

Abstract
Focal adhesions (FA) are large, multiprotein complexes that provide a mechanical link between the cytoskeletal contractile machinery and the extracellular matrix. FA exhibit mechanosensitive properties; they self-assemble and elongate upon application of pulling forces and dissociate when these forces are decreased. We propose a thermodynamic model for the mechanosensitivity of FA, according to which a molecular aggregate, subjected to pulling forces, tends to grow in the direction of force application by incorporating additional molecules. We demonstrate that this principle is consistent with the phenomenology of FA dynamics by considering a one-dimensional protein aggregate subjected to pulling forces and anchored to the substrate. Depending on the force level, force distribution along the aggregate, and the character of its anchoring to the substrate, the aggregate is predicted to exhibit distinct modes of assembly that are largely consistent with the experimentally observed FA behavior. We define here specific conditions that can lead to the different regimes of FA assembly, including growth, steady state, and disassembly.
Keywords: elasticity, mechanosensitivity, elastic stress, self-assembly
Mechanosensitive behavior of focal adhesions (FA) is an important component of cell ability to spread and move along substrates (1–3). The basic observation underlying FA mechanosensitivity is that alterations in the mechanical force applied to these adhesion sites either by the contractile machinery of the cell or after an external perturbation have a dramatic effect on FA properties (1). Although the molecular mechanism of this phenomenon is still obscure, the recent progress in characterizing the molecular organization and complexity of the integrin-mediated FA (4, 5) stimulated several attempts toward investigating the physical principles governing FA dynamics (6, 7, 32). The present work proposes a previously undescribed mechanism of FA mechanosensitivity based on thermodynamic properties of self-assembly under tension.
Phenomenology of FA Mechanosensitivity
FA are several-micrometer large multimolecular complexes, forming a mechanical link between the cytoskeleton and the extracellular matrix. A FA consists of a layer of transmembrane integrin molecules and a multiprotein “submembrane plaque.” Integrins are integral membrane heterodimeric proteins whose extracellular domains attach to the substrate while their intracellular domains provide docking sites for the assembly of the plaque. The plaque consists of a large number (>50) of different proteins (4, 5), including talin, α-actinin, filamin, vinculin, paxillin, and tensin, which are known to form the structural scaffold of the adhesion site, and others mediate signaling processes. The cytoplasmic aspect of the plaque serves as a platform for nucleation and development of stress fibers, the bundles of actin filaments containing myosin and various actin-binding proteins. The stress fibers are major generators of intracellular contractile forces (8, 9), which are transmitted via the FA plaque and the integrins to the substrate (for review, see refs. 1, 4, and 10).
Most FA are stationary structures and do not move with respect to the substrate. In some cases, however, FA become apparently mobile and start crawling in the direction of the force applied by the stress fibers (11–15). It should be emphasized that at the molecular level, both stationary and mobile FA are dynamic structures exhibiting a continuous exchange of components with the diffusible cytoplasmic pool, as evidenced by fluorescence recovery after photobleaching experiments (16, 17).
Mechanosensitivity of FA is manifested in the dependence of their shapes and dimensions on the applied forces (1, 3, 18). A developing FA usually acquires an elongated shape with a finite length, determined by the direction of force and its magnitude. In most cases the size of FA is proportional to the applied force (19).
The force-induced FA elongation is a reversible process. Impairment of the myosin contractile activity by chemical or natural (e.g., caldesmon) actomyosin inhibitors, reduces the pulling forces and leads to shrinkage of the FA (for review, see ref. 2).
Importantly, the FA mechanosensitive behavior proved to be largely independent of the origin of the pulling forces. Replacement of the actin–myosin contractile stresses by external forces applied either by using a micropipette attached to the cell surface or through flexible substrates results in FA dynamics similar to that occurring under natural conditions (18). Finally, it is noteworthy that the force-dependent growth of FA is due to protein self-assembly. This finding has been demonstrated by experiments showing a net addition of new fluorescently labeled plaque proteins to the growing, stressed FA (18, 19).
The Essence of the Present Work
Here, we demonstrate that the major features of the FA mechanosensitive behavior can be explained by a thermodynamic principle, which governs self-assembly of molecules into an aggregate subjected to pulling force.
