

Abstract
T细胞受体(TCR)与抗原肽的特异性结合在调节和介导免疫的过程中发挥着关键作用,为肿瘤疫苗设计提供了必要的基础。近年来的研究主要集中在主要组织相容性复合体(MHC)Ⅰ类抗原的TCR预测,针对MHC Ⅱ类抗原的TCR预测研究尚不充分,仍存在较大的提升空间。本研究利用ProtT5大模型进行MHC Ⅱ类抗原肽和TCR结合预测研究,探究其特征提取能力。此外,对模型进行微调,并构建前馈神经网络结构进行融合从而实现预测模型。实验结果显示,本研究提出的方法较传统方法表现更佳,预测准确度达到0.96,AUC达到0.93,验证了本文提出模型的有效性。
Keywords: MHC Ⅱ抗原肽-T细胞受体结合预测, 大模型, ProtT5模型, 免疫治疗
Abstract
The specific binding of T cell receptors (TCRs) to antigenic peptides plays a key role in the regulation and mediation of the immune process and provides an essential basis for the development of tumour vaccines. In recent years, studies have mainly focused on TCR prediction of major histocompatibility complex (MHC) class I antigens, but TCR prediction of MHC class II antigens has not been sufficiently investigated and there is still much room for improvement. In this study, the combination of MHC class II antigen peptide and TCR prediction was investigated using the ProtT5 grand model to explore its feature extraction capability. In addition, the model was fine-tuned to retain the underlying features of the model, and a feed-forward neural network structure was constructed for fusion to achieve the prediction model. The experimental results showed that the method proposed in this study performed better than the traditional methods, with a prediction accuracy of 0.96 and an AUC of 0.93, which verifies the effectiveness of the model proposed in this paper.
Keywords: Prediction of MHC Ⅱ antigen peptide-T cell receptors binding, Foundation model, ProtT5 model, Immunotherapy
0. 引言
肿瘤免疫治疗被认为是癌症治疗领域的一次革命性突破,有效的靶向免疫疗法需要准确预测可能触发T细胞免疫应答的肿瘤特异性表位。抗原依据其特性,分为肿瘤相关抗原与肿瘤特异性抗原,它们通过主要组织相容性复合体(major histocompatibility complex,MHC)呈现在肿瘤细胞表面,随后启动T细胞介导的免疫应答,以消灭肿瘤细胞[1]。T细胞受体(T cell receptor,TCR)的激活依赖于它与肽-MHC(pMHC复合物)的识别过程[2]。TCR是由两条不同肽链构成的二聚体,其胞外区由恒定区(C)和可变区(V)组成,V区又可分为三个互补决定区(CDR1-3),CDR1和CDR2负责识别MHC分子,而关键的CDR3区则是抗原的特异性识别部位[3]。CDR3的结构和功能在TCR与pMHC复合物的相互作用中至关重要,其特异性识别机制将影响免疫治疗策略的精确设计。
高通量测序的发展推动了抗原肽与TCR结合计算方法的开发。基于传统机器学习的方法,Gielis等[4]分析序列的物理化学性质,构建了基于随机森林的模型评估并预测结合概率。De Neuter等[5]通过训练随机森林分类器以探究TCR识别抗原的机制。基于深度学习的方法,NetTCR-2.0[6]利用卷积网络将肽和CDR3 β序列信息整合。ERGO[7]利用长短期记忆人工神经网络来提取序列特征,用于预测两者结合概率。本团队利用自构建编码方式AAPP,提出名为iTCep[8]的深度学习框架,预测MHC I类分子呈现的肽与TCR之间的相互作用。
尽管该研究领域展现出显著的发展,但仍有潜力进一步提升和完善。传统意义上认为CD8+ T细胞是主要的肿瘤杀伤细胞,但最近的临床试验表明CD4+ T细胞在肿瘤控制中也起到了重要作用。而现有研究主要集中于CD8+ T细胞与MHC Ⅰ类抗原结合预测,忽略了CD4+ T细胞与MHC Ⅱ类抗原的相互作用,模型性能仍有待提高。此外,现有模型多为单一模型,限制了从复杂数据中挖掘关联性的能力,鉴于生物学数据通常呈现高维特征,开发能够有效分析复杂数据集的模型变得尤为重要,因此本研究利用ProtT5模型对MHC Ⅱ类抗原肽与TCR的结合进行预测,希望能够提供有益的探索经验。
1. 材料与方法
1.1. 实验总体流程
为提升MHC Ⅱ类抗原肽-TCR结合预测模型的性能,本研究在现有研究基础上进行了改进。实验框架设计如图1所示,涵盖数据收集、特征编码与选择、模型构建和模型预测四个关键步骤。
图 1.

