

Abstract
Biliary atresia (BA) is a progressive inflammatory fibrosclerosing disease of the biliary system and a major cause of neonatal cholestasis. It affects 1:5,000–20,000 live births, with the highest incidence in Asia. The pathogenesis is still unknown, but emerging research suggests a role for ciliary dysfunction, redox stress and hypoxia. The study of the underlying mechanisms can be conceptualized along the likely prenatal timing of an initial insult and the distinction between the injury and prenatal and postnatal responses to injury. Although still speculative, these emerging concepts, new diagnostic tools and early diagnosis might enable neoadjuvant therapy (possibly aimed at oxidative stress) before a Kasai portoenterostomy (KPE). This is particularly important, as timely KPE restores bile flow in only 50–75% of patients of whom many subsequently develop cholangitis, portal hypertension and progressive fibrosis; 60–75% of patients require liver transplantation by the age of 18 years. Early diagnosis, multidisciplinary management, centralization of surgery and optimized interventions for complications after KPE lead to better survival. Postoperative corticosteroid use has shown benefits, whereas the role of other adjuvant therapies remains to be evaluated. Continued research to better understand disease mechanisms is necessary to develop innovative treatments, including adjuvant therapies targeting the immune response, regenerative medicine approaches, and new clinical tests to improve patient outcomes.
Introduction
Biliary atresia (BA) is a devastating inflammatory obliterative disease of the bile ducts. BA affects 1 in 5,000–20,000 newborns and has its highest incidence in Asia1,2. It is a heterogeneous, complex, multifactorial disorder with unknown pathogenesis3. Genetic and/or developmental susceptibility, injury by an environmental toxin or virus infection and immune–inflammatory dysregulation have been implicated. Irrespective of the initiating events, and often despite treatment, liver fibrosis and ultimately cirrhosis become the dominant pathologies4–6.
BA diagnosis is challenging, sometimes missed or made late, and can only be established by intraoperative cholangiography. Timely surgery by Kasai portoenterostomy (KPE), in which the obliterated extrahepatic bile duct (EHBD) is replaced with an intestinal conduit, restores bile flow in some patients. Failure to restore bile drainage is typically managed by liver transplantation, often within the first 2 years of life. However, even patients who have achieved clearance of jaundice (CoJ) frequently develop complications, including portal hypertension, cholangitis, hepatopulmonary syndrome and even malignancy due to progression of liver injury7–10.
Early diagnosis, centralization of resources and multidisciplinary management have led to improved rates of survival with the native liver11. The value of current adjuvant therapies remains debatable, highlighting the need to improve understanding of disease pathogenesis and to develop new treatment strategies12.
This Primer provides a comprehensive summary of up-to-date research on BA, covering its epidemiology, mechanisms and pathophysiology, diagnosis, management, quality of life, progression of liver injury and future directions for research.
Epidemiology
Incidence
BA is a rare disease that occurs in most, if not all, ethnic groups, but incidence rates differ considerably (Fig. 1). Incidence differences between ethnic groups within the same country or region suggest a genetic predisposition2,13. Studies in Western countries including Netherlands14, France15, Croatia16, Canada17, USA18, England and Wales19, Switzerland20, Finland21 and Sweden22 have found incidences between 1:14,000 and 1:22,000 per live births. BA is more common in East Asia, with reported incidences between 1:5000 and 1:9000 in Taiwan23, Japan24, Korea25, and Shanghai and Hong Kong, China2. In New Zealand, a striking difference in BA incidence was observed between the Māori population (1:5,000) and children of European origin (1:16,000)26. No published national data exist from Africa, South America, India or Australia.
Fig. 1 |. Global incidence of BA.
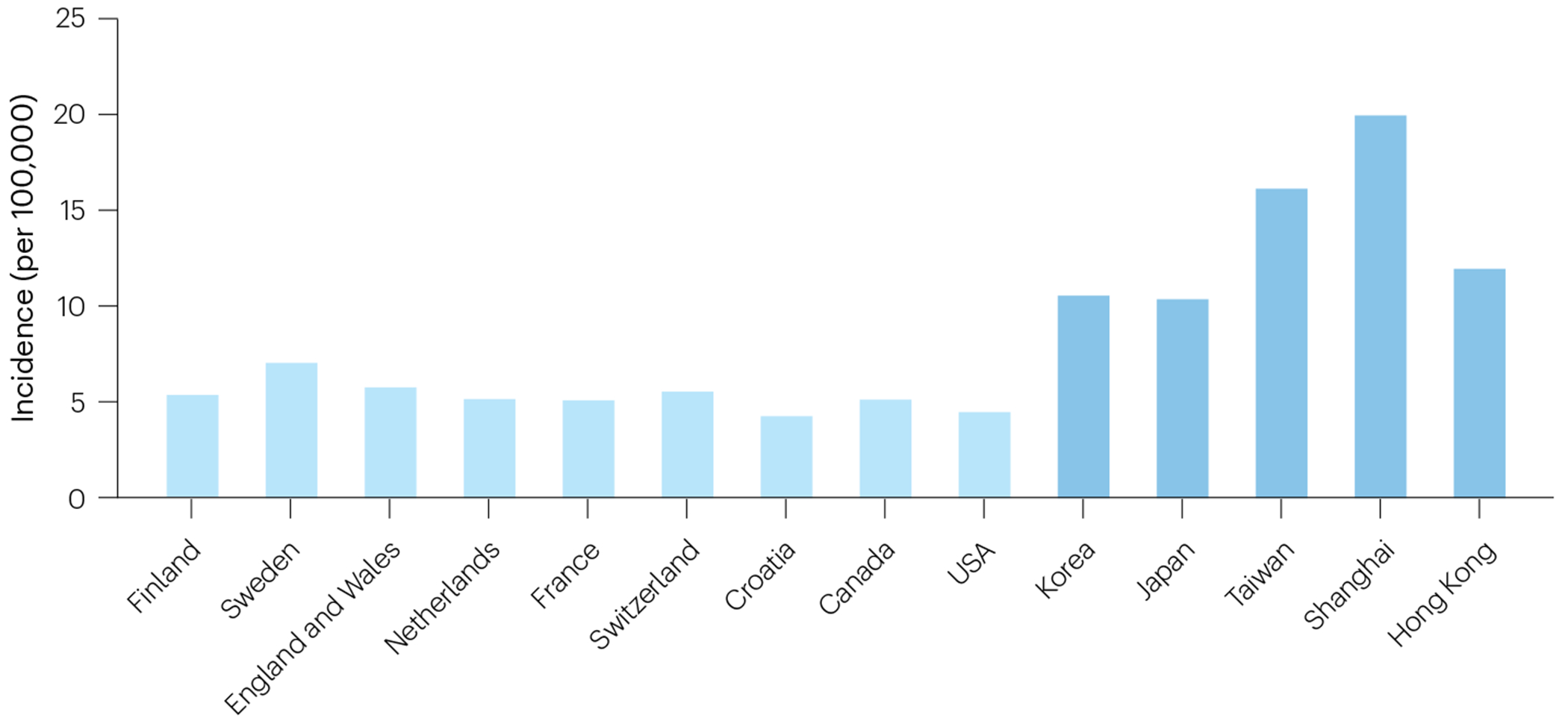
Incidences of BA reported in nationwide or multicentre studies. Overall, higher incidences were reported for East Asian than for Western countries/regions.
Demographic associations
Environmental factors.
Environmental factors are believed to be important in BA pathogenesis. However, results of studies analysing the effect of environmental exposures on BA incidence are inconclusive. The incidence of BA has been variably reported to be unchanged27 or higher in urban28 and rural settings14. Some studies report seasonal variations in the incidence of BA, with different seasonal peaks25,28,29, but most larger studies do not support seasonality18,19,30,31. Epidemiological data supporting a viral aetiology are also scarce. In Taiwan, decreasing BA incidence over time showed a statistically insignificant correlation with rotavirus vaccine coverage rates32. A European study showed that stricter COVID lockdowns were associated with larger decreases in BA incidence33.
Sex distribution.
The incidence of BA overall is higher in females, with a risk ratio of approximately 1.4 in most Western14,17–19,21 and Asian countries25, rising to 1.8 in Japan23,24. Inexplicably, female to male ratios of 1.1:1 and 0.7:1 have been reported in China34 and Sweden22, respectively. The sex distribution may also vary between different BA phenotypes, but evidence is scarce. An increased (2:1) female-to-male ratio was observed in patients with biliary atresia splenic malformation (BASM) compared with patients without BASM in a study in England and Wales35, whereas the ratios were similar in a study in Canada36.
Maternal factors.
Maternal diabetes mellitus during pregnancy was found to be ten times more likely to be associated with an infant with BASM than with an infant with BA without splenic malformation (isolated BA)35. Pregestational diabetes was associated with an increase in BA risk of more than twofold in two population-based studies37,38. In Sweden, no association with diabetes was seen, but maternal age >35 years and parity of four or more were associated with increases in BA risk of twofold to threefold22. Prenatal (third trimester) exposures to maternal intestinal and genitourinary infections were associated with increases in BA risk of sixfold and 1.6-fold, respectively39. First trimester anti-inflammatory asthma medication40 and maternal drug abuse37 were associated with increases in BA risk of more than threefold. Although not confirmed as a risk factor in population-based studies, in one cohort, 26% of 39 children with BA had been conceived through in vitro fertilization41.
Ethnicity.
The risk of BA is twofold to threefold higher in Asian countries and in children of Asian descent than in Caucasian populations18,26 (Fig. 1). Higher incidence rates in non-white than in white children have also been reported in England and Wales3, Atlanta and New York28,29. In addition, the relative proportions of different BA phenotypes differ between ethnicities, with BASM being much more common in Western countries3,14,15,24,25,34,36, whereas cytomegalovirus (CMV)-associated BA seems to be more frequent in Asia42–45.
Prematurity.
Twofold and fivefold higher BA incidence rates have been reported in children born prematurely at <37 weeks and at <32 weeks of gestation, respectively22,38,46,47. Premature children with BA present more often with associated malformations than children with BA born at term (19%47, 30%27 and 40%46). In a UK study among preterm babies with BA, a higher than expected proportion of twins was observed (48%)27. Preterm children with BA tend to be older at time of KPE38,47; however, they may reach similar CoJ and native liver survival rates to those in children with BA born at term27. Small for gestational age was found to be associated with an increase in BA risk of almost fivefold in a Swedish national study22.
CMV infection.
Human data implicate a relationship between CMV infection and BA42–45. Patients with BA and CMV infection are considered a special subgroup owing to their clinically distinguishable characteristics, including older age at time of KPE, lower CoJ rates, shorter native liver survival, and higher pretransplant mortality42–44,48–50. Infants with BA and higher anti-CMV IgM antibody levels have a worse prognosis following KPE44,51,52.
Syndromic biliary atresia and associated anomalies.
Syndromic BA usually refers to the BASM syndrome. In addition to asplenia, polysplenia or double spleen, patients with BASM may also present with other anomalies, including situs inversus (50% of patients), malrotation, a preduodenal portal vein or an absent intrahepatic vena cava (Fig. 2a,b). Some studies included patients with these laterality defects but without splenic malformations36, in contrast to the original definition of BASM35. BASM accounts for 12–16% of BA cases in Western countries3,7,14,15,36, but only 0–3% in Asia24,25,34,47. However, as the incidence of isolated BA is about threefold higher in Asia, the occurrence of BASM may be constant across continents. Patients with BASM typically have EHBDs that are more atrophic, a higher risk of hepatopulmonary syndrome31,53, and inferior native liver survival rates than those with isolated BA31,35. BA has also been reported in patients with other defined syndromes, such as cat-eye syndrome, Kabuki syndrome and Kartagener syndrome3,54,55.
Fig. 2 |. Aetiological heterogeneity of BA.

Biliary atresia splenic malformation syndrome showing a pre-duodenal portal vein (part a, arrow) and left-sided polysplenia (part b, circled). Cystic biliary atresia with cholangiogram (part c) showing contrast retention in the cyst, absence of flow into the intestine, and abnormally fine, interconnecting etiolated intrahepatic bile ducts. Isolated biliary atresia (part d) showing (1) mobilized atrophic gallbladder, (2) umbilical vein and (3) sling around right hepatic artery. Cut surface of the porta hepatis before Roux loop reconstruction (part e). Photomicrograph of cut surface showing scattered biliary ductules within a fibroinflammatory stroma (vimentin-positive) (part f). Part f reproduced with permission from La Pergola E et al. (2021)321.
The incidence of congenital heart disease in BA varies between cohorts without consistent geographical differences. Cardiac malformations are observed in 6–8% of patients with BA in the UK, Nordic countries, Korea and Canada7,25,36,56, in 3% in China and India34,57, and in 15–16% in Sweden and North America22,58. BASM has been reported in 50% and cat-eye syndrome in 11% of patients with cardiac malformations, and the co-existence of other congenital anomalies is also more common than in patients with isolated BA56. Patients requiring correction of heart defects should probably undergo cardiac surgery before KPE as this approach has been related to improved CoJ rates56.
Other congenital malformations observed in patients with BA include intestinal atresia (0.5–5%)3,57, anorectal malformations, Hirschsprung disease, abdominal wall defects, hypospadias (<1%)3,34, oesophageal atresia (1–3%)3,25,57 and duodenal atresia (1.5% in a UK series, with a higher incidence in patients with BASM than in those with isolated BA3, but none in cohorts from Korea, India and China, where BASM is uncommon25,34,57).
Mechanisms/pathophysiology
Injury and the response to injury in BA
We suggest that BA should be viewed as the combination of an injury and a response to injury, and we consider the pathophysiology of BA from this perspective, while noting that key mechanisms may contribute to both phases (Fig. 3).
Fig. 3 |. BA as a combination of injury and maladaptive response.

Biliary atresia (BA) is viewed as the result of an injury, probably prenatal, and the response to that injury, probably occurring in both the prenatal and postnatal periods. Agents that may contribute to injury include genetic causes (most notably genetic alterations that lead to abnormal primary cilia), environmental agents and developmental susceptibilities of the immature extrahepatic bile duct (EHBD). Other factors, such as sex and ethnicity, can predispose to BA. Dysregulated immune and wound healing responses comprise the response to injury and facilitate progression of the disease. Clinically detectable outcomes of the disease progress to BA and, subsequently, liver dysfunction and liver fibrosis. The potential for recovery after injury by regenerative or reparative healing is unknown and remains speculative. CMV, cytomegalovirus; ECM, extracellular matrix.
