

Abstract
Pleiotrophin (PTN) was found to regulate tyrosine phosphorylation of β-adducin through the PTN/receptor protein tyrosine phosphatase (RPTP)β/ζ signaling pathway. We now demonstrate that PTN stimulates the phosphorylation of serines 713 and 726 in the myristoylated alanine-rich protein kinase (PK) C substrate domain of β-adducin through activation of either PKC α or β. We also demonstrate that PTN stimulates translocation of phosphoserine 713 and 726 β-adducin either to nuclei, where it associates with nuclear chromatin and with centrioles of dividing cells, or to a membrane-associated site, depending on the phase of cell growth. Furthermore, we demonstrate that PTN stimulates the degradation of β-adducin in PTN-stimulated cells. Phosphorylation of serines 713 and 726 in β-adducin is known to markedly reduce the affinity of β-adducin for spectrin and actin and to uncouple actin/spectrin/β-adducin multimeric complexes needed for cytoskeletal stability. The data thus suggest that the PTN-stimulated phosphorylation of serines 713 and 726 in β-adducin disrupts cytoskeletal protein complexes and integrity, features demonstrated in both PTN-stimulated cells and of highly malignant cells that constitutively express the endogenous Ptn gene. The data also support the important conclusion that PTN determines the cellular location of β-adducin phosphorylated in serines 713 and 726 and raise the possibility that β-adducin functions in support of structure of heterochromatin and centrioles during mitosis.
Pleiotrophin (PTN the protein, Ptn the gene) is a secreted, highly conserved heparin-binding cytokine of 136 aa (1–3). Pleiotrophin is >50% identical in amino acid sequence and shares striking structural and functional similarities with midkine (MK the protein, Mk the gene). Together, PTN and MK constitute the only members of the Ptn/Mk developmental gene family (1, 3–5). Pleiotrophin stimulates proliferation of cultured fibroblasts (1, 3, 5), endothelial cells (6–9), epithelial cells (refs. 7 and 10–12, and N. Zhang, R. Zhong, Z. Y. Wang, and T.F.D., unpublished data). The product of the endogenous Ptn gene initiates the spontaneous lineage-specific differentiation that is characteristic of oligodendrocyte progenitors in primary culture through an autocrine mechanism (ref. 9 and H. J. Yeh, I. Silos-Santiago, M. Gurrieri, S. Jhaveri, Y. S. Li, and T.F.D., unpublished data).
The Ptn gene also is a proto-oncogene (12, 13). Its high-level importance in human malignancies is suggested because expression of the endogenous Ptn gene is seen frequently in many different highly malignant human neoplasms (14–22), and introduction of a dominant negative Ptn gene or targeted ribozymes into cells from malignancies with constitutive expression of the Ptn gene reverses the malignant phenotype to the phenotype of the premalignant cells both in vivo and in vitro, indicating that PTN signaling is responsible for the “switch” of the premalignant cell to the highly malignant phenotype (11, 12, 21, 23, 24).
Recent studies have uncovered clues to the mechanisms by which PTN initiates this striking diversity of phenotypes in PTN-stimulated cells. Pleiotrophin signals through two functionally and structurally independent signaling domains (refs. 5, 11, and 12 and N. Zhang, R. Zhong, Z. Y. Wang, and T.F.D., unpublished data), each of which binds heparin (25), and each of which signals very different phenotypes. The N-terminal domain, when expressed with the endogenous signal peptide, transforms murine fibroblasts, whereas the C-terminal domain does not; however, the C-terminal domain of PTN induces rapid growth and a striking angiogenic phenotype when it is expressed with the endogenous signal peptide in the premalignant cell. Thus, the separate domains of PTN function independently of each other, signal different phenotypes, and, a priori, need to signal through two separate receptor-like proteins (5, 11, 12).
One receptor that initiates PTN signaling is the transmembrane receptor protein tyrosine phosphatase (RPTP) β/ζ. The interaction of PTN with RPTPβ/ζ induces receptor dimerization and inactivates the endogenous tyrosine phosphatase activity of RPTPβ/ζ, thereby disrupting the balanced activity of RPTPβ/ζ and an unknown but constitutively active tyrosine kinase(s) on the mutual substrates of RPTPβ/ζ and the tyrosine kinase and thus initiating a sharp and rapid increase in the steady state levels of tyrosine phosphorylation (26). The first substrate of RPTPβ/ζ to be identified was β-catenin (26) and, more recently, β-adducin also was found to be a substrate of RPTPβ/ζ (47), thereby focusing attention on the cytoskeletal proteins as one target of the PTN/RPTPβ/ζ signaling pathway. In the case of β-catenin, the PTN-stimulated increase in tyrosine phosphorylation has led to the disruption of the association of β-catenin with the cadherin family, loss of adherins junction protein complexes, and loss of cell–cell adhesion (P.P.-P., Y. Chang, J. A. Vega, and T.F.D., unpublished data). β-Adducin was found to be interactive with the intercellular domain of the PTN receptor RPTPβ/ζ in a yeast two-hybrid system (47) and found to be a downstream target of the PTN/RPTPβ/ζ signaling pathway, suggesting that, together with β-catenin, β-adducin may have a role in the cytoskeletal rearrangements and fluidity characteristic of cells stimulated by PTN (unpublished data).
Here, we demonstrate that PTN activates protein kinase C (PKC) activity in PTN-stimulated cells, initiates phosphorylation of serines 713 and 726 in β-adducin through the PTN-dependent activated PKC activity, and redistributes β-adducin phosphorylated in serines 713 and 726 to different cellular sites, as well as targeting β-adducin for proteolysis. Because phosphorylation of serines 713 and 726 is known to markedly reduce the affinity of β-adducin for actin and spectrin, the data support the conclusion that PTN both stimulates cytoskeletal instability and plasticity through serine phosphorylation of β-adducin and redistributes serine phosphorylation β-adducin to determine its activity at different cellular sites. Furthermore, it is shown that β-adducin associates with heterochromatin and centrioles, raising the important possibility that β-adducin functions to stabilize these structures in addition to its well known functions in regulating the stability of the cell membrane associated cytoskeleton.