Framework diagram for the study of peptide-TCR binding prediction model
肽-TCR结合预测模型研究框架图
首先,本研究收集了公共数据库中的TCR与II类MHC提呈抗原肽的数据,并进行了过滤和预处理。随后,采用不同编码策略将抗原肽和TCR序列转换为相应的特征。为寻找最佳编码方式,使用机器学习交叉验证进行评估。在确定最优编码策略后,对ProtT5模型进行了微调,以更好地适应研究需求。为全面评估模型性能,用多种指标综合判断。最后,将微调后的模型应用于测试集,以评估其泛化能力。
1.2. 实验数据
随着测序技术的发展,越来越多的TCR序列及抗原肽数据被收集,现有数据库提供了TCR与抗原对接的信息,为人工智能预测TCR与抗原的结合奠定了数据基础。本研究使用的数据集源于公开的VDJdb[9]和McPAS-TCR[10]数据库,从中提取MHC II类抗原肽和TCR信息,其中阳性数据分别为782条和898条。为了构建阴性数据并模拟真实世界TCR与抗原肽的情况,从TCRdb[11]数据库中选取了来自健康供体的TCR β链序列共16 800条,使用的项目号为PRJNA390125(样本编号为SRR5676649、SRR5676658)。经合并去重后得到一个完整的数据集,划分训练集、验证集与测试集。
1.3. 数据预处理
本研究剔除了MHC Ⅰ类抗原肽段,以及VDJdb数据库中置信评分等于0的数据(0表示关键信息的缺失),着重研究MHC Ⅱ类抗原肽段;删除序列中出现错误的氨基酸(B、J、O、U、X、Z);删除不以半胱氨酸(C)开始或不以苯丙氨酸(F)结尾的CDR3序列。合并数据进行去重,最终得到有效阳性数据943条,阴性数据9 430条,共10 373条配对的抗原肽和TCR结合信息,其中MHC Ⅱ类分子呈递的抗原肽大多是13和20个氨基酸数目的肽段。
1.4. 基于ProtT5模型特征提取分析
ProtT5模型[12]是一种基于Transformer架构的蛋白质预训练模型,通过自监督方式在UniRef50数据集上(现包含6 600万个蛋白质序列)进行训练。模型从这些蛋白质序列中生成输入和标签,并随机掩盖输入中的15%氨基酸,通过这种方式训练得到的特征会捕捉到潜在的蛋白质信息。利用自注意力机制,ProtT5模型可以有效地识别和编码蛋白质序列中的远程交互依赖性。这使得ProtT5成为蛋白质相互作用预测等领域内一个强有力的工具。
在机器学习或深度学习模型的构建过程中,数据预处理环节涉及将原始数据编码为数值化的向量或张量,这一步骤被称为特征工程或特征表示学习。对于本研究中的TCR和Peptide序列数据,这一过程涉及到氨基酸残基的向量化或嵌入(embedding),将生物序列转换为机器学习算法可操作的数值形式,实现有效的模式识别和学习。
具体来说,在两个序列之间添加一个分隔符,然后将它们作为一个整体输入到模型中。ProtT5模型会处理这个长序列,并输出它的隐藏状态。提取出模型最后一层的隐藏嵌入,该层包含了一定数量的1 024维向量。这一处理过程旨在将抗原肽和TCR序列转换为高维度的特征向量表示,这些特征向量不仅包含了整体的序列信息,还涵盖了大量蛋白质功能等相关信息,具体流程如图2所示。此外,对三种不同的编码策略进行比较,分别是基于氨基酸理化性质的编码(Phychem)[13]、随机游走算法编码(Deep Walk)[14]和基于ProtT5大模型的编码,选取合适的特征提取方法是构建深度学习模型的重要一步。
图 2.

