

Abstract
A new approach to malarial chemotherapy based on quaternary ammonium that targets membrane biogenesis during intraerythrocytic Plasmodium falciparum development has recently been developed. To increase the bioavailability, nonionic chemically modified prodrugs were synthesized. In this paper, the pharmacological properties of a bisthiazolium salt (T3) and its bioprecursor (TE3) were studied. Their antimalarial activities were determined in vitro against the growth of P. falciparum and in vivo against the growth of P. vinckei in mice. Pharmacokinetic evaluations were performed after T3 (1.3 and 3 mg/kg of body weight administered intravenously; 6.4 mg/kg administered intraperitoneally) and TE3 (1.5 and 3 mg/kg administered intravenously; 12 mg/kg administered orally) administrations to rats. After intraperitoneal administration, very low doses offer protection in a murine model of malaria (50% efficient dose [ED50] of 0.2 to 0.25 mg/kg). After oral administration, the ED50 values were 13 and 5 mg/kg for T3 and TE3, respectively. Both compounds exerted antimalarial activity in the low nanomolar range. After TE3 administration, rapid prodrug-drug conversion occurred; the mean values of the pharmacokinetic parameters for T3 were as follows: total clearance, 1 liter/h/kg; steady-state volume of distribution, 14.8 liters/kg; and elimination half-life, 12 h. After intravenous administration, T3 plasma concentrations increased in proportion to the dose. The absolute bioavailability was 72% after intraperitoneal administration (T3); it was 15% after oral administration (TE3). T3 plasma concentrations (8 nM) 24 h following oral administration of TE3 were higher than the 50% inhibitory concentrations for the most chloroquine-resistant strains of P. falciparum (6.3 nM).
Malaria is still one of the most widespread parasitic tropical diseases, with its main impact being in sub-Saharan Africa, where at least 90% of all malaria-related deaths occur. The malaria situation has recently deteriorated, and the rate of mortality from this disease has increased, so this disease has a major effect on economic productivity and livelihood in areas of the world where malaria is endemic. This situation is partly explained by the fact that Plasmodium falciparum has developed resistance to cheap and effective drugs, such as chloroquine and sulfadoxine-pyrimethamine, and by the appearance of insecticide-resistant mosquitoes (14). Consequently, new antimalarial compounds, particularly those that are based on compounds structurally unrelated to existing antimalarial agents and that have new independent mechanisms of action, are needed in the battle against this major endemic disease (21, 24).
Phospholipid metabolism is now considered an attractive target for new malaria chemotherapy due to its vital importance for the parasite. Indeed, phospholipid metabolism is absent from normal mature human erythrocytes (28), but the erythrocyte phospholipid content increases by as much as 500% after infection, due to the parasite's metabolic pathways. Phosphatidylcholine is the major phospholipid and represents about 45% of total phospholipids (16, 26, 30, 32). A new approach to malaria chemotherapy that targets membrane biogenesis during intraerythrocytic P. falciparum development has thus been developed (33). This approach concerns monoquaternary ammonium compounds (2, 11) and bisquaternary ammonium compounds (10) that mimic choline and that alter the parasitic de novo biosynthesis of phosphatidylcholine. These compounds, now at the forefront of antimalarial research, have exceptional in vitro and in vivo antimalarial properties in both rodent and primate models of malaria and have shown high degrees of efficacy against multiresistant P. falciparum malaria (3, 4, 34, 35). It was recently shown that, in addition to their selective inhibition of de novo phosphatidylcholine biosynthesis, the potent antimalarial activities of bisquaternary ammonium compounds can also be attributed to the drug's compartmentalization into the parasite's food vacuole and, finally, their binding to ferriprotoporphyrin IX and/or to the growing malaria parasite pigment (hemozoin) (7).
One of the main drawbacks of these biscationic compounds is their low oral absorption due to their permanently charged quaternary ammonium moiety. To increase the bioavailability, nonionic chemically modified forms that possess thioester function have been synthesized. These prodrugs were shown to be quantitatively converted to active ionized bisthiazolium drugs in the presence of plasma. The bisthiazolium salt T3 is a lead compound that has been shown to have potent in vitro as well as in vivo antimalarial activities at low doses in the murine model. Its corresponding bioprecursor, TE3, was found to completely cure Plasmodium cynomolgi-infected Rhesus monkeys after one daily dose of 3 mg/kg of body weight for 4 consecutive days (34).
The main objective of this study is to report on the pharmacological properties of the TE3 prodrug and its related bisthiazolium salt, T3. The antimalarial activities of these compounds were determined in vitro against the growth of P. falciparum and in vivo against a Plasmodium vinckei petteri strain (279BY) in female Swiss mice. During the conventional drug development process, pharmacokinetic studies were carried out as early as possible with healthy animals to obtain information on absorption, distribution, metabolism, and excretion. So, in the present study, pharmacokinetic parameters were determined after T3 or TE3 administration to Sprague-Dawley rats. As limited numbers of plasma samples (one or two) were taken from each animal over a 36-h period, the nonlinear mixed-effect modeling approach was used to estimate the pharmacokinetic parameters of T3 and TE3. By using such an approach, the interindividual variability in pharmacokinetics can be estimated. Pharmacokinetic studies were performed with only single doses. The T3 compound was administered by the intravenous (i.v.) and intraperitoneal (i.p.) routes, while the TE3 compound was administered by the intravenous and oral routes.