Materials and Methods
Cell Culture. HeLa (American Type Culture Collection) cells were used in all experiments and were cultured in DMEM with 10% FBS unless otherwise noted.
Stimulation of HeLa Cells with Different Reagents in Culture. HeLa cells were grown to 70% confluence in 100-mm plates, serum starved for 24 h, and treated with 50 ng/ml PTN (R & D Systems) for different times, 0.1 μM phorbol 12-myristate 13-acetate (PMA) for 12 h, or 10 μM of a PKC α/β pseudosubstrate inhibitor of PKC (Calbiochem) for 1 h as indicated.
Western Blot Analysis. Lysates of cells were prepared in PBS (pH 7.2) with 1% SDS, 1 mM PMSF, 10 mg/ml leupeptin, and 1× Complete Protease Inhibition tablets (Roche Diagnostics), resolved by SDS/PAGE and transferred to a nitrocellulose membrane. The membranes were probed with rabbit anti-β-adducin antibodies (kindly provided by Vann Bennet, Duke University, Durham, NC) and mouse anti-phosphoserine 713 and 726 β-adducin-specific antibodies (Upstate, Charlottesville, VA) at a 1:1,000 dilution, mouse anti-phosphoadducin antibodies that react with phosphoserine 713 β-adducin and phosphoserine 724 α-adducin (Upstate) at a 1:1,000 dilution, and rabbit anti-phosphoserine 152 and 156 myristoylated alanine-rich PKC substrate (MARCKS)-specific antibodies (Upstate) at a 1:1,000 dilution, or mouse anti-actin antibodies at a 1:2,500 dilution as indicated. Immunoreactive proteins were detected with either goat anti-rabbit or goat anti-mouse horseradish peroxidase (HRP)-conjugated antibodies (Santa Cruz Biotechnology), and illuminated with the ECL detection system (Amersham Pharmacia).
Immunocytochemistry. HeLa cells were seeded on coverslips at either 0.5 × 106 or 9 × 106 cells per ml, as indicated, and cultured for 24 h. The cells were then serum starved for 24 h, treated with 50 ng/ml PTN for 60 min, washed twice in ice cold PBS (pH 7.2), and fixed for 30 min with 4% paraformaldehyde in PBS. The levels of nonspecific protein binding were reduced with 1% FBS in PBS with 0.1% Triton X-100 for 2 h, incubated with a 1:200 dilution of mouse anti-phosphoserine 713 and 726 β-adducin-specific antibodies (Upstate) overnight in PBS with 0.1% Triton X-100, washed three times for 10 min in PBS, and treated with goat anti-mouse FITC conjugated antibodies (diluted 1:1,000; Chemicon). Actin and nuclei were stained with 1 μM phalloidin and DAPI (1:15,000) (Sigma), respectively, for 10 min in 0.05% Triton X-100 in PBS, washed four times for 10 min in PBS, mounted by using ProLong Antifade kit (Molecular Probes) on slides, and visualized by confocal microscopy in a Nikon Eclipse E600 microscope.
PKC Activity Assay. PKC activity was measured by using a PKC Kinase Non-Radioactive Assay kit (Stressgen, Victoria, Canada) as follows: HeLa cells were grown to confluency in six-well plates, serum starved for 24 h, and treated in triplicate with 1 μM PMA for 1 h or with 50 ng/ml PTN for 25 min, washed twice with PBS at 4°C, and lysed in sample preparation lysis buffer (20 mM Mops, pH 7.2/1 mM sodium vanadate/5 mM EGTA/2 mM EDTA/1% Nonidet P-40/1 μM DTT/1 mM PMSF/10 μg/ml leupeptin and aprotinin in PBS) and centrifuged at 15,000 × g for 15 min. The supernatant protein concentration was determined with the standard Lowry assay. Eighteen micrograms of lysate protein was diluted into 30 μl of the kinase assay dilution buffer (Stressgen) and loaded on 96-well plates coated with a PKC substrate peptide. The PKC assay was initiated by the addition of 10 μl of ATP (diluted 1 mg/ml) to each well at 30°C and assayed as per the manufacturer's instructions, measuring incorporation of phosphate into the substrate peptide at 50 min. The wells were then washed twice with antibody dilution buffer (Stressgen), and 40 μl of phosphospecific substrate antibodies were added to each well and incubated for 1 h. Each well was subsequently washed three times for 10 min with wash buffer (Stressgen) and a 1:1,000 dilution of anti-rabbit IgG HRP-conjugated antibody preparation in dilution buffer and incubated for 30 min. The wells were washed three times, and 60 μl of tetramethylbenzidine substrate (Stressgen) was added and incubated in the wells for 45 min. The HRP reaction was quenched with the addition of 20 μl of acid stop solution (Stressgen), and absorbance at 450 nm of each well was measured. The reaction was found to be linear with protein concentration and time.
Results
Pleiotrophin Stimulates Increased Phosphorylation of Serines 713 and 726 in β-Adducin. We found that the C-terminal 191-aa fragment of β-adducin interacts with the intracellular domain of RPTPβ/ζ in a yeast two-hybrid system (47). Furthermore, it was found that β-adducin interacts with the active site containing (D1) domain of RPTPβ/ζ, that β-adducin is a substrate of RPTPβ/ζ, and that PTN stimulates sharp increases in tyrosine phosphorylation of β-adducin in PTN-stimulated cells, presumably through phosphorylation of a highly conserved tyrosine phosphorylation site at Y564 found also in adducins α and γ (Fig. 7, which is published as supporting information on the PNAS web site). The C-terminal 191 aa fragment of β-adducin contains important phosphorylation sites at serines 713 and 726 within the MARCKS domain of β-adducin, which also are highly conserved in adducins α and γ and in the MARCKS protein itself (see Figs. 7 and 8, which are published as supporting information on the PNAS web site) (27). When serines 713 and 726 in β-adducin are phosphorylated, the affinity of β-adducin with actin and spectrin is reduced manyfold (27). Phosphorylation of serines 713 and 726 in β-adducin thus destabilizes the spectrin/actin/β-adducin protein complexes and consequently also destabilizes the spectrin membrane associated network and its association with filamentous actin needed for cytoskeletal stability. The steady state levels of serines 713 and 726 phosphorylation thus are critical determinants of the relative plasticity of cytoskeletal structures.