Understanding the nature of the initial insult in BA requires understanding when it occurs. Although BA is diagnosed exclusively in neonates, most of whom are asymptomatic at birth, two studies in the USA including a combined 400,000 newborns demonstrated that elevated levels of conjugated bilirubin within the first few days of life were 100% sensitive for later development of BA59–61. This led to a paradigm shift in the concept of BA pathophysiology, suggesting that the injury leading to BA occurs in utero or (less likely) around birth, and that the postnatal period is a time of response to injury marked by rapid injury progression and maladaptive healing. Recovery of injured bile ducts in the neonatal period has never been observed, although animal studies as well as a small percentage of babies with elevated conjugated bilirubin levels who never develop evidence of any kind of liver disease raise this speculative possibility60–63.
In this section, we first outline the development and anatomy of the biliary tree, with a particular reference to differences between humans and rodents; then, because experimental models (particularly rodents) are critical to research into BA pathophysiology, we describe the major current models. We then discuss key aspects of the injury phase of BA, followed by a discussion of the response to injury (Fig. 3). Of note, many aspects (for example, genetic factors and immune dysregulation) undoubtedly contribute to both the initial injury and the progressive response to that injury; they are discussed according to their likely primary role.
Hepatobiliary development and anatomy
The biliary tree is a continuous and arborizing network consisting of two highly heterogeneous regions with different developmental origins: hepatocytes and intrahepatic bile ducts (IHBDs) are derived from the cranial liver bud and EHBDs and the gallbladder are derived from the caudal liver bud (pars cystica)64,65. Early BA primarily affects the extrahepatic and large intrahepatic/hilar bile ducts, although progressive disease affects the entire biliary tree and the effects on intrahepatic disease of the primary injury and the post-obstructive complications are not well understood. The EHBDs and large (distal) IHBDs are exposed to bile that may have been modified during passage through the intrahepatic ductal system, and have anatomical and biochemical features that may make them particularly susceptible to injury. Additionally, repair mechanisms in these structures are different: proximal IHBDs are surrounded by hepatocytes that function as a source of bipotential (hepatocyte and cholangiocyte) cells, whereas EHBDs (and distal IHBDs66) are surrounded by peribiliary glands that contribute to duct homeostasis and are mobilized to repair injured cholangiocyte monolayers67,68.
Human and rodent hepatobiliary trees develop with different relative time courses. Examples include the emergence of the ductal plate at weeks 7–8 of gestation in humans but after the midpoint of gestation in mice, and the apparent equivalence of the EHBD matrix of fetal humans and postnatal rodents64,69. Although the overall differences are poorly understood, they are essential to consider in using animal models.
Disease models
Various technology platforms for disease modelling have advanced our understanding of BA, with several new or improved in vivo and in vitro disease models introduced in recent years.
Rodent models.
The rhesus rotavirus (RRV) mouse model of BA, in which BALB/c mice are injected intraperitoneally with RRV in the first 24 h of life, mimics key features of human BA (including hyperbilirubinaemia and EHBD obstruction)70 and is the most commonly used animal model. It has yielded important insights into potential mechanisms of BA but is limited by a fatality rate >90% before the development of advanced fibrosis and other pathologies. Use of a rotavirus reassortant strain (RRV-TUCH) improves early survival to 63%71. Other models include common bile duct ligation in neonatal rats72, and BA-like disease in neonatal mice induced by the biliary toxin biliatresone63. No experimental animal models have yet generated BA in neonates after treatment of pregnant mothers, although a recent study showed abnormal bile acid synthesis in the pups of biliatresone-treated pregnant mice73.
Zebrafish.
Zebrafish (Danio rerio) and humans share substantial similarities in the development, anatomy and function of the biliary tree. The use of zebrafish in cholestasis research — in particular employing fluorescent dyes to identify biliary excretion in transparent larvae — has contributed to large-scale screening and characterization of potential biliary toxins74,75, validation of candidate genes from genetic studies76–79, and identification of the role of DNA hypomethylation in BA80,81.
In vitro models.
Liver derived organoids, which consist of cholangiocytes or hepatocytes in a spherical monolayer epithelium, are increasingly used in liver research82,83. Pluripotent stem cells, such as embryonic stem cells and induced pluripotent stem cells, are capable of generating hepatic progenitor cells and cholangiocyte organoids84–86, which have been used extensively in BA research to study mechanisms of cholangiocyte damage in BA. For example, breakdown in apical–basal polarity with perturbed cholangiocyte development occurred in organoids generated from the epithelial cell adhesion marker-expressing cells of patients with BA but not in organoids from cells of patients without BA87,88. In another study, CRISPR–Cas9-mediated knockout of the BA susceptibility genes ADD3 and GPC1 led to reductions in ductal structure formation in organoids89. Neonatal mouse EHBD explants in culture are also extensively used in BA research; following injury, these explants demonstrate BA-like cholangiocyte monolayer injury and collagen deposition90. Organs-on-a-chip have similarly been used, sometimes with human organoid-derived cells, as in vitro models of the neonatal EHBD91,92.
Critical to BA research in the future will be improved animal models (including large-animal and maternal-exposure models) as well as multicellular, human cell-based organoids and on-chip devices paired with specialized (and potentially patient-derived) extracellular matrix (ECM) scaffolds to study cell–cell and cell–ECM interactions.
Injury
Genetic susceptibility.
Despite low heritability of BA, recent large-scale genetic studies have unequivocally demonstrated that BA is a complex disorder with considerable contributions from common and rare genetic variants (Table 1). Genome-wide association studies (GWAS) identified a group of genes involved in hepatobiliary development and structure, including ADD3, EFEMP1, ARF6, GPC1, MAN1A2, AFAP1 and TUSC3 (refs. 13,77,93–97). Dysregulation of these genes may perturb embryogenesis of the biliary tree or disrupt ECM organization and integrity, thereby increasing susceptibility to extrahepatic biliary injury.
Table 1 |.
BA-associated genetic loci
| Loci | Odds ratio (95% confidence interval) | P value | Population | Mechanism | Ref. |
|---|---|---|---|---|---|
| Common variants | |||||
| ADD3 | 2.1 (1.4–3.2) | 6.9 × 10−9 | Chinese | Biliary morphogenesis | 13,93 |
| 1.7 (1.5–2.0) | 4.1 × 10−11 | Chinese | 318 | ||
| 1.7 (1.3–2.2) | 7.2 × 10−5 | Chinese | 319 | ||
| 1.7 (1.2–2.6) | 2.5 × 10−3 | Thai | 320 | ||
| 1.5 (1.1–1.9) | 2.0 × 10−3 | Caucasian | 108 | ||
| EFEMP1 | 1.5 (1.3–1.8) | 3.4 × 10−8 | Caucasian | ECM | 94 |
| AFAP1 | 1.4 (1.3–1.5) | 3.9 × 10−8 | Caucasian | Ciliogenesis | 97 |
| TUSC3 | 1.4 (1.3–1.5) | 1.3 × 10−7 | Caucasian | Ciliogenesis | 97 |
| MAN1A2 | 2.6a | 7.0 × 10−10 | Caucasian | Ciliogenesis | 95 |
| ARF6 | 2.7a | 4.2 × 10−8 | Caucasian | Biliary morphogenesis | 96 |
| Rare variants | |||||
| GPC1 (deletions) | 138.7a,b | 4.4 × 10−10 | Caucasian | Biliary morphogenesis | 77 |
| PKD1L1 (biallelic variants) | – | – | Caucasian | Ciliogenesis | 101 |
| Ciliary genes (PCNT, KIF3B, TTC17 and others) | 2.5 (1.2–6.07) | 0.034 | Chinese | Ciliogenesis | 78 |
BA, biliary atresia; ECM, extracellular matrix.
No standard error was provided.
Imprecise estimate due to a small number of cases.
Genetic data also suggest that polygenic predisposition to ciliary dysfunction underlies BA irrespective of the clinical subtype. Cholangiocyte cilia abnormalities, including misorientation and reduction in length and quantity, have been described in both syndromic and non-syndromic forms of BA98–100. Rare biallelic variants in a ciliary gene, PKD1L1, were first identified in five of 67 Caucasian patients with BASM101 and bile duct ligation in liver-specific Pkd1l1-deficient mice recapitulated features seen in patients, including reduced primary cilia, progressive cholangiocyte proliferation and peribiliary fibroinflammation102,103. A whole-exome sequencing study on 89 patients with non-syndromic BA and their parents showed a 2.6-fold increase in mutational burden of rare (de novo or biallelic) damaging mutations in ciliary genes in 31% of the patients78. Functional analyses showed the absence of cholangiocyte cilia in the patients with deleterious ciliary mutations. Non-motile primary cilia are at the hub of many signalling pathways, including Hedgehog, NOTCH, WNT and transforming growth factor-β (TGFβ)–bone morphogenetic protein pathways, which are essential for bile duct development and for detecting the flow, composition, osmolality and pH of the bile104–106. Defects in these signalling pathways could cause abnormal biliary development and contribute to the pathogenesis of BA4,64. Intriguingly, abnormally low nasal nitric oxide levels, a clinical screening test for primary ciliary dyskinesia, were detected in eight of ten patients with BA and ciliary mutations78. This phenotypic overlap supports an intrinsic ciliary defect in BA and is also consistent with the pleiotropic disease manifestations affecting multiple systems in some patients with non-syndromic BA.
A GWAS in 811 patients with BA requiring liver transplantation further showed that common variants associated with the ciliogenesis and planar polarity effector (CPLANE) complex may predispose to an increased risk of BA via ciliary dysgenesis97. The CPLANE regulatory network comprises a broad spectrum of proteins (~2,000 genes) essential for ciliogenesis and cilia-mediated patterning107. A polygenic risk score aggregating the effects of ~6,000 common variants in 102 CPLANE genes showed a strong correlation with BA (P = 5.5 × 10−15). Knockdown of the CPLANE genes AFAP1 and TUSC3 impeded the formation of motile cilia in mouse tracheal cells. Additionally, whole-genome sequencing of 100 patients with BA identified enrichment of rare variants of CPLANE genes97.
In line with the variable incidence of BA worldwide, locus heterogeneity is notable across ethnically diverse populations. Except for ADD3, BA candidate loci have been reported in either Caucasian patients or Asian patients but not both13,77,93–97,108. This may result from differences in underlying genetic architecture or reflect different levels of exposure (and, therefore, different susceptibilities) to injurious environmental agents. Nevertheless, the convergence on a limited number of functional pathways, particularly those related to ciliogenesis, supports a shared genetic predisposition across populations. Although BA-associated rare variants have reduced penetrance and common variants have modest effects individually (odds ratio (OR) 1.4–2.7; Table 1), they collectively contribute considerably to the overall risk of abnormal biliary development in BA.
Interestingly, animal models of BA demonstrate acquired ciliary defects — cholangiocyte cilia are abnormal in the RRV mouse model as well as in primary cholangiocytes treated with biliatresone74,99. Human organoids treated with biliatresone also demonstrate abnormal cilia, akin to those seen in organoids derived from patients109. Ciliary abnormalities may represent a common mechanism of disease between genetic and environmental injuries or, alternatively, defects in ciliary genes may predispose to cholangiocyte injury.
Environmental injury.
The injury agent in BA is not known. Viruses have long been suggested as possible aetiological factors for BA. Virus infection could cause defective bile duct development, direct injury to the bile duct epithelium, or a secondary autoimmune reaction1,110,111. CMV, Epstein–Barr virus, rotavirus and human papillomavirus have all been implicated112,113 but data from humans exist mostly for CMV44,48,114. CMV-associated BA has been reported in 10–30%, 60% and 78% of patients with BA in Europe42–44, China45 and South Africa115, respectively. However, studies vary in their definition of CMV positivity and diagnostic methods43–45,49,50. Proof of a causal relationship remains elusive.
Rotavirus is a double-stranded RNA virus of the Reoviridae family that can infect hepatocytes and cholangiocytes116,117. As noted above, RRV infection of neonatal mice is an established model of BA118. In one study, group C rotavirus RNA was detected in 50% of liver specimens from infants with BA but not in specimens from controls with other cholestatic diseases119. By contrast, other studies did not detect rotavirus RNA in liver specimens from infants with BA120 or increased rotavirus IgM in infants with BA compared with controls121.
In proof-of-concept studies, several toxins have been shown to have biliary toxicity potentially relevant to BA. Biliatresone was isolated from Australian plants ingested by pregnant livestock after several outbreaks of a BA-like disease in their newborns74,122. Biliatresone causes selective EHBD injury in larval zebrafish and newborn mice62,63,74. Chemically, it contains a reactive α-methylene ketone group and binds to reduced glutathione (GSH)123–125, the most abundant endogenous small-molecule antioxidant. Levels of GSH are particularly low in neonatal EHBD, which may partially explain the biliatresone mechanism of action75,90. Owing to its limited distribution, biliatresone is probably not relevant to human BA but provides evidence that prenatal exposure to a toxin could lead to BA in neonates while sparing their mother. Preliminary data suggest that microcystin-RR, found in harmful algal blooms, is a biliary toxin, and like biliatresone, is specific in causing redox stress in neonatal (as opposed to adult) cholangiocytes126.
An Egyptian study in infants with BA and their mothers found that BA can result from maternal exposure to the hepatocarcinogen aflatoxin B1 and its toxic metabolite aflatoxin B1-8,9-epoxide, which is normally detoxified by glutathione S-transferase Mu 1 (GSTM1)-mediated conjugation to GSH127,128. These unconfirmed data are intriguing because of the link to GSH.
The role of the maternal microbiome in BA is an emerging area of research. Treatment of pregnant mice with antibiotics during pregnancy and lactation led to a substantial reduction in the incidence of RRV-induced experimental BA in their pups, associated with colonic enrichment of the butyrate-producing bacteria Anaerococcus lactolyticus129. Butyrate administration to pregnant mice rendered their newborns resistant to experimental BA, with intestinal enrichment of Bacteroidetes and Clostridia and an increase in faecal glutamate and glutamine, which protected cholangiocytes against natural killer (NK) cell-mediated toxicity130. Notably Bacteroidetes and Clostridia were under-represented in the faecal microbiota of patients with BA with decreased glutamate and glutamine130,131. These findings may have broad therapeutic implications.
Developmental susceptibility to injury.
Oxidative stress is an important cause of teratogenesis and fetal injury132. Hypoxia is necessary for some stages of development, and the uterine environment during most of the first trimester is low in oxygen and energy metabolism is based on glycolysis. At the end of the first trimester, oxygen levels in the uterine environment increase and metabolism switches to oxidative phosphorylation, suggesting that this is potentially a time of redox stress if antioxidant production lags behind the increased production of reactive oxygen species (ROS)133. Birth and the immediate postnatal period are additional times of potential redox stress due to both ischaemia–reperfusion (from contractions) and the start of breathing with the switch to the postnatal circulation134. A systems biology study combining RNA sequencing (RNA-seq), GWAS and whole-exome sequencing of human BA tissue identified hypoxia signalling as a potential mediator of BA-like bile duct abnormalities; this study validated the potential role of hypoxia in bile duct abnormalities in zebrafish135. Immunostaining of human BA tissue showed expression of hypoxiainducible factor 1α (HIF1α) in hepatic and porta hepatis cholangiocytes; HIF1α activation may result from hypoxia and/or oxidative stress136.