The feature extraction process diagram of the ProtT5 model
ProtT5模型特征提取流程图
1.5. 预测模型构建
尽管ProtT5模型经预训练后积累了丰富的蛋白质信息,但在TCR与抗原肽的预测任务中并不完全适用。为更好地满足研究需求,本研究对ProtT5模型进行了训练和微调,显著提升了其性能和适用性,能够更有效地处理MHC II类抗原肽与TCR之间的复杂结合关系。
具体而言,本研究通过嵌入(Embedding)抗原肽和TCR序列以提取关键特征,获取对当前数据的映射,并将嵌入后的向量输入ProtT5模型进行训练。由于ProtT5模型的前层网络已能提供良好的底层特征,我们冻结了相对底层的权重,保留原有特征提取能力。为了更好地满足任务需求,接入一个前馈神经网络层,提取和组合底层特征,最终输出预测结果。网络模型结构示意图见图3。
图 3.

Structural diagram of the ProtT5 fine-tuned model
ProtT5模型微调结构图
模型训练过程中,采用学习率预热(warm-up)的策略,优势在于能够有效避免模型在训练初期出现的震荡问题。在初始几轮训练中,采用较小的学习率让模型逐渐稳定。当模型达到相对稳定状态时,逐步增加学习率至预设值,以加速模型的收敛速度并提高效果。
1.6. 针对不平衡数据集的训练策略
由于数据集中正负样本分布比例不平衡,分类器可能倾向于预测出现频率较高的类别,进而忽略出现频率较低的类别,导致训练效果低下。为此,选取Focal Loss[15]作为损失函数,扩展评估指标范围,而不仅仅局限于准确率。
Focal Loss(聚焦损失)是一种用于处理类别不平衡问题的损失函数,它具有调节正负样本权重和易分类与难分类样本权重的能力,通过增加困难样本的权重,降低简单样本的权重,使模型能够有针对性地学习难易样本之间的差异。Focal Loss公式如式(1)所示:
 |
1 |
Pt表示预测概率,α表示控制正负样本对loss的贡献,本研究我们选取α为0.5、γ为2作为训练的参数。
1.7. 实验环境和评价指标
本研究通过Python 3.9.7编写实验代码,基于PyTorch框架实现,硬件环境为Intel(R) Core(TM) i9-12900K,显卡为RTX 3090,操作系统为Ubuntu 20.04.6 LTS。
为了验证不同编码方式对模型性能的影响,我们采用准确率(Accuracy,ACC)、精准率(Precision)、召回率(Recall)、F1-score和受试者操作特征(receiver operating characteristic,ROC)曲线下面积(area under curve,AUC)五个测量指标进行评估。
准确率表示抗原肽与TCR预测正确的个数占预测所有数据个数的比例,计算公式如式(2)所示:
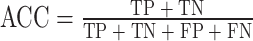 |
2 |
精准率表示抗原肽与TCR预测为结合的对子中,实际上为真实数据的比例,公式如式(3)所示:
 |
3 |
召回率又叫做查全率,直观地说是分类器找到所有正样本的能力,公式如式(4)所示:
 |
4 |
F1-score是一个综合评价分类模型性能的指标,适用于二分类问题,它同时考虑了模型的准确率和召回率,公式如式(5)所示:
 |
5 |
AUC表示ROC曲线下的面积,主要用于评估模型的泛化能力,即分类器效果的优劣。AUC作为一个数值指标,它比ROC更具可比性,能够进行量化比较。其值范围在0到1之间,越接近于1表示分类器性能越好。
2. 实验结果与分析
2.1. 不同特征提取方法的比较
为研究不同特征提取方法对模型性能的影响,对三种编码策略进行实验测试,包括氨基酸理化性质编码、随机游走编码和ProtT5模型生成的嵌入向量编码。随后,将原始数据集(训练集、验证集)分为五个相等大小的子集,结合不同编码策略和SVM模型,采用三次独立的五折交叉验证评估各特征对模型性能的影响。在实验中,将AUC作为主要评价指标,结果如图4所示。结果说明,在TCR与MHC II类抗原肽结合预测任务中,ProtT5模型展现出显著的优势,其AUC的平均值为85.33%,高于氨基酸理化性质(71.99%)和随机游走(77.06%)方法,表明了本文所提出模型的可靠性。
图 4.

