

Abstract
Linezolid is a new antimicrobial agent effective against drug-resistant gram-positive pathogens which are common causes of infections in hospitalized patients. Many such patients rely on the intravenous or enteral route for nutrition and drug administration. Therefore, the bioavailability of linezolid administered enterally in the presence of enteral feedings in hospitalized patients was examined. Eighteen subjects were assessed in a randomized single-dose crossover study; 12 received continuous enteral feedings, while 6 did not (controls). Both groups received linezolid 600 mg intravenously and orally (control) or enterally, with the alternate route of administration separated by a 24-h washout period. Pharmacokinetic parameters derived from noncompartmental and compartmental analysis incorporating linear and nonlinear elimination pathways were compared between groups: F, Ka, Vs, K23, K32, Vmax, Km, and K20 (bioavailability, absorption rate constant, volume of central compartment normalized to body weight, intercompartmental rate constants, maximum velocity, Michaelis-Menten constant, and elimination rate constant, respectively). Pharmacokinetic (PK) data were available from 17 patients. The linezolid oral suspension was rapidly and completely absorbed by either the oral or enteral route of administration. Bioavailability was unaltered in the presence of enteral feedings. PK estimates remain similar regardless of the model applied. At the therapeutic dose used, only slight nonlinearity in elimination was observed. A linezolid oral suspension may be administered via the enteral route to hospitalized patients without compromise in its excellent bioavailability and rapid rate of absorption. Compartmental pharmacokinetic analysis offers a more flexible study application, since bioavailability (F) can be estimated directly with intermixed intravenous/oral doses without a need for a washout period.
Gram-positive pathogens, such as Staphylococcus aureus and enterococci, have become the leading pathogens causing nosocomial infections. Many of these strains have acquired resistance against multiple classes of antimicrobial agents commonly prescribed for the treatment of infections. According to the most recent data from the National Nosocomial Infection Surveillance from the Centers for Disease Control, more than 50% of S. aureus strains causing infections in hospitalized patients are resistant to methicillin (MRSA), while enterococcal strains resistant to vancomycin (VRE) are endemic in many hospitals. In addition, infections involving MRSA are a growing problem not only in the acute and long-term care settings but in the community as well (3).
Linezolid is the first Food and Drug Administration-approved agent representing a new class of antibiotics, the oxazolidinones (9). It is active primarily against gram-positive organisms including MRSA and VRE. Linezolid has been demonstrated to be effective in the treatment of skin and skin structure infections as well as nosocomial and community-acquired pneumonia in clinical trials. It is available in both parenteral and oral dosage forms which allow administration of drug to both hospitalized and outpatient populations.
The pharmacokinetics (PK) of linezolid has been extensively evaluated in healthy volunteers and in patients enrolled in compassionate use programs (6, 7, 13, 14, 16, 18). Results of these analyses consistently demonstrate an essentially complete oral bioavailability for linezolid and without significant alteration by food intake. The excellent oral absorption characteristic of linezolid is particularly attractive as the enteral route is increasingly used by clinicians to administer both nutrition and drug therapy to hospitalized as well as long-term care residents in lieu of the parenteral route. In addition, conversion of intravenous (IV) to oral therapy facilitates early discharge of hospitalized patients and minimizes IV catheter-related complications and health care costs. However, drug absorption in the presence of enteral feedings (EF) may be altered from that observed in healthy volunteers. A significant decrease in bioavailability has been observed for certain medications when EF is concomitantly administered (1, 4, 8, 11, 12). For example, ciprofloxacin is a broad-spectrum antibiotic which chelates with divalent cations present in EFs, resulting in close to a 70% decrease in bioavailability (4). Thus far, the effect of EFs on linezolid absorption has not been examined. Considering that many of the hospitalized and long-term care patients receive enteral nutrition support via the nasogastric (NG) or gastrostomy tubes (GT) and are at increased risk for infections caused by drug-resistant pathogens, such as MRSA and VRE, absorption of linezolid in the presence of EFs is important to characterize. Therefore, the purpose of this investigation is to determine the impact of continuous enteral feedings on the bioavailability of linezolid in hospitalized patients.