MATERIALS AND METHODS
This research adhered to the Principles of Laboratory Animal Care (18a). The animal study was approved by the local animal use committee.
Drugs.
The bisthiazolium active compound T3 and its corresponding nonionic prodrug, TE3, were obtained from the Laboratoire des Aminoacides, Peptides et Proteines, CNRS UMR 5810 (Montpellier I and II University, Montpellier, France) (Fig. 1). Synthesis procedures and structural analyses of T3 and TE3 were recently described (31, 34). From in vitro experiments, during TE3-T3 bioconversion studies, a compound named mTE3 with only one positive charge was detected (34). This compound has been synthesized (Fig. 1), and its structure has been confirmed by liquid chromatography-mass spectrometry. All blood samples collected after TE3 administration were evaluated for the presence of mTE3.
FIG. 1.

Chemical structures of T3 (active compound; molecular weight, 614.7), mTE3 (intermediate metabolite; molecular weight, 577), and TE3 prodrug (molecular weight, 540.7).
In vitro and in vivo antimalarial activities.
The Nigerian strain of Plasmodium falciparum was maintained in culture in complete medium (RPMI 1640 containing 25 mM HEPES buffer, pH 7.4, and 10% type AB-positive human serum) by the method of Trager and Jensen (27). The antimalarial activities of the compounds against the in vitro growth of P. falciparum were determined as described by Desjardins et al. (12) by using [3H]hypoxanthine incorporation as an assessment of parasite growth. Stock drug solutions were prepared in complete medium (T3) or in 100% dimethyl sulfoxide (DMSO) (TE3) at a concentration of 10−4 mM and were serially diluted to the appropriate concentration with complete medium. Assays were performed in sterile 96-well microtiter plates. The final volume in each well was 200 μl, consisting of 50 μl of complete medium without (controls) or with drug and 150 μl of P. falciparum-infected erythrocyte suspension (1.5% final hematocrit and 0.6% parasitemia; DMSO concentration, <0.2%). After 48 h of incubation at 37°C, 30 μl of complete medium containing 0.7μCi [3H]hypoxanthine was added to each well. After 18 h at 37°C, the cells were lysed with an automatic cell harvester; and the parasite macromolecules, including the radioactive nucleic acids, were harvested onto glass fiber filters. The filters were counted for radioactivity in a liquid scintillation spectrometer. The radioactivity background was obtained by incubation of noninfected erythrocytes under the same conditions and was deducted. Each drug was tested in duplicate at least three times with different batches of cells, and the parasite growth was compared to that in the control wells, which constituted 100% parasite growth. Parasitic viability was expressed as the drug concentration that led to 50% parasite growth inhibition (IC50).
In vivo antimalarial activity against a P. vinckei petteri strain (279BY) was determined in female Swiss mice (3). The mice were inoculated intravenously with 107 parasitized red blood cells (resuspended in 200 μl of 0.9% sodium chloride). The T3 and TE3 compounds were administered both i.p. and orally, with each compound given once daily for 4 consecutive days. T3 was dissolved in 100 μl of 0.9% sodium chloride, and TE3 was dissolved in 100 μl of DMSO. Control mice received only the vehicle. The parasitemia levels were monitored in Giemsa-stained blood smears, and blood samples were collected for determination of parasitemia levels on a fluorescence-activated cell sorter (5). The 50% efficient dose (ED50), which is the dose that led to 50% parasite growth inhibition compared to the parasite growth in the control (treated with an equal volume of vehicle), was determined on the day following the last treatment. All results are expressed as means ± standard errors of the means (SEMs).
Pharmacokinetic study. (i) Animals.
Two hundred thirty-seven Sprague-Dawley rats (weight, 226 to 398 g) purchased from Charles River (L'Arbresle Cedex, France) at age 10 weeks were used. They were kept under conditions of constant temperature (21 to 25°C) and relative humidity of approximately 40 to 65% with a standard light-dark cycle. The animals were group housed in stainless steel cages with suspended wire-mesh floors (maximum of five rats per cage). The rats were fed a standard rodent diet (UAR sterile food; Usine d'Alimentation Rationnelle, Villemoisson, Epinay s/Orge, France) and allowed free access to tap water. All animals underwent a period of 2 weeks of observation and acclimatization before treatment. The rats were fasted overnight (12 h) before drug administration and then weighed.
(ii) Dosing.
The rats were randomly distributed into six experimental groups. In the first three groups, rats received the T3 compound at the following doses (expressed in the form of bis-charged compound): (i) 1.3 mg/kg of body weight by a short-term i.v. infusion (2 min), (ii) 3 mg/kg of body weight by a short-term i.v. infusion (2 min), and (iii) 6.4 mg/kg of body weight by the i.p. route. The last three groups received the TE3 prodrug by i.v. infusion (2 min) at 1.5 mg/kg (equivalent to 1.26 mg/kg of T3) and 3 mg/kg (equivalent to 2.52 mg/kg of T3) and by the oral route (12 mg/kg, equivalent to 10.1 mg/kg of T3). To obtain these doses, three different solutions of the T3 compound were prepared in 0.9% sterile isotonic saline on the day of administration: 0.75 and 1.5 mg/ml for i.v. administration and 1.28 mg/ml for i.p. administration. As the TE3 prodrug is poorly soluble in water, dimethyl sulfoxide (DMSO) was used to dissolve this drug, according to previously published data (17, 18). Thus, TE3 solutions were prepared in a mixture of DMSO-0.9% sterile isotonic saline (75/25; vol/vol) for i.v. administration (0.3 and 0.6 mg/ml) and in pure DMSO for oral administration (4 mg/ml). These solutions were used to treat the animals, and the volume administered was 2 to 5 ml/kg. Two minutes before drug administration, the rats were slightly anesthetized with diethyl ether.
