

Abstract
Hypothalamic growth hormone (GH)-releasing hormone (GHRH) stimulates the synthesis and release of GH from the pituitary gland. GHRH and its mRNA are also found in human cancers of the breast, ovary, prostate, lung, and other tumors, suggesting that GHRH is also a tumor growth factor. Various studies show that GHRH antagonists have antiproliferative effects in many tumor models; however, glioblastomas were examined only recently. Previous studies have demonstrated that s.c. administration of GHRH antagonist (JV-1-36) inhibited growth of s.c. U-87MG human glioblastomas and increased survival of nude mice with orthotopic implants of glioblastomas. Although treatment with JV-1-36 reduced tumorigenicity, it is not known whether peripherally administered GHRH antagonists can cross the blood-brain barrier. Brain endothelial cells joined by tight junctions form the blood-brain barrier, a “barrier” between the general circulation and the CNS. In this study, we administered a GHRH antagonist (JV-1-42) and showed that, after i.v. injection, iodinated JV-1-42 (131I-JV-1-42) enters the brain intact at a rate of 0.8514 μl/g per min with a serum half-life of 12.2 min. A one-site binding hyperbolic model indicated that the maximal percent of i.v. dose taken up per gram of brain was 0.41%. Coinjection of unlabeled JV-1-42 indicated that the transport from blood to brain is not saturable; however, transport from brain to blood is saturable and involves P-glycoprotein. Taken together, these results demonstrate that i.v.-administered 131I-JV-1-42 readily crosses the blood-brain barrier and accumulates in the brain. This finding indicates that GHRH antagonists could provide a potential treatment for malignant glioblastomas.
Keywords: cancer therapy, P-glycoprotein
Hypothalamic growth hormone (GH)-releasing hormone (GHRH) regulates the synthesis and release of GH in the pituitary gland. GHRH and its mRNA are also expressed in many human cancers and cancer cell lines and in some normal tissues, suggesting that GHRH may act as a tumor growth factor (1, 2). Although pituitary-type GHRH receptors cannot be detected in most human tumors by RT-PCR or radioligand-binding assays, the expression of tumor-specific splice variants of this receptor has been demonstrated (2-6). Numerous studies show that antagonistic analogs of GHRH have inhibitory effects on the growth of various human cancer cell lines xenografted into nude mice, including osteosarcomas (7), glioblastomas (8), small cell lung carcinomas (SCLC), non-SCLC (9), and ovarian (10), breast (11), colorectal (12), pancreatic (13), prostatic (14), and renal (5, 15) cancers.
Splice variants of tumoral GHRH receptors have been identified in various human cancer cell lines, including prostatic, pancreatic, breast and ovarian, colorectal, gastric, small cell lung carcinomas, and osteosarcomas, and also were detected in primary specimens of human prostate cancer (1, 3-7, 10, 14, 16). Radioligand-binding studies using a 125I-labeled GHRH antagonistic analog, JV-1-42, confirmed that specific high-affinity binding sites for GHRH and its synthetic antagonists were present in membrane fractions of 60% of the primary prostate tumors examined (4). In vivo, these antagonists have both direct and indirect effects on the secretion of insulin-like growth factor I (IGF-I) and -II (1, 2). GHRH antagonists can indirectly lower the levels of IGF-I in the serum by blocking the release of GH from the pituitary, an effect that reduces IGF-I synthesis in the liver and other tissues. These antagonists also have direct effects on GHRH receptors on tumors, an effect that may be associated with a reduction in IGF-I and -II in tumor tissue.
Glioblastomas are the most frequent primary brain tumor subtype in adults. The dominant therapeutic approach to treat these tumors is surgical removal. Because malignant gliomas are characterized by aggressive proliferation and expansion into surrounding brain tissue, it is difficult to completely remove the tumor (17). GHRH antagonists (MZ-5-156 and JV-1-36) have been shown to reduce the tumorigenicity and growth of U-87MG human glioblastomas transplanted onto mouse flanks (8). However, the clinical use of these analogs will remain limited unless it is shown that they can cross the blood-brain barrier in quantities sufficient to elicit a tumoricidal response within the CNS.
The brain capillary endothelial cells are joined together by tight junctions to form the blood-brain barrier. These tight junctions prevent significant paracellular diffusion of small molecules between blood and brain. Instead, entry of most molecules into the brain is mediated by membrane diffusion or specialized transport systems. Specific transport proteins exist for the movement of small hydrophilic molecules such as glucose, amino acids (18), thyroid hormone (19), and nucleosides (20) across the blood-brain barrier. Larger molecules such as leptin (21) and some cytokines (22) are also transported across the brain capillary endothelial cells by saturable processes. Membrane diffusion across the blood-brain barrier is directly correlated with the lipid solubility of a molecule.