MATERIALS AND METHODS
Patients.
This prospective, randomized, single-dose, crossover study was approved by the Institutional Review Boards of both Huntington Hospital and Western University. Each patient participated in the study on a voluntary basis and provided written informed consent. Two groups of hospitalized patients were recruited to participate in the study: those who receive continuous enteral feedings via the NG or GT and those who do not and are able to tolerate oral intake. The choice of tube feeding formula was determined by the attending physician according to the individual needs of the patient. Patients who had any one of the following conditions were excluded from the study: age of <18 years, known hypersensitivity or intolerance to linezolid, pregnancy or attempting to become pregnant, or nursing an infant, history of gastrointestinal disease, active diarrhea, receipt of prokinetic agents (e.g., metoclopramide, cisapride, or erythromycin), active severe liver disease (liver function tests more than three times the upper limit of normal), thrombocytopenia (platelet count, <200 mm3) or anemia (hemaglobin, <11 g/dl; hematocrit, 38%). In addition, patients receiving continuous enteral feedings were excluded if they had excessive residual gastric contents (>200 ml/4 h).
Treatment.
A total of 18 patients who met all study enrollment criteria were stratified into treatment and control groups based on receipt of continuous EF (n = 12) or no EFs (n = 6). The latter group served as controls. The enteral nutrition formulas administered included a fiber-fortified, high-nitrogen liquid formula (Jevity; n = 5), a reduced-carbohydrate, modified-, fiber-containing formula (Glucerna; n = 3), a high-protein liquid formula (Promote), a specialized renal formula (Nepro), and a high-calorie formula (Two-cal). The sample size was estimated to provide 80% power to detect a 35% difference in bioavailability of oral linezolid between patients receiving continuous EF and controls (α = 0.05; two-tailed), assuming 100% bioavailability in the control subjects with a coefficient of variation of 20% (18). The percent difference in bioavailability of 35% was deemed clinically significant and is similar in magnitude to the difference observed with other antibiotics when administered with continuous enteral feedings in hospitalized patients (4, 10).
Patients in the EF group were randomly assigned to receive a single dose of linezolid, either 600 mg intravenously (2 mg/ml) over 30 min or an oral suspension of 600 mg (100 mg/5 ml) via the NG or GT. The feeding tube was flushed with 50 ml of water before and after the drug was administered. An alternate route of drug administration was separated by a washout period of 24 h. Patients in the control group followed the same randomization scheme as shown in Fig. 1. A linezolid oral suspension was administered by mouth in the control group.
FIG. 1.

Study design.
Pharmacokinetic and analytical methods. (i) Sample collection and handling.
Blood samples (5 ml) were collected at the following times with respect to the linezolid doses: immediately prior to administration, 0, 0.5, 1, 2, 4, 8, and 12 h after initiation of the intravenous infusion and immediately prior to administration, and 0.5, 1, 2, 4, 8, and 12 h after administration of the oral suspension. Plasma was separated by centrifugation for 15 min at 14,000 rpm and stored at −70°C until the drug assay was performed.
(ii) Linezolid HPLC assay.
Plasma concentrations of linezolid were assayed using a high-performance liquid chromatography (HPLC) method modified from two previously published methods (2, 17). Briefly, plasma samples of 100 μl were diluted with an equal volume of water and then subjected to solid-phase extraction using a C2 (100 mg) cartridge (Alltech). The injection volume was 50 μl. Samples were separated using an Ultrasphere C8 column (Beckman), 125 mm by 4 mm. The mobile phase consisted of 17% acetonitrile in 20 mM NH4H2PO4 (vol/vol), where the flow rate was set at 1.5 ml/min for a total of 15 min. All HPLC experiments were performed on an Agilent 1100 HPLC system linked with a diode array UV detector set at 251 nm.