For i.v. administration, the dose was administered into the penis vein. For oral administration, the drug was given by gavage through stomach tubing by using a polypropylene catheter.
Doses were chosen according to the 50% lethal dose (LD50) of the T3 compound after i.v. and i.p. administration (i.e., 5.3 and 18 mg/kg, respectively, expressed in the form of doubly charged species).
(iii) Blood sampling.
Blood samples were obtained at the following times: 5, 10, and 30 min and 1, 2, 4, 8, 12, 24, and 36 h after dosing. At each of these times, samples were collected from three to six rats for each dose. One or two blood samples were drawn per animal. The samples were collected from the tongue vein or after the animal was killed by sectioning of the carotid artery, and the samples were placed in heparinized polypropylene tubes (0.1 ml lithium heparinate per tube) and then immediately placed on ice. The blood samples were centrifuged (3,000 × g) at 4°C for 6 min. The plasma was separated and then immediately stored at −80°C until it was assayed. Special attention was paid to sample handling after TE3 administration. Indeed, in blood at 37°C, rapid TE3-T3 conversion occurred (initial conversion half-life [t1/2], about 5 min; after 30 min, about 94% of the TE3 had been converted to T3) (34). The conversion rate decreased at a lower temperature. At 4°C the conversion half-life was almost 3 h; after 15 min, the mean percent recovery was 92 to 97% (20). Thus, blood must be collected in tubes, placed in an ice water bath, and then immediately centrifuged at 4°C (20). Thereafter, acidification of plasma samples with trifluoroacetic acid avoided the TE3-T3 conversion during the freezing and thawing steps.
Three to six untreated animals per experiment group were killed to provide predose blood samples.
(iv) Preliminary pharmacokinetic analysis.
A preliminary independent analysis was carried out from the average concentration values at each time point by using Pk-fit software (13, 23). Such an analysis allowed estimation of (i) the pharmacokinetic model and (ii) initial pharmacokinetic parameters that will be used in the population analysis. Moreover, the area under the mean plasma drug concentration-versus-time curve extrapolated to infinity (AUC0-∞) was estimated by using the log-linear trapezoidal method. Bioavailability was computed from the ratio (AUC0-∞)i.p. or per os./(AUC0-∞)i.v. after normalization to the same administered dose.
(v) Population pharmacokinetic analysis.
Individual pharmacokinetic parameters were then estimated by using an empirical Bayes methodology (25). In this analysis, the population characteristics of the parameters to be estimated were used as prior information to estimate each individual pharmacokinetic parameter. Plasma concentrations less than the limit of quantitation were omitted from the analysis.
The population analysis was performed by the nonlinear mixed-effects modeling approach, as implemented in the NONMEM computer program (6) with the graphical interface Visual-NM (22). The population characteristics of the pharmacokinetic parameters (fixed and random effects) were estimated by using the subroutines ADVAN-4 and TRANS-1 from the library of programs provided with the NONMEM-PREDPP package. Both, first-order and first-order conditional estimation methods were used to estimate population pharmacokinetic parameters. The estimation was markedly improved by use of the first-order method. The following structural models with first-order absorption and elimination rate constants were tested: (i) a two-compartment model and (ii) a three-compartment model. A model that included T3 formation from TE3 was also tested. The structural model was chosen on the basis of changes in the −2 log likelihood and qualitative assessment of diagnostic plots. Because the −2 log likelihood is approximately χ2 distributed and the addition of one compartment increases the degrees of freedom by 2, a change of 5.99 in the −2 log likelihood was required at the 5% significance level for selection of the more complex model.
Deviations of pharmacokinetic parameters of the jth individual animal from the estimated population average values were modeled with the use of an exponential interindividual variability error model:
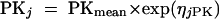 |
(1) |
where PKj is the required pharmacokinetic parameter in the jth individual rat, and ηjPK is a random variable distributed with zero mean and variance of ω2PK about the average value (PKmean) for the population. NONMEM also estimated the residual variance among pairs of observed and model-predicted data. Various error models were tested. The error for the concentration measurements of individual animal j was best described by a combined additive and proportional model for the T3 compound, described as
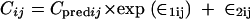 |
(2) |
and by a proportional model for the TE3 prodrug, described as
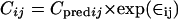 |
(3) |
where Cij is the ith observed concentration for the jth individual, Cpredij is the plasma concentration predicted by the pharmacokinetic model, and ∈ij (the difference between Cij and Cpredij) is a randomly distributed variable with zero mean and a variance of σ2. Such error arises from factors such as assay variability, model misspecification, inaccurate recording of dosing or sampling times, and intraindividual pharmacokinetic variability.
The uncertainty (coefficient of variation) in the estimation of the fixed and random parameter values was determined by expressing the standard error of estimation (calculated in NONMEM) as a percentage of the estimated value.
The individual predicted plasma concentrations (CIPREDs) were computed for each individual by using the empirical Bayes estimate of the pharmacokinetic parameters with the POSTHOC option in the NONMEM program.