The redox state of the human EHBD during development is not known. The EHBDs of rodents and zebrafish have low levels of GSH in the neonatal and larval stages compared with the adult stage and with the IHBDs, suggesting susceptibility to redox stress75,90,126. Evidence of redox stress in liver tissue is a negative prognostic factor in patients with BA, whereas upregulation of genes involved in GSH metabolism is correlated with increased transplant-free survival137,138. Consistent with fetal and neonatal EHBD susceptibility to redox stress as a risk factor for BA, toxins and viral infections (including CMV infection) cause redox stress139,140. Gestational diabetes and preterm birth, both linked to BA, are similarly associated with redox stress132,141. In support of a causal association between redox stress and BA, the antioxidant N-acetyl cysteine (NAC) reduced RRV-associated EHBD damage in mouse pups138, and a small pilot study of NAC treatment in babies with BA showed improved bile flow, decreased ROS production and decreased inflammation142.
In addition to low levels of GSH, the neonatal mouse EHBD has immature tight junctions that predispose to bile leakage, a collagenpoor submucosa that facilitates spread of leaked bile, and a sparse, immature epithelial apical glycocalyx that predisposes to injury from hydrophobic bile acids143,144. Neonatal EHBDs with these developmental immaturities seem to be particularly sensitive to bile-acid-mediated injury145. The bile acid pool changes considerably between prenatal and perinatal periods146,147, but the relative toxicity of prenatal, perinatal and postnatal bile has not been established.
Susceptibility factors with unknown mechanisms.
Sex, ethnicity, maternal diabetes and prematurity are susceptibility factors with undetermined mechanisms.
β-Amyloid deposition.
β-Amyloid (Aβ) deposition has been identified in the liver of patients with BA, the RRV mouse model and BA organoids87. Importantly, addition of exogenous Aβ to normal control liver organoids was sufficient to cause BA-like morphological changes. Thus, BA could be grouped under diseases with Aβ deposition, such as Alzheimer disease and cerebral amyloid angiopathy; however, in contrast to the extracellular deposits in other amyloid diseases, Aβ deposition in BA seems to be intracellular. Aβ deposition is a novel pathobiological feature of BA and may provide new diagnostic biomarkers (Aβ42/Aβ40) and adjuvant therapies (anti-Aβ).
Taken together, multiple factors — genetic, environmental, developmental and others, as-yet undefined — may contribute, alone or in combination, to the initial injury that probably occurs in the prenatal period.
Response to injury
Dysregulated and aberrant immune responses.
A leading hypothesis in the pathogenesis of BA is that, in the genetically predisposed infant with immature or altered biliary development, cholangiocyte injury leads to exaggerated autoimmune and autoinflammatory responses targeting cholangiocytes, resulting in progressive bile duct damage, fibrosis and obliteration5. In BA, there is abundant evidence that adaptive immune cells (T cells148–152 and B cells152–155) and innate immune cells (NK cells130,148,156, macrophages157–160, dendritic cells157,161 and neutrophils142,162–164) contribute to biliary injury and disease pathogenesis (Fig. 4).
Fig. 4 |. Hypothetical contribution of immune dysregulation to the pathogenesis of biliary atresia.

Injury by virus infection or toxins of altered cholangiocytes triggers a cascade of early innate (autoinflammatory) and subsequent adaptive (autoimmune) responses that proceed unchecked in the setting of regulatory T (Treg) cell deficits. The chronic inflammation further damages cholangiocytes and activates stellate cells, resulting in progressive biliary fibrosis. EHBD, extrahepatic bile duct; IHBD, intrahepatic bile duct; ILC2, type 2 innate lymphoid cell; NET, neutrophil extracellular traps; NK, natural killer; ROS, reactive oxygen species.
In addition, various inflammatory pathways contribute to biliary injury in BA. Extensive investigations on the role of T cells showed that early in biliary injury a T helper 1 (TH1) (that is, IFNγ and TNF)-predominant response occurs165–167 followed by a subsequent TH2 (that is, IL-33) response that is associated with bile duct proliferation and fibrosis150,168,169. IL-33 activates a type 2 innate lymphoid cell (ILC2) that shows plasticity and includes the subsets of natural ILC2s (nILC2) and inflammatory ILC2s (iILC2). IL-33-activated nILC2s produce IL-13, leading to cholangiocyte hyperplasia and epithelial repair150,170. When the nILC2 IL-13–IL-4Rα–STAT6 pathway (regulated by amphiregulin) is inhibited, iILC2s predominate and the full experimental BA phenotype ensues170.
The role of B cells in BA pathogenesis has also been investigated. Liver immune profiling in infants with BA by single-cell RNA-seq (scRNA-seq)152 confirmed previous findings of cytotoxic CD8+ T cell expansion and aberrant NK cell and macrophage responses, and revealed aberrant hepatic B cell lymphopoiesis. In the RRV BA mouse model, administration of anti-CD20 B cell-depleting antibodies152 or use of B cell receptor-deficient mice155 resulted in decreased liver CD4+ and CD8+ T cell levels and increased survival. In addition, B cell activation was associated with accumulation of non-specific IgG autoantibodies152 and cholangiocyte-targeted IgM autoantibodies that correlated with worse outcomes153.
Cholangiocyte proteins may be identified as foreign by immune system components owing to alterations by the disease agent or exposure of sequestered proteins. Alternatively, there may be molecular mimicry, whereby viral proteins or toxic moieties have a high sequence homology with cholangiocyte proteins, with cross-reactivity resulting in adaptive ‘autoimmune’ responses171. Deficits in the CD4+FOXP3+ regulatory T (Treg) cells that are responsible for inhibiting autoreactive T cells172 in both human BA and animal models resulted in autoreactive T cells targeting cholangiocytes and contributing to disease pathogenesis173–177. Epigenetic modifications of FOXP3 (ref. 178) could cause functional Treg cell deficiencies in BA, and decreased Treg cell frequency was associated with higher bilirubin levels in infants after KPE179. Another potential trigger of an ‘autoimmune’ response in BA is maternal microchimerism, whereby maternal cells entering the fetus may alter its response to self-antigens and trigger autoimmunity180. Many investigators have identified increased numbers of maternal cells in the liver of patients with BA compared with the numbers in the liver of control patients without BA181–184. Whether or not these maternal cells are pathogenic or simply bystanders related to inflammation is unclear. A working theory is that the maternal microchimeric cells initially function as effector T cells causing biliary injury in utero (similar to a graft-versus-host response). Postnatally, the retained maternal cellular proteins are seen as foreign targets, eliciting anautoimmune-like response (host-versus-graft response)185.
BA mimics autoinflammatory diseases, which are defined by dysregulation or disturbance of the innate immune system, with macrophages and neutrophils as the main effector cells186. Macrophages were found to be recruited to the liver via a dendritic cell–TH17 cell–macrophage axis and contributed to cholangiocyte injury in a mouse model of BA157. An effector mechanism of liver macrophages within murine BA includes the NLRP3 inflammasome, a potent producer of pro-inflammatory cytokines and cytotoxic molecules187. scRNA-seq demonstrated that the livers of infants with BA have deficits within a regulatory (anti-inflammatory) macrophage subset, which would promote chronic macrophage activation160.
In murine BA, CD177+ neutrophil extracellular trap (NET)-forming neutrophils resulted in cholangiocyte injury, whereas CD177−/− mice did not develop BA163, and patients with BA demonstrated a predominance of CD177+ neutrophils in peripheral blood that led to increased NET production and resulted in cholangiocyte apoptosis in in vitro studies162. Similarly in patients, increasing levels of plasma IL-8 (neutrophil activator), neutrophil elastase and NETs positively correlated with bilirubin levels 60 days after KPE, whereas rising IL-8 levels were associated with an increased risk of transplantation or death 1 year after KPE179, suggesting that persistent neutrophil activation promotes biliary injury and fibrosis.
Inflammation and bile acid metabolism.
The relationship between bile acids and inflammation is dichotomous: on the one hand, bile acid-mediated cellular injury can lead to subsequent activation of inflammation and, on the other hand, inflammation directly altering bile acid metabolism could result in bile acid retention188. In the RRV mouse model of BA, early inflammatory responses resulted in persistent downregulation of multiple bile acid transporters and nuclear receptors, despite high bile acid concentrations in the liver189. A potential cause of the inflammation-induced downregulation of bile acid transport may be an epigenetic phenomenon, with production of microRNAs that specifically target bile acid transporters and/or nuclear receptors190. By contrast, low levels of PPARα and FXR (regulators of bile acid formation and transport) were associated with upregulation of inflammatory and fibrotic pathways in BA, suggesting bile-acid-induced inflammation191.
Taken together, these studies suggest that a sustained induction of adaptive and innate immune responses in the setting of dysregulated immunity results in chronic biliary injury, bile acid retention and fibrosis, contributing to the pathogenesis of BA.
The ECM in disease progression
The ECM is a complex and dynamic cellular environment that provides biochemical and biomechanical cues as part of wound healing and fibrosis. The ECM of the fetal EHBD in humans and other species has minimal collagen but large amounts of hyaluronic acid (HA). Interestingly, wound healing in fetal sheep and neonatal mice features considerable HA, rather than collagen, deposition69,143. As fetal wound healing192 is scarless (regenerative rather than reparative), this raises questions about the effects of the switch to an adult wound healing programme in late gestation and the perinatal appearance of actively collagen-secreting fibroblasts in the EHBD submucosa143. Fibrotic remnants excised during KPE from patients with BA show that type I collagen is outside the thickened HA ring that surrounds epithelial remnants (obliterated bile duct lumen), raising the possibility that swelling of the highly hygroscopic HA could contribute to duct obstruction143,193.
Multiple studies in patients with BA also support a key role for collagen degradation in disease progression. Expression of matrix metalloproteinase 7 (MMP7), which breaks down ECM proteins, is elevated in cholangiocytes of patients with BA194, and MMP7 is a biomarker for the disease195. In the liver from patients with BA, MMP7 was mainly produced by hepatocytes, bile duct epithelial cells and some Kupffer cells, and elevated MMP7 expression was detected not only in these cells but also in interstitial fibrous tissue196, suggesting that alterations in the ECM niche are linked to disease progression.
Outcome
The response phase after the initial injury involves dysregulation of immunity, inflammation and the ECM. These factors underpin BA pathogenesis and disease progression, and are critical in determining CoJ and native liver and overall survival. Identifying suitable therapeutic targets and optimal timing for new interventions is the next challenge in improving patient outcomes.
Diagnosis, screening and prevention
Diagnosis
Up to 50% of newborns experience transient jaundice after birth, but only around one in 2,500 full-term neonates develops cholestatic jaundice, characterized by increased conjugated bilirubin (>17 μmol/l; ≥1 mg/dl)197. Although some asymptomatic infants in whom conjugated hyperbili-rubinaemia is detected on neonatal screening may not develop BA, persistent neonatal cholestasis should be considered pathological, and BA is the most common cause (25–40%)198. Prompt diagnostic evaluation is necessary in all infants who remain jaundiced beyond 2 weeks of age if formula-fed or 3 weeks if breastfed197. Differential diagnosis includes monogenic aetiologies (25%), such as Alagille syndrome, other congenital causes, such as ɑ1-antitrypsin deficiency, cystic fibrosis, tyrosinaemia, choledochal cyst and others, total parenteral nutrition-associated cholestasis and so-called idiopathic neonatal hepatitis. The antenatal history is rarely specific for BA, but cystic BA may be detected during maternal ultrasonography, with a differential of a choledochal cyst, together with some of the associated anomalies (particularly cardiac anomalies) typically seen in BASM (Fig. 2).
Serum liver biochemistry changes, imaging results and liver biopsy findings may suggest BA, but these are not specific for the disease. Features on ultrasonographic scans that suggest BA include the presence of a hypoplastic or atrophic gallbladder and fibrosis of the bile ducts in the porta hepatis (triangular cord sign). Sometimes the cystic element of cystic BA may be visible2,197 ,199,200. Radioisotope hepatobiliary scans may show absence of hepatic isotope excretion but this finding may not be discriminatory197,201. Percutaneous liver biopsy has been the mainstay of preoperative diagnosis of BA for some time, particularly in North America and the UK. Histology may show portal inflammation, bile plugs within the duct lumen, bile duct proliferation, ductular reaction (DR) and fibrosis. The estimated diagnostic accuracy of a needle liver biopsy is ~90% (95% CI 85–95%)202,203. The risk of a false-negative biopsy is increased in infants <30 days old, due to immature liver histopathology202,204. The need to wait for 4–7 days for the histological diagnosis of BA has led some centres in Asia to advocate for diagnostic laparoscopy followed immediately by KPE if the operative diagnosis is BA.
Definitive diagnosis is achieved on laparotomy or laparoscopy with possible intraoperative cholangiography. BA is usually classified according to the level of proximal obstruction: types I, II and III are characterized by atresia at the levels of the common bile duct, common hepatic duct and porta hepatis, respectively1. Absence of bile in the gallbladder is highly suggestive of BA, whereas presence of bile in the gallbladder almost always excludes a diagnosis of BA, with the possible exception of type I BA. Nevertheless, results of intraoperative cholangiography are not always conclusive. Contrast flowing from the gallbladder to the intestine does not rule out a more proximal obstruction, and visualization of the intrahepatic ducts may require pressure on the common bile duct. A hypoplastic but patent biliary tree may be a feature of Alagille syndrome6.
New, less-invasive diagnostic methods have been developed with the aim of improving accuracy. Serum MMP7 seems to have high discriminatory potential with an area under the curve reaching 0.90–0.99 (refs. 194,205,206). The optimal cut-off concentration of MMP7 depends on the analytical method, and further research is warranted before routine clinical application207 ,208. Plasma Aβ levels have been reported to enhance diagnostic accuracy in combination with liver biochemistry209, and increased liver stiffness (measurements of >7.7 kPa in infants up to the age of 90 days and >8.8 kPa in infants older than 90 days) has shown promising accuracy in differentiating between BA and non-BA cholestasis210. However, large multicentre studies are needed to confirm these observations211.
Screening
BA fulfils most of the criteria of a condition suitable for newborn screening212. The original screening method used in Japan, Taiwan, and some parts of Canada, Germany and Switzerland involved distribution of stool colour cards (SCC) to parents, with subsequent evaluation in those shown to be pale. Digital versions are now available212. SCC screening resulted in earlier age at time of KPE and improved native liver survival rates213. Although universal use and long-term evidence of the benefit of SCC screening have not been established214–216, in a study in China, this method led to earlier diagnosis and better prognosis217.