The reproducibility of the assay, expressed as the percentage coefficient of variation, was <7% after repeated assay of samples (n = 5) containing 0.5, 1, 2, 5, 10, and 20 mg/liter. The correlation between drug concentration and peak height was good (r = 1.0), demonstrating linearity over the range of 0.5 to 20 mg/liter. The interday accuracy, expressed as percentage error, was 5.3% (2 mg/ml). The lower level of detection for linezolid was established at 0.5 mg/ml.
(iii) Pharmacokinetic analysis.
Pharmacokinetic parameters were first derived using noncompartmental methods (PK functions for Microsoft Excel; Department of Pharmacokinetics and Drug Metabolism, Allergan Corporation, Irvine, CA). The area under the plasma linezolid concentration-time curve (AUCt) from 0 to the last sampling time at which a measurable drug concentration was detected (Clast) was determined using the linear trapezoidal method. The apparent terminal elimination rate constant (λz) was determined by linear least-squares regression of the semilogarithmic concentration versus time data using the last three data points. AUC0-12 was extrapolated to infinity (AUC0--∞) by adding Clast/λz to AUCt. Bioavailability (F) was calculated based on the ratio of AUC0--∞ from the oral, NG, or GT to that of the IV. Clearance was determined by (Dose · F)/AUC0--∞. The volume of distribution (Varea) was calculated as CL/λz. The maximal concentration (Cmax) and time to maximum concentration (Tmax) were determined from visual inspection of the plasma concentration-time curve.
Population pharmacokinetic analysis was performed using the nonparametric adaptive grid algorithm (NPAG) (USC*PACK pharmacokinetic program, version 11.91; University of Southern California, Los Angeles, CA). The model utilized in the fitting (see Fig. 2) enabled simultaneous analysis of data from separate intravenous and oral studies on all subjects and explicit determination of F. Plasma concentrations were fit to one- and two-compartment linear and nonlinear models. Explicit determination of F was achieved by the addition of an absorptive compartment. The input of drug into the absorptive compartment is a function of dose and F as an initial condition of this absorptive compartment. F is provided by a function hard-coded into the NPAG program. Essentially, at each bolus event, the amount in the absorptive compartment is increased by the bolus amount multiplied by F. The estimate of F occurs, as with the other random parameters of the model, in the course of the iterative expectation maximization algorithm. First-order absorption and lag times were also evaluated in the fitting of the oral data. Due to the rapid absorption noted, lag times were fixed at 0 in the models. Plasma concentration data were weighted according to the inverse of the assay error using a linear variance model (standard deviation [SD] = 0.5 + 0.1 [concentration]). Parameters estimated in the model included the F, absorption rate constant (Ka), volume of the central compartment normalized to body weight (Vs), intercompartmental rate constants (K23 and K32), maximum velocity (Vmax) versus Michaelis-Menten (MM) constant (Km) ratio (Vmax/Km) for the nonlinear elimination pathway, and the elimination rate constant (K20) for the linear elimination pathway. Interoccasion stationarity of all PK parameters was assumed. Model discrimination was determined using final log-likelihood values, Akaike's information criterion, goodness of fit (r2), bias, and precision. The lowest Akaike's information criterion value was used to finally select the best model. The median parameter values from the NPAG analysis were then used in the maximum a posteriori (MAP) Bayesian analysis to determine the individualized pharmacokinetic parameters.
FIG. 2.

Structural models for pharmacokinetic analysis. Model equations are as follows.
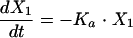
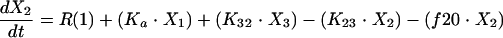
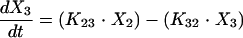
For linear models:
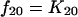
For nonlinear models:
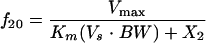
D, dose; F, bioavailability; Ka, absorption rate constant; R(1), rate of infusion into central compartment; Ti, infusion time; Vs, volume of central compartment normalized to body weight; K23, rate constant from central to peripheral compartment; K32, rate constant from peripheral to central compartment. For nondistributive models, K23 and K32 were fixed at 0.0. Nonlinear elimination: Vmax, maximum velocity; Km, Michaelis-Menten constant; linear elimination, K20 (elimination rate constant).
