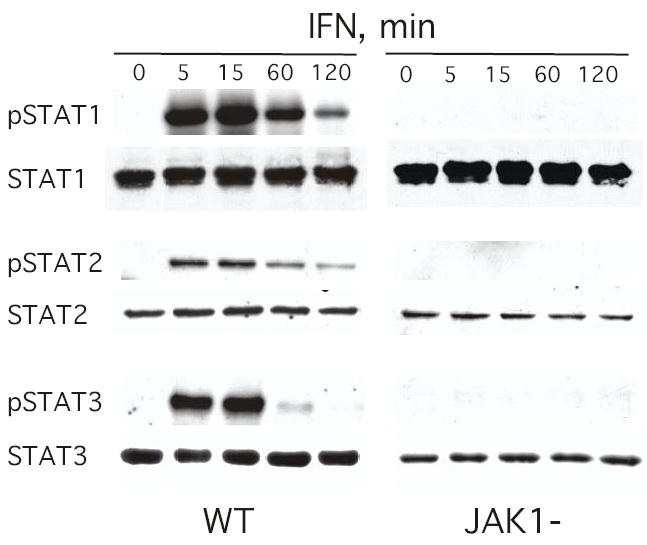
Lysates prepared from control or IFNα-treated (1000 IU/ml) cells were resolved by SDS-PAGE, transferred onto PVDF membranes and probed with the indicated anti-STAT or anti-phosphoSTAT Ab. Blots were visualized by ECL.
IFN induces NF-κB DNA binding activity in parental 2fTGH cells and JAK1− mutant fibrosarcoma cells.
In diverse cells IFNα/β promotes NF-κB DNA-binding activity with cell-type differences in the specific composition of the IFN-induced Rel/NF-κB complexes (4). For example, p50/c-Rel complexes are induced by IFN in human lymphoblastoid cells, while p50/p65 complexes are induced in mouse 3T3 fibroblast cells. To determine whether IFNs also promoted NF-κB activation in human fibrosarcoma cells, cells were stimulated with IFNα, nuclear extracts were prepared and NF-κB activation was examined by EMSA. Nuclear extracts from untreated cells show little constitutive binding to a consensus κB oligonucleotide probe (Fig. 2). However, IFN induced NF-κB activity in both 2fTGH and JAK1− cells. DNA binding to the probe was not detected in the presence of excess NF-κB oligonucleotide, indicating that the binding to the DNA probe was specific. Supershift assays with Rel-specific antisera demonstrated that p50 and p65, but not c-Rel, were present in the IFN-induced NF-κB complexes in both parental and JAK1− cells (Fig. 2). These results were somewhat surprising since JAK1− cells are nonresponsive to IFN as determined by STAT tyrosine phosphorylation and DNA-protein binding assays.
FIGURE 2. IFN induces NF-κB DNA binding activity in parental 2fTGH cells and JAK1−fibrosarcoma cells.