The recognition that BA is present at birth in most infants59 led to an increase in studies involving measurement of fractionated bilirubin in whole-blood samples, and in dried blood spots in a two-stage screening programme as a marker of cholestasis60,218–220. Such screening programmes have decreased the age at KPE and improved surgical outcomes60,213. Elevated conjugated bilirubin in early whole-blood samples has been shown to be a sensitive screening tool for BA, but technical difficulties remain in developing consistent conjugated bilirubin screening using dried blood samples for large-scale screening in different laboratories.
Prevention
Prevention of BA remains elusive. Some epidemiological data link maternal intestinal and genitourinary infections39, first-trimester anti-inflammatory asthma medication40 and maternal drug abuse with an increased risk of BA37. Experimental studies have suggested benefits of altering the maternal gut microbiome in BA prevention129–131. However, no clinical data exist that show whether intervention on these risk factors can prevent the development of BA in susceptible infants.
Management
Kasai portoenterostomy
Open Kasai portoenterostomy.
Modern surgery for BA dates from the 1950s and 1960s. Kasai transformed BA from a mostly untreatable and lethal condition into a treatable and chronic disease with the introduction of portoenterostomy. In a KPE, the obliterated bile duct remnant is sectioned in the porta hepatis and reconstructed using a jejunal Roux loop (Fig. 5). Microscopic examination shows patent biliary ductules at the site of anastomosis retaining a connection with the intrahepatic biliary system8,221. The nature of the IHBDs can sometimes be directly appreciated in patients with cystic BA when the cholangiogram shows a myriad of intercommunicating etiolated ductules (Fig. 2c). Since the 1970s, KPE has become the global standard treatment for BA. Ohi extended the level of hilar dissection with the upper limit being flush with the liver capsule but not breaching the liver parenchyma222. This radical dissection achieves complete excision of the extrahepatic biliary system exposing the widest possible portal plate for reconstruction. Typically, the Roux loop is 30–40 cm long and passes behind the colon (Fig. 5). Subsequent modifications of this procedure, including the creation of a ‘valve’ from partial intussusception of the mucosa, have not shown additional benefit223.
Fig. 5 |. Kasai portoenterostomy.

The mobilized jejunum is transected, and its proximal end is anastomosed to the transected portal plate (portoenterostomy) to enable bile drainage from the intrahepatic biliary system into the intestine via remnant patent biliary ductules. The proximal limb of the small intestine (downstream of the duodenum) is anastomosed to the 30–40 cm retrocolic Roux loop (jejunojejunostomy).
Laparoscopic Kasai portoenterostomy.
Early attempts to replicate the open procedure in a laparoscopic approach had little success. In the past 10 years, surgeons in high-volume centres, particularly in China, have reinvigorated laparoscopic KPE224,225. However, compared with open KPE, the extent of the dissection is fairly restrained and similar to Kasai’s original description without the subsequent extended dissection proposed by Ohi. This is predominantly because a safe extended dissection is much more difficult to accomplish with current laparoscopic technology.
KPE can also be accomplished using robotic technology (three arms, four ports). However, in a report from China226, hospital stay was prolonged to ~10 days, compared with 6–7 days for open KPE typically seen in the West227. This is obviously contradictory to one of the purported benefits of laparoscopy. In one of the largest published experiences of laparoscopic KPE225, patients remained in hospital for 30 days, largely for intravenous corticosteroid therapy. These techniques seem to have limited real advantages beyond cosmesis. Real outcomes data from prospective studies vary considerably and suggest rates of CoJ ranging from 41%228 to >80%229. The only long-term study involving laparoscopic KPE showed a dramatic difference in 10-year native liver survival: 45% (5/11) after laparoscopic KPE and 85% (17/20) after open KPE (P = 0.03)230.
Outcomes of Kasai portoenterostomy.
Multiple national series and large consortia reports on the outcomes of KPE have been published7,9,10,15,20,23,213,231–238 (Table 2). These are predominantly from Japan, Taiwan and Europe with few from the Americas, South Asia or Africa. The key outcome measures are time to KPE, CoJ to normal levels, native liver survival and true survival. Reports with the lowest time to KPE either come from regions with some form of screening programme (for example, Taiwan23,213) or from regions with centralized care (for example, England and Wales238). Similarly, the highest CoJ rates have been reported from Asia (for example, Taiwan and Japan9,213) or in a centralized European series (England and Wales, the Nordic consortium7,238), suggesting that high patient volumes positively influence outcomes. By contrast, the lowest CoJ rate (~24%) has been reported in high-income countries where care is not centralized (for example, Germany232).
Table 2 |.
Outcomes of KPE
| Country | Age at KPE (days) | Number of patients | Study period | Clearance of jaundice (%) | Native liver survival (%) | Ref. |
|---|---|---|---|---|---|---|
| Asia | ||||||
| Taiwan | NA | 102 | 2004–2010 | 61 | 57 (3 years) | 213 |
| Japan | NA | 3,160 | 1989–2015 | 58 | 49 (10 years) | 9 |
| Taiwan | 58–53 (mean) | 540 | 1997–2011 | NA | 65 (no time point) | 23 |
| Europe (centralized care) | ||||||
| England and Wales | 51–48 (median) | 867 | 1999–2019 | 61 | 47 (10 years) | 238 |
| Nordic consortium | 64 (median) | 158 | 2005–2016 | 64 | 53 (5 years) | 7 |
| Europe (non-centralized care) | ||||||
| France | 59 (median) | 1,428 | 1997–2015 | 39 | 35 (10 years) | 15 |
| Netherlands | 59 (median) | 214 | 1987–2008 | 34 | 46 (4 years) | 231 |
| Switzerland | 68 (median) | 48 | 1994–2004 | 40 | 33 (4 years) | 20 |
| Germany | NA | 173 | 2010–2014 | 24a | 29 (2 years) | 232 |
| Other | ||||||
| Saudi Arabia | 70 (median), | 204 (diagnosed), 146 (KPE) | 2000–2018 | 45 (KPE) | 26 (10 years) 73 (10 years overall survival) |
233 |
| Canada | 64 (median) | 230 | 1992–2002 | NA | 39 (4 years) | 234 |
| New Zealand | 63 (median) | 95 | 2002–2014 | NA | 30 (5 years) | 26 |
| North American consortia | ||||||
| USA (nine centres; BARC) | 61 (mean) | 104 | 1997–2000 | 40 | 52 (2 years) | 10 |
| USA (16 centres; PROBE) | 58 (mean) | 137 | 2004–2011 | 50 | 53 (2 years) | 235 |
| USA and Canada (16 centres; PROBE) | 65 (median) | 237 (136 underwent surgical drainage) | 2004–2010 | 46 | 47 (2 years) | 236 |
| USA Western Surgical Consortium (nine centres) | 64 (mean) | 223 | 2009–2017 | NA | 41 (no time point) | 237 |
KPE, Kasai portoenterostomy; NA, not available.
Jaundice-free native liver.
Liver transplantation
Liver transplantation is the treatment for real or expected KPE failures. In a retrospective cohort study in 3,438 patients with BA listed for liver transplantation, 15% were listed for primary transplantation (no prior KPE), 17% for salvage transplantation after early failure (at <1 year of age) and 67% after late failure (at >1 year of age)239,240. For many, particularly in the USA, liver transplantation as a primary procedure has become increasingly attractive239, owing to poor immediate and long-term KPE outcomes. In a review from 2024, the inevitability of KPE failure in the natural history of the disease was noted as an argument for primary liver transplantation, whereas an argument against the procedure is that some patients do have excellent long-term outcomes following KPE, and that the real problem is the inability to predict the outcome at the time of presentation241. Primary transplantation clearly has value in some patients. In the UK, <5% of all infants presenting with BA undergo the procedure238. Typically, these are patients who present late (for example, >100 days old) and have obvious liver cirrhosis and associated ascites or portal hypertension238.
Prognosis at time of KPE
Important prognostic factors for BA can be divided into intrinsic factors related to the natural history and nature of the disease and external factors related to its management242.
Intrinsic factors.
The effect of age in isolated BA is undeniable: those <30 days old at KPE have CoJ rates of >70%, whereas those >100 days old at KPE have CoJ rates of <40%238. The type of extrahepatic anatomy is also crucial. Effective surgery for cystic BA (5–10%), which is usually type I (with bile in the cyst), should still involve a radical resection and is associated with CoJ of >80%; however, some evidence suggests that late failure of this surgery is particularly evident during adolescence238. BASM-associated and CMV-infection-associated BAs are associated with inferior outcomes compared with isolated BA243.
Liver biomarkers at time of KPE can predict outcome, but their precision and clinical usefulness are limited. The best known biomarker is the APRi, a composite of aspartate aminotransaminase and platelet levels, and a surrogate for liver fibrosis. Large series have shown significantly different long-term native liver survival depending on the initial APRi quartile50. A large Chinese series also showed a negative effect of having a low γ-glutamyl transferase (<300 IU/l) at the time of KPE244. Serum MMP7 has only recently been evaluated, not only as a diagnostic biomarker but also for its prognostic value245.
Evaluation of the degree of histological liver fibrosis at KPE has been studied but its prognostic value has been disappointing. The Childhood Liver Disease Research Network (ChiLDReN)202 reviewed 316 liver biopsy samples using an array of histological features (for example, fibrosis, portal oedema and others) and found none to have any relationship with CoJ, although some relatively small differences emerged in terms of native liver survival. For example, the hazard ratio for the need for transplantation was 1.73 (95% CI 1.21–2.48) times greater in patients with stage 3 or 4 fibrosis than in those with stage 0–2 fibrosis (Scheuer system).
New liver or serum biomarkers have been suggested to have prognostic value, although most have not yet entered routine clinical practice. For example, secretin receptor mRNA expression in the liver correlated strongly with both CoJ and native liver survival246. Serum levels and hepatic expression of IL-8 is high in infants with BA and low levels have been associated with improved native liver survival247. Similarly, serum FGF19 levels were associated with ductular proliferation but not liver fibrosis, and predicted KPE outcomes, with those with levels >109 pg/ml having markedly diminished native liver survival248. These high levels are believed to originate from the liver, with aberrant hepatic expression induced by the high levels of bile acids in cholestasis seen in BA. Finally, Ki67, a marker for cellular proliferation, may be more actively expressed in the liver from patients with BA, and higher expression has been associated with worse native liver survival249.
Extrinsic factors.
The obvious extrinsic factor is the expertise of the surgical team and the technique adopted. Most studies investigating centre experience and case-mix have found a good correlation between larger centre size and improvements in outcome, although some found satisfactory results for smaller centres234. This finding led to a policy of centralization and concentration of KPE and transplant resources for BA, initially implemented in England and Wales in 1999 (ref. 11). Subsequent outcome studies showed a dramatic improvement in national KPE outcomes with a reduction in the time to KPE to <50 days and improvement in CoJ rates238. This policy has now been adopted by several other (predominantly Northern European) countries7,250.
Clearly, there are major geographical obstacles to centralization, such as in North America and Australia236,251. The nature of the health-care system may preclude centralization where there is competition between potential providers (for example, in Germany and the USA)232.
Adjuvant therapies
The use of adjuvant treatments following KPE is controversial with an arguably marginal evidence base and many institutions sticking with outdated protocols in the absence of certainty. Nevertheless, the field may be entering a period of increased optimism. As BA is a rare disease, incontrovertible trial-based evidence of the benefit of these therapies remains slim. The merits of each pharmacological agent should be assessed individually.
Antibiotics and ursodeoxycholic acid.
The use of both agents is widespread and uncontentious but with virtually no actual evidence. Cholangitis is a common postoperative problem — it occurs in up to 80% of patients after KPE in some recent series252 — and its treatment usually involves intravenous antibiotics for 1–2 weeks. Bacterial cultures are rarely positive, but one study showed a prevalence of the Gram-positive Enterococcus faecium and the Gram-negative organisms Escherichia coli, Enterobacter cloacae and Klebsiella pneumoniae252. In one Chinese multicentre (n = 14) open-label study involving 211 children, the severity of cholangitis was graded as ‘mild’, ‘moderate’ and ‘severe’, and each grade was randomized to a different regimen253. The duration of fever was the primary outcome. Meropenem alone was found to be equivalent to cefoperazone in mild and moderate cholangitis and the addition of intravenous immunoglobulin improved the efficacy of meropenem in severe cholangitis. Prophylactic antibiotic regimens of variable duration (1–12 months) have been designed; however, little scientific evidence favours one over another according to one systematic analysis254. Ursodeoxycholic acid use is prevalent but only one study showed biochemical improvement in a withdrawal–reintroduction format in 16 children with stable disease255.
Anti-inflammatory therapy: corticosteroids and immunoglobulin.
Inflammation has a key role in the early pathophysiology of some infants with BA, after which fibrosis takes over. Over the past 20 years, corticosteroids have been explored as postoperative adjuvant therapy to modulate this inflammatory response. There have been two prospective randomized controlled trials (RCTs), and a number of large prospective open-label trials256,257 (Table 3). For example, a RCT trial in the UK using a low-dose regimen (2 mg/kg/day oral prednisolone) showed little clinical difference but significantly reduced bilirubin levels in the corticosteroid arm256,258. A multicentre RCT (START trial by ChiLDReN) using intravenous or oral 4 mg/kg/day prednisolone was powered to detect a difference of 25% in CoJ, and showed a difference of 15% in CoJ (compared with placebo) in patients <70 days old at KPE, but without achieving statistical significance259. A follow-on prospective study from the original UK study using an increased dose (5 mg/kg/day oral prednisolone) in infants <70 days old at KPE showed the same difference in CoJ but because of a larger number of participants the results were statistically significant256. A larger, Chinese single-surgeon trial showed significant effects on CoJ and native liver survival with a 10-week regimen of intravenous or oral 4 mg/kg/day prednisolone257.
Table 3 |.
Adjuvant corticosteroid trials
| Country | Number of patients | Regimen | Outcome | Ref. |
|---|---|---|---|---|
| Randomized placebo-controlled trials | ||||
| UK | 71 | Oral 2 mg/kg/day (low-dose) prednisolone 4 weeks | ↓Bilirubin at 3 months (P = 0.06) Improved with lower age at KPE |
258 |
| North America | 140 | IV or oral 4 mg/kg/day prednisolone 9 weeks | CoJ 59 vs 49 at 6 months (P = 0.43) Improved with lower age at KPE No difference in transplant-free survival |
259 |
| Prospective open-label trials | ||||
| UK | 153 | Oral 2 mg/kg/day (n = 18) prednisolone Oral 5 mg/kg/day (n = 44) prednisolone |
All infants <70 days of age at KPE. CoJ (no corticosteroids) 52% vs (corticosteroids) 66% (P = 0.037) at 6 months |
256 |
| China | 200 | IV or oral 4 mg/kg/day prednisolone 10–12 weeks | ↑CoJ at 6 months 54% vs 31% (P < 0.001) ↑NLS (12 months) 66% vs 50% (P = 0.02) |
|
CoJ, clearance of jaundice; IV, intravenous; KPE, Kasai portoenterostomy; NLS, native liver survival.