Five-fold cross-validation results of model training with three encoding features
三种编码特征的模型训练五折交叉验证的结果
接下来,在完全独立的测试集中评估模型的泛化能力,该测试集未参与原模型的训练,用于在训练结束后对原模型进行测试,以对比和分析不同编码方法的性能表现,结果见表1。由表1结果可知,仅依赖氨基酸理化性质特征和随机游走的方法并不能充分捕捉到抗原肽与TCR序列的信息,相比之下,基于ProtT5模型的编码方法在不同特征中依然表现出显著的优越性。
表 1. Comparison of the performance of each embedding method on Testing data.
各编码方式在测试集上的性能比较
| 编码方法 | ACC | Precision | Recall | F1-score | AUC |
| Phychem | 0.90 | 0.73 | 0.50 | 0.49 | 0.67 |
| Deep Walk | 0.91 | 0.93 | 0.53 | 0.54 | 0.72 |
| ProtT5 | 0.92 | 0.95 | 0.61 | 0.66 | 0.81 |
2.2. 基于ProtT5的深度学习预测模型结果分析
为了对比研究不同的深度学习模型在TCR和MHC II类抗原肽结合预测任务中的表现,我们选用机器学习算法以及已发表的工具进行比较,包括K近邻(KNN)、AttnTAP[16]和ATM-TCR[17]。为了保证公平性,使用相同的训练集对模型进行训练,通过多次实验,综合考察各模型在测试集上的准确度、召回率等性能指标,结果如表2所示。
表 2. Comparison of the performance for different classification algorithms on Testing set.
不同分类算法在测试集的性能比较
| 方法 | ACC | Precision | Recall | F1-score |
| KNN | 0.92 | 0.77 | 0.63 | 0.66 |
| AttnTAP | 0.93 | 0.71 | 0.52 | 0.59 |
| ATM-TCR | 0.94 | 0.88 | 0.56 | 0.74 |
| ProtT5-finetune | 0.96 | 0.88 | 0.71 | 0.78 |
结果显示,ATM-TCR和本研究的模型均表现良好。进一步分析比较两者的AUC和平均精度(average precision,AP)(见图5),可以观察到本研究模型在区分正负样本上具有明显优势(AUC = 0.93,AP = 0.83),优于其他分类器。高AP值还反映出模型在极度不平衡的肽-TCR阳性和阴性样本中具备出色的适应性和鲁棒性,能更有效地识别肽-TCR的真实相互作用,减小数据不平衡带来的偏差。
图 5.

Comparison of AUC and AP values for predicting MHC class II peptide TCR interactions using different classifiers on Testing set
不同分类器在测试集预测MHC II类肽-TCR相互作用的AUC、AP值比较
2.3. 模型可解释性
在当前的数据集中,筛选出与TCR匹配对数最多的肽以及相应的TCR序列,将两段序列拼接后输入ProtT5模型以提取对应嵌入特征。同样地,选取另一种简单的编码方案——氨基酸理化特性作为比较,它将输入序列中的每个氨基酸转化为多个特征向量表示(有疏水性、β结构偏好、亲水性等)。
接下来,使用t分布随机邻居嵌入(t-distributed stochastic neighbor embedding,t-SNE)算法将这些高维向量降至二维,以便更直观地展示它们之间的关系(见图6a~b)。结果显示,相较于氨基酸理化特性(见图6b),经ProtT5嵌入后(见图6a)的阳性和阴性肽-TCR显著分离,尤其是在右下角形成了一个明显的阳性肽-TCR富集区。这表明,经过ProtT5模型嵌入后,已经实现了良好的分类效果,这一结果也说明ProtT5预训练模型在提取生物序列特征方面具有潜在优势。
图 6.