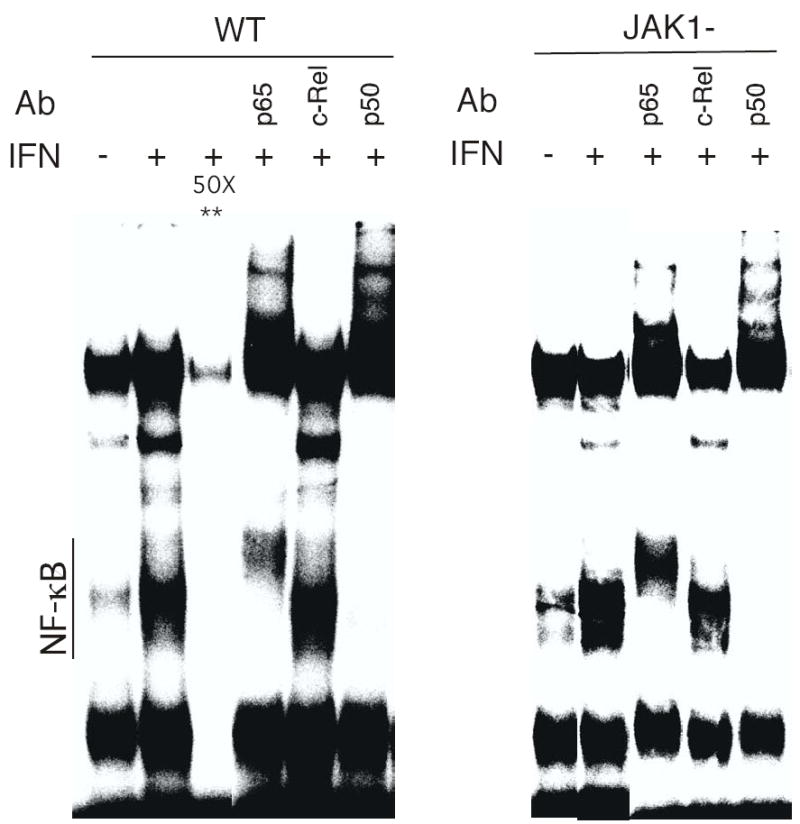
Nuclear extracts from control or IFN-treated cells were incubated with a [32P]-labeled probe derived from the κB binding sequence in the immunoglobulin gene promoter. To demonstrate binding specificity, extracts were incubated with a 50-fold excess of unlabeled κB oligonucleotide probe (**). To define specific Rel proteins, extracts were preincubated with anti-Rel antibodies prior to EMSA.
To further characterize the induction of NF-κB activity in the JAK1− cells, we examined the time course of IFN induction of NF-κB binding activity. As shown in Figure 3A, nuclear extracts from untreated cells (0 time) show low constitutive binding to a consensus κB oligonucleotide probe. IFN increased NF-κB binding in JAK1− cells within 5 min after IFN treatment, with high DNA binding observed from 15 min to 60 min after IFN addition that returned to near basal levels by 120 min. The time course of NF-κB activation is highly similar to that previously reported for IFN mediated NF-κB activation in human lymphoblastoid cells and in mouse embryo fibroblasts (4,6). IFN has biologic efficacy at extremely low concentrations, for example 1 U/ml (~10−13 M) IFN is defined by the ability to inhibit virus infection by 50%. Therefore, we next examined the IFN concentration-dependent induction of NF-κB activation in the JAK1− cells. As shown in Figure 3B, a clear induction of DNA binding activity is observed at 10 U/ml of IFN and a dose-dependent increase in DNA binding activity was observed at the range of IFN concentrations tested. These results show that the induction of NF-κB activity in the JAK1− cells occurs at physiologically relevant IFN concentrations.
FIGURE 3. Time course, dose-dependence and effect of protein kinase inhibitors on IFN induced NF-κB DNA binding activity in JAK1− fibrosarcoma cells.
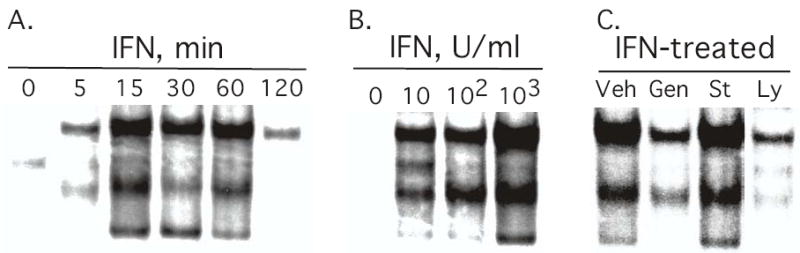
U4A cells were either (Panel A) treated with IFN (1000 IU/ml) for the indicated times; (Panel B) treated with the indicated concentrations of IFN for 30 min; or (Panel C) pretreated with either DMSO as vehicle (Veh), the tyrosine kinase inhibitor genistein (100 μM), the PI-3K inhibitor LY294002 (10 μM), or the general protein kinase inhibitor staurosporin (100 nM) for 1 hr prior to IFN addition (1000 IU/ml, 30 min). Nuclear extracts were subjected to EMSA with a [32P]-labeled κB oligonucleotide probe.
The role of tyrosine and serine protein kinases in IFN-dependent NF-κB activation in U4A cells.