Most large centres in Europe, China and Japan use high-dose corticosteroids in a variety of regimens and some American centres, despite the negative results of the START trial, also use corticosteroid-based regimens260,261. In these centres, CoJ rates of >60% may be achieved. It is also important to recognize that age at KPE is an important factor in the efficacy of corticosteroids262. An interesting question is whether corticosteroid therapy should be repeated for disease relapses. This use is common practice in Japanese centres, which have the best overall results worldwide. Of note, corticosteroids may have adverse effects. A post hoc analysis of child growth following the START trial showed impaired length, weight and head circumference growth trajectories for at least 6 months after KPE263.
The anti-inflammatory potential of intravenous immunoglobulin administration has been proven in several autoimmune and inflammatory disorders. However, a ChiLDReN phase Ib/II clinical trial264 of intravenous immunoglobulin in BA failed to show a beneficial effect compared with the placebo arm of the START trial.
Antiviral therapy.
Specific antiviral therapy against CMV has been used in small, single-centre studies265,266. At least one study showed remarkably improved outcomes for CoJ and reduced need for transplantation267 in patients with BA who were CMV-positive. Currently, oral valganciclovir is favoured based on ease of use and efficacy42,267.
Targeting bile acid metabolism.
Three major randomized trials investigating targeting bile acid metabolism are currently in progress. Maralixibat (NCT04524390)268 and odevixibat (NCT04336722)269 are ileal bile acid transporter (IBAT) inhibitors that reduce the re-uptake of bile acids in the terminal ileum, and could diminish bile acid accumulation after KPE270. Obeticholic acid (NCT05321524)271 is a modified primary bile acid and a potent agonist of nuclear farnesoid X receptor (FXR). FXR activation may reduce endogenous bile acid production, promote bile acid export from hepatocytes and inhibit a variety of NF-κB mediated inflammatory precursors (such as TNF) and may limit the severity of fibrosis272.
The two IBAT inhibitors have shown promising results with reductions in serum bile acids and pruritus in other allied neonatal cholestatic conditions, such as Alagille syndrome273,274. However, early unpublished results from the phase II maralixibat trial suggest no significant effect on the primary and secondary outcomes according to a press release.
Potential for regeneration
Regenerative medicine aims to restore or replace damaged tissues and organs using various methods, including stem cells, gene therapy, tissue engineering and organoids, and is emerging as a potential solution for various liver diseases including BA275 (Fig. 6).
Fig. 6 |. Potential regenerative strategies in biliary atresia treatment.

Pluripotent stem cells derived from reprogramming of primary somatic cells from patients or embryonic stem cells can be differentiated into various cell types useful for liver regeneration as cellular or tissue engineering therapies. Progenitors could also be used for mobilization of patients’ cells which can help regeneration through modulation of inflammation and fibrosis.
Stem cell therapy.
Stem cells generated from induced pluripotent stem cells, embryonic stem cells or directly from tissue-derived cells can be injected into the liver or the bile ducts to promote regeneration and repair of damaged tissues. Liver-derived and cholangiocyte-derived organoids are most promising because they can replace both the damaged epithelium and niche276. Stem cells can also act by modulating responses to inflammation and fibrosis. In preclinical models, autologous bone-marrow-derived mononuclear cells (BMMNC), haematopoietic stem cells, mesenchymal stem cells and endothelial progenitors improved liver cirrhosis by reducing hepatic fibrosis and inducing recovery of liver function, but limited clinical data are currently available. An early study from New Delhi using BMMNC injected via the hepatic artery and portal vein showed improvements in postoperative biochemistry but patient survival at 1 year was low, limiting the relevance of the study277. A clinical trial from Hanoi testing a regimen of two autologous BMMNC administrations in children with BA and liver cirrhosis showed no change in fibrosis or histology on liver biopsy, or in endoscopic features, but bilirubin levels and the Paediatric End-Stage Liver Disease values decreased278. Injection of granulocyte colony stimulating factor to mobilize haematopoietic stem cells has also been studied in infants after KPE279. This study showed a reasonable safety profile and the desired haematopoietic response, but with very limited differences in outcomes apart from less frequent cholangitis compared with controls.
Tissue engineering.
Tissue engineering using scaffolds, bioreactors or 3D printing can create artificial or bioengineered bile ducts that mimic the natural ones. This methodology can be very effective in replacing EHBDs, but there are limitations to the engineering of intrahepatic ducts even though preliminary data showed their ability to repair human biliary epithelium12,280. Future treatments of liver failure may also benefit from using tissue-engineered liver which takes advantage of cells, such as organoids, ECM and signalling molecules82,83,275. Functional engineered livers using both synthetic and natural biomaterials such as decellularized livers have been transplanted in large-animal models, but 3D bioprinting may offer a more tailored regenerative option for patients with BA in the future281. These regenerative therapies are still at the experimental stage and face considerable challenges, including safety, efficacy, scalability and ethical issues, before they may become available in practice.
Quality of life
The most important clinical manifestations of progressive liver disease and indications for liver transplantation include portal hypertension, cholangitis and advanced cirrhosis associated with synthetic liver failure, recurring cholestasis and ascites, which greatly affect patients’ quality of life282,283. Few long-term studies in patients with BA are available. KPE outcomes seem to be better in highly specialized centres in East Asia, but the only study comparing an Asian and a UK centre, published in 2001, showed no difference in objective scoring284.
Although successful KPE resolves histological cholestasis, DR and liver fibrosis persist, reflected by expansion of cholangiocytes and activated portal myofibroblasts with increased expression of cytokeratin 7 (K7)/KRT7, a cholangiocyte marker, and α-smooth muscle actin/ACTA, a marker of activated hepatic stellate cells21,285,286. DR is a central pathological feature in cholangiopathies in which expansion of reactive neocholangiocytes is thought to promote liver fibrogenesis by activation of myofibroblasts287. DR initially decreases in patients in whom bilirubin levels normalize after KPE285,286,288, but it increases after unsuccessful KPE. Progression of DR is also associated with the extent of liver fibrosis and transplant-free survival both at and after KPE288. The key biliary elements of DR, bile duct expansion and K7+ hepatocytes, are closely correlated with serum bile acid levels, which predict transplant-free survival following KPE288,289. The underlying cholangiopathy advances variably, and nearly 50% of patients surviving with their native liver at age 2 years require liver transplantation by adulthood290,291.
By school age, 43% of children undergoing protocol follow-up liver biopsies following successful KPE have cirrhosis; 79–97% adult native liver survivors also show histological, clinical and/or ultrasonographic signs of cirrhosis292–294 (Table 4). Chronic advanced liver fibrosis predisposes to development of both benign hepatic nodules (6%) and hepatocellular carcinoma (HCC) (1%) in native liver survivors295,296. Surveillance with biannual ɑ-fetoprotein measurement and abdominal ultrasonography is recommended, and tumour biopsy should be obtained on suspicion of malignancy295–297. HCC diagnosis warrants prompt evaluation for liver transplantation.
Table 4 |.
Main complications and follow-up of chronic liver disease in paediatric and adult long-term native liver survivors
| Complication | Prevalence in children (%) | Prevalence in adults (%) | Surveillance measures | Ref. |
|---|---|---|---|---|
| Cirrhosis | 40–50 | 80–90 | Serum liver biochemistry, APRi, abdominal ultrasonography, liver stiffness, liver biopsy | 292–294 |
| Portal hypertension | 60–70 | 50–70 | Spleen size, platelets, liver stiffness, echo cardiography | 282,292–294,298,299,301 |
| Oesophageal varices | 50–60 | 20–30 | Endoscopic primary/secondary prophylaxis | 298,299,302,303 |
| Bleeding | 10–15 | 20–30 | ||
| Cholangitis | 60–70 | 50–60 | Antibiotic prophylaxis, MRCP and scintigraphy if recurrent | 254,293,294,300,304,306,307 |
| Liver malignancy | <1 | >1 | 6-monthly ultrasonography and serum AFP | 295,296,302,304 |
| Impaired growth | <5 | 0–20% | Anthropometric and biometric surveillance | 293,300,304 |
| Bone fractures, osteoporosis, osteopenia | 10–20 | ? | Serum vitamin D levels, bone mineral density surveillance | 300,309 |
| Neurodevelopmental and/or educational shortcomings | 0–50 | 0–30 | Surveillance for neurocognitive and motor skills | 293,301,310–313,316 |
| Impaired quality of life | 0–30 | 0–30 | Psychosocial assessment | 293,301,314–316 |
AFP, ɑ-fetoprotein; APRi, aspartate aminotransferase to platelets ratio index; MRCP, magnetic resonance cholangiopancreatography.
Portal hypertension affects most patients, typically developing at a young age paralleling the progression of liver fibrosis, and is frequently complicated by oesophageal varices292. By a median age of 8.5 years, 62% of 348 paediatric native liver survivors had developed splenomegaly, thrombocytopenia or both298. Oesophageal varices were verified at a median age of 8.6 years in 60% of native liver survivors who underwent endoscopy299. Ascites is a less common manifestation of portal hypertension, detected in <10% of paediatric native liver survivors300. At a mean age of 19 years, 69% and 64% of native liver survivors had portal hypertension and oesophageal varices, respectively301, and 46–70% of adult native liver survivors demonstrated clinical, ultrasonographic or endoscopic signs of portal hypertension293,294. In paediatric native liver survivors presenting with splenomegaly and thrombocytopenia at a median age of 8.5 years, the 5-year cumulative incidence of variceal bleeding exceeded 10%298, and a systematic review reported gastrointestinal bleeding in 21% of adults302. Although primary endoscopic prophylaxis of oesophageal varices remains controversial in children with BA, variceal bleeding is preventable by endoscopic surveillance and eradication, particularly in patients with a functioning KPE299,303. Infrequent pulmonary complications of portal hypertension include hepatopulmonary syndrome and portopulmonary hypertension, both of which are indications for listing for liver transplantation to avoid the development of irreversible cardiopulmonary complications that would be a contraindication for transplantation282,304. Hepatopulmonary syndrome affects 5–7% of children surviving with their native liver and is characterized by hypoxia and intrapulmonary vasodilatation282,300. Portopulmonary hypertension is diagnosed in <1% of children with end-stage liver disease, predominantly female adolescents305. It is asymptomatic until increased pulmonary artery pressures cause hypoxia, dyspnoea and, eventually, right-sided heart failure305.
Cholangitis episodes negatively affect native liver survival282,306. Two-thirds of 219 paediatric native liver survivors aged 5–18 years had experienced cholangitis, 17% during the preceding year, while 31% of adult native liver survivors faced cholangitis aged 17–30 years293,300. Although widely used, oral antibiotic prophylaxis fails to reduce the incidence of cholangitis episodes according to a meta-analysis254. Approximately 10–15% of patients with BA develop intrahepatic bile lakes, predisposing to recurrent cholangitis by bile stagnation307. The presence of bile lakes should be assessed with MRI so that they can be effectively drained to reduce cholangitis episodes and maintain native liver survival307. Future research should focus on development of accurate diagnostic tools and efficient prevention and treatment of cholangitis308.
Although most older patients experience stable growth, undergo normal pubertal development, and reach normal adult height and weight, growth and biometrics of nutritional status should be regularly assessed to avoid malnutrition300,304. Vitamin D deficiency is common even after successful KPE despite enteral supplementation, and predisposes to rickets and osteoporosis309. Although the risk of metabolic bone disease is highest in patients in whom jaundice does not clear, fractures have been reported in 12–15% after successful KPE301,309. Infants with BA are at risk of neurodevelopmental delays, particularly after unsuccessful KPE310. Most North American native liver survivors aged 3–12 years showed normal neurodevelopment, whereas two smaller European national studies, which also included liver-transplanted patients, found impaired motor skills and decreased intelligence quotient, which were associated with advanced liver disease311–313. Compared with healthy controls, all domains of patient and parent-proxy reported health-related quality of life were decreased at a mean age of 9.7 years in 221 children surviving with their native liver, with the greatest differences occurring in school functioning314. Adult native liver survivors, particularly women, have reported decreased general health perception, which is correlated with complications of liver disease315,316. Based on limited data, most adult native liver survivors demonstrate educational levels and employment rates comparable to those in the general population293,301,316.
Patients with BA require lifelong structured multidisciplinary follow-up in specialist centres to optimize safe transition of care to adult practice and timing of liver transplantation, as adult liver disease scores are not appropriate for these patients282,297,304,317 (Fig. 7). For successful transition, health professionals caring for the adult population should be familiarized with the long-term liver disease complications and the challenging timing of liver transplantation, and young adult patients with BA deserve a holistic management approach that does not overlook psychosocial aspects and comprehensive patient education about their condition282,297,304.
Fig. 7 |. Postoperative follow-up of native liver survivors.

For optimal timing of liver transplantation, native liver survivors should be monitored and managed by multidisciplinary teams in specialist centres throughout their life. Evaluation for liver transplantation should occur promptly after failed Kasai portoenterostomy (KPE), as most patients develop rapidly progressing end-stage liver disease within 1–2 years. Despite successful KPE, the underlying cholangiopathy advances variably increasing the proportion of transplanted patients over time. Successful transition of young patients to adult health services requires special cooperative efforts between paediatric and adult specialists and comprehensive patient education. BIL, bilirubin; LT, liver transplantation.
Outlook
The global burden of BA remains substantial. CoJ with timely KPE is achievable in 50–70% of patients with BA in centres with relevant expertise, and this approach should, therefore, become the benchmark for less-developed communities to target in the short term. However, in the medium and long term, further improvements in patient outcomes are likely to require multiple innovative measures inspired by a comprehensive understanding of the complex mechanisms of disease causation and progression (Fig. 8).
Fig. 8 |. Future research directions and effects on patient outlook.