Thermodynamics predict that elastic stresses generated within a protein complex subject to pulling forces decrease the chemical potential of the aggregated molecules as compared with the pool of nonassembled molecules (20). This effect means that self-assembly of proteins is favored when pulling forces act on the aggregate and is disfavored when these forces are relaxed. Considering various types of linkage between the aggregate and the substrate, we determine different modes of FA assembly and disassembly and show that the suggested model accounts for the major regimes of FA behavior observed experimentally. We determine the conditions upon which the protein aggregate subjected to pulling forces reaches a steady state and analyze the dependence of the dimension of the steady-state aggregate on the force applied.
Qualitative Considerations
To grasp the essence of the suggested mechanism, consider first a simplified case. Whereas a plaque of focal contact is composed of a variety of different protein molecules, we will consider it to consist of similar building blocks, which, for simplicity, will be called “FA proteins.” Assume that the FA is represented by a protein aggregate subject to a pulling force. The edge of the aggregate pointing in the direction of pulling will be referred to as the front edge, and the opposite one will be called the rear edge. In this section, we assume that FA is attached to the substrate only at the rear edge and the pulling force is applied only to the front edge (Fig. 1a). The aggregate exchanges constituent molecules with the cytoplasm, which act as a molecular reservoir. As a result of pulling force application, the aggregate undergoes stretching deformation (Fig. 1b), which is accompanied by accumulation of the elastic energy. Further evolution of the aggregate is governed by the tendency of the total free energy of the system to decrease.
Fig. 1.

Schematic representation of self-assembly upon pulling force. (a) The protein aggregate and the free proteins in the surrounding medium. (b) Application of the pulling force results in the aggregate stretching and the related accumulation of the elastic stresses within the aggregate. (c) Insertion of new proteins into the aggregate results in stress relaxation.
The total free energy, Gtot, includes the elastic energy of the aggregate, Felast, the free energy of nonassembled protein molecules in the cytoplasm, Ffree, and the free energy of the contractile machinery, referred to below as the energy of the pulling force, Fpull,
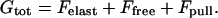 |
[1] |
To understand qualitatively the aggregate evolution, we dissect it into sequential steps. Right after the force is applied to its front edge, the aggregate undergoes an elastic elongation ΔL in the direction of force action (Fig. 1b). This elongation is favorable in terms of the energy of the pulling force, which decreases by
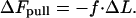 |
[2] |
Conversely, the elastic energy of the aggregate, Felast, increases, ΔFelast > 0, due to the stretching (Fig. 1b). At the second step, new molecules from the solution insert into the aggregate and fully, or partially, relieve the stresses (Fig. 1c), thus lowering the elastic energy, ΔFelast → 0. Importantly, the stress relaxation is induced only by incorporation of the molecules in the direction of the pulling force. Joining of molecules to the aggregate from the sides would not contribute to the stress relaxation and, hence, is not favorable. Therefore, the insertion of new molecules gives rise to unidirectional growth of the aggregate in the direction of the force.
As a result of the two steps, the sum of the elastic and the pulling force energies decreases as compared with the initial value, Δ(Felast + Fpull) ≈ ·fΔL < 0. The energy of the nonassembled molecules, Ffree, may go up as a result of transition of some amount of them into the aggregate leading to a decrease of entropy. However, provided that the pulling force f is sufficiently large, increase in Ffree does not overcompensate for the decrease in Fpull, and the total free energy decreases, Δ(Felast + Fpull + Ffree) < 0, meaning that the whole sequence of events is energetically favorable.
At the next stage, the aggregate with the newly added molecules is stretched again by the pulling force giving rise to continuous aggregate growth. Abortion of the pulling force results in disintegration of the aggregate. The qualitative consideration above has been confirmed by Monte Carlo simulations (data not shown).
There may be two different modes of action of the contractile machinery on the aggregate. In the first, dimension-controlled mode, the pulling force acts only until the aggregate reaches a certain dimension. In the second, force-controlled mode, the contractile machinery does not stop pulling. According to the existing data the force-controlled mode is more plausible because the FA behavior upon persistent pulling by an external experimentally controlled force is similar to that driven by the intracellular contractile machinery (18). In the following, we assume the force-controlled mode of FA self-assembly.
In the following, we present a rigorous thermodynamic analysis of the suggested model for a one-dimensional (1D) aggregate where both the attachment points to the pulling stress fibers and the links between the aggregate and the substrate are distributed along the aggregate.