Because phosphorylation of serines 713 and 726 in β-adducin is required for the known disruption of cytoskeletal complexes and loss of homophilic cell–cell adhesion, which is a characteristic of cells stimulated by PTN, to seek a connection between these two phenomena, we treated HeLa cells with 50 ng/ml PTN for different times, and the levels of phosphoserines 713 and 726 in β-adducin were measured in Western blots of lysates of PTN-treated and control, nontreated, cells probed with antiphosphoserine 713 and 726 β-adducin antibodies. Fig. 1 demonstrates that the levels of phosphorylation of serines 713 and 726 β-adducin increase progressively with time of exposure to PTN in PTN-stimulated cells; a significant increase in phosphorylation of serines 713 and 726 β-adducin was seen within 5 min of stimulation (Fig. 1, lane 2); a further increase in the phosphorylation was seen at 20 min (Fig. 1, lane 3), and the levels then remained essentially constant to 1 h (Fig. 1, lane 4). Pleiotrophin thus stimulates a rapid, time-dependent, and readily detectable increase in the levels of phosphoserine 713 and 726 β-adducin in HeLa cells. In contrast, the levels of phosphoserine 713 and 726 β-adducin in the cells not stimulated with PTN remained constant for the 60-min period of incubation (data not shown).
Fig. 1.

Western blot of lysates from PTN-stimulated HeLa cells probed with anti-phosphoserine 713 and 726 β-adducin antibodies. (Upper) Lanes 1, untreated; 2, 5 min after PTN treatment; 3, 20 min after PTN treatment; 4, 60 min after PTN treatment. (Lower) Reprobe of Western blot with anti-actin antibodies.
Interestingly, HeLa cells that were not stimulated by PTN also have readily detectable levels of phosphoserine 713 and 726 β-adducin when lysates were analyzed in Western blots (Fig. 1, lane 1), suggesting that, in nonstimulated cells, an equilibrium of phosphorylated and nonphosphorylated serines 713 and 726 β-adducin may determine the relative cytoskeletal fluidity, and that the endogenous PTN/RPTPβ/ζ signaling pathway may be a central regulator of the fluidity of the cytoskeleton in cells that have not been stimulated with exogenous PTN.
Pleiotrophin Stimulates Redistribution of Phosphoserine 713 and 726 β-Adducin to Different Cellular Sites. We also examined PTN-stimulated cells stained with f luorescent-tagged anti-phosphoserine 713 and 726 β-adducin antibodies with confocal microscopy. In a control study to confirm an optimal time for cells to be examined after stimulation with PTN, HeLa cells in log phase growth and at confluence were first stimulated with 50 ng/ml PTN for 60 min. Cells both in log phase growth and at confluence stimulated with PTN were found to have readily detected increases of phosphorylation of serines 713 and 726 in β-adducin in lysates when examined in Western blots probed with anti-phosphoserine 713 and 726 antibodies (data not shown). When non-PTN-stimulated, nonconfluent HeLa cells were treated with anti-phosphoserine 713 and 726 β-adducin antibodies and examined by confocal microscopy, the fluorescent signal generated by the anti-phosphoserine 713 and 726 β-adducin antibodies was nearly exclusively limited to nuclei (Fig. 2B). However, when these cells were stimulated with PTN, a sharp increase in the levels of the fluorescent signal generated by the anti-phosphoserine 713 and 726 β-adducin antibodies was found in the cytosol (Fig. 2D, compared to B, arrows); the increase in the cytoplasmic signal in PTN-stimulated nonconfluent cells stained with anti-phosphoserine 713 and 726 β-adducin antibodies, DAPI, and phalloidin is seen more clearly (Fig. 2C, compared to A, arrows). The β-adducin phosphorylated in serines 713 and 726 is diffusely spread throughout the cytosol and appears to be localized in small endocytic vesicles (Fig. 2 C and D), suggesting that PTN may signal the degradation of β-adducin in PTN-stimulated nonconfluent HeLa cells as well as its redistribution. The increase in serine 713 and 726 β-adducin in PTN-stimulated cells in cytosol is presumed to be derived by translocation from nucleus; however, the density of the nuclear signal was sufficiently high that it was not possible to correlate a loss of the nuclear signal with the increase in cytosolic fluorescent staining that was observed.
Fig. 2.

Cellular localization of β-adducin in confluent and nonconfluent non-PTN-treated (control) and PTN-treated HeLa cells. (A) Non-PTN-treated sparse HeLa cells stained with phalloidin (red), anti-phosphoserine 713 and 726 β-adducin antibodies (green), and DAPI (blue). (B) Non-PTN-treated sparse HeLa cells stained with anti-phosphoserine 713 and 726 β-adducin antibodies. (C) PTN-treated (50 ng/ml) sparse HeLa cells for 60 min stained with phalloidin, anti-phosphoserine 713 and 726 β-adducin antibodies, and DAPI. (D) PTN-treated (50 ng/ml) sparse HeLa cells for 60 min stained with anti-phosphoserine 713 and 726 β-adducin antibodies. (E) Non-PTN-treated confluent HeLa cells stained with phalloidin, anti-phosphoserine 713 and 726 β-adducin antibodies, and DAPI. (F) Non-PTN-treated confluent HeLa cells stained with anti-phosphoserine 713 and 726 β-adducin antibodies. (G) PTN-treated (50 ng/ml) confluent HeLa cells for 60 min stained with phalloidin, anti-phosphoserine 713 and 726 β-adducin antibodies, and DAPI. (H) PTN-treated (50 ng/ml) confluent HeLa cells for 60 min stained with anti-phosphoserine 713 and 726 β-adducin antibodies.