At each step of the model building, diagnostic plots were analyzed for their closeness to and randomness along the line of identity on the observed (DV) versus the predicted (PRED) and individual predicted (IPRED) concentration plots, as well as for their randomness along the residual (DV-PRED) and weighted residual zero line on the predicted concentration or time versus residual or weighted residual plot. PRED concentrations were computed based on population parameter estimates; IPRED concentrations were computed based on individual parameter estimates. The model was accepted when (i) the plots showed no systematic pattern and (ii) descriptive statistics did not show any systematic deviation from the initial hypothesis.
Assay method.
T3, TE3, and mTE3 plasma levels were determined by a liquid chromatography-electrospray ionization mass spectrometry method (19, 20). Verapamil was used as the internal standard. The precision ranged from 3.2 to 10.5%, and the accuracy was between 90 and 114%. The lower limits of quantitation were 6.4 ng/ml for T3, 10 ng/ml for TE3, and 20 ng/ml for mTE3.
Samples from the animals were processed by use of a standard curve, and quality control samples were included in each analytical sequence to verify the stability of the study samples during storage and the accuracy and the precision of the analysis.
RESULTS
In vitro and in vivo antimalarial activities.
The T3 and TE3 compounds were tested in vitro against the Nigerian strain of P. falciparum. Figure 2 shows the dose-response curves, obtained by monitoring [3H]hypoxanthine uptake as an index of growth. Both compounds exerted potent inhibition of the growth of this human parasite. Inhibition was complete and occurred in a narrow range of concentrations that covered less than 2 log units of drug concentrations. The mean IC50 values (±SEMs) that indicate the drug concentrations that reduce cell growth by up to 50% of that of the untreated control following one cycle were 2.4 ± 0.12 (n = 9) and 2.78 ± 0.07 nM (n = 5) for the T3 drug and the TE3 prodrug, respectively. We have already reported that the intrinsic antimalarial activity of the prodrug ingredient is absent at these concentrations and that the antimalarial activity of the prodrug is due to its rapid quantitative conversion into the drug in the presence of the plasmatic esterases (35). The compounds are more active than chloroquine, the most used antimalarial (Fig. 2), and potent like artemisinin or artemisinin-like compounds, halofantrine and atovaquone (data not shown).
FIG. 2.

Comparative in vitro antimalarial activities against P. falciparum (Nigerian strain) of the bisthiazolium compound (T3), its bioprecursor (TE3), and chloroquine (CQ). The parasites were incubated with T3, TE3, or chloroquine for 48 h before the addition of [3H]hypoxanthine to determine cell viability. Values are the means ± SEMs of three independent experiments, each performed in duplicate.
We next examined the antimalarial activities of T3 and TE3 in vivo in mice infected with Plasmodium vinckei. Figure 3 shows the dose-response curves obtained after i.p. and oral treatment with one daily dose of T3 or TE3 for 4 consecutive days. Both compounds exerted potent inhibition of the in vivo growth of this rodent parasite. Similarly to the in vitro activity, inhibition occurred in a very narrow range of concentrations, covering less than 2-log dose concentrations. After i.p. administration, T3 and TE3 had outstanding antimalarial activities (ED50s, 0.2 to 0.25 mg/kg). After oral administrations, the ED50 values were 14 ± 1.7 (n = 3) and 5.00 ± 0.28 mg/kg (n = 3) for the T3 drug and the TE3 prodrug, respectively. DMSO alone did not alter the malarial infection in mice.
FIG. 3.

In vivo antimalarial activities of T3 and TE3 against P. vinckei-infected mice. Mice were infected i.v. with 107 parasites on day 0. Treatments started on day 1 and consisted of either a once-daily i.p. dose (filled symbols) or oral dose (open symbols) for 4 consecutive days. The results presented here are those of a typical experiment, and the values are the means ± SEMs for at least four mice per dosage. Each curve is representative of two independent tests. Squares, T3; circles, TE3.
Pharmacokinetic parameters. (i) Preliminary pharmacokinetic analysis.
Semilogarithmic plots of the mean (±standard deviation) T3 plasma concentration-time profiles are illustrated in Fig. 4A and B after T3 and TE3 administrations, respectively. When the concentrations were below the lower limit of quantitation of the analytical method, the mean values were obtained from values above the limit of quantitation, with the other values taken as zero.
FIG. 4.

Mean (± standard deviation) T3 plasma concentration-versus-time curve after T3 (A) (▪, 1.3 mg/kg i.v.; ▴, 3 mg/kg i.v.; •, 6.4 mg/kg i.p.) and TE3 (B) (▪, 1.5 mg/kg i.v.; ▴ 3 mg/kg i.v.; •, 12 mg/kg orally) administration to rats.
Five minutes after intravenous administrations of the T3 compound (1.3 and 3 mg/kg), plasma concentrations were 3,040 ± 886 and 4,770 ± 1,570 ng/ml, respectively. After intraperitoneal administration (6.4 mg/kg), the mean maximum concentration in plasma (Cmax) was 4,819 ± 952 ng/ml, and the time required to achieve Cmax was 10 min.
After TE3 administration, whatever the route of administration, rapid biotransformation of prodrug-drug occurred. TE3 and mTE3 were detected at concentrations below the lower limit of quantitation of the analytical method in the first two blood samples collected after intravenous administration. These two compounds were never detected after oral administration. The T3 plasma concentrations observed 5 min after intravenous administration (1.5 and 3 mg/kg) were 2,141 ± 489 and 5,740 ± 2,906 ng/ml, respectively. After oral administration (12 mg/kg), the Cmax of 5,916 ± 2,947 ng/ml was reached after 10 min.