The Model
Description of the System. As shown above, the force-induced incorporation of new building blocks into the aggregate proceeds unidirectionally. Therefore, to account for the major features of the system, we consider a 1D aggregate consisting of identical molecules. The aggregate is anchored to the substrate by links and is subjected to forces pulling along the aggregate axis (Fig. 2). To describe the positions along the aggregate, we choose the axis X originating at the aggregate rear and directed toward its front (Fig. 2). The aggregate length will be denoted by L.
Fig. 2.

A 1D aggregate subject to pulling forces f and anchored to substrate, showing an illustration of the model. The points of force application and the points of anchoring are distributed along the aggregate surface, each with its own density.
We assume that the molecular exchange between the aggregate and the surrounding medium can occur at every point along the aggregate length. Whereas the current data do not provide unambiguous support for this assumption, the rapid fluorescence recovery after photobleaching of paxilin (21) and motion of vinculin within FA¶ demonstrate that at least for the plaque proteins our assumption is plausible.
The pulling forces are applied to the aggregate in discrete points. The value of the force in each of such points, referred to below as the elementary force, is denoted by f. One of the points of force application is located at the front edge of the aggregate (x = L), whereas the others are distributed along the aggregate with a constant linear density ϕf (Fig. 2).
The anchors are distributed along the aggregate with linear density ϕA, and one of the anchors is situated at the rear edge (x = 0) (Fig. 2).
Energy and Criteria for Self-Assembly. The self-assembly in the system will be described in terms of the chemical potential μ (see e.g., ref. 23), which equals the derivative of free energy with respect to the number of particles in the system. The chemical potential equals the Gibbs free energy per particle (23).
The chemical potential of the nonassembled FA building molecules distributed in the cytosol, μfree, accounts for their translational entropy and the energy of interaction of each of them with the surrounding. The chemical potential of the aggregated molecules, μagg, takes into account the attractive interaction between them enabling the aggregation and other factors such as the elastic stresses (see below). Aggregation occurs when the chemical potential in the aggregate is smaller than that in the cytosol, μagg < μfree. In the opposite case, μagg > μfree, the aggregate must disintegrate.
In the absence of pulling forces, the chemical potentials of aggregated and nonaggregated molecules will be denoted by μagg and μfree and referred to as the standard chemical potentials. We will assume that the chemical potential of the free molecules μfree does not change in the course of the aggregation process. This assumption implies that the number of the nonaggregated proteins in the cytosol is large enough and remains essentially constant despite exchange with the aggregate.
Pulling forces produce elastic stress, γ, within the aggregate, which can depend on the position along the aggregate, γ(x). The change of the chemical potential of the aggregated molecules, dμagg, resulting from the stress change, dγ, is given by the Gibbs–Dühem relationship (23, 24). In the 1D case it has a form (25)
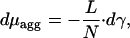 |
[3] |
where L is the length of the aggregate and N is the number of the aggregated molecules so that l = L/N is a molecular length. To obtain from Eq. 3 an explicit expression for the chemical potential μagg, we assume a linear relationship (Hooke law) between the stress γ and the molecular length l
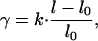 |
[4] |
where l0 is the molecular length at zero stress and k is the stretching rigidity of the aggregate. The relationship in Eq. 4 is expected to be valid for small deformations, |l – l0|/l0 « 1. Integration of Eq. 3 accounting for Eq. 4 gives at any point along the aggregate (25)
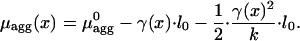 |
[5] |
Note that whereas, according to Eq. 5, the stress γ decreases the chemical potential, it can be readily shown that the corresponding change of the elastic energy per particle increases by the amount (1/2)·{[γ(x)2]/k}·l0. This distinction reflects the fundamental difference between the elastic energy, which describes the aggregate itself, and the notion of the chemical potential accounting also for the work performed by the source of the stress-producing force.
In the following, we will consider a rigid aggregate, meaning that the stretching rigidity, k, is large compared with the stress, γ/k « 1, and we neglect the elastic contribution to the chemical potential given by the last term in Eq. 5. The latter assumption does not change qualitative predictions of the model but simplifies considerably the calculations. Note that the chemical potential μagg(x) may be different in different points of the aggregate, meaning that we do not limit our consideration by equilibrium states of the system.