t-SNE plot for the peptide-TCR
关于肽-TCR的t-SNE图
a. 经ProtT5嵌入后;b. 经氨基酸理化特性嵌入后。阳性TCR(标注为1)以橙色显示,阴性TCR(标注为0)以淡蓝色显示
a. after ProtT5 embedding; b. after embedding with amino acid physicochemical properties. Orange indicates positive TCR (labeled as 1), and light blue indicates negative TCR (labeled as 0)

我们猜测,ProtT5模型是通过大规模的蛋白质序列进行预训练得到的,包括一级、二级结构以及氨基酸间的交互,这些都是传统的人工筛选特征难以捕捉的,可能是ProtT5嵌入特征性能优异的一个重要原因。其次,ProtT5模型利用注意力机制的优势来处理蛋白质序列的复杂性,能够关注蛋白质序列中的关键部分,在不依赖于多序列比对的情况下可能实现准确的预测。
3. 讨论与结论
已有研究表明,CD4+ T细胞识别的MHC II分子对新发肿瘤的影响可能大于MHC I类,且它们也是CD8+ T细胞至关重要的辅助因子,在肿瘤发生中起着核心作用[18-19]。缺乏对MHC II类抗原免疫原性准确的预测,可能会导致很多患者对免疫治疗无反应,即使这些患者肿瘤被预测含有免疫原性的MHC I抗原或具有有利的肿瘤突变负荷。
本研究构建了基于ProtT5模型的深度学习方法,利用预训练蛋白质语言模型提取的特征作为输入参数,并集成前馈全连接神经网络模块,以实现对TCR与MHC II类抗原肽结合的预测。通过多组实验验证,证明了模型在不同数据集上的稳定性和泛化能力,可以更准确地捕捉TCR与抗原肽序列的信息,有效地预测潜在的、可激活CD4+ T细胞的抗原靶点,缩小临床上实验验证新抗原的范围,提升靶点预测效率。
本研究虽然在TCR与MHC Ⅱ类呈递抗原肽的免疫原性预测研究方面取得一定进展,但仍存在一些局限性。肿瘤的免疫反应是一个复杂的生物过程,其中涉及多肽的降解、蛋白酶的剪切加工、TAP的转运以及MHC的结合呈递等因素影响[20]。但由于训练数据的有限性,这些影响抗原肽免疫原性的特征因素并未完全考虑到模型中。其次,大模型通常由数以亿计的参数组成,使得解释模型的决策过程变得更加困难。因此,准确地鉴定具有免疫原性的CD4+ T细胞表位仍然是一个技术瓶颈。但已有大量工作表明,深度学习方法可以用来解决这些问题,这在之前的工作中也得到了初步探索并验证[8, 21]。我们相信随着对肿瘤免疫行为的深入解析以及精准医学时代下高质量数据的不断累积,结合高通量的TCR免疫组库测序技术[22],深度学习算法会不断得到升级拓展,可有效帮助科研人员发现肿瘤治疗的潜在通用靶点。
总体而言,本研究提出的模型准确预测出MHC Ⅱ类抗原肽和TCR的识别,有效地鉴定出高质量候选抗原肽,这一进展为开发更精确的TCR抗原预测模型奠定了基础,并有望促进肿瘤免疫疗法的优化和新治疗策略的探索。本文模型训练的数据集和代码可在如下地址查看:https://gitee.com/rxumin/MHC-TCR。
重要声明
利益冲突声明:本文全体作者均声明不存在利益冲突。
作者贡献说明:许珉瑞主要负责整体实验的设计、算法修改及论文撰写;张斯文、鲁曼曼主要负责医学背景指导及论文修订;高媛、张梦欢主要负责数据处理方案设计及算法指导;林勇、谢鹭负责整体实验的设计指导、算法技术指导与论文修订。
Funding Statement
国家自然科学基金(31870829);上海市卫健委协同创新集群项目(2019CXJQ02)
Contributor Information
勇 林 (Yong LIN), Email: yong_lynn@usst.edu.cn.
鹭 谢 (Lu XIE), Email: xielu@sibpt.com.