When confluent, non-PTN-stimulated HeLa cells were examined by confocal microscopy, the fluorescent signal generated with anti-phosphoserine 713 and 726 antibodies was distinctly localized to regions of cell–cell contact (Fig. 2F); however, when these cells were stimulated with PTN, a striking increase in the fluorescent signal generated by anti-phosphoserine 713 and 726 β-adducin antibodies was found in nuclei, accompanied by a sharp loss of the fluorescent signal from regions of cell–cell contact. The fluorescent signal in the PTN-stimulated confluent cells was found diffusely spread in cytoplasm (Fig. 2H). Importantly, when PTN-stimulated cells were stained with anti-phosphoserine 713 and 726 β-adducin, DAPI, and phalloidin, phosphoserine 713 and 726 β-adducin was seen to colocalize with nucleus but also was found diffusely spread in cytoplasm (Fig. 2G).
Rapid Loss of β-Adducin Protein in PTN-Stimulated Cells. Because of the apparent degradation of β-adducin in log phase growing HeLa cells stimulated with PTN, lysates of HeLa cells in log phase growth stimulated with PTN at different times were analyzed in Western blots probed with anti-β-adducin antibodies. In Fig. 3, a time-dependent loss of β-adducin in nonconfluent HeLa cells is demonstrated, whereas no loss of the levels of the actin control were found, supporting strongly the view that PTN not only redistributes phosphoserine 713 and 726 β-adducin in both log phase and confluent HeLa cells, but also signals a rapid loss of β-adducin protein.
Fig. 3.

Western blot of lysates of the time-dependent PTN-stimulated nonconfluent HeLa cells probed with anti-β-adducin antibodies.
β-Adducin Phosphorylated at Serines 713 and 726 Is Associated with Nuclear Chromatin and Centrioles. The nuclear fluorescent staining generated by anti-phosphoserine 713 and 726 β-adducin antibodies in confocal microscopy was surprising, because only a single reference of β-adducin associated with nuclei in mammalian cells was found in searches of different databases (28); in that study, immunoreactive β-adducin was seen in nuclei of mouse oocytes; it was suggested that β-adducin was associated with nuclear chromatin. Phosphoserine 713 and 726 β-adducin in nuclei in our studies appeared also to associate with nuclear chromatin (Fig. 2G). To confirm this impression, images of cells were again stained with DAPI (Fig. 4A) and anti-phosphoserine 713 and 726 β-adducin (Fig. 4B) and superimposed with images of the same cells (Fig. 4C). We found that phosphoserine 713 and 726 β-adducin associates directly with nuclear chromatin in the superimposed image.
Fig. 4.
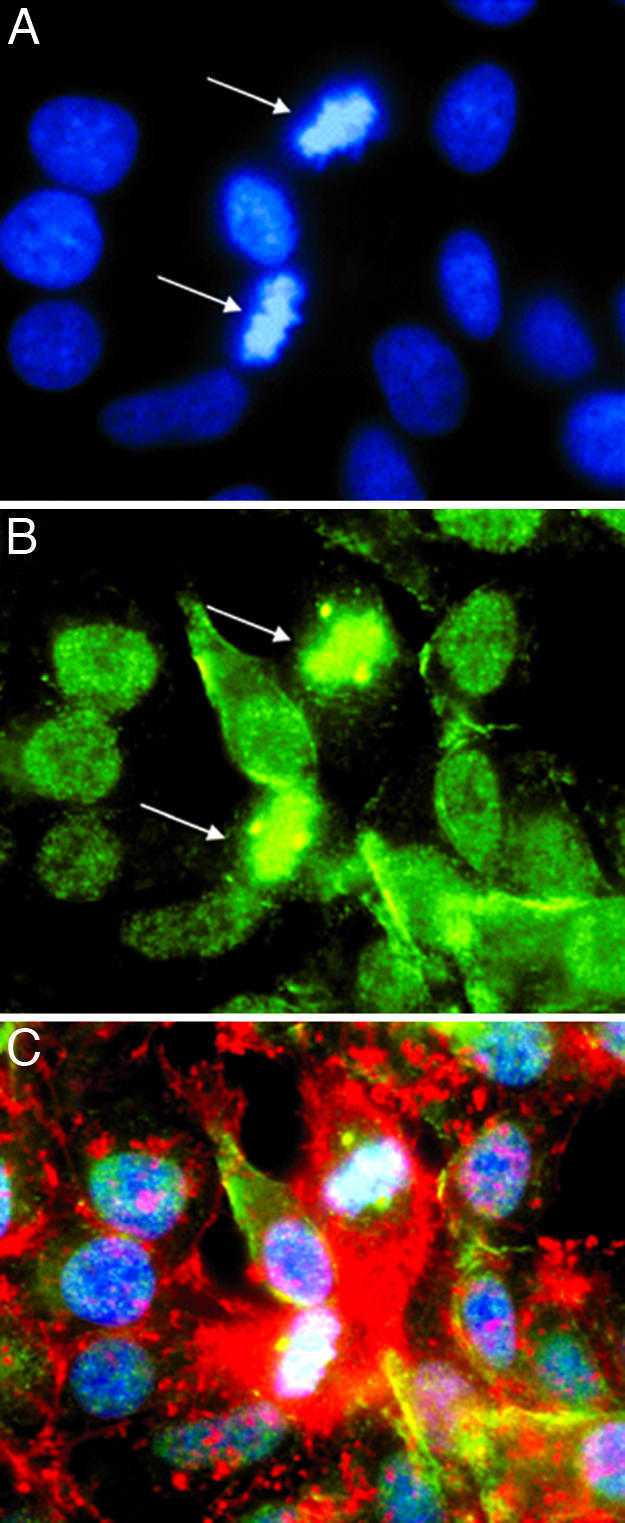
Localization of β-adducin in centrioles of PTN-treated dividing HeLa cells. (A) PTN-treated (50 ng/ml) HeLa cells stained with DAPI (blue). (B) PTN-treated (50 ng/ml) HeLa cells stained with anti-phosphoserine 713 and 726 β-adducin antibodies (green). (C) PTN-treated (50 ng/ml) HeLa cells stained with phalloidin (red), anti-phosphoserine 713 and 726 β-adducin antibodies (green), and DAPI (blue). Note the localization of phosphoserine 713 and 726 β-adducin to chromatin and the centrioles during metaphase (arrows).