The JAK-mediated tyrosine phosphorylation pathway is an integral element in IFN-mediated signal transduction in general. However, serine phosphorylation events are also critical for the biological response to IFNα (2,13–15). IFNα activates PI-3K by inducing the rapid tyrosine phosphorylation of its regulatory 85-kDa (p85) subunit (16,17), which leads to the subsequent activation of the serine-threonine kinase Akt/PKB (5). The PI-3K/Akt pathway is involved in NF-κB activation by IFN in human lymphoblastoid cells (5). To characterize the role of protein kinases in JAK1-independent NF-κB activation, U4A cells were treated with the tyrosine kinase inhibitor genistein, the PI-3K inhibitor LY294002, or protein kinase C inhibitor staurosporin. As shown in Figure 3C treatment with either genistein or LY294002 blocked induction of NF-κB activity by IFN in JAK1-defective cells. However, staurosporin did not block NF-κB activation by IFN. This result is surprising since staurosporin blocks IFN-induced biological events mediated not only by protein kinase C but also by tyrosine kinases (15,18,19). None of these inhibitors had any effect on the low basal level of NF-κB expression in U4A cells (data not shown).
The effect of JAK1-independent signaling on NF-κB and SIE-dependent reporter constructs.
To determine the functional consequences of IFN-induced STAT and NF-κB activation, we examined the effect of IFN on the transcriptional activity of NF-κB and SIE-dependent CAT reporter constructs. 2fTGH and U4A mutant cells were transfected with either the IRF-1 3X GAS-CAT reporter construct containing three copies of the SIE site from the IRF-1 gene, the pUX-CAT 3XHLAκB CAT reporter construct containing three tandemly repeated copies of the NF-κB site from the HLA-B7 gene, or the corresponding promoter-less CAT construct. At 2 days after transfection the cells were treated with IFN and assayed for CAT activity. As shown in Figure 4, while IFN treatment had no effect on the control CAT reporter construct (EV), IFN treatment of 2fTGH cells resulted in a marked enhancement in the transcriptional activity of both the IRF-1 3X GAS-CAT and 3XHLAκB reporter constructs, 4- and 6-fold respectively. In contrast, IFN treatment of U4A cells resulted in enhanced transcriptional activity of 3XHLAκB reporter (~3.5-fold) but not of the IRF-1 3X GAS-CAT reporter construct. These results demonstrate that IFN induced NF-κB dependent gene transcription in JAK1− cells, but did not affect STAT-dependent gene transcription in these mutant cells. Therefore, IFN induced NF-κB-driven gene transcription is JAK1-independent, while IFN induced SIE-driven gene transcription is JAK1-dependent. These results parallel our findings on DNA binding activity, i.e. IFN induced NF-κB activity is JAK1-independent, but IFN induced SIE activity is JAK1-dependent.
FIGURE 4. The effect of JAK1-independent signaling on NF-κB and SIE-dependent reporter constructs.
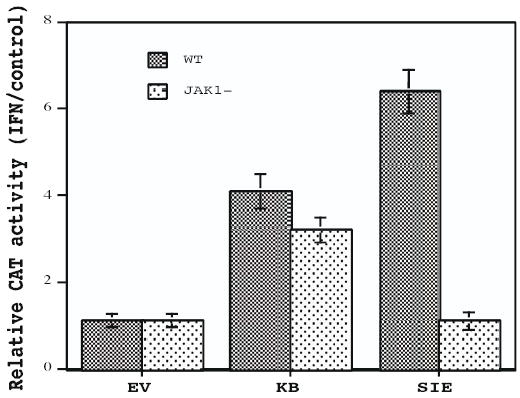
2fTGH and JAK1− cells were transiently transfected by electroporation with the promoter-less CAT construct, with the pUX-CAT 3XHLAκB CAT reporter construct (κB) containing three copies of the κB site from the HLA-B7 gene, or the IRF-1 3X SIE-CAT reporter plasmid containing three copies of the SIE site from the IRF-1 gene. After 48 hr the cells were treated with IFN (1000 IU/ml) for 4 hr and assayed for CAT activity. Acetylated and unacetylated [14C] chloramphenicol were separated by thin layer chromatography and quantified by phosphorimaging. The results of three separate experiments performed in duplicate on IFN-induced κB and SIE-driven gene transcription were averaged (error bars shown). The data are expressed as CAT activity in IFN-treated cells relative to control cells.