Rapid absorption occurred after both intraperitoneal administration of T3 and oral administration of TE3.
The bioavailability was 72% after intraperitoneal administration of the T3 compound and 16% after oral administration of the TE3 prodrug. The elimination half-life (t1/2elim) of the active bisthiazolium compound, T3, was 12 h.
(ii) Population analysis.
After administration of T3 and TE3, a two-compartment model with first-order absorption and elimination was optimal for modeling of the data. Substantial misfit of the predicted concentrations versus the individual observed concentrations occurred, as determined by using a structural model that included T3 formation from TE3. The six dimensional vectors, θ's, of the pharmacokinetic parameters considered in the population analysis consisted of clearance (θ1 = CL), initial volume of distribution (θ2 = V1), transfer rate constants (θ3 = k12 and θ4 = k21), the absorption rate constant (θ5 = ka), and bioavailability (θ6 = F).
After T3 administration, a total of 131 concentrations were used to compute population parameters. The results are presented in Table 1. V1 and CL were estimated to be 0.34 liter/kg and 1.1 liters/h/kg, respectively. The following secondary pharmacokinetic parameters, C2 min (end of infusion), Cmax (model-predicted maximum concentration after intraperitoneal administration), AUC0-∞, V (steady-state volume of distribution), and t1/2elim were calculated from the primary pharmacokinetic parameters (CL, V1, k12, k21, ka, and F) and are given in Table 2.
TABLE 1.
Population pharmacokinetic parameters of the T3 compound after T3 and TE3 administrationsa
| Parameter | T3 administration
|
TE3 administration
|
||
|---|---|---|---|---|
| Mean value | CV (%) | Mean value | CV (%) | |
| CL (liter/h) | 0.30 (7.50) | 24.6 (33.7) | 0.26 (15.1) | 37.6 (18.5) |
| V1 (liter) | 0.0853 (14.0) | 37.3 (40.8) | 0.103 (8.8) | 47.1 (19.1) |
| k12 (h−1) | 1.80 (14.3) | 50.3 (32.8) | 2.44 (10.0) | 42.5 (27.5) |
| k21 (h−1) | 0.076 (15.0) | 58.1 (22.1) | 0.107 (15.2) | 50.0 (37.0) |
| ka (h−1) | 2.20 (19.5) | 37.2 (31.2) | 4.71 (14.7) | 67.2 (21.8) |
| F | 0.72 (20.6) | 49.4 (15.3) | 0.15 (3.3) | 60.1 (31.3) |
T3 was administered by the intravenous and intraperitoneal routes, while TE3 was administered by the intravenous and oral routes. CV, coefficient of variation. The standard errors of the estimates (expressed as the coefficient of variation [in percent]) are given in parentheses. The intraindividual variabilities were 23.4% and 9.3% for T3 and 16.6% for TE3. The values of the objective function were 1,384 and 1,799 for T3 and TE3, respectively.
TABLE 2.
Secondary parameters calculated by Bayesian estimation from primary parameters after T3 administrationa
| Parameter | Result after administration of the following dose:
|
||
|---|---|---|---|
| 1.3 mg/kg, i.v. | 3 mg/kg, i.v. | 6.4 mg/kg, i.p. | |
| C2 min (μg/liter) | 4,417 ± 1,342 | 7,889 ± 1,367 | |
| Cmax (μg/liter) | 5,390 ± 1,086 | ||
| AUC0-∞ (mg · h/liter) | 1.40 ± 0.38 | 2.60 ± 0.30 | 5.0 ± 0.88 |
| t1/2elim (h) | 12.1 ± 2.92 | 12.8 ± 2.51 | 13.3 ± 2.82 |
| V (liters/kg) | 16.3 ± 3.85 | 15.2 ± 3.35 | 14.3 ± 2.53 |
T3 was administered by the i.v. and i.p. routes. The values are presented as means ± standard deviations.
After TE3 administration, a total of 138 T3 concentrations were used to compute population parameters. The results are presented in Table 1. The pharmacokinetic parameters were similar to those computed after T3 administration. Total clearance was 0.9 liter/h/kg; V1 was 0.4 liter/kg. The mean values of the secondary pharmacokinetic parameters, calculated by Bayesian estimation, are given in Table 3.
TABLE 3.
Secondary parameters calculated by Bayesian estimation from primary parameters after TE3 administrationa
| Parameter | Result after administration of the following dose:
|
||
|---|---|---|---|
| 1.5 mg/kg, i.v. | 3 mg/kg, i.v. | 12 mg/kg, per os | |
| C2 min (μg/liter) | 3,310 ± 696 | 7,497 ± 1,248 | |
| Cmax (μg/liter) | 5,916 ± 2,947 | ||
| AUC0-∞ (mg · h/liter) | 1.60 ± 0.27 | 3.35 ± 0.44 | 2.23 ± 0.98 |
| t1/2elim (h) | 11.2 ± 2.80 | 11.0 ± 2.50 | 12.5 ± 2.39 |
| V (liters/kg) | 12.4 ± 3.26 | 16.2 ± 2.70 | 14.2 ± 2.45 |
TE3 was administered by the i.v. and oral routes. The values are presented as means ± standard deviations.
(iii) Model acceptance.