Remarkably, the fluorescent stain representing phosphoserine 713 and 726 β-adducin was prominently localized in centrioles as well as nuclear chromatin (Fig. 4, arrows denote centrioles). To the best of our knowledge, the identification of phosphoserine 713 and 726 β-adducin in centrioles in mammalian cells has not been described, raising the distinct possibility that β-adducin may stabilize the centriole protein complexes during mitosis.
Pleiotrophin Stimulates Activation of PKC. Different studies demonstrate that phosphorylation of serine 713 and 726 β-adducin is catalyzed by a PKC (27). Lysates from the PMA-stimulated and PTN-stimulated confluent HeLa cells were therefore analyzed in Western blots probed with a different antibody preparation from that used in Fig. 1. This antibody (see Materials and Methods above) reacts with phosphoserine 713 of β-adducin and 724 of α-adducin, and was used to confirm the results obtained in Fig. 1 and, furthermore, to determine whether α-adducin is also phosphorylated in PTN-stimulated cells. It was found that PMA increased the phosphorylation of serine 713 β-adducin (Fig. 5 Upper, lower band) and 724 in α-adducin (Fig. 5 Upper, upper band) in lysates of PMA-stimulated cells (Fig. 5, lane 4) to levels similar to those found in lysates of PTN-stimulated cells (Fig. 5, lane 3). The PMA- and PTN-stimulated increases in levels of phosphoserines 713 in β-adducin and 724 α-adducin were markedly reduced when the pseudosubstrate peptide inhibitor of PKC α/β was included in cultures stimulated with either PMA (Fig. 5, lane 5) or PTN (Fig. 5, lane 6), supporting the conclusions that PTN and, likely, PMA activate either PKC α or β (or both) to catalyze the PMA- and PTN-stimulated increase in phosphoserine 713 in β-adducin and 724 in α-adducin.
Fig. 5.

Both PTN and PMA enhance phosphorylation of β-adducin residue 713 and α-adducin residue 724. (Upper) Western blot of lysates from PTN-treated (50 ng/ml, for 15 and 60 min), PMA-tretaed (1 μM, 60 min), or PKCα/β inhibitor-treated (60 min before PTN and PMA treatment) HeLa cells probed with anti-phosphoserine 713 β-adducin and anti-phosphoserine 724 α-adducin antibodies. (Lower) Reprobe of Western blot with an anti-actin-specific antibody.
We also tested the ability of PTN to stimulate serines 152 and 156 of a second important cytoskeleton protein and known PKC substrate, the MARCKS protein (29). The MARCKS protein contains the important regulatory “MARCKS” domain homologous with the MARCKS domain in β-adducin. Lysates from HeLa cells stimulated with 50 ng/ml PTN and PMA for 30 and 60 min were prepared and analyzed in Western blots probed with anti-phosphoserine 152 and 156 MARCKS antibodies. Both PMA and PTN stimulated a marked increase in phosphorylation of serine 152 and 156 MARCKS. The increase in phosphorylation of serine 152 and 156 MARCKS is both PTN concentration- and time-dependent to 60 min. Levels of phosphoserine 152 and 156 MARCKS remained constant in cells that were not stimulated with PTN throughout the experiment (Fig. 9, which is published as supporting information on the PNAS web site).
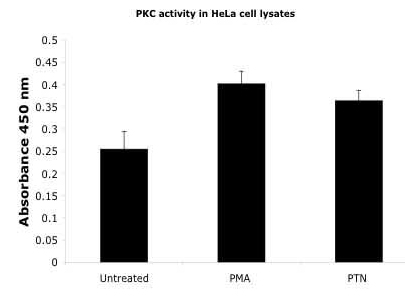
The levels of PKC(s) activity in non-PTN-stimulated (control) and in PTN- and PMA-stimulated confluent HeLa cells were also directly measured (see Materials and Methods above). The PKC activity in lysates from PTN-stimulated cells lysates was ≈42% above the endogenous activity in non-PMA-stimulated cells, and that in lysates from PMA-stimulated cells was ≈62% above the endogenous activity in non-PMA-stimulated cells (Fig. 10, which is published as supporting information on the PNAS web site). The data make clear that the baseline PKC activity of HeLa cells is high, a finding that is consistent with the high baseline levels of phosphorylation of serines 713 and 726 in β-adducin both in logarithmic and conf luent non-PTN-stimulated HeLa cells. The data also make clear that PTN activates one or more isoforms of PKC; however, the level of the PTN-dependent increase of PKC activity is masked because of high endogenous PKC activity in nonstimulated cells. The data thus suggest that the endogenous PKC activity functions to maintain the steady-state levels of serine 713 and 726 phosphorylation and point to the possibility that the endogenous PTN/RPTPβ/ζ pathway may regulate the stability of β-adducin, spectrin, and actin complexes and that this pathway is a principle regulator of MARCKS domain containing cytoskeletal proteins.