The molecular exchange between the aggregate and cytosol is determined by the corresponding difference in the chemical potentials, Δμ(x) = μagg – μfree, which is given within our assumptions by
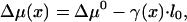 |
[6] |
where 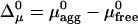 . The nonassembled molecules tend to join the aggregate if Δμ < 0 and leave the aggregate if Δμ > 0.
. The nonassembled molecules tend to join the aggregate if Δμ < 0 and leave the aggregate if Δμ > 0.
Note that, because of the nonvanishing differences in the protein chemical potentials, the system is not in an equilibrium but rather in a kinetic state, which can be characterized by molecular fluxes J from the surrounding medium to the aggregate. Positive, J > 0, and negative, J < 0, fluxes correspond, respectively, to the molecule entering and exiting the aggregate. The local flux, dJ, to a segment of the aggregate of length dx is proportional to the local difference of the chemical potentials
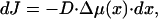 |
[7] |
where D is a positive coefficient accounting for kinetic limitations of the protein exchange between the aggregate and the surrounding medium. The total flux of the proteins to the aggregate is given by
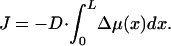 |
[8] |
Although an extensive description of the aggregate kinetics requires determination of the time dependence of the aggregate dimension, the present work will address only the steady states of the system, which is sufficient to find the major regimes of the system behavior. Therefore, the specific value of D is not essential for our analysis and will not be discussed.
Strategy of Analysis. Our aim is to analyze the total flux J of the proteins to the cluster for various values of the system parameters, which are the difference of standard chemical potentials, Δμ0, the densities of the points of force application, ϕf, and of the anchors, ϕA, and the elementary pulling force, f. The positive total flux, Jtot > 0, corresponds to the aggregate growth; negative flux, Jtot < 0, indicates aggregate disintegration; and, finally, vanishing total flux, Jtot = 0, describes a constant steady-state dimension of the aggregate. To perform this analysis, we determine the stress distribution within the aggregate, γ(x), by considering mechanical interplay between the pulling forces on one hand and the resisting forces developed by the anchors on the other. Inserting the result into Eqs. 7 and 8, we compute the total flux of proteins to the aggregate as a function of the system parameters and determine conditions for different types of the system behavior including the steady-state regime. We further analyze the dependence of the aggregate steady-state length, Lst, on the elementary force value, f.
Results
Stationary Anchoring of FA: Possible Regimes of FA Behavior. If the attachment of the anchors to the substrate is sufficiently strong, they do not move upon pulling. In this case, the anchors develop elastic force acting on the aggregate in the direction opposite to the pulling forces (Fig. 2). We assume the aggregate to have a stretching rigidity much larger than that of the anchors, so that all anchors develop the same force, fA.
The condition of mechanical equilibrium throughout the aggregate requires that in any cross-section with coordinate x the stress γ(x) is equal to the difference between the sum of the pulling forces f and the resisting forces of the anchors fA accumulated between the front of the aggregate and the cross-section under consideration. As a result, the stress is given by
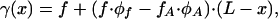 |
[9] |
where the first term accounts for the elementary force applied to the front of the aggregate (x = L). Equilibrium at the rear (x = 0) requires that the tension at this boundary is equal to the force exerted by the last anchor γ(x = 0) = fA. By using Eq. 9, we obtain the relationship between fA and f
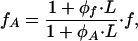 |
[10] |
and the distribution of stresses along the aggregate
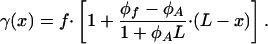 |
[11] |
Based on Eqs. 6 and 11, for each point of the aggregate the difference of the chemical potentials between the aggregated and nonaggregated molecules is given by
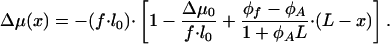 |
[12] |
According to Eqs. 7 and 12, the local flux varies linearly along the aggregate and may even change sign, being negative in one and positive in the other part of the protein assembly.
Based on Eqs. 8 and 12, the total flux of proteins to the aggregate is given by
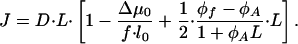 |
[13] |
Analysis of the total flux in Eq. 13 as a function of the aggregate length L shows that the mode of self-assembly is determined by the value of the parameter
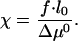 |
[14] |
There can be four different regimes of self-assembly, whose criteria are determined by two characteristic values of the parameter χ
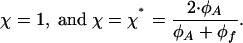 |
[15] |
The two extreme regimes are an unlimited growth and unrestricted disintegration of the aggregate. They correspond, respectively, to small and large values of the elementary force f and, hence, also of the parameter χ (Eq. 13).