References
- 1.Xie N, Shen G, Gao W, et al Neoantigens: promising targets for cancer therapy. Signal Transduct Target Ther. 2023;8(1):9. doi: 10.1038/s41392-022-01270-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joglekar A V, Li G T cell antigen discovery. Nat Methods. 2021;18(8):873–880. doi: 10.1038/s41592-020-0867-z. [DOI] [PubMed] [Google Scholar]
- 3.Chen J, Zhao B, Lin S, et al TEPCAM: Prediction of T-cell receptor-epitope binding specificity via interpretable deep learning. Protein Sci. 2024;33(1):e4841. doi: 10.1002/pro.4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gielis S, Moris P, Bittremieux W, et al Detection of enriched T cell epitope specificity in full T cell receptor sequence repertoires. Front Immunol. 2019;10:2820. doi: 10.3389/fimmu.2019.02820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Neuter N, Bittremieux W, Beirnaert C, et al On the feasibility of mining CD8+ T cell receptor patterns underlying immunogenic peptide recognition. Immunogenetics. 2018;70(3):159–168. doi: 10.1007/s00251-017-1023-5. [DOI] [PubMed] [Google Scholar]
- 6.Montemurro A, Schuster V, Povlsen H R, et al NetTCR-2. 0 enables accurate prediction of TCR-peptide binding by using paired TCRα and β sequence data. Commun Biol. 2021;4(1):1060. doi: 10.1038/s42003-021-02610-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Springer I, Besser H, Tickotsky-Moskovitz N, et al Prediction of specific TCR-peptide binding from large dictionaries of TCR-peptide pairs. Front Immunol. 2020;11:1803. doi: 10.3389/fimmu.2020.01803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Jian X, Xu L, et al iTCep: a deep learning framework for identification of T cell epitopes by harnessing fusion features. Front Genet. 2023;14:1141535. doi: 10.3389/fgene.2023.1141535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shugay M, Bagaev D V, Zvyagin I V, et al VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 2018;46(D1):D419–D427. doi: 10.1093/nar/gkx760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tickotsky N, Sagiv T, Prilusky J, et al McPAS-TCR: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics. 2017;33(18):2924–2929. doi: 10.1093/bioinformatics/btx286. [DOI] [PubMed] [Google Scholar]
- 11.Chen S Y, Yue T, Lei Q, et al TCRdb: a comprehensive database for T-cell receptor sequences with powerful search function. Nucleic Acids Res. 2021;49(D1):D468–D474. doi: 10.1093/nar/gkaa796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elnaggar A, Heinzinger M, Dallago C, et al ProtTrans: Toward understanding the language of life through self-supervised learning. IEEE Trans Pattern Anal Mach Intell. 2022;44(10):7112–7127. doi: 10.1109/TPAMI.2021.3095381. [DOI] [PubMed] [Google Scholar]
- 13.Kidera A, Konishi Y, Oka M, et al Statistical analysis of the physical properties of the 20 naturally occurring amino acids. J Protein Chem. 1985;4(1):23–55. [Google Scholar]
- 14.Bi J, Zheng Y, Yan F, et al Prediction of epitope-associated TCR by using network topological similarity based on deepwalk. IEEE Access. 2019;7:151273–151281. doi: 10.1109/ACCESS.2019.2948178. [DOI] [Google Scholar]
- 15.刘桂霞, 裴志尧, 宋佳智 基于深度学习的蛋白质-ATP结合位点预测. 吉林大学学报(工学版) 2022;52(01):187–194. [Google Scholar]
- 16.Xu Y, Qian X, Tong Y, et al AttnTAP: A dual-input framework incorporating the attention mechanism for accurately predicting TCR-peptide binding. Front Genet. 2022;13:942491. doi: 10.3389/fgene.2022.942491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai M, Bang S-J, Zhang P, et al. ATM-TCR: TCR-epitope binding affinity prediction using a multi-head self-attention model. Front Immunol, 2022, 13: 893247.
- 18.Alspach E, Lussier D M, Miceli A P, et al MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574(7780):696–701. doi: 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marty Pyke R, Thompson W K, Salem R M, et al. Evolutionary pressure against MHC class II binding cancer mutations. Cell, 2018, 175(2): 416-428. e13.
- 20.王广志, 李雨雨, 谢鹭 个性化肿瘤新抗原疫苗中抗原肽预测研究进展. 生物化学与生物物理进展. 2019;(5):8. [Google Scholar]
- 21.Lu M, Xu L, Jian X, et al dbPepNeo2. 0: A database for human tumor neoantigen peptides from mass spectrometry and TCR recognition. Front Immunol. 2022;13:855976. doi: 10.3389/fimmu.2022.855976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.赵静静, 简星星, 王广志, 等 TCR组库及其在肿瘤免疫中的应用前景与挑战. 生命科学. 2021;33(4):6. [Google Scholar]