In the present study, we tested the ability of a radioactively labeled GHRH antagonist (JV-1-42) to cross the blood-brain barrier. We found that peripherally administered iodinated JV-1-42 rapidly crosses the blood-brain barrier in therapeutically relevant quantities, indicating this compound holds great potential for the treatment of malignant gliomas.
Methods
Radioiodination of GHRH Antagonist JV-1-42. The GHRH antagonist JV-1-42 was radioactively labeled with 131I using a modified version of the chloramine-T method that was described previously (4). First, 10 μg of JV-1-42 was dissolved in 10 μl of 0.01 M aqueous acetic acid. This was then added to 80 μl of 0.1 M NH4OAc in 50% (vol/vol) aqueous acetonitrile (pH = 6.5-7). Next, 10 μg of chloramine-T [dissolved in a solution of 0.1 M NH4OAc in 50% (vol/vol) aqueous acetonitrile] was added. Approximately 2 mCi (1 Ci = 37 GBq) of 131I (PerkinElmer) was added to the mix and, after 45 sec at room temperature, the reaction was stopped by adding 50 μg of l-cysteine dissolved in 10 μl 0.01 M HCl. Radioactively labeled JV-1-42 (131I-JV-1-42) was separated from free 131I by reverse-phase HPLC (Shimadzu) on a Vydac 214TP54 column (P. J. Cobert and Associates, St. Louis). The radioactively labeled JV-1-42 was eluted with a linear gradient of 45-65% solvent B [0.1% aqueous trifluoroacetic acid in 70% (vol/vol) aqueous acetonitrile] over a period of 50 min against solvent A [0.1% (vol/vol) aqueous trifluoroacetic acid in HPLC grade water]. Fractions were collected in polypropylene tubes that were siliconized by using Sigmacote (Sigma), because the radioiodinated derivative adsorbs strongly to the surface of borosilicate glass and polypropylene tubes. Fifty 1-ml fractions were collected and the radioactivity measured by counting 1-μl fractions in a gamma counter. The four fractions corresponding to the monoiodinated compound were identified by elution position as well as a peak in radioactivity. Fractions were dried for 1 h and then stored at -70°C.
Radioactive Labeling of Albumin with 99mTc. The reaction mixture consisted of 0.12 mg of stannous tartrate, 1 mg of BSA, and 500 μl of distilled water. The pH of this mixture was adjusted to 2.2-3.3 with 0.2 M HCl. Approximately 1 mCi of 99mTc in NaOH was added to the mixture along with 500 μl of distilled water. The mixture was allowed to incubate at room temperature for 20 min before total radioactivity was measured in a gamma counter.
Determination of Octanol/Buffer Partition Coefficient. Lipid solubility of 131I-JV-1-42 was determined by adding 1 × 105 cpm of 131I-JV-1-42 to 0.5 ml of 0.25 M chloride-free sodium phosphate buffer (pH = 7.5) and 0.5 ml of octanol. This solution was vigorously mixed for 1 min. The two phases were separated by centrifugation at 5,000 × g for 10 min. Aliquots of 100 μl were taken in triplicate from each phase, and total radioactivity was measured for each aliquot in a gamma counter. The partition coefficient was calculated from the following formula:
 |
Determination of Rate of Clearance of 131I-JV-1-42 from the Serum. Male CD-1 mice (Charles River Breeding Laboratories) weighing 20-25 g were anesthetized with 0.2 ml of i.p. urethane. Next, the skin from the neck was removed to expose the jugular vein and carotid artery. Each mouse was given an injection into the jugular vein of 0.2 ml of lactated Ringer's solution (LR) (Baxter Health Care, Deerfield, IL) containing 1% BSA (LR-BSA) and 3 × 105 cpm of 131I-JV-1-42. Blood was collected from the carotid artery 2-120 min after i.v. injection, the mouse was decapitated, and the brain removed and weighed. The whole arterial blood was centrifuged at 5,000 × g for 10 min and the level of radioactivity determined in the serum and brain. To determine rate of clearance of 131I-JV-1-42 from the serum after i.v. injection, the log of the injected dose in each milliliter of serum (log %Inj/ml) was plotted against time (min).
Determination of Blood-to-Brain Influx Rate. The rate of uptake of 131I-JV-1-42 from the blood to the brain was determined by using multiple time regression analysis (23, 24). For the purpose of this analysis, a graph was created in which the brain/serum ratios for time points ranging from 2 to 60 min after i.v. injection were plotted against their respective exposure times (Expt). The slope of the linear portion of this relation represents the unidirectional influx rate (Ki), and the y intercept represents the initial volume of distribution (Vi). Experimental clock time was reexpressed as exposure time (Expt) to correct for clearance of 131I-JV-1-42 from the serum. Expt was calculated from the following formula:
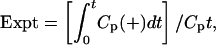 |
where t is time, Cp represents the level of radioactivity in the serum, and Cpt is the level of radioactivity in the serum at time t. Without this correction, Ki would be overestimated.