Statistical analysis.
The Mann-Whitney test was used to compare patient characteristics and pharmacokinetic parameters (derived from MAP Bayesian analysis) between the EF and control groups. A probability value of <0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism, version 4.00 for Windows (GraphPad Software, San Diego, California).
RESULTS
Eighteen subjects completed the study protocol. Sufficient data to determine pharmacokinetic parameters were available from 17 patients: 11 in the EF group and 6 in the control group. One patient was excluded from the analysis due to the presence of interfering substances on the chromatogram. Patient characteristics are shown in Table 1. Compared to controls, patients receiving EF were older and exhibited slightly reduced renal function (age-related decline). The mean (± standard deviation) feeding rate was 62.3 ± 18.6 ml/min. Gastric residuals were <5 ml/h with the exception of one patient, who exhibited residuals of 60 ml for 2 h.
TABLE 1.
Patient characteristics
| Variable | Mean value (SD)
|
P value | |
|---|---|---|---|
| Oral (control) | Enteral | ||
| No. of patients | 6 | 11 | |
| Age | 64.5 (16.5) | 83.1 (13.4) | 0.05 |
| Height (cm) | 172 (13.5) | 165 (10.1) | 0.37 |
| Weight (kg) | 80.7 (30.7) | 69.8 (15.5) | 0.80 |
| Gendera | 4 M, 2 F | 3 M, 8 F | |
| Body mass index (kg/m2) | 26.6 (6.8) | 25.7 (5.9) | 0.88 |
| Estimated creatinine clearance (ml/min/1.73 m2)b | 81.6 (30.5) | 58.1 (20.8) | 0.15 |
M, male; F, female.
Using method of Jelliffe (5).
Pharmacokinetics. (i) Noncompartmental analysis.
Plasma pharmacokinetic parameters for patients receiving oral (control) and enteral administration of linezolid are shown in Table 2. Data for one oral patient were omitted due to an obvious outlier (parameters greater than two standard deviations from the mean). No statistically significant differences were observed between the two administration routes. The Cmax and Tmax values differed by less than 10% between study groups. The AUC appeared larger in patients receiving oral linezolid than for those receiving enteral administration of linezolid; however, a statistically significant difference could not be demonstrated due to the large interpatient variability. The overall extent of absorption was nearly complete in both groups.
TABLE 2.
Noncompartmental pharmacokinetic parameters
| Parameter | Mean value (SD)
|
P value | |
|---|---|---|---|
| Oral (control)a | Enteral | ||
| Cmax (mg/liter) | 11.9 (4.51) | 11.1 (5.31) | 0.91 |
| Tmax (h) | 1.56 (0.816) | 1.51 (0.737) | 0.82 |
| AUC0-12 (mg · h/liter) | 73.8 (47.6) | 62.3 (26.5) | 0.82 |
| AUC0-∞ (mg · h/liter) | 109 (87.3) | 73.8 (38.9) | 0.43 |
| F | 0.884 (0.143) | 0.990 (0.247) | 0.21 |
| CL (ml/min/70 kg) | 147 (113) | 152 (43.5) | 0.50 |
| Varea (liter/70 kg) | 60.2 (45.4) | 58.2 (33.8) | 0.57 |
| λz (h−1) | 0.176 (0.161) | 0.181 (0.061) | 0.23 |
| Half-life (h) | 6.02 (3.09) | 4.33 (1.63) | 0.21 |
n = 5, one outlier omitted.
(ii) Compartmental analysis.
Population pharmacokinetic parameter estimates were generated from both linear and nonlinear two-compartment modeling of the combined intravenous and either oral or enteral data (Table 3). In this population of elderly hospitalized patients, the oral and/or enteral absorption of linezolid was found to be relatively fast (Ka, >1) and complete (F, ∼1) for most patients regardless of the PK model applied. The volume of distribution was linearly correlated with total body weight for all models tested. Creatinine clearance was not found to correlate with any model parameter.
TABLE 3.