Weighted residual plots did not show any pattern, and plots representing population or individual predictions versus the observed data did not show either any substantial or systematic deviations from the line of identity (Fig. 5 and 6 for the data obtained after T3 and TE3 administration, respectively). Overall, the proposed model correctly described the data. The mean weighted residual values of −0.006 (T3 administration) and −0.015 ng/ml (TE3 administration) were not significantly different from 0 (Student t test). In a last step, the individual predicted concentrations were compared to the observed concentrations by computing the bias. The results are as follows: (i) for T3 administration, bias, 9.87 ng/ml; 95% confidence interval, −14.1 to 33.8; (ii) for TE3 administration, bias, 4.03 ng/ml; 95% confidence interval, −10.3 to 18.4.
FIG. 5.

Model performance and diagnostic plots after T3 administration. (A) Individual predicted concentrations (○) (slope, 0.990 [95% confidence interval, 0.97 to 1.01]; intercept, 3.64 ng/ml [95% confidence interval, −24.2 to 31.5]) and predicted concentrations (•) versus observed concentrations. The solid line represents the line of identity. (B) Weighted residuals versus model-predicted concentrations based on population parameter estimates.
FIG. 6.

Model performance and diagnostic plots after TE3 administration. (A) Individual predicted concentrations (□) (slope, 0.991 [95% confidence interval, 0.98 to 1.03]; intercept, 2.22 ng/ml [95% confidence interval, −13.2 to 17.7]) and predicted concentrations (▪) versus observed concentrations. The solid line represents the line of identity. (B) Weighted residuals versus model-predicted concentrations based on population parameter estimates.
DISCUSSION
In the battle against malaria, a novel pharmacological approach that uses choline analogues that interfere with the de novo CDP-choline-dependent phosphatidylcholine biosynthetic pathway has been developed by our group. Thus, quaternary ammonium salts that mimic the choline structure and that target plasmodial phosphatidylcholine biosynthesis of the erythrocytic stage of P. falciparum were optimized for their antimalarial activities. We discovered that these compounds also interact with malarial pigments, thus enhancing the antimalarial effect (7). Our studies led to the development of novel classes of compounds that have in vitro antimalarial activity in the low nanomolar range and that possess potent antimalarial activity in both rodent and primate models of malaria (at doses lower than 1 mg/kg) (3, 4, 10). One prominent feature of these compounds was their ability to specifically accumulate inside their target cells. However, these compounds have low oral bioavailabilities. Oral administration appears to be essential for dispensaries in countries where malaria is endemic that often do not have adequate facilities to safely give drug injections and is also required for prophylactic or curative treatments for travelers. Therefore, we recently synthesized neutral prodrugs that are rapidly converted to the active drugs in vivo (34).
In the present study, (i) we determined the in vitro and in vivo antimalarial activities of two compounds from this new series of drugs, a bisthiazolium compound (T3) and its corresponding prodrug (TE3), and (ii) we studied the pharmacokinetic profiles of the two compounds in Sprague-Dawlay rats after single i.v. or i.p. administration (T3) and after single i.v. or oral administration (TE3). Based on the concentrations that inhibited P. falciparum growth by 50%, T3 and TE3 had antimalarial activities more potent than that of chloroquine. Evaluation of the activities of the compounds against the lethal rodent malaria, Plasmodium vinckei, revealed ED50 values as low as 0.3 mg/kg after i.p. administration and 13 and 5 mg/kg for the T3 drug and the TE3 prodrug after oral administration, respectively. Thus, T3 and TE3 possess very potent antimalarial activities that were higher than those of the currently used antimalarial drugs (34).
During this study the pharmacokinetic profiles of T3 and TE3 were investigated in rats. One to two blood samples were drawn per animal. In this condition, a naive pooled data approach can be used to estimate pharmacokinetic parameters, but under the misleading assumption that all data come from the same animal. So it is not possible to draw conclusions on the interindividual variability in pharmacokinetic parameters (8). Thus, in this context, the nonlinear mixed-effects approach provides a valuable alternative to classical noncompartmental pharmacokinetic analysis (1, 9) for suitable estimation of the values of the pharmacokinetic parameters for these two compounds. This mixed-effect modeling approach may appear to be complex compared with the complexities of conventional methods; but the method has several advantages, such as (i) the possibility of the application of this method on the basis of either rich or sparse data, (ii) the ability to estimate individual parameters by using Bayesian feedback (15), and (iii) the ability to obtain an accurate estimate of interindividual variability. In addition, all of this information can be used to interpret toxicological and pharmacological end points, to compare the characteristics of products that are following in the development, and to integrate the results with those of other in vitro-in vivo studies to supply a comprehensive pharmacokinetic behavior before the first use of the compounds in studies with humans.