The percent of the injected dose taken up by each gram of brain tissue (%Inj/g) was calculated from the following formula:
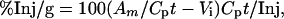 |
where Am/Cpt represents the brain/serum ratio at time t, and Inj is the dose of 131I-JV-1-42 injected intravenously. Subtracting Vi from the brain/serum ratio corrects for 131I-JV-1-42 trapped in the vascular space of the brain. This ensures that quantities expressed represent only the 131I-JV-1-42 that has entered brain tissue. The values for %Inj/g were plotted against time (min).
To determine whether brain uptake of 131I-JV-1-42 was a saturable process, 1 μg per mouse of unlabeled JV-1-42 was included in the i.v. injection. Brain and serum samples were collected 10 min after i.v. injection, and results were expressed as brain/serum ratios.
Determination of Brain-to-Blood Efflux Rate. The method has been used previously to quantify the rate of transport from brain to blood (25). First, mice were anesthetized with an i.p. injection of 0.2 ml of urethane. After removal of the scalp, a hole through the cranium (1.0 mm lateral and 0.5 mm posterior to bregma) was created with a 26-gauge needle. The needle was covered with polyethylene-10 (PE-10) tubing, with the final 2.5-3.0 mm of the tip exposed. This ensured that the needle did not penetrate the floor of the ventricle. One microliter of LR-BSA containing 5 × 103 cpm of 131I-JV-1-42 was then injected into the lateral ventricle of the brain with a 1.0-μl Hamilton syringe.
Mice were decapitated at 2, 5, 10, and 20 min after injection, the brain was removed, and the amount of radioactivity was measured by a gamma counter. The amount of radioactivity in the brain at t = 0 was estimated in mice overdosed with urethane. These mice were killed and 15 min later injected with 131I-JV-1-42. Brains were removed from these mice 10 min after injection. The log of the mean level of radioactivity in each brain was plotted against time (in minutes). Two sets of three mice were used at each of the five time points, and the mean of the three mice in each set was used. Efflux curves were calculated with n = 10 based on triplicates.
To determine whether efflux of 131I-JV-1-42 was saturable, 100 ng of unlabeled JV-1-42 per mouse was included in the injection. To determine the role of P-glycoprotein (P-gp), 100 ng of cyclosporin A per mouse or 5 nM verapamil per mouse was included in the injection per mouse to test for ability to inhibit influx. Brain samples were collected 10 min after injection, and results are expressed as the mean percent of the intracerebroventricular (i.c.v.) injected dose taken up per gram of brain (%Inj/Brain).
Capillary Depletion With and Without Vascular Washout. The capillary depletion method (26) as modified for mice (27) was performed to separate the cerebral capillaries from the parenchymal tissue so the compartmental distribution of 131I-JV-1-42 could be determined. Male CD-1 mice were anesthetized by i.p. injection of 0.2 ml of urethane. Each mouse received an injection of 3 × 105 cpm of 131I-JV-1-42 and 3 × 105 cpm 99mTc-albumin in 0.8 ml of LR-BSA into the jugular vein. Mice were then divided into two groups (washout or no washout). Next, the abdomen was opened, and arterial blood was collected from the abdominal aorta at either 10, 30, or 60 min after i.v. injection. For the washout group, the descending thoracic aorta was clamped to cut off the circulation to the lower extremities. Both jugular veins were then severed, and a 20-ml syringe with an 18-gauge needle was inserted into the left ventricle of the heart. The 20 ml of lactated Ringer's solution (LR) was infused into the mouse over a period of 1-2 min. This infusion circulates through the brain microvessels, washes out the vascular space of the brain, and drains from the severed jugular veins.