Population-pharmacokinetic parameter estimates comparing two-compartment linear versus nonlinear modelinga
| Parameter | Linear mean (SD), median (interquartile range) | Nonlinear mean (SD), median (interquartile range) |
|---|---|---|
| Ka (h−1) | 7.30 (16.1), 0.751 (0.750-3.99) | 4.37 (11.7), 0.755 (0.500-2.00) |
| F | 0.874 (0.164), 0.878 (0.800-0.995) | 0.994 (0.118), 1.02 (0.935-1.08) |
| K20 (h−1) | 0.400 (0.203), 0.315 (0.245-0.610) | NA |
| Vc (liters/kg) | 0.340 (0.116), 0.348 (0.244-0.397) | 0.394 (0.111), 0.405 (0.325-0.480) |
| Vmax (mg/h) | NA | 82.0 (42.2), 79.5 (46.5-85.5) |
| K23 (h−1) | 1.68 (1.51), 1.28 (0.489-2.33) | 0.816 (1.13), 0.475 (0.200-1.10) |
| K32 (h−1) | 3.19 (1.83), 2.42 (1.68-4.99) | 0.821 (1.21), 0.485 (0.230-1.05) |
Km fixed at 3.6 mg/liter.
Analysis by the NPAG algorithm demonstrated excellent predictions for two-compartment distributive models either with MM or with linear elimination. Administration of the same dose in all experiments restricted the actual analysis of MM parameters to the Vmax/Km ratio (Km was fixed at 3.6 mg/liter, which is in the range of values determined in a prior study) (7). Slightly less bias and greater precision were noted in predicted concentrations based on individual parameter estimates (posterior, from MAP Bayesian analysis) using the MM (mean error = −0.104, mean squared error = 0.900) when compared with the linear model (mean error = 0.151, mean squared error = 0.990). Although the predictions given by the individualized PK parameters were excellent (r2 = >0.97), median parameters (prior; NPAG analysis) predicted individual observed levels well (r2 = >0.80) in both MM and linear two-compartment models (Fig. 3). A plasma concentration versus time curve with the combined intravenous and enteral data for a representative patient demonstrates the goodness of fit using the linear two-compartment model (Fig. 4).
FIG. 3.

NPAG plot of predicted versus observed linezolid concentrations based on (A) population medians (prior) and (B) individual parameter estimates (posterior). The solid line is the line of identity.
FIG. 4.

Plasma concentration-time curve for a representative patient using linear two-compartment model.
A comparison of the pharmacokinetic parameter estimates (using the linear two-compartment model) between patients receiving oral versus enteral administration is shown in Table 4. The pharmacokinetics in these two groups demonstrated remarkable similarity, with no significant differences in any of the parameter estimates noted; however, these findings require confirmation given the small sample size.
TABLE 4.
Comparative pharmacokinetics of oral versus enteral administration of linezolid derived from linear two-compartment model
| Parameter | Median value (interquartile range)
|
P value | |
|---|---|---|---|
| Oral (control) | Enteral | ||
| Ka (h−1) | 0.750 (0.250-29.0) | 0.761 (0.750-1.72) | 0.63 |
| F | 0.845 (0.676-0.943) | 0.904 (0.839-1.04) | 0.34 |
| K20 (h−1) | 0.270 (0.145-0.705) | 0.325 (0.269-0.605) | 0.70 |
| Vc (liter/kg) | 0.339 (0.253-0.393) | 0.348 (0.240-0.497) | 0.77 |
| K23 (h−1) | 1.63 (0.450-3.65) | 1.13 (0.474-2.33) | 0.66 |
| K32 (h−1) | 3.52 (1.22-5.02) | 2.42 (1.25-5.00) | 0.92 |
A significant gender effect was noted for several pharmacokinetic parameters (Table 5). The F and Vc values (and volume of distribution at steady state = 0.543 versus. 0.484 liters/kg; P = 0.01) were significantly greater, while the K20 and K23 values were significantly less, for women than for men. The difference in elimination can be attributed to differences in volume of distribution, since the clearance was not significantly different between the two groups (0.129 versus. 0.120 liters/h; P = 0.54). An examination of subject characteristics revealed that the women were significantly older (84.7 versus 63.7 years; P = 0.01) and shorter (157 versus 177 cm; P < 0.01) than the men. In addition, the women exhibited significantly less renal function (estimated creatinine clearance, 40.6 versus 94.3 ml/min/1.73 m2; P < 0.01).