Discovery of disease mechanisms and identification of gene sets for patient stratification could lead to early diagnosis and new interventions at several important time points to improve the outcomes in patients with biliary atresia (BA). Currently, patients undergo Kasai portoenterostomy (KPE) (85–95%) at ~2 months of age, or primary liver transplantation (LT) (5–15%) because of late diagnosis. Improved screening with new biomarkers would enable diagnosis of emerging BA in early infancy (7–60 days), providing the first preventative and/or therapeutic window for earlier KPE and avoidance of primary LT. Introduction of neoadjuvant therapies targeting the injury phase of BA, such as the antioxidant N-acetylcysteine in the expanded window before KPE, might improve clearance of jaundice rate and reduce early (at <1 year of age) LT incidence (currently 17–40%). After KPE, patients experience complications leading to progression of liver pathology requiring late (at >1 year of age) LT (currently 30–67%). During this second therapeutic window, disease mechanism-inspired novel drugs, targeting the dysregulated immune-inflammation and ECM response phase could reduce recalcitrant cholangitis and fibrosis responsible for late LT, and increase long-term native liver survival (NLS) (currently only 40%). For the remaining patients currently requiring LT, regenerative medicine might reduce or replace the need for LT.
Early diagnosis will be an important first step in the new management paradigm. New biomarkers, probably in combination, informed by big data and analysed with artificial intelligence, may enable improved newborn screening and a diagnosis to be made in the first few weeks of life. Expediting the diagnosis will provide a larger window for neoadjuvant therapy before KPE. The distinction between prenatal and perinatal insult to the extrahepatic biliary tree and postnatal hepatic response to biliary obstruction will enable more targeted medical interventions in different phases of disease progression. For example, neonatal susceptibility to redox stress and hypoxia suggests that antioxidants, such as NAC, may be more appropriate for neoadjuvant therapy. Likewise, testing of butyrates (that modulate glutamate and glutamine) and ciliary dysfunction remedies (repurposing drug targets developed for other primary ciliopathies) may be warranted during the early injury phase of BA.
Surgery will continue to be the cornerstone of BA management. Centralization of surgery and optimizing medical interventions for complications after KPE, such as cholangitis, will improve patient outcomes and provide a sufficiently large patient pool to better understand early (at <1 year of age) and late (at >1 year of age) KPE failures and their underlying mechanisms.
Emerging adjuvant therapies targeting immune response to obstruction, bile acid homeostasis and fetal wound healing are promising and should be evaluated with robust multicentre prospective randomized trials. It is possible that some emerging therapies may work better in either the injury or the response phase, whereas others may be beneficial in both phases, which exist as a continuum. An additional question is whether the emerging therapies and existing therapies (for example, corticosteroids) would work better alone or in combination.
Regenerative medicine approaches are a speculative strategy for BA. Human stem cell and organoid research provides a preclinical platform for disease modelling and drug screening, capable of diagnostic and therapeutic breakthroughs275. Stem cells modulating inflammatory or immune dysregulation or liver fibrosis progression may be tested as adjuvants to KPE and tissue engineering may ultimately replace liver transplantation. With focused academia–industry collaboration, some of these emerging research directions might be successful and transform this devastating disease into a curable condition.
Acknowledgements
P.K.H.T., C.S.M.T. and V.C.H.L. disclose funding support for the research for this work from Theme-based Research Scheme (T12-712/21-R), CERG (17105119), HMRF (09201836) and Commissioned HMRF (PR-HKU-1) from Hong Kong SAR Government; P.K.H.T. was also supported by Macau FDCT0011/2023/AKP. R.G.W. was supported by NIH R01 DK119290 and Children’s Hospital of Philadelphia-Fred and Suzanne Biesecker Paediatric Liver Center.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Hartley JL, Davenport M & Kelly DA Biliary atresia. Lancet 374, 1704–1713 (2009). [DOI] [PubMed] [Google Scholar]
- 2. Chung PHY, Zheng S & Tam PKH Biliary atresia: East versus West. Semin. Pediatr. Surg 29, 150950 (2020). A review summarizing the geographical differences in BA aetiology and treatment.
- 3. Davenport M, Muntean A & Hadzic N Biliary atresia: clinical phenotypes and aetiological heterogeneity. J. Clin. Med 10, 5675 (2021). A description of different BA phenotypes and associated malformations.
- 4.Lendahl U, Lui VCH, Chung PHY & Tam PKH Biliary atresia – emerging diagnostic and therapy opportunities. EBioMedicine 74, 103689 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bezerra JA et al. Biliary atresia: clinical and research challenges for the twenty-first century. Hepatology 68, 1163–1173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vij M & Rela M Biliary atresia: pathology, etiology and pathogenesis. Future Sci. OA 6, FSO466 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pakarinen MP et al. Outcomes of biliary atresia in the Nordic countries – a multicenter study of 158 patients during 2005-2016. J. Pediatr. Surg 53, 1509–1515 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Davenport M, Yamataka A and Nio M in 50 Landmark Papers Every Pediatric Surgeon Should Know Ch. 25 (eds Davenport M, Aldeiri B & Davidson J) 74–76 (CRC Press, 2023). [Google Scholar]
- 9.Nio M Japanese biliary atresia registry. Pediatr. Surg. Int 33, 1319–1325 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Shneider BL et al. A multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J. Pediatr 148, 467–474 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Davenport M et al. Seamless management of biliary atresia in England and Wales (1999-2002). Lancet 363, 1354–1357 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Tam PKH, Yiu RS, Lendahl U & Andersson ER Cholangiopathies – towards a molecular understanding. EBioMedicine 35, 381–393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng G et al. Common genetic variants regulating ADD3 gene expression alter biliary atresia risk. J. Hepatol 59, 1285–1291 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Nomden M et al. A higher incidence of isolated biliary atresia in rural areas: results from an epidemiological study in the Netherlands. J. Pediatr. Gastroenterol. Nutr 72, 202–209 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Fanna M et al. Management of biliary atresia in France 1986 to 2015: long-term results. J. Pediatr. Gastroenterol. Nutr 69, 416–424 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Grizelj R, Vuković J, Novak M & Batinica S Biliary atresia: the Croatian experience 1992-2006. Eur. J. Pediatr 169, 1529–1534 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Schreiber RA et al. Biliary atresia: the Canadian experience. J. Pediatr 151, 659–665.e1 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Hopkins PC, Yazigi N & Nylund CM Incidence of biliary atresia and timing of hepatoportoenterostomy in the United States. J. Pediatr 187, 253–257 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Livesey E et al. Epidemiology of biliary atresia in England and Wales (1999-2006). Arch. Dis. Child. Fetal Neonatal Ed 94, F451–F455 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Wildhaber BE et al. Biliary atresia: Swiss national study, 1994-2004. J. Pediatr. Gastroenterol. Nutr 46, 299–307 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Hukkinen M et al. Treatment policy and liver histopathology predict biliary atresia outcomes: results after national centralization and protocol biopsies. J. Am. Coll. Surg 226, 46–57.e1 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Fischler B, Haglund B & Hjern A A population-based study on the incidence and possible pre- and perinatal etiologic risk factors of biliary atresia. J. Pediatr 141, 217–222 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Lin JS et al. Reduction of the ages at diagnosis and operation of biliary atresia in Taiwan: a 15-year population-based cohort study. World J. Gastroenterol 21, 13080–13086 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nio M et al. Five- and 10-year survival rates after surgery for biliary atresia: a report from the Japanese biliary atresia registry. J. Pediatr. Surg 38, 997–1000 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Lee KJ, Kim JW, Moon JS & Ko JS Epidemiology of biliary atresia in Korea. J. Korean Med. Sci 32, 656–660 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans HM et al. Ethnic disparity in the incidence and outcome of biliary atresia in New Zealand. J. Pediatr. Gastroenterol. Nutr 66, 218–221 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Durkin N, Deheragoda M & Davenport M Prematurity and biliary atresia: a 30-year observational study. Pediatr. Surg. Int 33, 1355–1361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caton AR, Druschel CM & McNutt LA The epidemiology of extrahepatic biliary atresia in New York State, 1983-98. Paediatr. Perinat. Epidemiol 18, 97–105 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Yoon PW, Bresee JS, Olney RS, James LM & Khoury MJ Epidemiology of biliary atresia: a population-based study. Pediatrics 99, 376–382 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Wada H et al. Insignificant seasonal and geographical variation in incidence of biliary atresia in Japan: a regional survey of over 20 years. J. Pediatr. Surg 42, 2090–2092 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Chardot C et al. Prognosis of biliary atresia in the era of liver transplantation: French national study from 1986 to 1996. Hepatology 30, 606–611 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Lin YC et al. Decreasing rate of biliary atresia in Taiwan: a survey, 2004-2009. Pediatrics 128, e530–e536 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Nomden M et al. Incidence of isolated biliary atresia during the COVID lockdown in Europe: results from a collaborative project by RARE-liver. J. Clin. Med 12, 775 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhan J, Feng J, Chen Y, Liu J & Wang B Incidence of biliary atresia associated congenital malformations: a retrospective multicenter study in China. Asian J. Surg 40, 429–433 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Davenport M et al. The biliary atresia splenic malformation syndrome: a 28-year single-center retrospective study. J. Pediatr 149, 393–400 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Guttman OR et al. Biliary atresia with associated structural malformations in Canadian infants. Liver Int. 31, 1485–1493 (2011). [DOI] [PubMed] [Google Scholar]
- 37. Chang CM et al. Maternal risk factors associated with offspring biliary atresia: population-based study. Pediatr. Res 93, 1064–1071 (2023). The key study revealing an association between maternal infections and BA development in the offspring.
- 38. Cavallo L et al. The epidemiology of biliary atresia: exploring the role of developmental factors on birth prevalence. J. Pediatr 246, 89–94.e2 (2022). A large population-based study describing risk factors for BA during fetal development.
- 39.Wang WH, Chiu FY, Kuo TT & Shao YJ Maternal prenatal infections and biliary atresia in offspring. JAMA Netw. Open 7, e2350044 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howley MM et al. Asthma medication use and risk of birth defects: national birth defects prevention study, 1997-2011. J. Allergy Clin. Immunol. Pract 8, 3490–3499.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Costa CM et al. In vitro fertilization: an unexpected finding in a cohort of patients with biliary atresia. Braz. J. Med. Biol. Res 56, e12671 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fischler B et al. Incidence, impact and treatment of ongoing CMV infection in patients with biliary atresia in four European centres. J. Clin. Med 11, 945 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rauschenfels S et al. Incidence of hepatotropic viruses in biliary atresia. Eur. J. Pediatr 168, 469–476 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Zani A, Quaglia A, Hadzic N, Zuckerman M & Davenport M Cytomegalovirus-associated biliary atresia: an aetiological and prognostic subgroup. J. Pediatr. Surg 50, 1739–1745 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Xu Y et al. The perinatal infection of cytomegalovirus is an important etiology for biliary atresia in China. Clin. Pediatr 51, 109–113 (2012). [DOI] [PubMed] [Google Scholar]
- 46.van Wessel DBE et al. Preterm infants with biliary atresia: a nationwide cohort analysis from The Netherlands. J. Pediatr. Gastroenterol. Nutr 65, 370–374 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Chiu CY et al. Biliary atresia in preterm infants in Taiwan: a nationwide survey. J. Pediatr 163, 100–103.e1 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Kemme S et al. Cytomegalovirus in biliary atresia is associated with increased pretransplant death, but not decreased native liver survival. Hepatol. Commun 7, e0175 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao Y, Xu X, Liu G, Yang F & Zhan J Prognosis of biliary atresia associated with cytomegalovirus: a meta-analysis. Front. Pediatr 9, 710450 (2021). The largest analysis on CMV-associated BA demonstrating an association with inferior outcomes after KPE.
- 50.Muntean A, Kronfli R, Makin E & Davenport M The AST-to-platelet ratio index (APRi) at Kasai portoenterostomy: standing the test of time. J. Pediatr. Surg 58, 2347–2351 (2023). [DOI] [PubMed] [Google Scholar]
- 51.Fischler B, Ehrnst A, Forsgren M, Orvell C & Nemeth A The viral association of neonatal cholestasis in Sweden: a possible link between cytomegalovirus infection and extrahepatic biliary atresia. J. Pediatr. Gastroenterol. Nutr 27, 57–64 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Shen C, Zheng S, Wang W & Xiao XM Relationship between prognosis of biliary atresia and infection of cytomegalovirus. World J. Pediatr 4, 123–126 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Nio M, Wada M, Sasaki H, Tanaka H & Watanabe T Long-term outcomes of biliary atresia with splenic malformation. J. Pediatr. Surg 50, 2124–2127 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Tralongo P et al. Biliary atresia in an infant presenting with Kabuki syndrome: an autopsy report and review of the literature. Pediatr. Dev. Pathol 26, 318–320 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allotey J et al. Congenital bile duct anomalies (biliary atresia) and chromosome 22 aneuploidy. J. Pediatr. Surg 43, 1736–1740 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Aldeiri B et al. Cardiac-associated biliary atresia (CABA): a prognostic subgroup. Arch. Dis. Child 106, 68–72 (2021). [DOI] [PubMed] [Google Scholar]
- 57.Gupta L & Bhatnagar V A study of associated congenital anomalies with biliary atresia. J. Indian. Assoc. Pediatr. Surg 21, 10–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwarz KB et al. Extrahepatic anomalies in infants with biliary atresia: results of a large prospective North American multicenter study. Hepatology 58, 1724–1731 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harpavat S, Finegold MJ & Karpen SJ Patients with biliary atresia have elevated direct/conjugated bilirubin levels shortly after birth. Pediatrics 128, e1428–e1433 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Harpavat S et al. Diagnostic yield of newborn screening for biliary atresia using direct or conjugated bilirubin measurements. JAMA 323, 1141–1150 (2020). A representative paper on newborn screening for BA.
- 61.Kastenberg ZJ et al. Fractionated bilirubin among 252 892 Utah newborns with and without biliary atresia: a 15-year historical birth cohort study. J. Pediatr 257, 113339 (2023). [DOI] [PubMed] [Google Scholar]
- 62.Schmidt HC et al. Biliatresone induces cholangiopathy in C57BL/6J neonates. Sci. Rep 13, 10574 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang Y et al. The synthetic toxin biliatresone causes biliary atresia in mice. Lab. Invest 100, 1425–1435 (2020). [DOI] [PubMed] [Google Scholar]
- 64.Lemaigre FP Development of the intrahepatic and extrahepatic biliary tract: a framework for understanding congenital diseases. Annu. Rev. Pathol 15, 1–22 (2020). [DOI] [PubMed] [Google Scholar]
- 65.de Jong IEM, van den Heuvel MC, Wells RG & Porte RJ The heterogeneity of the biliary tree. J. Hepatol 75, 1236–1238 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Jong IEM & Wells RG In utero extrahepatic bile duct damage and repair: implications for biliary atresia. Pediatr. Dev. Pathol 10.1177/10935266241247479 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Jong IEM et al. Peribiliary glands are key in regeneration of the human biliary epithelium after severe bile duct injury. Hepatology 69, 1719–1734 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh S et al. Heterogeneous murine peribiliary glands orchestrate compartmentalized epithelial renewal. Dev. Cell 58, 2732–2745.e5 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. de Jong IEM et al. A fetal wound healing program after intrauterine bile duct injury may contribute to biliary atresia. J. Hepatol 79, 1396–1407 (2023). Experimental and clinical evidence of an injury response typical of fetal wound healing in BA.