After perfusion (washout group) or collection of arterial blood (no washout group), the brain was removed, weighed, and placed in a glass homogenizer containing 0.8 ml of physiologic buffer (10 mM Hepes/141 mM NaCl/4 mM KCl/2.8 mM CaCl2/1 mM MgSO4/1 mM NaH2PO4/10 mM d-glucose; this buffer was then adjusted to pH = 7.4). After 10 strokes with the glass pestle in the homogenizer, 1.6 ml of the physiologic buffer containing 26% dextran was added to the homogenate, vortexed, and then homogenized a second time (three strokes). All homogenization steps were performed on ice. The homogenate was centrifuged at 5,400 × g for 15 min at 4°C in a Beckman Allegra-21R centrifuge with a swinging bucket rotor. The supernatant containing the brain parenchyma was separated from the pellet containing the brain microvasculature, and the level of 131I-JV-1-42 was determined for each fraction in a gamma counter. Volume of distribution (VD) values were expressed as tissue (parenchyma or capillary)/serum ratios and were calculated for both fractions with the equation:
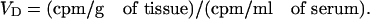 |
Determination of Uptake into the CSF. Male CD-1 mice weighing 20-25 g were given an i.p. injection of 0.2 ml of urethane anesthesia. Samples were collected 30 min after i.v. injection of 0.2 ml of LR-BSA with 5 × 105 cpm of 131I-JV-1-42 into the jugular vein. To collect CSF, skin from the top of the head was removed to expose the skull, and a 30-gauge needle connected to a 25-cm length of PE-10 tubing was inserted ≈5 mm into the base of the skull at the level of the posterior fossa. Once the needle was inserted, CSF from the posterior fossa flowed into the PE-10 tubing by capillary action. After obtaining the CSF, the needle was removed and arterial blood collected from the carotid artery. Finally, the mouse was decapitated and the whole brain removed. The total amount of CSF collected was determined (in microliters) by measuring the length of PE-10 tubing filled with CSF (in centimeters) and multiplying this value by 0.668 (the volume in microliters present in 1 cm of PE-10 tubing). Only absolutely clear CSF samples were used for this analysis to avoid overestimation due to serum contamination. Total radioactivity present in the brain, serum, and CSF for each mouse was quantified in a gamma counter, and the results were expressed in the following ratios: brain/serum (μl/g), brain/CSF (ml/g), and CSF/serum (μl/ml). The formula used to determine these ratios is the same as the brain/serum ratio formula used for analysis in the capillary depletion experiment.
HPLC of Brain and Serum Samples 30 Min After i.v. Injection of 131I-JV-1-42. This procedure was used to determine whether the radioactivity in brain and blood represented intact 131I-JV-1-42.
Brain samples were removed and weighed 30 min after the i.v. injection of 131I-JV-1-42. Cold ammonium acetate solution (4 ml; 10% ammonium acetate with 0.1% BSA in water, pH = 6.87) was added and the brain homogenized with a Polytron homogenizer (Kinematica, Lucerne, Switzerland) on setting 26 for 60 sec. The homogenate was allowed to stand on ice for 2 h and then centrifuged at 5,400 × g for 20 min. The supernatant was removed, lyophilized, and stored until rehydrated 10 min before characterization by HPLC with a Vydac 214 TP54 column. Blood from the carotid artery collected 30 min after injection of 131I-JV-1-42 in 0.2 ml of LR-BSA was allowed to clot at room temperature for 20-30 min before centrifugation at 5,400 × g for 10 min. The serum was collected, and a quantity of 0.3 ml was added to 4 ml of cold ammonium acetate solution. This mixture was vortexed for 15 sec and allowed to incubate on ice for 2 h. After incubation, the serum was treated with the same procedure used for brain homogenate. The control sample consisted of brain and serum from a mouse that had not been injected. Instead, 131I-JV-1-42 was added to the brain and serum ex vivo. These control samples were used to correct for any degradation that may have occurred as a result of processing. For comparison, 131I-JV-1-42 was used as a standard to characterize the elution peak for the labeled peptide analog. Results were expressed as the percent per fraction of total cpm eluted.
Forty 1-ml fractions were collected for each sample. The amount of radioactivity present in each of these fractions was measured in a gamma counter. Fractions 1-5 were excluded from the analysis, because these represented void volume.
Statistical Analysis. Unpaired t tests or one-way ANOVA with the Newman-Keuls multiple comparison test were calculated by using prism 4.0 (GraphPad, San Diego). Means are reported with their standard errors. Regression lines were calculated by the least-squares method.
Results
Clearance of 131I-JV-1-42 from Serum. The early phase of clearance from the serum for 131I-JV-1-42 followed first-order kinetics. Linear regression analysis showed a significant correlation between log (%Inj/ml) and time (t), P < 0.01, for the first 10 min after i.v. injection (Fig. 1A Inset). Half-life in the serum was calculated from the inverse of the slope of this relation to be ≈12.2 min. The initial rapid decline was followed by a steady state that remained unchanged between 30 and 120 min.
Fig. 1.

Kinetics of blood-to-brain transport of 131I-JV-1-42 after i.v. administration. (A) The initial phase of clearance from serum follows first-order kinetics. 131I-JV-1-42 has a serum half-life of 12.2 min. (B) 131I-JV-1-42 readily entered the brain tissue. The maximal percent of the injected dose taken up by each gram of brain was 0.41%, indicating that brain uptake was high. (C) Brain uptake values were corrected for exposure time and plotted against their respective brain/serum ratios. The slope of the resulting graph revealed that 131I-JV-1-42 enters the brain at a rate of 0.8514 μl/g per min (n = 1-2 per time point for B and C). (D) Coinjection of 131I-JV-1-42 with unlabeled JV-1-42 did not saturate blood-to-brain transport of 131I-JV-1-42. Instead, it produced a paradoxical increase in the amount of 131I-JV-1-42 detected in the brain (t = 10 min after i.v. injection; n = 6 per group; *, P < 0.05).