NF-κB activation in JAK1-deficient cells is TYK2-dependent.
Since JAK1− mutant cells contain TYK2, one possibility is that NF-κB is activated through a TYK2-dependent pathway. To determine the role of TYK2 in NF-κB activation by IFN, TYK2-deficient U1A fibrosarcoma cells were treated with IFNα, nuclear extracts were prepared and NF-κB activation was examined by EMSA. As shown in Figure 5 IFN treatment failed to promote NF-κB activation in these TYK2-mutant cells. In contrast, IFN induced NF-κB activity in JAK1- U4A cells. These results taken together demonstrate that TYK2, but not JAK1 is required for IFN induced NF-κB activation.
FIGURE 5. IFN does not induce NF-κB DNA binding activity in TYK2− fibrosarcoma cells.
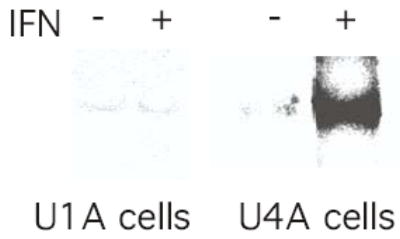
Nuclear extracts from control or IFN-treated (1000 IU/ml, 15 min) TYK2− (U1A) fibrosarcoma cells were incubated with a [32P]-labeled probe derived from the κB binding sequence in the immunoglobulin gene and analyzed by EMSA. The analysis of nuclear extracts from control and IFN-treated U4A cells is shown for reference.
To further analyze the role of TYK2 in JAK1-independent NF-κB activation, U4A cells were transiently-transfected with TYK2 or a kinase-inactive form of TYK2 (KD-TYK2), in which the invariant lysine at position 930 of the ATP binding site was substituted with an arginine. At 48 hr after transfection, the U4A cells were stimulated with IFNα, nuclear extracts were prepared and NF-κB activation was examined by EMSA. As shown in Figure 6A, IFN induces NF-κB activation in U4A cells transfected with either empty vector or a vector encoding TYK2. In contrast, transfection with the kinase-inactive K930R (KD-TYK2) construct completely abrogated IFN-dependent NF-κB activation in JAK1-deficient cells. In addition, extracts from transiently transfected cells were immunoblotted with Abs against TYK2 and phospho-TYK2. As shown in Figure 6B although KD-TYK2 and TYK2 were expressed at similar levels in transiently-transfected U4A cells, IFN induced tyrosine phosphorylation of TYK2 in cells overexpressing TYK2 but not in cells overexpressing KD-TYK2. Moreover, the low endogenous level of IFN-induced TYK2 tyrosine phosphorylation was abrogated by expression of KD-TYK2 (compare IFN-induced tyrosine phosphorylation of TYK2 in EV-transfected cells versus KD-TYK2-transfected cells in the upper panel of Figure 6B), demonstrating that KD-TYK2 exhibits dominant negative effects on endogenous TYK2. These results demonstrate that NF-κB activation by IFN in JAK1-deficient cells is TYK2-dependent.
FIGURE 6. The role of TYK2 in JAK1-independent NF-κB activation by IFN.