- If χ is smaller than the both critical values in Eq. 15,
the total flux J is negative for any value of the aggregate length L, as illustrated in Fig. 3a. This result means that the aggregate does not start to self-assemble. Alternatively, if the force was initially large but has been dropped to a small value satisfying Eq. 16, the aggregate disintegrates.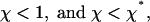
[16] - If χ is larger than the two critical values,
the total flux J is positive for any length (Fig. 3b) and the aggregate undergoes unlimited growth. The two other regimes correspond to intermediate values of the pulling force f.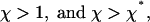
[17] - If the parameter χ satisfies
the total flux is negative for the aggregate length smaller than a certain value L* but becomes positive for L > L*, as illustrated in Fig. 3c. This result means that the aggregate does not self-assemble spontaneously for small lengths, but in case it overcomes L* due to fluctuations or some additional factors, which are not accounted for by the present model, the force-induced self-assembly takes over, and the aggregate starts to grow unlimitedly. This regime is possible if the density of the anchors is smaller than that of the points of force application, ϕA < ϕf.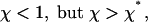
[18] - Finally, in the case where
the total flux J is positive for the small lengths L of the aggregate, meaning that the pulling force initiates self-assembly. However, when the length reaches a particular value Lst, the flux vanishes and for L > Lst becomes negative (Fig. 3d). This result means that the aggregate reaches a finite length L = Lst and stops growing. Hence, Lst is the finite steady-state length. Moreover, as follows from Fig. 3d, this steady state is stable.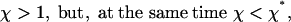
[19]
Fig. 3.

Four regimes of self-assembly determined by the parameter χ = (f·l0)/(Δμ0). The curves represent the total protein flux, J(arbitrary units), to the aggregate as a function of the aggregate length, L(arbitrary units). (a) Atχ < 1, and χ < χ*, the flux is negative, meaning that the aggregate undergoes disintegration. (b) At χ > 1, and χ > χ*, the flux is positive, meaning that the aggregate undergoes unlimited growth. (c) At χ < 1 but χ > χ*, the flux is negative until the aggregate reaches a certain length where the flux changes sign and starts increasing with the aggregate length. This regime corresponds to unlimited growth after overcome of a critical length. (d) At χ > 1 and χ <χ*, the flux remains positive until the aggregate reaches a critical length. For length larger than the critical values, the flux is negative. This state is the steady-state regime, where the critical length corresponds to the stable steady-state length of the aggregate.
The regime of the stable finite steady-state length of the aggregate is possible if the density of the anchors exceeds that of the points of force application, ϕA > ϕf.
The dependence of the steady-state length on the pulling force is given by
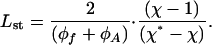 |
[20] |
According to ref. 20, the steady-state length starts from zero at χ = 1 and increases to infinitely large values at χ approaching χ*.
The four possible regimes and the corresponding ranges of the parameters can be presented as a phase diagram (Fig. 4).
Fig. 4.

Phase diagram of the system representing the different regimes of the aggregate assembly–disassembly corresponding to different ranges of the system parameters, which are the density of the points of force application along the aggregate length ϕf, the density of the points of the aggregate anchoring to the substrate ϕA, and the dimensionless parameter χ = (f·l0)/(Δμ0), characterizing the ratio between the molecular energy provided by an elementary pulling force, f·l0, and the difference of the protein standard chemical potentials in the aggregated and free states, Δμ0.
Mobile FA. When the attachment of the anchoring molecules to the substrate is not sufficiently stable, the action of the pulling forces may cause the aggregate to begin a crawl-like motion. We suggest that in this case each anchor produces an effective friction force, τA, pointing against the motion and, consequently, against the direction of pulling. The simplest assumption about the value of the friction force is that it is proportional to the velocity of crawling, ν,
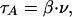 |
[21] |
where β is the friction coefficient.
We further suppose that in the course of crawling, the aggregate remains in the state of internal mechanical equilibrium. Therefore, the distribution of stresses within the mobile aggregate has the same character as in the previous case of a stationary FA. As a result, the assembly–disassembly behavior of the aggregate remains exactly the same as in the previous case with the only difference being that it occurs during crawling with velocity
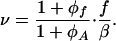 |
[22] |
According to the criteria in Eqs. 16–19, the regimes of the aggregate crawling with unlimited growths, crawling with disintegration, crawling with growth after overcoming a critical size, and, finally, crawling with a finite steady-state length are possible depending on the values of f, Δμ0, ϕf, and ϕA.