Brain Uptake (Influx) of 131I-JV-1-42 from the Blood. An octanol/buffer partition coefficient of 78.31 ± 5.72 indicates that 131I-JV-1-42 is highly lipid-soluble. Brain uptake for the first 10 min after i.v. injection was rapid (Fig. 1B), with an influx rate (Ki) of 0.8514 μl/g per min and a Vi of 23.87 μl/g (Fig. 1C). A one-site binding model was used to fit a hyperbola to the values in Fig. 1B. According to this model, the maximal percent of the injected dose taken up by each gram of brain is 0.41%. There were regional differences in brain uptake of 131I-JV-1-42, with the thalamus taking up a significantly lower percentage of the i.v.-injected dose than both the pons/medulla and the cerebellum (Fig. 5, which is published as supporting information on the PNAS web site).
Coinjection of unlabeled peptide did not significantly decrease influx of 131I-JV-1-42 but instead produced a paradoxical increase in the brain/serum ratio (P < 0.05) (Fig. 1D).
Stability in Blood and Brain. HPLC analysis found no degradation in either brain or serum processing controls or in 10-min experimental samples (Fig. 2 A and C, respectively). All samples were processed identically before separation by HPLC. For brain and serum samples obtained 30 min after i.v. injection, 69.5% of counts for brain and 68.2% of counts for serum eluted as intact 131I-JV-1-42. The remaining radioactivity eluted as multiple peaks (Fig. 2B).
Fig. 2.

Stability of 131I-JV-1-42 in blood and brain after i.v. injection. HPLC analysis revealed that the majority of 131I-JV-1-42 was recovered in all samples. Percentage of total radioactivity recovered in the peak was 86.2% and 49.27% in the brain at 10 and 30 min after i.v. injection, respectively. Percentage of total radioactivity recovered in the peak was 90.1% and 42.04% in the serum at 10 and 30 min after i.v. injection, respectively. For the processing controls, the percentage of total radioactivity recovered in the peak was 97% in the brain and 95.48% in the serum.
Capillary Depletion. Capillary depletion was performed with and without washout of the vascular space. The results in Fig. 3A show that conducting vascular washout did not significantly reduce the percentage of 131I-JV-1-42 measured in the parenchyma. This indicates that any 131I-JV-1-42 detected in the brain was not loosely adhering to the luminal surface of the brain capillaries but was either internalized into the capillaries or had completely crossed the blood-brain barrier to enter brain parenchyma.
Fig. 3.

Distribution of intravenously administered 131I-JV-1-42 taken up by brain into the parenchyma, capillaries, and CSF. (A) Capillary depletion was preceded with or without washout of the vascular space. Because vascular washout did not significantly reduce the percentage of 131I-JV-1-42 in the parenchyma, this indicates that 131I-JV-1-42 was not loosely adhering to the vascular lumen. Therefore, 131I-JV-1-42 detected in the brain had either internalized into the capillary endothelial cells or had completely crossed the blood-brain barrier and entered the parenchyma (t = 10 min; n = 4-7 per group). (B) i.v. 131I-JV-1-42 was detected in the brain and CSF. However, the mean CSF/serum and brain/serum ratios were not significantly different, suggesting that 131I-JV-1-42 enters the CSF as readily as it enters the brain (t = 30 min; n = 2 per group).
Uptake into CSF. Brain, serum, and CSF samples were collected with a mean of 11.5 μl of CSF obtained from the posterior fossa for each mouse. Analysis by two-tailed t test revealed that the mean CSF/serum ratio (45.37 ± 10.58 μl/ml) was not significantly different (P < 0.05) from the mean brain/serum ratio (45.37 ± 0.57 μl/g), and the mean CSF/brain ratio was ≈1.0 (1.03 ± 0.25 ml/g) (Fig. 3B). This suggests that 131I-JV-1-42 can equally penetrate the CSF and the brain tissue.
Brain-to-Blood Transport (Efflux) of 131I-JV-1-42. Labeled antagonist injected into the ventricular system was effluxed from the brain with a half-life of 17.91 min (Fig. 4A). Coinjection of unlabeled JV-1-42 inhibited 131I-JV-1-42 efflux, showing that brain-to-blood efflux occurs by a saturable mechanism (Fig. 4B). Coinjection of cyclosporin A (P-gp substrate) significantly increased the amount of 131I-JV-1-42 present in the brain; however, coinjection of a different P-gp substrate, verapamil, had an opposite effect (Fig. 6, which is published as supporting information on the PNAS web site), indicating that efflux may be mediated by P-gp.
Fig. 4.