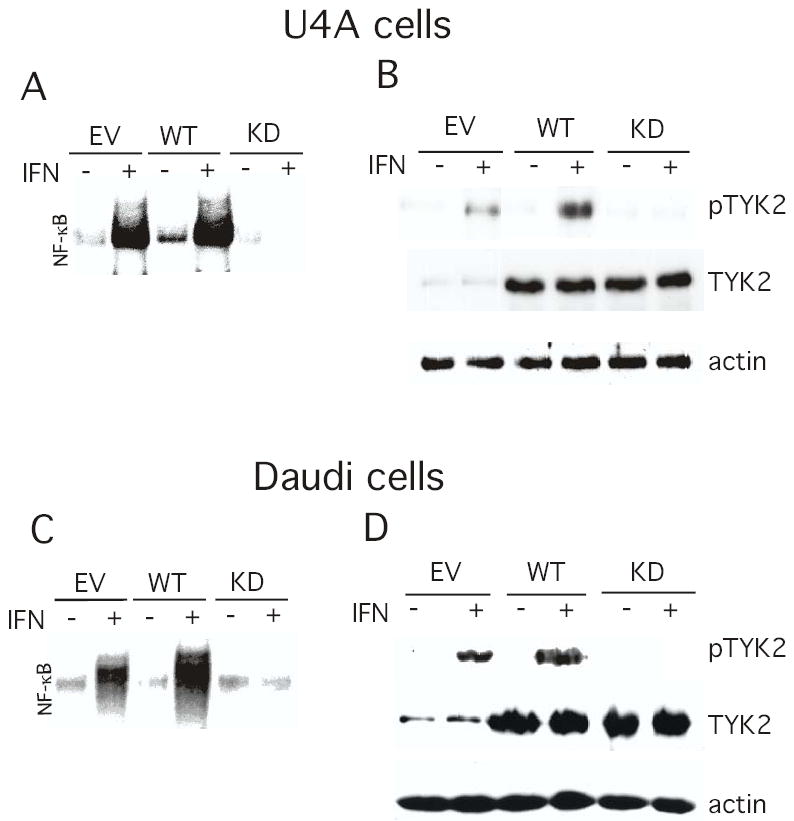
JAK1− U4A cells (Panels A and B) and Daudi cells (Panels C and D) were transiently transfected by electroporation with either empty vector (EV), TYK2 (WT) or the K930R TYK2 mutant (KD). After 48 hr the cells were treated with IFN (1000 IU/ml) for 30 min and either total cell lysates were prepared analyzed by immunoblotting for phospho-TYK2, TYK2 and actin (Panels A and C), or nuclear extracts were prepared and subjected to EMSA with a [32P]-labeled κB oligonucleotide probe (Panels B and D).
NF-κB activation in Daudi lymphoblastoid cells is TYK2-dependent.
We previously demonstrated that IFN induces an NF-κB -dependent cell survival pathway in the highly IFN-sensitive Daudi lymphoblastoid cell line (4,5). To further probe the role of TYK2 in NF-κB activation by IFN, Daudi cells were transfected with TYK2 or KD-TYK2. As shown in Figure 6C and consistent with the findings in U4A cells, IFN induces NF-κB activation in cells transfected with either empty vector or TYK2. However, transfection with KD-TYK2 completely blocked IFN induced NF-κB activation in Daudi cells. Moreover, KD-TYK2 blocked the induction by IFN of TYK2 phosphorylation (Fig. 6D), and of STAT-dependent DNA binding activity in Daudi cells (data not shown), indicating that KD-TYK2 functions in a dominant-negative manner for several signaling events induced by IFN.
DISCUSSION
The major conclusion of the present work is that IFN produces biologically relevant signals that are independent of the “classical’ JAK-STAT signaling pathway. We show that IFN activates NF-κB through a TYK2-dependent but a JAK1-independent pathway. We have previously shown that IFNα/β activates NF-κB in diverse cell types derived from several animal species, and have extensively characterized the pathway involved in NF-κB activation by IFN. In human lymphoblastoid cells NF-κB activation by IFN protects the cells from death induced by apoptotic stimuli including that of IFN itself (4). The IFN signaling pathway leading to NF-κB activation involves the tyrosine phosphorylation-dependent association of STAT3 with the IFN receptor, resulting in the activation of a serine kinase cascade through PI-3K and Akt (5). Consistent with these findings are the present findings that the pharmacologic inhibitors of PI-3K and tyrosine kinases block the JAK1-independent activation of NF-κB by IFN.