- 70.Riepenhoff-Talty M et al. Group A rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatr. Res 33, 394–399 (1993). [DOI] [PubMed] [Google Scholar]
- 71. Mohanty SK et al. Rotavirus reassortant-induced murine model of liver fibrosis parallels human biliary atresia. Hepatology 71, 1316–1330 (2020). This report introduces an improved RRV mouse model for study of experimental BA and hepatic fibrosis.
- 72.Garrido M et al. Bile duct ligature in young rats: a revisited animal model for biliary atresia. Eur. J. Histochem 61, 2803 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gupta K et al. Low-dose biliatresone treatment of pregnant mice causes subclinical biliary disease in their offspring: evidence for a spectrum of neonatal injury. PLoS ONE 19, e0301824 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lorent K et al. Identification of a plant isoflavonoid that causes biliary atresia. Sci. Transl. Med 7, 286ra267 (2015). A zebrafish study providing a proof-of-concept demonstration that a toxin can initiate experimental BA.
- 75.Zhao X et al. Glutathione antioxidant pathway activity and reserve determine toxicity and specificity of the biliary toxin biliatresone in zebrafish. Hepatology 64, 894–907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cheung Y et al. Deletion of interleukin enhancer binding factor 2 (ILF2) resulted in defective biliary development and bile flow blockage. J. Pediatr. Surg 56, 352–359 (2021). [DOI] [PubMed] [Google Scholar]
- 77.Cui S et al. Evidence from human and zebrafish that GPC1 is a biliary atresia susceptibility gene. Gastroenterology 144, 1107–1115.e3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lam WY et al. Identification of a wide spectrum of ciliary gene mutations in nonsyndromic biliary atresia patients implicates ciliary dysfunction as a novel disease mechanism. EBioMedicine 71, 103530 (2021). This paper highlights ciliary dysfunction as a major genetic susceptibility factor in patients with non-syndromic BA, and implicates BA as a more generalized ciliopathy.
- 79.Tang V et al. Loss of a candidate biliary atresia susceptibility gene, add3a, causes biliary developmental defects in zebrafish. J. Pediatr. Gastroenterol. Nutr 63, 524–530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cofer ZC et al. Methylation microarray studies highlight PDGFA expression as a factor in biliary atresia. PLoS ONE 11, e0151521 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Matthews RP et al. DNA hypomethylation causes bile duct defects in zebrafish and is a distinguishing feature of infantile biliary atresia. Hepatology 53, 905–914 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guan Y et al. Human hepatic organoids for the analysis of human genetic diseases. JCI Insight 2, e94954 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huch M et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 160, 299–312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sato K et al. Organoids and spheroids as models for studying cholestatic liver injury and cholangiocarcinoma. Hepatology 74, 491–502 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roos FJM et al. Cholangiocyte organoids from human bile retain a local phenotype and can repopulate bile ducts in vitro. Clin. Transl. Med 11, e566 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soroka CJ et al. Bile-derived organoids from patients with primary sclerosing cholangitis recapitulate their inflammatory immune profile. Hepatology 70, 871–882 (2019). [DOI] [PubMed] [Google Scholar]
- 87. Babu RO et al. Beta-amyloid deposition around hepatic bile ducts is a novel pathobiological and diagnostic feature of biliary atresia. J. Hepatol 73, 1391–1403 (2020). A key study establishing human BA organoids as a valid disease model and identifying β-amyloid as a novel pathobiological factor.
- 88.Amarachintha SP et al. Biliary organoids uncover delayed epithelial development and barrier function in biliary atresia. Hepatology 75, 89–103 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tian L et al. Biliary atresia relevant human induced pluripotent stem cells recapitulate key disease features in a dish. J. Pediatr. Gastroenterol. Nutr 68, 56–63 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Waisbourd-Zinman O et al. The toxin biliatresone causes mouse extrahepatic cholangiocyte damage and fibrosis through decreased glutathione and SOX17. Hepatology 64, 880–893 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Du Y et al. A bile duct-on-a-chip with organ-level functions. Hepatology 71, 1350–1363 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Du Y et al. Human vascularized bile duct-on-a chip: a multi-cellular micro-physiological system for studying cholestatic liver disease. Biofabrication 16, 015004 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Garcia-Barceló MM et al. Genome-wide association study identifies a susceptibility locus for biliary atresia on 10q24.2. Hum. Mol. Genet 19, 2917–2925 (2010). Benchmark study showing that genetic predisposition by common regulatory variants increases the risk of BA.
- 94.Chen Y et al. A genome-wide association study identifies a susceptibility locus for biliary atresia on 2p16.1 within the gene EFEMP1. PLoS Genet. 14, e1007532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.So J et al. Biliary-atresia-associated mannosidase-1-alpha-2 gene regulates biliary and ciliary morphogenesis and laterality. Front. Physiol 11, 538701 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ningappa M et al. The role of ARF6 in biliary atresia. PLoS ONE 10, e0138381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Glessner JT et al. Biliary atresia is associated with polygenic susceptibility in ciliogenesis and planar polarity effector genes. J. Hepatol 79, 1385–1395 (2023). This study highlights common variants dysregulating ciliary genes predisposing to a higher risk of developing BA.
- 98.Chu AS, Russo PA & Wells RG Cholangiocyte cilia are abnormal in syndromic and non-syndromic biliary atresia. Mod. Pathol 25, 751–757 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Karjoo S et al. Extrahepatic cholangiocyte cilia are abnormal in biliary atresia. J. Pediatr. Gastroenterol. Nutr 57, 96–101 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Frassetto R et al. Intrahepatic bile duct primary cilia in biliary atresia. Hepatol. Res 48, 664–674 (2018). [DOI] [PubMed] [Google Scholar]
- 101. Berauer JP et al. Identification of polycystic kidney disease 1 like 1 gene variants in children with biliary atresia splenic malformation syndrome. Hepatology 70, 899–910 (2019). A key study identifying rare damaging mutations in ciliary genes linked to BASM.
- 102.Hellen DJ et al. Liver-restricted deletion of the biliary atresia candidate gene Pkd1l1 causes bile duct dysmorphogenesis and ciliopathy. Hepatology 77, 1274–1286 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lim YZ et al. Pkd1l1-deficiency drives biliary atresia through ciliary dysfunction in biliary epithelial cells. J. Hepatol 10.1016/j.jhep.2024.02.031 (2024). [DOI] [PubMed] [Google Scholar]
- 104.Anvarian Z, Mykytyn K, Mukhopadhyay S, Pedersen LB & Christensen ST Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol 15, 199–219 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mill P, Christensen ST & Pedersen LB Primary cilia as dynamic and diverse signalling hubs in development and disease. Nat. Rev. Genet 24, 421–441 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mansini AP et al. The cholangiocyte primary cilium in health and disease. Biochim. Biophys. Acta Mol. Basis Dis 1864, 1245–1253 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Toriyama M et al. The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat. Genet 48, 648–656 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tsai EA et al. Replication of a GWAS signal in a Caucasian population implicates ADD3 in susceptibility to biliary atresia. Hum. Genet 133, 235–243 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hai-Bing Y et al. Environmental toxin biliatresone-induced biliary atresia-like abnormal cilia and bile duct cell development of human liver organoids. Toxins 16, 144 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Barnes BH et al. Cholangiocytes as immune modulators in rotavirus-induced murine biliary atresia. Liver Int. 29, 1253–1261 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mack CL The pathogenesis of biliary atresia: evidence for a virus-induced autoimmune disease. Semin. Liver Dis 27, 233–242 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Averbukh LD & Wu GY Evidence for viral induction of biliary atresia: a review. J. Clin. Transl. Hepatol 6, 410–419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Saito T et al. Evidence for viral infection as a causative factor of human biliary atresia. J. Pediatr. Surg 50, 1398–1404 (2015). [DOI] [PubMed] [Google Scholar]
- 114.Zhao D et al. Effects of cytomegalovirus infection on the differential diagnosis between biliary atresia and intrahepatic cholestasis in a Chinese large cohort study. Ann. Hepatol 23, 100286 (2021). [DOI] [PubMed] [Google Scholar]
- 115.Moore SW, Zabiegaj-Zwick C & Nel E Problems related to CMV infection and biliary atresia. S Afr. Med. J 102, 890–892 (2012). [DOI] [PubMed] [Google Scholar]
- 116.Gilger MA et al. Extraintestinal rotavirus infections in children with immunodeficiency. J. Pediatr 120, 912–917 (1992). [DOI] [PubMed] [Google Scholar]
- 117.Clemente MG, Patton JT, Anders RA, Yolken RH & Schwarz KB Rotavirus infects human biliary epithelial cells and stimulates secretion of cytokines IL-6 and IL-8 via MAPK pathway. Biomed. Res. Int 2015, 697238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Petersen C et al. Progress in developing animal models for biliary atresia. Eur. J. Pediatr. Surg 8, 137–141 (1998). [DOI] [PubMed] [Google Scholar]
- 119.Riepenhoff-Talty M et al. Detection of group C rotavirus in infants with extrahepatic biliary atresia. J. Infect. Dis 174, 8–15 (1996). [DOI] [PubMed] [Google Scholar]
- 120.Bobo L et al. Lack of evidence for rotavirus by polymerase chain reaction/enzyme immunoassay of hepatobiliary samples from children with biliary atresia. Pediatr. Res 41, 229–234 (1997). [DOI] [PubMed] [Google Scholar]
- 121.Clemente MG et al. Prevalence of groups A and C rotavirus antibodies in infants with biliary atresia and cholestatic controls. J. Pediatr 166, 79–84 (2015). [DOI] [PubMed] [Google Scholar]
- 122.Harper P, Plant JW & Unger DB Congenital biliary atresia and jaundice in lambs and calves. Aust. Vet. J 67, 18–22 (1990). [DOI] [PubMed] [Google Scholar]
- 123.Koo KA et al. Biliatresone, a reactive natural toxin from Dysphania glomulifera and D. littoralis: discovery of the toxic moiety 1,2-diaryl-2-propenone. Chem. Res. Toxicol 28, 1519–1521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Estrada MA et al. Synthesis and structure-activity relationship study of biliatresone, a plant isoflavonoid that causes biliary atresia. ACS Med. Chem. Lett 9, 61–64 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Koo KA, Waisbourd-Zinman O, Wells RG, Pack M & Porter JR Reactivity of biliatresone, a natural biliary toxin, with glutathione, histamine, and amino acids. Chem. Res. Toxicol 29, 142–149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gupta K, Chen D & Wells RG Microcystin-RR is a biliary toxin selective for neonatal cholangiocytes. bioRxiv 10.1101/2023.08.09.552661 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kotb MA Aflatoxins in infants with extrahepatic biliary atresia. Med. J. Cairo Univ 83, 207–210 (2015). [Google Scholar]
- 128.Kotb MA & Kotb A Glutathione S transferase M1 polymorphism in extrahepatic biliary atresia. Med. J. Cairo Univ 83, 109–112 (2015). [Google Scholar]
- 129.Jee J et al. Cxcr2 signaling and the microbiome suppress inflammation, bile duct injury, and the phenotype of experimental biliary atresia. PLoS ONE 12, e0182089 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Jee JJ et al. Maternal regulation of biliary disease in neonates via gut microbial metabolites. Nat. Commun 13, 18 (2022). This study demonstrates a key role for the microbiome and butyrate metabolism in BA.
- 131.Wang J et al. Gut microbial profile in biliary atresia: a case-control study. J. Gastroenterol. Hepatol 35, 334–342 (2020). [DOI] [PubMed] [Google Scholar]
- 132. Hansen JM & Harris C Redox control of teratogenesis. Reprod. Toxicol 35, 165–179 (2013). A review of teratogens that induce ROS and oxidative injury, their teratogenic mechanisms and the rationale for targeted interventions.
- 133.Jauniaux E, Watson A & Burton G Evaluation of respiratory gases and acid-base gradients in human fetal fluids and uteroplacental tissue between 7 and 16 weeks’ gestation. Am. J. Obstet. Gynecol 184, 998–1003 (2001). [DOI] [PubMed] [Google Scholar]
- 134.Turner JM, Mitchell MD & Kumar SS The physiology of intrapartum fetal compromise at term. Am. J. Obstet. Gynecol 222, 17–26 (2020). [DOI] [PubMed] [Google Scholar]
- 135.Min J et al. Systems analysis of biliary atresia through integration of high-throughput biological data. Front. Physiol 11, 966 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Quelhas P et al. HIF-1alpha-pathway activation in cholangiocytes of patients with biliary atresia: an immunohistochemical/molecular exploratory study. J. Pediatr. Surg 58, 587–594 (2023). [DOI] [PubMed] [Google Scholar]
- 137.Wang D et al. Identifying and validating molecular subtypes of biliary atresia using multiple high-throughput data integration analysis. Front. Immunol 13, 1008246 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Luo Z, Shivakumar P, Mourya R, Gutta S & Bezerra JA Gene expression signatures associated with survival times of pediatric patients with biliary atresia identify potential therapeutic agents. Gastroenterology 157, 1138–1152.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Foo J, Bellot G, Pervaiz S & Alonso S Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 30, 679–692 (2022). [DOI] [PubMed] [Google Scholar]
- 140.Speir E, Shibutani T, Yu ZX, Ferrans V & Epstein SE Role of reactive oxygen intermediates in cytomegalovirus gene expression and in the response of human smooth muscle cells to viral infection. Circ. Res 79, 1143–1152 (1996). [DOI] [PubMed] [Google Scholar]
- 141.Buonocore G et al. Oxidative stress in preterm neonates at birth and on the seventh day of life. Pediatr. Res 52, 46–49 (2002). [DOI] [PubMed] [Google Scholar]
- 142.Ye R et al. Single cell RNA-sequencing analysis reveals that N-acetylcysteine partially reverses hepatic immune dysfunction in biliary atresia. JHEP Rep. 5, 100908 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Khandekar G et al. Coordinated development of the mouse extrahepatic bile duct: implications for neonatal susceptibility to biliary injury. J. Hepatol 72, 135–145 (2020). This study identified key differences in the extrahepatic mouse bile duct between neonates and adults, suggesting that the neonatal EHBD is particularly susceptible to injury and fibrosis.