Brain-to-blood efflux of 131I-JV-1-42. (A) Administration by i.c.v. injection indicates that 131I-JV-1-42 is transported from brain to blood with a half-life in the brain of 17.91 min (n = 3 per point). (B) Coinjection of JV-1-42 with 131I-JV-1-42 resulted in a 1.5-fold increase in the mean %Inj Dose/Brain, indicating that efflux of 131I-JV-1-42 is saturable (t = 10 min after i.c.v. injection, n = 10 per group). (C) Efflux of 131I-JV-1-42; 131I-JV-1-42 coinjected with unlabeled JV-1-42 and 131I-JV-1-42 coinjected with cyclosporin A. Coadministration of 131I-JV-1-42 with cyclosporin A, a P-gp substrate, resulted in a 2.8-fold increase in the percent of 131I-JV-1-42 taken per gram of brain, indicating that efflux of 131I-JV-1-42 from the brain is mediated by P-gp (t = 10 min after i.c.v. injection; n = 8-10 per group; *, P < 0.05, **, P < 0.001).
Discussion
Brain tumors are the second and fourth leading causes of cancer mortality in children and young adults, respectively (17). GHRH antagonists have already shown promise as a potential therapy for malignant gliomas; however, their ability to access the CNS after peripheral administration may be impeded at the blood-brain barrier. CNS access is limited by the blood-brain barrier for chemotherapeutic drugs by two mechanisms: (i) poor penetration of the blood-brain barrier and (ii) brain-to-blood efflux, especially by the P-gp transporter (28). In the present study, we have examined the kinetics of blood-brain barrier transport for radioactively labeled JV-1-42, a GHRH antagonistic analog, and found it largely overcame both of these difficulties.
The initial volume of distribution into the serum at t = 0 (antilog of the y intercept) was 23.8 μl/g, a value greater than the usual estimate of the vascular space (Vi), which is 8-12 μl/g. However, because the half-life of the peptide in the serum is characteristic of a peptide of this size, any binding to serum proteins may not be of a high affinity. It is also possible that a transporter exists in the peripheral tissues that can rapidly bind and take up the peptide. Previous studies have indicated that tissue uptake of this compound is high (5). Multiple time regression analysis found the rate of entry of the peptide into the brain (slope of the line, or Ki) is 0.8514 ml/g per min. This rapid rate of entry corresponds with the high lipophilicity found for 131I-JV-1-42 (octanol/buffer partition coefficient of 78.31 ± 5.72). Lipid solubility is a strong predictor of blood-brain barrier permeability because of the high lipid content of cell membranes. A one-site binding hyperbolic model was used to determine the maximal percent of i.v. dose taken up per gram of brain. This value was 0.41%, indicating that brain uptake of 131I-JV-1-42 is high. By comparison, brain uptake of 131I-JV-1-42 is 20-fold greater than morphine (29) and 5-fold higher than IL-1α (30), two compounds known to cross the blood-brain barrier and exert CNS effects (31).
We also conducted a study designed to competitively inhibit the influx of the labeled antagonist from blood into the brain (as represented by brain/serum ratios, an expression of the volume of distribution). Interestingly, results from this study revealed that coinjection of unlabeled peptide paradoxically increased the brain/serum ratios for the labeled peptide. One possible explanation for this paradoxical increase is that the labeled peptide is rapidly transported out of the brain. In this case, addition of the unlabeled peptide would saturate the efflux mechanism and thus increase the amount of time the labeled peptide is present in the brain parenchyma. Administration of 131I-JV-1-42 by i.c.v. injection with and without unlabeled JV-1-42 confirmed saturable efflux of 131I-JV-1-42.
P-gp is a drug efflux pump present at the abluminal side of the blood-brain barrier. It is associated with resistance to chemotherapy, because it is a ubiquitous transporter that prevents the passage and accumulation of many drugs from the CNS. P-gp also targets endogenous molecules such as the glucocorticoid cortisol (32) and cholesterol (33), as well as the β amyloid peptide 1-42 (30). Substrates for P-gp tend to be peptides that are lipophilic or amphipathic; therefore, it is not surprising that 131I-JV-1-42 efflux is modulated by P-gp. Inhibition of the efflux when cyclosporin A, a P-gp substrate, is coinjected with radioactively labeled JV-1-42 indicates that 131I-JV-1-42 is likely a P-gp substrate. However, brain efflux of 131I-JV-1-42 was not inhibited by verapamil, another P-gp substrate. This indicates that the association of 131I-JV-1-42 with P-gp may be somewhat complicated. Furthermore, this association with P-gp does not appear so robust as to exclude 131I-JV-1-42 from brain uptake.