TABLE 5.
Gender effects on pharmacokinetics of linezolid
| Parameter | Median value (interquartile range)
|
P value | |
|---|---|---|---|
| Males (n = 7) | Females (n = 10) | ||
| Ka (h−1) | 0.750 (0.250-6.25) | 0.895 (0.75-25.7) | 0.23 |
| F | 0.774 (0.618-0.878) | 0.923 (0.865-1.04) | 0.02 |
| K20 (h−1) | 0.605 (0.315-0.685) | 0.272 (0.205-0.415) | 0.04 |
| Vc (liters/kg) | 0.267 (0.195-0.330) | 0.370 (0.298-0.499) | 0.01 |
| K23 (h−1) | 2.33 (1.53-4.98) | 0.711 (0.3-1.55) | 0.01 |
| K32 (h−1) | 2.27 (2.12-4.87) | 3.70 (0.791-5.05) | 0.47 |
DISCUSSION
Linezolid is a relatively new agent proven effective in the treatment of infections caused by antibiotic-resistant gram-positive pathogens. However, the bioavailability of linezolid following enteral administration in the presence of enteral feedings has not been studied, and its determination is important considering that the enteral route is increasingly utilized in the hospitalized patient population to minimize IV-related complications.
Overall, the pharmacokinetic data from this study are in agreement with previously published data (15). The elderly, hospitalized patient population represented in this study is comparable to that described by Meagher et al., which included 318 patients in a compassionate use treatment program. In that study, a parametric iterative two-stage Bayesian approach was used to determine the pharmacokinetics of linezolid based on sparse sampling (average = 4; range, 2 to 10). The final model reported to best describe the data was a two-compartment model with parallel linear and nonlinear elimination pathways. While differences in the structural model preclude a direct comparison of the pharmacokinetic parameter estimates, the volume of the central compartment is smaller (22 versus 39.6 liters/65 kg) and clearance larger (8.8 versus 6.9 liters/h/65 kg) in our study than that reported by Meagher et al. (7). One potential reason for the difference in the volume of distribution may be related to differences in body composition. Our population is significantly older and perhaps exhibited less lean body mass. The Cmax values in our study (∼11 mg/liter) are comparable to those of previously published studies, indicating that the volume of distribution is likely similar (15). Finally, individual variability, at least in this group of patients, seems to be small enough to make the population mean parameter estimates useful for individual prediction.
In this study we performed both noncompartmental and compartmental pharmacokinetic analysis. Bioavailability estimates are commonly derived from ratio of AUCs obtained from studies requiring administration of a single- or multiple-dose regimen of alternate dosage forms separated by a washout period. Such study design may be limited primarily to studies with healthy volunteers within a research study unit, since the inclusion of a washout period would not be appropriate for studying patients who may be receiving the agent for therapeutic purposes. In contrast, the compartmental pharmacokinetic modeling approach offers the advantage of explicitly determining F from the model, which offers the ability to perform bioavailability assessments for patients with intermixed IV/oral dosage regimens.
In general, the data on linezolid oral absorption in this study are consistent with published reports showing that oral absorption of linezolid is essentially complete in most patients (F = ∼1) (18). Our results add new data demonstrating that the bioavailability of oral linezolid is unaltered in the presence of continuous EF. We found the bioavailability estimates from both analyses to be largely in agreement with one another, demonstrating very rapid and complete absorption of linezolid in patients with and without enteral feedings. The slightly greater bioavailability estimates from the noncompartmental analysis may have been overestimated due to a lag time in absorption. Our sampling scheme was not dense enough during the absorption phase to give good estimates of both Tlag and Ka, and consequently in the final analysis Tlag was fixed at 0. Nevertheless, the analysis shows that in most cases, the oral and/or enteral absorption of linezolid was found to be rapid (Ka = >1), regardless of PK model applied. Surprisingly, no difference in Ka was observed between enterally fed or control patients receiving oral suspension. Analysis of F, which was directly incorporated into the compartmental model, gave results essentially identical to those of a conventional comparison of AUC ratios (noncompartmental model approach). The lack of good precise lag-time estimates will influence the assessment of F but to a lesser degree for compartmental than for noncompartmental analysis.