In common with a wide variety of cytokines and growth factors IFNα/β activates a JAK-STAT intracellular signaling pathway. IFN activation of JAKs results in the tyrosine phosphorylation of IFN receptor subunits to which the STATs bind. In turn STATs become tyrosine phosphorylated, dimerize and translocate into the nucleus to regulate gene transcription. However, our results demonstrate a dichotomy between the signaling components required in the classical JAK-STAT pathway and in the NF-κB signaling pathway. In the well-described IFN signaling pathway leading to STAT activation both JAK1 and TYK2 are required for signaling, while NF-κB activation appears to be TYK2-dependent but JAK1-independent. In the parental 2fTGH fibrosarcoma cell line IFN induces the activation of STAT1, STAT2 and STAT3 as determined by DNA binding assays and by immunoblotting with phosphospecific antibodies. However, in JAK1−cells these STAT proteins are not activated in DNA binding assays or in immunoblots. The strict requirement for both JAK1 and TYK2 is consistent with previous studies (7,20).
In contrast NF-κB is activated by IFN in parental 2fTGH cells and JAK1-deficient mutant cells, indicating that there is a JAK1-independent pathway leading to NF-κB activation. NF-κB activation by IFN in JAK1-deficient cells was demonstrated by EMSA with a κB-specific oligonucleotide probe and by using a κB -dependent reporter construct. The time-course of induction of NF-κB and dependence on IFN concentration was similar to previous reports using human and murine cells treated with IFN. This suggests that the JAK1-independent NF-κB signaling pathway activated by IFN may function in many cell types. Although NF-κB activation is JAK1-independent, this pathway appears to be tyrosine kinase and PI-3K dependent, since pharmacological inhibitors of these pathways blocked it.
The role of PI-3K and tyrosine kinase in IFN-induced NF-κB activation has been previously demonstrated in human lymphoblastoid cells. Since JAK1− mutant cells contain TYK2, we examined whether NF-κB is activated through a TYK2-dependent pathway. Expression of the catalytically-inactive K930R mutant of TYK2 (9) in JAK1-deficient U4A cells or in the highly IFN-sensitive Daudi cell line abrogated the induction of NF-κB activity by IFN. These results demonstrate that catalytically active TYK2 is required not only in cells which are deficient in JAK1, but also in cells in which NF-κB has been previously demonstrated to function in a cell survival pathway. Moreover, our results demonstrate that KD-TYK2 functions as a dominant negative for IFN signal transduction involving NF-κB, as well as the activation of STAT proteins.
Under diverse experimental conditions IFNα has been found to efficiently induce cell death, as well as protect cells against apoptotic signals (4,5,21,22). The JAK/STAT tyrosine phosphorylation-signaling pathway plays a critical role in IFN-regulated gene expression, and in the antiviral and antiproliferative actions of IFN (3). However, while the molecular mechanisms underlying the anti-apoptotic/apoptotic signals induced by IFN are unclear, both the JAK-STAT and the NF-κB signaling pathways have been implicated (4,5,23). In human lymphoblastoid cells, NF-κB activation is mediated by the tyrosine phosphorylation-dependent association of STAT3 with the IFN receptor, and the subsequent activation of PI-3K (5). It is tempting to speculate that this IFN-induced PI-3K/Akt signaling pathway leading to NF-κB-mediated cell survival requires TYK2 but is JAK1-independent. Nonetheless, taken together our results demonstrate a dichotomy between the signaling components required in the classical JAK-STAT pathway and in the NF-κB/anti-apoptotic pathway. In the signaling pathway leading to STAT activation JAK1 is required, while NF-κB activation appears to be JAK1-independent but TYK2-dependent.
Acknowledgments
National Institute of Health Grant CA73753 (L.M.P) supported this work. We thank Drs. Lawrence Blatt, Howard Young, Sandra Pellegrini, George Stark, and Jan Vilcek for generously providing recombinant IFN, cell lines and plasmids.