Based on previous experience, it is likely these findings in the mouse are applicable to humans, because many aspects of blood-brain barrier permeability, including predictions of penetration based on lipid solubility and the functions of the P-gp efflux system, are conserved across species (34-38). Because of their strong structural similarity, other GHRH antagonists are expected to cross the blood-brain barrier as well. Therefore, GHRH antagonistic analogs show great promise for the treatment of cancer, because they bind specifically and with high affinity to splice variants of the GHRH receptor present in many malignant tumors and produce direct antiproliferative effects in these tumor cells (1, 2, 4-6). Previous studies have shown that GHRH antagonists can block tumorigenesis and growth of human malignant gliomas xenografted into nude mice and extend the survival of animals with orthotopic tumor implants (8). The present study extends the range of applications for these analogs, by demonstrating that peripherally administered JV-1-42 can readily penetrate into CNS tissue. Our findings suggest that GHRH antagonists may have potential as a treatment for primary tumors such as malignant glioblastomas and possibly metastases to the brain of tumors that respond to GHRH antagonists, such as breast cancers.
Supplementary Material
Acknowledgments
We thank Dr. Gabor Halmos for technical advice. This work was supported by the Medical Research Service of the Department of Veterans Affairs (A.V.S. and W.A.B.), by a grant from Zentaris (Frankfurt/Main, Germany) to Tulane University (A.V.S.), and by National Institutes of Health Grants RO1 NS41863 and RO1 AA12743 (to W.A.B.) and GM08306 (to L.B.J.).
Author contributions: W.A.B. and A.V.S. designed research; L.B.J. performed research; W.A.B., J.L.V., and A.V.S. contributed new reagents/analytic tools; L.B.J., W.A.B., and A.V.S. analyzed data; and L.B.J., W.A.B., and A.V.S. wrote the paper.
Abbreviations: GH, growth hormone; GHRH, GH-releasing hormone; IGF-I/II, insulin-like growth factor-I/II; LR, lactated Ringer's solution; LR-BSA, LR containing 1% BSA; PE-10, polyethylene-10; P-gp, P-glycoprotein; i.c.v., intracerebroventricular.
References
- 1.Schally, A. V., Comaru-Schally, A. M., Nagy, A., Kovacs, M., Szepeshazi, K., Plonowski, A., Varga, J. L. & Halamos, G. (2001) Front. Neuroendocrinol. 22, 248-291. [DOI] [PubMed] [Google Scholar]
- 2.Schally, A. V. & Varga, J. L. (1999) Trends Endocrinol. Metab. 10, 383-393. [DOI] [PubMed] [Google Scholar]
- 3.Plonowski, A., Schally, A. V., Busto, R., Krupa, M., Varga, J. L. & Halmos, G. (2002) Peptides 23, 1127-1133. [DOI] [PubMed] [Google Scholar]
- 4.Halmos, G., Schally, A. V., Czompoly, T., Krupa, M., Varga, J. L. & Rekasi, Z. (2002) J. Clin. Endocrinol. Metab. 87, 4707-4714. [DOI] [PubMed] [Google Scholar]
- 5.Halmos, G., Schally, A. V., Varga, J. L., Plonowski, A., Rekasi, Z. & Czompoly, T. (2000) Proc. Natl. Acad. Sci. USA 97, 10555-10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rekasi, Z., Czompoly, T., Schally, A. V. & Halmos, G. (2000) Proc. Natl. Acad. Sci. USA 97, 10561-10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braczkowski, R., Schally, A. V., Plonowski, A., Varga, J. L., Groot, K., Krupa, M. & Armatis, P. (2002) Cancer 95, 1735-1745. [DOI] [PubMed] [Google Scholar]
- 8.Kiaris, H., Schally, A. V. & Varga, J. L. (2000) Neoplasia 2, 242-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiaris, H., Schally, A. V., Varga, J. L., Groot, K. & Armatis, P. (1999) Proc. Natl. Acad. Sci. USA 96, 14894-14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatzistamou, I., Schally, A. V., Varga, J. L., Groot, K., Armatis, P. & Halmos, G. (2001) J. Clin. Endocrinol. Metab. 86, 2144-2152. [DOI] [PubMed] [Google Scholar]