Over the sampling times monitored, plasma concentration-versus-time profiles were compatible with a two-compartment model and first-order pharmacokinetics. Rapid prodrug-active drug conversion occurred after TE3 administration (i.v. or oral). TE3 and mTE3 were detectable only at concentrations below the lower limit of quantitation of the analytical method in the first blood samples drawn after i.v. administration. This rapid conversion has also been observed in vitro. Indeed, from in vitro experiments carried out in human plasma diluted to 80% with 0.16 M sodium phosphate buffer (pH 7.4), TE3 (15 μM) decreased concomitantly with the appearance of T3, with an initial conversion half-life of about 5 min. After 30 min, about 94% of the TE3 had been converted (34). After i.v. administration of the T3 and TE3 compounds, similar pharmacokinetic profiles were noted. The T3 distribution was rapid (mean half-life, 7 min); and the AUC under the distributive phase represented about 35% of the total AUC. After i.p. administration of the T3 compound, rapid absorption occurred, the mean absorption half-life (t1/2, ka) was 19 min, with 72% bioavailability. After oral administration of the TE3 prodrug, the absorption was rapid (t1/2, ka = 9 min) and the Cmax of the T3 compound of 5,916 ng/ml was reached 10 min after administration, with a mean bioavailability of 15%. T3 plasma concentrations (8 nM) 24 h following oral administration of TE3 are higher than the IC50 values of chloroquine-resistant strains of P. falciparum (6.3 nM) (34). The mean values of the pharmacokinetic parameters for T3, the bisthiazolium active compound, in rats were as follows: CL, 1 liter/h/kg; V, 14.8 liters/kg; and t1/2elim, 12 h. After intravenous administration of the range of doses studied, the pharmacokinetics appeared to be linear.
In conclusion, this study generated substantial information about the pharmacology of a compound of this class of antimalarial in rat. As this study was conducted with healthy animals, it was not possible to correlate plasma levels of this drug with a pharmacological response. Future studies will explore the pharmacodynamic-pharmacokinetic relationships in infected animals.
Acknowledgments
This study was supported by the European Community (QLK2-CT-2000-01166), Ministère de l'Education Nationale et Recherche Scientifique (PAL+).
REFERENCES
- 1.Aarons, L. 1993. Sparse data analysis. Eur. J. Drug Metab. Pharmacokinet. 1:97-100. [DOI] [PubMed] [Google Scholar]
- 2.Ancelin, M. L., M. Calas, J. Bompart, G. Cordina, D. Martin, M. Ben Bari, T. Jei, P. Druilhe, and H. J. Vial. 1998. Antimalarial activity of 77 phospholipid polar head analogs: close correlation between inhibition of phospholipid metabolism and in vitro Plasmodium falciparum growth. Blood 91:1426-1437. [PubMed] [Google Scholar]
- 3.Ancelin, M. L., M. Calas, A. Bonhoure, S. Herbute, and H. J. Vial. 2003. In vivo antimalarial activities of mono- and bis quaternary ammonium salts interfering with Plasmodium phospholipid metabolism. Antimicrob. Agents Chemother. 47:2598-2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ancelin, M. L., M. Calas, V. Vidal-Sailhan, S. Herbute, P. Ringwald, and H. J. Vial. 2003. Potent inhibitors of Plasmodium phospholipid metabolism with a broad spectrum of in vitro antimalarial activities. Antimicrob. Agents Chemother. 47:2590-2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barkan, D., H. Ginsburg, and J. Golenser. 2000. Optimisation of flow cytometric measurement of parasitaemia in Plasmodium-infected mice. Int. J. Parasitol. 30:649-653. [DOI] [PubMed] [Google Scholar]
- 6.Beal, S. L., and L. B. Sheiner. 1994. NONMEM user's guide. University of California at San Francisco, San Francisco.
- 7.Biagini, A. G., E. Richier, P. G. Bray, M. Calas, H. Vial, and S. A. Ward. 2003. Heme binding contributes to antimalarial activity of bis-quaternary ammoniums. Antimicrob. Agents Chemother. 47:2584-2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bois, F. Y. 1999. Analysis of PBPK models for risk characterization, p. 317-337. In A. J. Bailer, C. Maltoni, J. C. Bailar III, F. Belpoggi, J. V. Braizer, and M. Soffrintti (ed.), Uncertainty in the risk assessment of environmental and occupational hazards. New York Academy of Sciences, New York, N.Y. [DOI] [PubMed]
- 9.Burtin, P., F. Mentré, J. Van Bree, and J. L. Steimer. 1996. Sparse sampling for assessment of drug exposure in toxicological studies. Eur. J. Metab. Pharmacokinet. 21:105-111. [DOI] [PubMed] [Google Scholar]
- 10.Calas, M., M. L. Ancelin, G. Cordina, P. Portefaix, G. Piquet, V. Vidal-Sailhan, and H. Vial. 2000. Antimalarial activity of compounds interfering with Plasmodium falciparum phospholipid metabolism: comparison between mono- and bisquaternary ammonium salts. J. Med. Chem. 43:505-516. [DOI] [PubMed] [Google Scholar]
- 11.Calas, M., G. Cordina, J. Bompart, M. Ben Bari, T. Jei, M. L. Ancelin, and H. Vial. 1997. Antimalarial activity of molecules interfering with Plasmodium falciparum phospholipid metabolism. Structure-activity relationship analysis. J. Med. Chem. 40:3557-3566. [DOI] [PubMed] [Google Scholar]
- 12.Desjardins, R. E., C. J. Canfield, J. D. Haynes, and J. D. Chulay. 1979. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 16:710-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farenc, C., J. R. Fabreguette, and F. Bressolle. 2000. Pk-fit: a pharmacokinetic/pharmacodynamic and statistical data analysis software. Comput. Biomed. Res. 33:315-330. [DOI] [PubMed] [Google Scholar]
- 14.Greenwood, B., and T. Mutabingwa. 2002. Malaria in 2002. Nature 415:670-672. [DOI] [PubMed] [Google Scholar]
- 15.Hing, J. P., S. G. Woolfrey, P. Greenslade, and P. M. C. Wright. 2001. Is mixed effect modeling or naïve pooled data analysis preferred for interpretation of single sample per subject toxicokinetic data? J. Pharmacokinet. Biopharm. 28:193-210. [DOI] [PubMed] [Google Scholar]
- 16.Holz, G. G. 1977. Lipids and the malaria parasite. Bull. W. H. O. 55:237-248. [PMC free article] [PubMed] [Google Scholar]
- 17.Mottu, F., A. Laurent, D. A. Rufenacht, and E. Doelker. 2000. Organic solvents for pharmaceutical parenterals and embolic liquids: a review of toxicity data. PDA J. Pharm. Sci. Technol. 54:456-469. [PubMed] [Google Scholar]
- 18.Mottu, F., M. J. Stelling, D. A. Rufenacht, and E. Doelker. 2001. Comparative hemolytic activity of undiluted organic water-miscible solvents for intravenous and intra-arterial injection. PDA J. Pharm. Sci. Technol. 55:16-23. [PubMed] [Google Scholar]
- 18a.National Institutes of Health. 1985. Principles of laboratory animal care. NIH publication 85-23, revised. National Institutes of Health, Bethesda, Md.