References
- 1.Friedman RL, Stark GR. Nature. 1985;314:637–639. doi: 10.1038/314637a0. [DOI] [PubMed] [Google Scholar]
- 2.Larner AC, Chaudhuri A, Darnell JEJ. J Biol Chem. 1986;261:453–459. [PubMed] [Google Scholar]
- 3.Darnell JEJ, Kerr IM, Stark GR. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 4.Yang CH, Murti A, Basu L, Kim JG, Pfeffer LM. Proc Natl Acad Sci USA. 2000;97:13631–13636. doi: 10.1073/pnas.250477397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang CH, Murti A, Pfeffer SR, Kim JG, Donner DB, Pfeffer LM. J Biol Chem. 2001;276:13756–13761. doi: 10.1074/jbc.M011006200. [DOI] [PubMed] [Google Scholar]
- 6.Pfeffer LM, Kim JG, Pfeffer SR, Carrigan DJ, Baker DP, Wei L, Homayouni R. J Biol Chem. 2004;279:31304–31311. doi: 10.1074/jbc.M308975200. [DOI] [PubMed] [Google Scholar]
- 7.Muller M, Briscoe J, Laxton C, Guschin D, Ziemiecki A, Silvennoinen O, Harpur AG, Barbieri G, Witthuhn BA, Schindler C, Pellegrini S, Wilks AF, Ihle JN, Stark GR, Kerr IM. Nature. 1993;366:129–135. doi: 10.1038/366129a0. [DOI] [PubMed] [Google Scholar]
- 8.Pellegrini S, John J, Shearer M, Kerr IM, Stark GR. Mol Cell Biol. 1989;9:4605–4612. doi: 10.1128/mcb.9.11.4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gauzzi MC, Velazquez L, McKendry R, Mogensen KE, Fellous M, Pellegrini S. J Biol Chem. 1996;271:20494–20500. doi: 10.1074/jbc.271.34.20494. [DOI] [PubMed] [Google Scholar]
- 10.Yang CH, Shi W, Basu L, Murti A, Constantinescu SN, Blatt L, Croze E, Mullersman JE, Pfeffer LM. J Biol Chem. 1996;271:8057–8061. doi: 10.1074/jbc.271.14.8057. [DOI] [PubMed] [Google Scholar]
- 11.Yang CH, Murti A, Pfeffer LM. Proc Natl Acad Sci USA. 1998;95:5568–5572. doi: 10.1073/pnas.95.10.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliviera IC, Mukaida N, Matsushiam K, Vilcek J. Mol Cell Biol. 1994;14:5300–5308. doi: 10.1128/mcb.14.8.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faltynek CR, Princler GL, Gusella GI, Varesio L, Radzioch D. J Biol Chem. 1989;264:14305–14311. [PubMed] [Google Scholar]
- 14.Reich NC, Pfeffer LM. Proc Natl Acad Sci USA. 1990;87:8761–8765. doi: 10.1073/pnas.87.22.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfeffer LM, Eisenkraft BL, Reich NC, Improta T, Baxter G, Daniel-Issakani S, Strulovici B. Proc Natl Acad Sci USA. 1991;88:7988–7992. doi: 10.1073/pnas.88.18.7988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uddin S, Yenush L, Sun XJ, Sweet ME, White MF, Platanias LC. J Biol Chem. 1995;270:15938–15941. doi: 10.1074/jbc.270.27.15938. [DOI] [PubMed] [Google Scholar]
- 17.Pfeffer LM, Mullersman JE, Pfeffer SR, Murti A, Shi W, Yang CH. Science. 1997;276:1418–1420. doi: 10.1126/science.276.5317.1418. [DOI] [PubMed] [Google Scholar]
- 18.Strehlow I, Schindler C. J Biol Chem. 1998;273:28049–28056. doi: 10.1074/jbc.273.43.28049. [DOI] [PubMed] [Google Scholar]
- 19.Haspel RL, Darnell JE., Jr Proc Natl Acad Sci U S A. 1999;96:10188–10193. doi: 10.1073/pnas.96.18.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Velazquez L, Fellous M, Stark GR, Pellegrini S. Cell. 1992;70:313–322. doi: 10.1016/0092-8674(92)90105-l. [DOI] [PubMed] [Google Scholar]
- 21.Einhorn S, Grander D. J Interferon Cytokine Res. 1996;16:275–281. doi: 10.1089/jir.1996.16.275. [DOI] [PubMed] [Google Scholar]
- 22.Marrack P, Kappler J, Mitchell T. J Exp Med. 1999;189:521–530. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schindler C. Trends Cell Biol. 1998;8:97–98. doi: 10.1016/s0962-8924(98)01233-1. [DOI] [PubMed] [Google Scholar]