Discussion
β-Adducin is an important cytoskeletal protein; it belongs to a family of proteins that preferentially bind to actin–spectrin junctions (30–32) and functions to stabilize the growing actin filaments and actin–spectrin networks (33, 34) through the formation of complexes between spectrin and actin and the promotion of the association of spectrin with actin filaments (32) (Fig. 6). The affinity of β-adducin for this complex is greatest at the fast growing ends of actin filaments, where β-adducin contributes to actin filament stability. Adducin α and γ isoforms interact together with β-adducin to create heterotetrameric complexes at the growing ends of actin filaments and actin–spectrin junctions (32). Each isoform of adducin is comprised of an N-terminal globular head domain (39 kDa) that allows for tetramerization, a small neck domain (9 kDa), and the protease-sensitive tail domain containing a MARCKS like domain (32) that binds to actin and spectrin and thus contributes to the stability of adducin/actin/spectrin junctions as well as growing actin filaments. β-adducin is thus critically placed in the cytoskeleton in such a position that its regulation is required for it to function effectively in membrane stability and fluidity at different phases of the cell cycle.
Fig. 6.

Proposed model for PTN-stimulated dissociation of β-adducin from F-actin and spectrin. Modified from Bennet and Bainnes (32). β-Adducin forms complexes between spectrin and actin promoting the association of spectrin with actin filaments. Pleiotrophin-induced phosphorylation of serines 713 and 726 β-adducin reduces the affinity of β-adducin for filamentous actin dissociating these cytoskeletal structures and increasing cleavage of β-adducin by caspase-3 and degradation of β-adducin.
The present experiments demonstrate that PTN stimulates phosphorylation of serines 713 and 726 in the MARCKS domain of β-adducin (and serine 724 in α-adducin) and serines 152 and 156 in the MARCKS protein itself through the activation of either PKC α or β and perhaps other PKC(s) isoforms. These studies also demonstrate that PTN stimulates the translocation of phosphoserine 713 and 726 β-adducin from a nuclear localization to cytosol in nonconfluent HeLa cells and from a plasma membrane-associated site to nuclei in PTN-stimulated confluent HeLa cells. Furthermore, it is shown that PTN stimulates the degradation of β-adducin itself. Finally, these studies have uncovered the presence of β-adducin in nuclei, associated with heterochromatin and, in dividing cells, in centrioles.
These findings may be very important, because the characteristic features of PTN-stimulated cells and the malignant cells that have acquired an activated endogenous Ptn gene during tumor progression include disruption of cytoskeletal protein complexes and cytoskeletal stability. These features fit well with the known property of phosphorylation of serines 713 and 726 in β-adducin to greatly reduce the affinity of β-adducin to filamentous actin, the actin–spectrin junctions, and the fast growing ends of actin, resulting in dissociation of these cytoskeletal structures, loss of integrity of actin–spectrin complexes (32), and the cleavage of β-adducin by caspase-3 and degradation of β-adducin through the proteosome degradation pathway (36) (Fig. 6). Thus, PTN stimulation may not only decrease the association of actin, spectrin, β-adducin complexes, but also reduce the cellular pool of β-adducin, which may additionally add to loss of actin cytoskeletal integrity in PTN-stimulated cells. In support of our findings, the ability of β-adducin to contribute to cytoskeletal integrity is known to be regulated by PKCs (27), and PKC α, β, and γ are known to phosphorylate the consensus RTPS sequences (serines 713 and 726) sites within the highly conserved 22-aa residues at the extreme C-terminal MARCKS domain of β-adducin (and adducin α and γ) (27).
In other studies, we demonstrated that PTN stimulates tyrosine phosphorylation of β-catenin in PTN-stimulated cells, and, as a consequence, the affinity of β-catenin with the intracellular domains of E-cadherin and filamentous actin is reduced and adherens junctions complexes are dissolved, leading to disruption of cytoskeletal integrity and loss of homophilic cell–cell adhesion (ref. 26 and P.P.-P., Y. Chang, J. A. Vega, and T.F.D., unpublished data). β-Catenin links the catenin family and the highly conserved cytoplasmic domain of cadherins with the actin cytoskeleton (37–41), and it is known that levels of tyrosine phosphorylation of β-catenin correlate directly with increased perturbation of the link of the cadherins and actin filaments and with decreased cell–cell adhesion (37, 42–44). The previous findings that PTN regulates tyrosine phosphorylation of β-catenin, coupled with the striking modifications in β-adducin in PTN-stimulated cells, fit well with the loss of cytoskeletal structure and cell–cell adhesion and the increases in membrane plasticity that are characteristic of PTN-stimulated cells. These studies also fit well with similar features that are characteristic of the different human malignant cells with mutations that initiate constitutive activation of the endogenous Ptn gene.
It is also likely to be important that PTN translocates phosphoserine 713 and 726 β-adducin to different cellular sites, depending on whether cells are confluent; thus, PTN regulates the sites at which phosphoserine 713 and 726 β-adducin is located and where β-adducin protein complexes are less stable. Previous studies (45) demonstrated that, when Fyn and β-adducin are cotransfected, β-adducin was phosphorylated in tyrosine and translocated to the cell membrane. Whether the PTN-dependent increase in tyrosine phosphorylation is the signal to initiate this translocation is not clear; however, we have found that PTN increases tyrosine phosphorylation of Fyn (48), and this finding may link the studies of Shima et al. (45) with the studies presented here.
In these studies, β-adducin was also found in nucleus, presumably in association with nuclear chromatin and with centrioles in cells during cell division. Only a single report identifying β-adducin (through immunoreactivity with anti-β-adducin antibodies) in nuclei of cells in mammalian species was found (28). The C-terminal region of β-adducin shares sequence homology with other nuclear localization signals, supporting the view that the nuclear localization of phosphoserine 713 and 726 β-adducin is not artifactual. A C-terminal nuclear bipartite nuclear targeting consensus sequence (KKKKKFRTPSFLKKSK) has been identified at the C-terminal domain of β-adducin that may direct β-adducin to nuclei. We speculate that the translocation of phosphoserine β-adducin in response to PTN possibly results from phosphorylation of serines 713 and 726, which may unmask the nuclear recognition sequence of β-adducin from the known association of the MARCKS domain of β-adducin with calmodulin (46), and the displacement of calmodulin when serines 713 and 726 of β-adducin are phosphorylated by PKC.