- 11.Kahan, Z., Varga, J. L., Schally, A. V., Rekasi, Z. & Armatis, P. (2000) Breast Cancer Res. Treat. 60, 71-79. [DOI] [PubMed] [Google Scholar]
- 12.Szepeshazi, K., Schally, A. V., Groot, K., Armatis, P., Halmos, G., Hebert, F., Szende, B., Varga, J. L. & Zarandi, M. (1999) Br. J. Cancer 82, 1724-1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szepeshazi, K., Schally, A. V., Groot, K., Armatis, P., Hebert, F. & Halmos, G. (1999) Eur. J. Cancer 36, 128-136. [DOI] [PubMed] [Google Scholar]
- 14.Letsch, M., Schally, A. V., Busto, R., Bajo, A. M. & Varga, J. L. (2003) Proc. Natl. Acad. Sci. USA 100, 1250-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jungwirth, A., Schally, A. V., Pinski, J., Groot, K., Armatis, P. & Halmos, G. (1997) Proc. Natl. Acad. Sci. USA 94, 5810-5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busto, R., Schally, A. V., Varga, J. L., Garcia-Fernandez, M. O., Groot, K., Armatis, P. & Szepeshazi, K. (2002) Proc. Natl. Acad. Sci. USA 99, 11866-11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Behin, A., Hoang-Xuan, K., Carpentier, A. F. & Delattre, J. Y. (2003) Lancet 361, 323-331. [DOI] [PubMed] [Google Scholar]
- 18.Boado, R. J., Li, J. Y. & Pardridge, W. M. (2004) Pediatr. Res. 55, 557-560. [DOI] [PubMed] [Google Scholar]
- 19.Tohyama, K., Kusuhara, H. & Sugiyama, Y. (2004) Endocrinology 145, 4384-4391. [DOI] [PubMed] [Google Scholar]
- 20.Isakovic, A. J., Abbott, N. J. & Redzic, Z. B. (2004) J. Neurochem. 90, 272-286. [DOI] [PubMed] [Google Scholar]
- 21.Banks, W. A., Kastin, A. J., Huang, W., Jaspan, J. B. & Maness, L. M. (1996) Peptides 17, 305-311. [DOI] [PubMed] [Google Scholar]
- 22.Banks, W. A. (2005) Curr. Pharmacol. Des. 11, 973-984. [DOI] [PubMed] [Google Scholar]
- 23.Blasberg, R. G., Fenstermacher, J. D. & Patlak, C. S. (1983) J. Cereb. Blood Flow Metab. 3, 8-32. [DOI] [PubMed] [Google Scholar]
- 24.Patlak, C. S., Blasberg, R. G. & Fenstermacher, J. D. (1983) J. Cereb. Blood Flow Metab. 3, 1-7. [DOI] [PubMed] [Google Scholar]
- 25.Banks, W. A., Kastin, A. J., Fischman, A. J., Coy, D. H. & Strauss, S. L. (1986) Am. J. Physiol. 251, E477-E482. [DOI] [PubMed] [Google Scholar]
- 26.Triguero, D., Buciak, J. & Pardridge, W. M. (1990) J. Neurochem. 54, 1882-1888. [DOI] [PubMed] [Google Scholar]
- 27.Guitierrez, E. G., Banks, W. A. & Kastin, A. J. (1993) J. Neuroimmunol. 47, 169-176. [DOI] [PubMed] [Google Scholar]
- 28.Thomas, H. & Coley, H. M. (2003) Cancer Control 10, 159-165. [DOI] [PubMed] [Google Scholar]
- 29.Banks, W. A. & Kastin, A. J. (1994) Peptides 15, 23-29. [DOI] [PubMed] [Google Scholar]
- 30.Lam, F. C., Liu, R., Lu, P., Shapiro, A. B., Renior, J. M., Sharom, F. J. & Reiner, P. B. (2001) J. Neurochem. 76, 1121-1128. [DOI] [PubMed] [Google Scholar]
- 31.Banks, W. A., Farr, S. A., La Scola, M. E. & Morley, J. E. (2001) J. Pharmacol. Exp. Ther. 299, 536-541. [PubMed] [Google Scholar]
- 32.Yates, C. R., Chang, C., Kearbey, J. D., Yasuda, K., Schuetz, E. G., Miller, D. D., Dalton, J. T. & Swaan, P. W. (2003) Pharmacol. Res. 20, 1794-1803. [DOI] [PubMed] [Google Scholar]
- 33.Troost, J., Lindenmaier, H., Haefeli, W. E. & Weiss, J. (2004) Mol. Pharmacol. 66, 1332-1339. [DOI] [PubMed] [Google Scholar]
- 34.Begley, D. J. (2004) Curr. Pharmcol. Des. 10, 1295-1312. [DOI] [PubMed] [Google Scholar]
- 35.Chikale, E. G., Burton, P. S. & Borchardt, R. T. (1995) J. Pharmacol. Exp. Ther. 273, 298-303. [PubMed] [Google Scholar]
- 36.Fellay, J., Marzolini, C., Meaden, E. R., Black, D. J., Buclin, T., Chave, J. P., Decosterd, L. A., Furrer, H., Opravil, M., Pantaleo, G., et al. (2002) Lancet 359, 30-36. [DOI] [PubMed] [Google Scholar]
- 37.Löscher, W. & Potschka, H. (2002) J. Pharmacol. Exp. Ther. 30, 7-14. [DOI] [PubMed] [Google Scholar]
- 38.Taylor, E. M. (2002) Clin. Pharmacokinet. 41, 81-92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.