Discussion
Thermodynamic Mechanism Underlying FA Mechanosensitivity. FA subject to pulling forces exhibit mechanosensitive behavior (for review, see ref. 1). A stationary FA self-assembles and reaches a finite size as long as pulling forces are acting on its surface. A decrease of pulling results in reduction of the FA size while the ratio between the total force and the FA area remains approximately constant (19). Complete blockage of the intracellular contractile machinery leads to FA disassembly (26, 27). Also the mobile FA can exhibit growing, shrinking or a constant length in the course of their movement with respect to the substrate (11, 12, 15).
The present work suggests that the mechanosensitive behavior of stationary and mobile FA can be understood based on a thermodynamic principle, according to which self-assembly can be driven by the stresses generated within an FA by pulling forces. These stresses promote insertion of new building blocks into FA in such a way that the FA dimension increases in the direction of pulling. The same principle predicts the phenomenon of the force-driven self-assembly of actin filament mediated by a processive capper such as formin (28).
The proposed model predicts several regimes of FA assembly– disassembly. Occurrence of any specific regime is determined by the value of an elementary pulling force f and by the ratio between the densities of the points of force application ϕf and the anchoring points ϕA. This ratio determines how effectively the anchors screen the stress produced by the pulling forces along the aggregate. If ϕA > ϕf, the screening is strong, the average stress is low, and, consequently, the aggregate growth can be prevented. This state is necessary, for example, for existence of the steady-state regime. In an opposite case of ϕA < ϕf, screening is weak, which results in unlimited aggregate growth for any value of the elementary pulling force. However, for small forces the onset of unlimited growth occurs after overcoming a critical aggregate length.
The regime of the steady-state length deserves special discussion. The essence of this regime consists in a mutual compensation of local fluxes (Eq. 7) of proteins to and from the aggregate. According to Eqs. 7 and 12, in the steady-state regime (Eq. 19), the local fluxes to the front part of the aggregate are positive, whereas the fluxes to the rear part are negative. The net flux vanishes so that the length of the aggregate does not change, but there is a persistent turnover of the proteins between the aggregate and the cytoplasm. Depending on intermolecular interactions within the aggregate and the mode of interaction between the aggregate and the anchors, this turnover can result in treadmilling-coupled displacement of the aggregate with respect to the substrate or in treadmilling-type molecular rearrangement inside a stationary aggregate.
Displacement will take place if the outflux of the proteins results in detachment of the aggregate from the anchors at the rear edge, whereas the protein influx in the front part leads to recruitment of new anchors. The velocity of the displacement will be determined by the rate of the protein turnover.
The molecular rearrangement within stationary FA is expected if the connections between its molecules and the anchors are dynamic, allowing for the effective movement of the proteins within the aggregate against the direction of the force application. This treadmilling-type movement from the front to the rear of FA will compensate for the molecular turnover so that the aggregate as a whole will not shift with respect to the substrate.
In migrating FA, the displacement or the internal rearrangement are predicted to occur on top of the aggregate motion. In case the displacement occurs, it may contribute to the FA motility.
Studies of dynamics of FA components by the method of fluorescence recovery after photobleaching revealed an extensive rearrangement of integrin molecules (16, 17). However, verification of our model requires more detailed studies of the internal FA dynamics, for example, by the fluorescence speckle microscopy technique. The recently observed inter-FA movement of vinculin molecules (24) may represent the first indication to the treadmilling-type movement of the plaque proteins within the FA.
Specific Mechanisms of FA Regulation. The physical background of the FA mechanosensitive behavior suggested here has a generic character. The only essential requirement to these mechanisms is a reversible relaxation of the stresses after lifting the pulling forces. At the same time, several models have been discussed of hypothetical force-dependent conformational changes of specific molecules associated with FA (6). These conformational changes may provide a mechanism of deformations of the FA building blocks and, in addition, trigger signaling pathways regulating the FA assembly. The role of such force-sensing proteins may be played by integrins, which can exist in two distinct conformational states characterized by different ability to bind both the extracellular matrix and plaque proteins (29, 30). Another candidate for force sensing is one of the FA major components, vinculin, which undergoes a regulated conformational change upon its interaction with talin (31). Finally, the force-driven opening of membrane ionic channels may contribute to the mechanosensing (22).