The function of β-adducin in the nucleus and in centrioles of dividing cells remains to be determined, but suggests the possibility that β-adducin may stabilize chromatin and centriolar structure. Thus, phosphorylation of serines 713 and 726 in β-adducin may be a critical step in destabilization of these structures at critical times in the cell cycles. In this context, indirect support for this possibility was found in human U87-MG glioblastoma cells into which a dominant negative Ptn gene was introduced; these cells have a high degree of anaploidy and tetraploidy (Y. Chang and T.F.D., unpublished data), raising the possibility that the failure of PTN-dependent phosphorylation of serines 713 and 726 in β-adducin may have reduced the levels of β-adducin phosphorylated at serines 713 and 726 needed to destabilize critical structures associated with nuclear-chromatin and centrioles for chromosomal separation. In Fig. 4, β-adducin appears to be localized to chromosomes during metaphase, supporting the hypothesis that β-adducin and phosphoserine 713 and 726 β-adducin may act to critically regulate stabilization of chromatin and chromosomal separation.
Supplementary Material
Acknowledgments
We thank Dr. Vann Bennet for the generous gift of anti-β-adducin antisera. This work was supported by National Institues of Health Grants CA88440, CA66029, and DK 53557 and National Heart, Blood, and Lung Institute Grant 31102. This is manuscript 17147-MEM from The Scripps Research Institute.
Author contributions: H.P., G.H., L.E., P.P.-P., and T.F.D. designed research; H.P. performed research; H.P. and T.F.D. contributed new reagents/analytic tools; H.P., G.H., L.E., P.P.-P., and T.F.D. analyzed data; and H.P., G.H., L.E., P.P.-P., and T.F.D. wrote the paper.
Abbreviations: PMA, phorbol 12-myristate 13-acetate; MARCKS, myristoylated alanine-rich PKC substrate; RPTP, receptor protein tyrosine phosphatase.
References
- 1.Milner, P. G., Li, Y. S., Hoffman, R. M., Kodner, C. M., Siegel, N. R. & Deuel, T. F. (1989) Biochem. Biophys. Res. Commun. 165, 1096–1103. [DOI] [PubMed] [Google Scholar]
- 2.Rauvala, H. (1989) EMBO J. 8, 2933–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li, Y. S., Milner, P. G., Chauhan, A. K., Watson, M. A., Hoffman, R. M., Kodner, C. M., Milbrandt, J. & Deuel, T. F. (1990) Science 250, 1690–1694. [DOI] [PubMed] [Google Scholar]
- 4.Kha, H., Li, Y. S. & Deuel, T. F. (1996) Genomics 37, 242–244. [DOI] [PubMed] [Google Scholar]
- 5.Zhang, N. & Deuel, T. F. (1999) Curr. Opin. Hematol. 6, 44–50. [DOI] [PubMed] [Google Scholar]
- 6.Courty, J., Dauchel, M. C., Caruelle, D., Perderiset, M. & Barritault, D. (1991) Biochem. Biophys. Res. Commun. 180, 145–151. [DOI] [PubMed] [Google Scholar]
- 7.Fang, W., Hartmann, N., Chow, D. T., Riegel, A. T. & Wellstein, A. (1992) J. Biol. Chem. 267, 25889–25897. [PubMed] [Google Scholar]
- 8.Laaroubi, K., Delbe, J., Vacherot, F., Desgranges, P., Tardieu, M., Jaye, M., Barritault, D. & Courty, J. (1994) Growth Factors 10, 89–98. [DOI] [PubMed] [Google Scholar]
- 9.Yeh, H. J., He, Y. Y., Xu, J., Hsu, C. Y. & Deuel, T. F. (1998) J. Neurosci. 18, 3699–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delbe, J., Vacherot, F., Laaroubi, K., Barritault, D. & Courty, J. (1995) J. Cell Physiol. 164, 47–54. [DOI] [PubMed] [Google Scholar]
- 11.Zhang, N., Zhong, R. & Deuel, T. F. (1999) J. Biol. Chem. 274, 12959–12962. [DOI] [PubMed] [Google Scholar]
- 12.Deuel, T. F., Zhang, N., Yeh, H. J., Silos-Santiago, I. & Wang, Z. Y. (2002) Arch. Biochem. Biophys. 397, 162–171. [DOI] [PubMed] [Google Scholar]
- 13.Chauhan, A. K., Li, Y. S. & Deuel, T. F. (1993) Proc. Natl. Acad. Sci. USA 90, 679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aigner, A., Brachmann, P., Beyer, J., Jager, R., Raulais, D., Vigny, M., Neubauer, A., Heidenreich, A., Weinknecht, S., Czubayko, F. & Zugmaier, G. (2003) Ann. Oncol. 14, 1525–1529. [DOI] [PubMed] [Google Scholar]
- 15.Brockmann, M. A., Ulbricht, U., Gruner, K., Fillbrandt, R., Westphal, M. & Lamszus, K. (2003) Neurosurgery 52, 1391–1399; discussion, 1399. [DOI] [PubMed] [Google Scholar]
- 16.Mentlein, R. & Held-Feindt, J. (2002) J. Neurochem. 83, 747–753. [DOI] [PubMed] [Google Scholar]
- 17.Muramatsu, T. (2002) J. Biochem. (Tokyo) 132, 359–371. [DOI] [PubMed] [Google Scholar]