Significant gender effects were noted in several pharmacokinetic parameters. The larger volume of distribution in the women suggests a different body composition from that of the men. Based on the differences in age and gender, less muscle mass is expected in the EF group. Given the similarity in body mass index, the elderly women likely have more adipose and water, which could account for the increased volume of distribution and faster distribution to the tissues. The slower elimination can be explained by the larger volume of distribution in the women given that there was no significant difference in clearance between the two groups. The larger F value for the women is not as easily explained but could be a result of a slower intestinal transit time.
Since linezolid exhibits predominantly linear elimination but with an additional nonlinear (Michaelis-Menten) pathway, we modeled the data using multiple-compartment models which incorporated both linear and nonlinear elimination (15). The greater bias and less precision observed with the linear model in our study suggest that the elimination of linezolid is indeed saturable at high plasma levels, which are present initially before distribution equilibrium is achieved, but becomes close to linear at levels usually present after distribution equilibrium. Our data are in agreement with the findings from a dose-escalating trial evaluating 375 mg, 500 mg, or 625 mg of linezolid twice daily. The clearance values for linezolid at steady state were 10 to 30% lower at the higher dose following oral administration and 11 to 19% lower following intravenous administration (16). These data indicated a slight degree of nonlinearity with doses used clinically (600 mg twice daily). Our analysis did not contain any dose escalation data, and thus, we were only able to estimate the Vm/Km ratio. Km was fixed at 3.6 mg/ml, which is in the range of values identified in a recent study (7). Further analyses with other fixed Km values up to 12 mg/liter gave slightly improved fit quality, an indication that the actual Km might be higher. In general, nondistributive models gave substantially poorer model fits of our data. Interestingly, for such models, linear elimination gave superior fits compared to MM elimination, which also suggests that the degree of nonlinear elimination is small in the dose examined in this study.
Conclusion. Linezolid oral suspension may be administered via the enteral route to hospitalized patients without compromise in its excellent bioavailability and rapid rate of absorption. Compartmental pharmacokinetic analysis offers more flexible study application, since F can be estimated directly with intermixed IV/oral doses without a need for washout period.
Acknowledgments
This study was supported by a research grant from Pharmacia Co.
Beringer, Nguyen, Hoem, Louie, Gill, and Gurevitch have no conflict of interest; Wong-Beringer has received research funding from Pharmacia Co.