We propose that the nonspecific thermodynamic mechanism suggested in the present work provides the driving force common for all particular manifestations of the FA mechanosensitive behavior. At the same time, the specific signaling mechanisms such as those mentioned above serve for time and space modulation of the FA mechanosensitivity according to various biological requirements.
Model Implications. The suggested model is based on essential assumptions on coupling between FA and the force-generating machinery and on properties of interprotein bonds within FA. The force has to be transmitted to the aggregate through dynamic junctions, which do not break during the aggregate assembly-disassembly. This kind of junction may be mediated by such proteins as formins, which perform processive capping of actin filaments (28). The intermolecular bonds have to be sufficiently stable to prevent aggregate rupture, a scenario alternative to the elastic stress-induced self-assembly. At the same time, proteins have to be able to join the aggregate, which is accompanied by bond rearrangements. This process can be realized by a fusion-like mechanism of molecule insertion into the aggregate, where the new intermolecular bonds form before the previous ones break. Such a delicate mechanism of bond reorganizations may require concerted conformational changes of several proteins accompanying their insertion into FA. A possible candidate for this kind of mechanism may be the recently described conformational transition resulting from the complex formation between two major FA components, vinculin and talin (31).
Ways of Experimental Validation of the Model. Both the assumptions underlying the suggested model and the predictions it makes require experimental verification. The most important issues to be checked are as follows:
Persistent turnover of FA proteins across the whole area of the plaque and the related treadmilling-type internal FA dynamics, as implied by the model.
Difference in the character of protein exchange with the surrounding cytoplasm between the front and the rear region of FA.
The dependence of the regime of FA behavior on the pulling force developed in actin filaments by modulating the myosindriven contractility of the filaments. Although it has been established experimentally that impairment of the myosin-driven contractility results in decrease or even disassembly of the FA (19, 26, 27), our model predicts quantitatively the range of the pulling force, where FA are stabilized, and describes the dependence of the aggregate length on the force value.
Influence of the density of links between the plaque and the substrate on FA self-assembly. Increase in the density of integrin molecules underneath the plaque is expected to favor the steadystate regime and to determine the finite length of the FA.
Acknowledgments
We thank Sam Safran, Robin Bruinsma, and Michael Urbakh for discussions. B.G. is the E. Neter Professor for Cell and Tumor Biology and was supported by the Cell Migration Consortium of the National Institutes of Health and the Minerva Foundation (Munich). A.D.B. holds the Joseph Moss Chair of Biomedical Research and was supported by the Israel Science Foundation, the Binational United States–Israel Foundation, the Minerva Foundation, and the Clore Center for Biological Physics. M.M.K. was supported by the Israel Science Foundation, the Binational United States–Israel Foundation, and the Human Frontier Science Program Organization.
Author contributions: B.G., A.D.B., and M.M.K. designed research; T.S. and M.M.K. performed research; and T.S., B.G., A.D.B., and M.M.K. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office. Abbreviation: FA, focal adhesions.
Footnotes
Hu, K., Ji, L., Ginsberg, M., Danuser, G. & Waterman-Storer, C. M. (2004) Mol. Biol. Cell 15, Suppl., 177a (abstr.).