- 18.Riegel, A. T. & Wellstein, A. (1994) Breast Cancer Res. Treat. 31, 309–314. [DOI] [PubMed] [Google Scholar]
- 19.Nakagawara, A., Milbrandt, J., Muramatsu, T., Deuel, T. F., Zhao, H., Cnaan, A. & Brodeur, G. M. (1995) Cancer Res. 55, 1792–1797. [PubMed] [Google Scholar]
- 20.Jager, R., Noll, K., Havemann, K., Pfluger, K. H., Knabbe, C., Rauvala, H. & Zugmaier, G. (1997) Int. J. Cancer 73, 537–543. [DOI] [PubMed] [Google Scholar]
- 21.Zhang, N., Zhong, R., Wang, Z. Y. & Deuel, T. F. (1997) J. Biol. Chem. 272, 16733–16736. [DOI] [PubMed] [Google Scholar]
- 22.Klomp, H. J., Zernial, O., Flachmann, S., Wellstein, A. & Juhl, H. (2002) Clin. Cancer Res. 8, 823–827. [PubMed] [Google Scholar]
- 23.Czubayko, F., Schulte, A. M., Berchem, G. J. & Wellstein, A. (1996) Proc. Natl. Acad. Sci. USA 93, 14753–14758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Czubayko, F., Riegel, A. T. & Wellstein, A. (1994) J. Biol. Chem. 269, 21358–21363. [PubMed] [Google Scholar]
- 25.Kilpelainen, I., Kaksonen, M., Avikainen, H., Fath, M., Linhardt, R. J., Raulo, E. & Rauvala, H. (2000) J. Biol. Chem. 275, 13564–13570. [DOI] [PubMed] [Google Scholar]
- 26.Meng, K., Rodriguez-Pena, A., Dimitrov, T., Chen, W., Yamin, M., Noda, M. & Deuel, T. F. (2000) Proc. Natl. Acad. Sci. USA 97, 2603–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuoka, Y., Li, X. & Bennett, V. (1998) J. Cell Biol. 142, 485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinto-Correia, C., Goldstein, E. G., Bennett, V. & Sobel, J. S. (1991) Dev. Biol. 146, 301–311. [DOI] [PubMed] [Google Scholar]
- 29.Brooks, S. F., Herget, T., Broad, S. & Rozengurt, E. (1992) J. Biol. Chem. 267, 14212–14218. [PubMed] [Google Scholar]
- 30.Gardner, K. & Bennett, V. (1987) Nature 328, 359–362. [DOI] [PubMed] [Google Scholar]
- 31.Coleman, T. R., Fishkind, D. J., Mooseker, M. S. & Morrow, J. S. (1989) Cell Motil. Cytoskeleton 12, 248–263. [DOI] [PubMed] [Google Scholar]
- 32.Bennett, V. & Baines, A. J. (2001) Physiol. Rev. 81, 1353–1392. [DOI] [PubMed] [Google Scholar]
- 33.Bennett, V. (1989) Biochim. Biophys. Acta 988, 107–121. [DOI] [PubMed] [Google Scholar]
- 34.Hughes, C. A. & Bennett, V. (1995) J. Biol. Chem. 270, 18990–18996. [DOI] [PubMed] [Google Scholar]
- 35.Li, X. & Bennett, V. (1996) J. Biol. Chem. 271, 15695–15702. [DOI] [PubMed] [Google Scholar]
- 36.van de Water, B., Tijdens, I. B., Verbrugge, A., Huigsloot, M., Dihal, A. A., Stevens, J. L., Jaken, S. & Mulder, G. J. (2000) J. Biol. Chem. 275, 25805–25813. [DOI] [PubMed] [Google Scholar]
- 37.Daniel, J. M. & Reynolds, A. B. (1997) Bioessays 19, 883–891. [DOI] [PubMed] [Google Scholar]
- 38.Ferreira-Cornwell, M. C., Veneziale, R. W., Grunwald, G. B. & Menko, A. S. (2000) Exp. Cell Res. 256, 237–247. [DOI] [PubMed] [Google Scholar]
- 39.Castano, J., Raurell, I., Piedra, J. A., Miravet, S., Dunach, M. & Garcia de Herreros, A. (2002) J. Biol. Chem. 277, 31541–31550. [DOI] [PubMed] [Google Scholar]
- 40.Kim, K. & Lee, K. Y. (2001) Cell Biol. Int. 25, 421–427. [DOI] [PubMed] [Google Scholar]
- 41.Roura, S., Miravet, S., Piedra, J., Garcia de Herreros, A. & Dunach, M. (1999) J. Biol. Chem. 274, 36734–36740. [DOI] [PubMed] [Google Scholar]
- 42.Behrens, J., Vakaet, L., Friis, R., Winterhager, E., Van Roy, F., Mareel, M. M. & Birchmeier, W. (1993) J. Cell Biol. 120, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamaguchi, M., Matsuyoshi, N., Ohnishi, Y., Gotoh, B., Takeichi, M. & Nagai, Y. (1993) EMBO J. 12, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kypta, R. M., Su, H. & Reichardt, L. F. (1996) J. Cell Biol. 134, 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shima, T., Okumura, N., Takao, T., Satomi, Y., Yagi, T., Okada, M. & Nagai, K. (2001) J. Biol. Chem. 276, 42233–42240. [DOI] [PubMed] [Google Scholar]
- 46.Matsuoka, Y., Hughes, C. A. & Bennett, V. (1996) J. Biol. Chem. 271, 25157–25166. [DOI] [PubMed] [Google Scholar]
- 47.Pariser, H., Perez-Pinera, P., Ezquerra, L., Herradon, G. & Devel, T. F. (2005) Biochem. Biophys. Res. Commun., in press. [DOI] [PubMed]
- 48.Pariser, H., Ezquerra, L., Herradon, G., Parez-Pinera, P. & Devel, T. F. (2005) Biochem. Biophys. Res. Commun. 332, 664–699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}