REFERENCES
- 1.Bass, J., M. V. Miles, M. B. Tennison, B. J. Holcombe, and M. D. Thorn. 1989. Effects of enteral tube feeding on the absorption and pharmacokinetic profile of carbamazepine suspension. Epilepsia 30:364-369. [DOI] [PubMed] [Google Scholar]
- 2.Borner, K., E. Borner, and H. Lode. 2001. Determination of linezolid in human serum and urine by high-performance liquid chromatography. International J. Antimicrob. Agents 18:253-258. [DOI] [PubMed] [Google Scholar]
- 3.Chambers, H. F. 2001. The changing epidemiology of Staphylococcus aureus? Emerg. Infect. Dis. 7:178-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Healy, D. P., M. C. Brodbeck, and C. E. Clendening. 1996. Ciprofloxacin absorption is impaired in patients given enteral feedings orally and via gastrostomy and jejunostomy tubes. Antimicrob. Agents Chemother. 40:6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jelliffe, R., and S. Jelliffe. 1972. A computer program for estimation of creatinine clearance from unstable serum creatinine levels, age, sex and weight. Math. Biosci. 14:17-24. [Google Scholar]
- 6.MacGowan, A. P. 2003. Pharmacokinetic and pharmacodynamic profile of linezolid in healthy volunteers and patients with Gram-positive infections. J. Antimicrob. Chemother. 51:ii17-ii25. [DOI] [PubMed] [Google Scholar]
- 7.Meagher, A. K., A. Forrest, C. R. Rayner, M. C. Birmingham, and J. J. Schentag. 2003. Population pharmacokinetics of linezolid in patients treated in a compassionate-use program. Antimicrob. Agents Chemother. 47:548-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mimoz, O., V. Binter, A. Jacolot, A. Edouard, M. Tod, O. Petitjean, and K. Samii. 1998. Pharmacokinetics and absolute bioavailability of ciprofloxacin administered through a nasogastric tube with continuous enteral feeding to critically ill patients. Intensive Care Med. 24:1047-1051. [DOI] [PubMed] [Google Scholar]
- 9.Moellering, R. C. 2003. Linezolid: the first oxazolidinone antimicrobial. Ann. Intern. Med. 138:135-142, 144. [DOI] [PubMed] [Google Scholar]
- 10.Mueller, B. A., D. G. Brierton, S. R. Abel, and L. Bowman. 1994. Effect of enteral feeding with Ensure on oral bioavailabilities of ofloxacin and ciprofloxacin. Antimicrob. Agents Chemother. 38:2101-2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodman, D. P., T. L. Stevenson, and T. R. Ray. 1995. Phenytoin malabsorption after jejunostomy tube delivery. Pharmacotherapy 15:801-805. [PubMed] [Google Scholar]
- 12.Rosemurgy, A. S., S. Markowsky, S. E. Goode, K. Plastino, and R. E. Kearney. 1995. Bioavailability of fluconazole in surgical intensive care unit patients: a study comparing routes of administration. J. Trauma Injury Infect. Crit. Care 39:445-447. [DOI] [PubMed] [Google Scholar]
- 13.Sisson, T. L., G. L. Jungbluth, and N. K. Hopkins. 2002. Age and sex effects on the pharmacokinetics of linezolid. Eur. J. Clin. Pharmacol. 57:793-797. [DOI] [PubMed] [Google Scholar]
- 14.Slatter, J. G., D. J. Stalker, K. L. Feenstra, I. R. Welshman, J. B. Bruss, J. P. Sams, M. G. Johnson, P. E. Sanders, M. J. Hauer, P. E. Fagerness, R. P. Stryd, G. W. Peng, and E. M. Shobe. 2001. Pharmacokinetics, metabolism, and excretion of linezolid following an oral dose of [(14)C]linezolid to healthy human subjects. Drug Metab. Dispos. 29:1136-1145. [PubMed] [Google Scholar]
- 15.Stalker, D. J., and G. L. Jungbluth. 2003. Clinical pharmacokinetics of linezolid, a novel oxazolidinone antibacterial. Clin. Pharmacokinet. 42:1129-1140. [DOI] [PubMed] [Google Scholar]
- 16.Stalker, D. J., G. L. Jungbluth, N. K. Hopkins, and D. H. Batts. 2003. Pharmacokinetics and tolerance of single- and multiple-dose oral or intravenous linezolid, an oxazolidinone antibiotic, in healthy volunteers. J. Antimicrob. Chemother. 51:1239-1246. [DOI] [PubMed] [Google Scholar]
- 17.Tobin, C. M., J. Sunderland, L. O. White, and A. P. MacGowan. 2001. A simple, isocratic high-performance liquid chromatography assay for linezolid in human serum. J. Antimicrob. Chemother. 48:605-608. [DOI] [PubMed] [Google Scholar]
- 18.Welshman, I. R., T. A. Sisson, G. L. Jungbluth, D. J. Stalker, and N. K. Hopkins. 2001. Linezolid absolute bioavailability and the effect of food on oral bioavailability. Biopharm. Drug Dispos. 22:91-97. [DOI] [PubMed] [Google Scholar]