References
- 1.Bershadsky, A. D., Balaban, N. Q. & Geiger, B. (2003) Annu. Rev. Cell Dev. Biol. 19, 677–695. [DOI] [PubMed] [Google Scholar]
- 2.Geiger, B. & Bershadsky, A. (2001) Curr. Opin. Cell Biol. 13, 584–592. [DOI] [PubMed] [Google Scholar]
- 3.Geiger, B. & Bershadsky, A. (2002) Cell 110, 139–142. [DOI] [PubMed] [Google Scholar]
- 4.Zamir, E. & Geiger, B. (2001) J. Cell Sci. 114, 3583–3590. [DOI] [PubMed] [Google Scholar]
- 5.Zamir, E. & Geiger, B. (2001) J. Cell Sci. 114, 3577–3579. [DOI] [PubMed] [Google Scholar]
- 6.Nicolas, A., Geiger, B. & Safran, S. (2004) Proc. Natl. Acad. Sci. USA 101, 12520–12525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Novak, I. L., Slepchenko, B. M., Mogilner, A. & Loew, L. M. (2004) Phys. Rev. Lett. 93, 268109. [DOI] [PubMed] [Google Scholar]
- 8.Bresnick, A. R. (1999) Curr. Opin. Cell Biol. 11, 26–33. [DOI] [PubMed] [Google Scholar]
- 9.Somlyo, A. P. & Somlyo, A. V. (2000) J. Physiol. 522, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geiger, B., Bershadsky, A., Pankov, R. & Yamada, K. M. (2001) Nat. Rev. Mol. Cell. Biol. 2, 793–805. [DOI] [PubMed] [Google Scholar]
- 11.Smilenov, L. B., Mikhailov, A., Pelham, R. J., Marcantonio, E. E. & Gundersen, G. G. (1999) Science 286, 1172–1174. [DOI] [PubMed] [Google Scholar]
- 12.Zamir, E., Katz, M., Posen, Y., Erez, N., Yamada, K. M., Katz, B. Z., Lin, S., Lin, D. C., Bershadsky, A., Kam, Z. & Geiger, B. (2000) Nat. Cell Biol. 2, 191–196. [DOI] [PubMed] [Google Scholar]
- 13.Wehrle-Haller, B. & Imhof, B. (2002) Trends Cell Biol. 12, 382–389. [DOI] [PubMed] [Google Scholar]
- 14.Webb, D. J., Parsons, J. T. & Horwitz, A. F. (2002) Nat. Cell Biol. 4, E97–E100. [DOI] [PubMed] [Google Scholar]
- 15.Small, J. V. & Kaverina, I. (2003) Curr. Opin. Cell Biol. 15, 40–47. [DOI] [PubMed] [Google Scholar]
- 16.Ballestrem, C., Hinz, B., Imhof, B. A. & Wehrle-Haller, B. (2001) J. Cell Biol. 155, 1319–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuruta, D., Gonzales, M., Hopkinson, S. B., Otey, C., Khuon, S., Goldman, R. D. & Jones, J. C. (2002) FASEB J. 16, 866–868. [DOI] [PubMed] [Google Scholar]
- 18.Riveline, D., Zamir, E., Balaban, N. Q., Schwarz, U. S., Ishizaki, T., Narumiya, S., Kam, Z., Geiger, B. & Bershadsky, A. D. (2001) J. Cell Biol. 153, 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balaban, N. Q., Schwarz, U. S., Riveline, D., Goichberg, P., Tzur, G., Sabanay, I., Mahalu, D., Safran, S., Bershadsky, A., Addadi, L. & Geiger, B. (2001) Nat. Cell Biol. 3, 466–472. [DOI] [PubMed] [Google Scholar]
- 20.Hill, T. L. (1987) Linear Aggregation Theory in Cell Biology (Springer, New York).
- 21.von Wichert, G., Haimovich, B., Feng, G. S. & Sheetz, M. P. (2003) EMBO J. 22, 5023–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munevar, S., Wang, Y. L. & Dembo, M. (2004) J. Cell Sci. 117, 85–92. [DOI] [PubMed] [Google Scholar]
- 23.Reif, F. (1965) Fundamentals of Statistical and Thermal Physics (McGraw, New York).
- 24.Gibbs, J. W. (1961) The Scientific Papers (Dover, New York).
- 25.Hill, T. L. & Kirschner, M. W. (1982) Int. Rev. Cytol. 78, 1–125. [PubMed] [Google Scholar]
- 26.Helfman, D. M., Levy, E. T., Berthier, C., Shtutman, M., Riveline, D., Grosheva, I., Lachish-Zalait, A., Elbaum, M. & Bershadsky, A. D. (1999) Mol. Biol. Cell 10, 3097–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chrzanowska-Wodnicka, M. & Burridge, K. (1996) J. Cell Biol. 133, 1403–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozlov, M. M. & Bershadsky, A. D. (2004) J. Cell Biol. 167, 1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Springer, T. A. & Wang, J. H. (2004) Adv. Protein Chem. 68, 29–63. [DOI] [PubMed] [Google Scholar]
- 30.Hynes, R. O. (2002) Cell 110, 673–687. [DOI] [PubMed] [Google Scholar]
- 31.Izard, T., Evans, G., Borgon, R. A., Rush, C. L., Bricogne, G. & Bois, P. R. (2004) Nature 427, 171–175. [DOI] [PubMed] [Google Scholar]
- 32.Bruinsma, R. (2005) Biophys. J. 89, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]