- 19.Nicolas, O., C. Farenc, M. Calas, H. Vial, and F. Bressolle. 2005. Quantitation of antimalarial bis-thiazolium compounds and neutral bioprecursor in plasma by liquid chromatography-electrospray mass spectrometry. Clin. Chem. 51:593-603. [DOI] [PubMed] [Google Scholar]
- 20.Nicolas, O., D. Margout, N. Taudon, M. Calas, H. Vial, and F. Bressolle. 2005. Liquid chromatography-electrospray mass spectrometry determination of a bis-thiazolium compound with potent antimalarial activity and its neutral bioprecursor in human plasma, whole blood and red blood cells. J. Chromatogr. B 820:83-93. [DOI] [PubMed] [Google Scholar]
- 21.Olliaro, P. L., and Y. Yuthavong. 1999. An overview of chemotherapeutic targets for antimalarial drug discovery. Pharmacol. Ther. 81:91-110. [DOI] [PubMed] [Google Scholar]
- 22.Research Development in Population Pharmacokinetics. 1998. Visual-NM user's manual, version 5.1. Research Development in Population Pharmacokinetics, Montpellier, France.
- 23.Research Development in Population Pharmacokinetics. 2000. Pk-fit software, version 2.1. Research Development in Population Pharmacokinetics, Montpellier, France.
- 24.Ridley, R. G. 2002. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature 415:686-693. [DOI] [PubMed] [Google Scholar]
- 25.Sheiner, L. B., B. Rosenberg, and K. L. Melmon. 1972. Modelling of individual pharmacokinetics for computer-aided drugs dosage. Comput. Biomed. Res. 5:411-459. [DOI] [PubMed] [Google Scholar]
- 26.Sherman, L. 1979. Biochemistry of Plasmodium (malarial parasites). Microbiol. Rev. 43:453-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trager, W., and J. B. Jensen. 1976. Human malaria parasites in continuous culture. Science 193:673-675. [DOI] [PubMed] [Google Scholar]
- 28.Van Deenen, L. L. M., and J. De Gier. 1975. Lipids of the red cell membrane, p. 147-211. In G. Surgenor (ed.), The red blood cell. Academic Press, Inc., New York, N.Y.
- 29.Vial, H. 1996. Recent developments and rationale towards new strategies for malarial chemotherapy (a large review). Parasite 3:3-23. [DOI] [PubMed] [Google Scholar]
- 30.Vial, H., and M. L. Ancelin. 1998. Malarial lipids, p. 159-175. In I. W. Sherman (e.d.), Malaria: parasite biology, biogenesis, protection. ASM Press, Washington, D.C.
- 31.Vial, H., M. Calas, J. J. Bourguignon, M. L. Ancelin, V. Vidal, and E. Rubi. Jan. 2001. Précurseurs de sels de bis-ammonium quaternaire et leurs applications comme prodrogues ayant une activité antiparasitaire. Patent 99 09471 applied by CNRS, 21 July 1999, WO 0105742 A1, published 25 January 2001.
- 32.Vial, H., P. Eldin, L. Tielens, and J. van Hellemond. 2003. Phospholipids in parasitic protozoa. Mol. Biochem. Parasitol. 126:143-154. [DOI] [PubMed] [Google Scholar]
- 33.Vial, H. J., and M. Calas. 2001. Inhibitors of phospholipid metabolism, p. 347-365. In P. J. Rosenthal (ed.), Antimalarial chemotherapy, mechanism of action, modes of resistance, and new directions in drug development. Humana Press, Totowa, N.J.
- 34.Vial, H. J., S. Wein, C. Farenc, C. Kocken, O. Nicolas, M. L. Ancelin, F. Bressolle, A. Thomas, and M. Calas. 2004. Prodrugs of bisthiazolium salts are orally potent antimalarials. Proc. Natl. Acad. Sci. USA 101:15458-15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wengelnik, K., V. Vidal, M. L. Ancelin, A. M. Cathiard, J. L. Morgat, C. H. Kocken, M. Calas, S. Herrera, A. W. Thomas, and H. J. Vial. 2002. A class of potent antimalarials and their specific accumulation in infected erythrocytes. Science 295:1311-1314. [DOI] [PubMed] [Google Scholar]