- 144.Sollier J & Cimprich KA Breaking bad: R-loops and genome integrity. Trends Cell Biol. 25, 514–522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dotan M et al. Periductal bile acid exposure causes cholangiocyte injury and fibrosis. PLoS ONE 17, e0265418 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Colombo C, Zuliani G, Ronchi M, Breidenstein J & Setchell KD Biliary bile acid composition of the human fetus in early gestation. Pediatr. Res 21, 197–200 (1987). [DOI] [PubMed] [Google Scholar]
- 147.Hardy KJ, Hoffman NE, Mihaly G, Sewell RB & Smallwood RA Bile acid metabolism in fetal sheep; perinatal changes in the bile acid pool. J. Physiol 309, 1–11 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shivakumar P, Sabla GE, Whitington P, Chougnet CA & Bezerra JA Neonatal NK cells target the mouse duct epithelium via Nkg2d and drive tissue-specific injury in experimental biliary atresia. J. Clin. Invest 119, 2281–2290 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shivakumar P, Mourya R & Bezerra JA Perforin and granzymes work in synergy to mediate cholangiocyte injury in experimental biliary atresia. J. Hepatol 60, 370–376 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li J et al. Th2 signals induce epithelial injury in mice and are compatible with the biliary atresia phenotype. J. Clin. Invest 121, 4244–4256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mack CL et al. Oligoclonal expansions of CD4+ and CD8+ T-cells in the target organ of patients with biliary atresia. Gastroenterology 133, 278–287 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang J et al. Liver immune profiling reveals pathogenesis and therapeutics for biliary atresia. Cell 183, 1867–1883.e26 (2020). [DOI] [PubMed] [Google Scholar]
- 153.Luo Y et al. Unique cholangiocyte-targeted IgM autoantibodies correlate with poor outcome in biliary atresia. Hepatology 73, 1855–1867 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bednarek J et al. Cytokine-producing B cells promote immune-mediated bile duct injury in murine biliary atresia. Hepatology 68, 1890–1904 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Feldman AG, Tucker RM, Fenner EK, Pelanda R & Mack CL B cell deficient mice are protected from biliary obstruction in the rotavirus-induced mouse model of biliary atresia. PLoS ONE 8, e73644 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Qiu Y et al. HMGB1-promoted and TLR2/4-dependent NK cell maturation and activation take part in rotavirus-induced murine biliary atresia. PLoS Pathog. 10, e1004011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lages CS et al. The dendritic cell-T helper 17-macrophage axis controls cholangiocyte injury and disease progression in murine and human biliary atresia. Hepatology 65, 174–188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Antala S et al. Single-cell sequencing of a novel model of neonatal bile duct ligation in mice identifies macrophage heterogeneity in obstructive cholestasis. Sci. Rep 13, 14104 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang J et al. NLRP3 inflammasome activation promotes liver inflammation and fibrosis in experimental biliary atresia. Dig. Liver Dis 56, 458–467 (2023). [DOI] [PubMed] [Google Scholar]
- 160.Taylor SA et al. Transcriptional profiling of pediatric cholestatic livers identifies three distinct macrophage populations. PLoS ONE 16, e0244743 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Saxena V et al. Dendritic cells regulate natural killer cell activation and epithelial injury in experimental biliary atresia. Sci. Transl. Med 3, 102ra194 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang R, Huang J, Shan J, Chen Y & Xia H Peripheral blood CD177+ cells as an early diagnostic marker for biliary atresia: a prospective multicentre study in pediatric patients with cholestasis. J. Hepatol 77, 1714–1716 (2022). [DOI] [PubMed] [Google Scholar]
- 163.Zhang R et al. CD177+ cells produce neutrophil extracellular traps that promote biliary atresia. J. Hepatol 77, 1299–1310 (2022). [DOI] [PubMed] [Google Scholar]
- 164.Ye C et al. Single-cell and spatial transcriptomics reveal the fibrosis-related immune landscape of biliary atresia. Clin. Transl. Med 12, e1070 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shivakumar P et al. Obstruction of extrahepatic bile ducts by lymphocytes is regulated by IFN-γ in experimental biliary atresia. J. Clin. Invest 114, 322–329 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shivakumar P et al. Preferential TNFɑ signaling via TNFR2 regulates epithelial injury and duct obstruction in experimental biliary atresia. JCI Insight 2, e88747 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mack CL et al. Biliary atresia is associated with CD4+ Th1 cell-mediated portal tract inflammation. Pediatr. Res 56, 79–87 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liu J et al. Correlation of interleukin-33/ST2 receptor and liver fibrosis progression in biliary atresia patients. Front. Pediatr 7, 403 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Behairy OG et al. Clinical value of serum interleukin-33 biomarker in infants with neonatal cholestasis. J. Pediatr. Gastroenterol. Nutr 70, 344–349 (2020). [DOI] [PubMed] [Google Scholar]
- 170.Russi AE, Shivakumar P, Luo Z & Bezerra JA Plasticity between type 2 innate lymphoid cell subsets and amphiregulin expression regulates epithelial repair in biliary atresia. Hepatology 78, 1035–1049 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Feldman AG & Mack CL Biliary atresia: cellular dynamics and immune dysregulation. Semin. Pediatr. Surg 21, 192–200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rajendeeran A & Tenbrock K Regulatory T cell function in autoimmune disease. J. Transl. Autoimmun 4, 100130 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mack CL et al. Cellular and humoral autoimmunity directed at bile duct epithelia in murine biliary atresia. Hepatology 44, 1231–1239 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tucker RM, Feldman AG, Fenner EK & Mack CL Regulatory T cells inhibit Th1 cell-mediated bile duct injury in murine biliary atresia. J. Hepatol 59, 790–796 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Brindley SM et al. Cytomegalovirus-specific T-cell reactivity in biliary atresia at the time of diagnosis is associated with deficits in regulatory T cells. Hepatology 55, 1130–1138 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lages CS, Simmons J, Chougnet CA & Miethke AG Regulatory T cells control the CD8 adaptive immune response at the time of ductal obstruction in experimental biliary atresia. Hepatology 56, 219–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Miethke AG et al. Post-natal paucity of regulatory T cells and control of NK cell activation in experimental biliary atresia. J. Hepatol 52, 718–726 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Li K et al. Foxp3 promoter methylation impairs suppressive function of regulatory T cells in biliary atresia. Am. J. Physiol. Gastrointest. Liver Physiol 311, G989–G997 (2016). [DOI] [PubMed] [Google Scholar]
- 179.Kim S et al. Correlation of immune markers with outcomes in biliary atresia following intravenous immunoglobulin therapy. Hepatol. Commun 3, 685–696 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Muraji T et al. New insights in understanding biliary atresia from the perspectives on maternal microchimerism. Front. Pediatr 10, 1007987 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Suskind DL et al. Maternal microchimerism in the livers of patients with biliary atresia. BMC Gastroenterol. 4, 14 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kobayashi H et al. Maternal microchimerism in biliary atresia. J. Pediatr. Surg 42, 987–991 (2007). discussion 991. [DOI] [PubMed] [Google Scholar]
- 183.Hayashida M et al. The evidence of maternal microchimerism in biliary atresia using fluorescent in situ hybridization. J. Pediatr. Surg 42, 2097–2101 (2007). [DOI] [PubMed] [Google Scholar]
- 184.Muraji T et al. Maternal microchimerism in underlying pathogenesis of biliary atresia: quantification and phenotypes of maternal cells in the liver. Pediatrics 121, 517–521 (2008). [DOI] [PubMed] [Google Scholar]
- 185.Muraji T Maternal microchimerism in biliary atresia: are maternal cells effector cells, targets, or just bystanders. Chimerism 5, 1–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Al-Hakim A, Mistry A & Savic S Improving diagnosis and clinical management of acquired systemic autoinflammatory diseases. J. Inflamm. Res 15, 5739–5755 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Yang L et al. Regulation of epithelial injury and bile duct obstruction by NLRP3, IL-1R1 in experimental biliary atresia. J. Hepatol 69, 1136–1144 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bertolini A, Fiorotto R & Strazzabosco M Bile acids and their receptors: modulators and therapeutic targets in liver inflammation. Semin. Immunopathol 44, 547–564 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Yang H et al. Inflammation mediated down-regulation of hepatobiliary transporters contributes to intrahepatic cholestasis and liver damage in murine biliary atresia. Pediatr. Res 66, 380–385 (2009). [DOI] [PubMed] [Google Scholar]
- 190.Azeltine MW et al. Inflammation brives microRNAs to limit hepatocyte bile acid transport in murine biliary atresia. J. Surg. Res 256, 663–672 (2020). [DOI] [PubMed] [Google Scholar]
- 191.Ma Y et al. Reduced peroxisome proliferator-activated receptor-ɑ and bile acid nuclear receptor NR1H4/FXR may affect the hepatic immune microenvironment of biliary atresia. Front. Immunol 13, 875593 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Larson BJ, Longaker MT & Lorenz HP Scarless fetal wound healing: a basic science review. Plast. Reconstr. Surg 126, 1172–1180 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Trampert DC & Beuers U A beneficial response of fetal wound healing gone bad in the bile duct: the overarching cause of biliary atresia. J. Hepatol 80, 387–389 (2024). [DOI] [PubMed] [Google Scholar]
- 194.Lertudomphonwanit C et al. Large-scale proteomics identifies MMP-7 as a sentinel of epithelial injury and of biliary atresia. Sci. Transl. Med 9, eaan8462 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Jiang J et al. Serum MMP-7 in the diagnosis of biliary atresia. Pediatrics 144, e20190902 (2019). This report highlights the value of serum MMP-7 as a diagnostic biomarker.
- 196.Huang CC et al. Matrilysin (MMP-7) is a major matrix metalloproteinase upregulated in biliary atresia-associated liver fibrosis. Mod. Pathol 18, 941–950 (2005). [DOI] [PubMed] [Google Scholar]
- 197.Fawaz R et al. Guideline for the evaluation of cholestatic jaundice in infants: joint recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J. Pediatr. Gastroenterol. Nutr 64, 154–168 (2017). [DOI] [PubMed] [Google Scholar]
- 198.Feldman AG & Sokol RJ Neonatal cholestasis: emerging molecular diagnostics and potential novel therapeutics. Nat. Rev. Gastroenterol. Hepatol 16, 346–360 (2019). [DOI] [PubMed] [Google Scholar]
- 199.Zhou W & Zhou L Ultrasound for the diagnosis of biliary atresia: from conventional ultrasound to artificial intelligence. Diagnostics 12, 51 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Caponcelli E, Knisely AS & Davenport M Cystic biliary atresia: an etiologic and prognostic subgroup. J. Pediatr. Surg 43, 1619–1624 (2008). [DOI] [PubMed] [Google Scholar]
- 201.Chan WK, Chung PHY & Wong KKY The value of hepatic scintigraphy in the diagnosis of biliary atresia. Front. Pediatr 10, 874809 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Russo P. et al. Key histopathologic features of liver biopsies that distinguish biliary atresia from other causes of infantile cholestasis and their correlation with outcome: a multicenter study. Am. J. Surg. Pathol 40, 1601–1615 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Russo P. et al. Design and validation of the Biliary Atresia Research Consortium histologic assessment system for cholestasis in infancy. Clin. Gastroenterol. Hepatol 9, 357–362.e2 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Azar G. et al. Atypical morphologic presentation of biliary atresia and value of serial liver biopsies. J. Pediatr. Gastroenterol. Nutr 34, 212–215 (2002). [DOI] [PubMed] [Google Scholar]
- 205.He L. et al. Biomarkers for the diagnosis and post-Kasai portoenterostomy prognosis of biliary atresia: a systematic review and meta-analysis. Sci. Rep 11, 11692 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pandurangi S. et al. Diagnostic accuracy of serum matrix metalloproteinase-7 as a biomarker of biliary atresia in a large North American cohort. Hepatology 10.1097/HEP.0000000000000827 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Jiang J. et al. Protocol for a diagnostic accuracy study to develop diagnosis algorithm for biliary atresia using MMP-7 (DIABA-7 study): a study recruiting from Chinese Biliary Atresia Collaborative Network. BMJ Open. 11, e052328 (2021). [Google Scholar]
- 208.Davenport M. Serum matrix metalloproteinase-7 (MMP-7): as good as it gets? Hepatology 10.1097/HEP.0000000000000835 (2024). [DOI] [PubMed] [Google Scholar]
- 209.Lyu H. et al. Plasma amyloid-beta levels correlated with impaired hepatic functions: an adjuvant biomarker for the diagnosis of biliary atresia. Front. Surg 9, 931637 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Boo YA et al. Diagnostic performance of transient elastography in biliary atresia among infants with cholestasis. Hepatol. Commun 5, 882–890 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wagner ES et al. Use of shear wave elastography for the diagnosis and follow-up of biliary atresia: a meta-analysis. World J. Gastroenterol 28, 4726–4740 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Rabbani T, Guthery SL, Himes R, Shneider BL & Harpavat S Newborn screening for biliary atresia: a review of current methods. Curr. Gastroenterol. Rep 23, 28 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lien TH et al. Effects of the infant stool color card screening program on 5-year outcome of biliary atresia in Taiwan. Hepatology 53, 202–208 (2011). [DOI] [PubMed] [Google Scholar]
- 214.Schreiber RA et al. Home-based screening for biliary atresia using infant stool colour cards: a large-scale prospective cohort study and cost-effectiveness analysis. J. Med. Screen 21, 126–132 (2014). [DOI] [PubMed] [Google Scholar]
- 215.Mogul D, Zhou M, Intihar P, Schwarz K & Frick K Cost-effective analysis of screening for biliary atresia with the stool color card. J. Pediatr. Gastroenterol. Nutr 60, 91–98 (2015). [DOI] [PubMed] [Google Scholar]
- 216.Matsui A & Dodoriki M Screening for biliary atresia. Lancet 345, 1181 (1995). [DOI] [PubMed] [Google Scholar]
- 217.Zheng J, Ye Y, Wang B & Zhang L Biliary atresia screening in Shenzhen: implementation and achievements. Arch. Dis. Child 105, 720–723 (2020). [DOI] [PubMed] [Google Scholar]
- 218.Powell JE, Keffler S, Kelly DA & Green A Population screening for neonatal liver disease: potential for a community-based programme. J. Med. Screen 10, 112–116 (2003). [DOI] [PubMed] [Google Scholar]
- 219.Harpavat S, Garcia-Prats JA & Shneider BL Newborn bilirubin screening for biliary atresia. N. Engl. J. Med 375, 605–606 (2016). [DOI] [PubMed] [Google Scholar]
- 220.Schreiber RA Newborn screening for biliary atresia. JAMA 323, 1137–1138 (2020). [DOI] [PubMed] [Google Scholar]
- 221.Kasai M & Suzuki M A new operation for “non-correctable” biliary atresia – portoenterostomy. Shijitsu 13, 733–739 (1959). [Google Scholar]
- 222.Ohi R. A history of the Kasai operation: hepatic portoenterostomy for biliary atresia. World J. Surg 12, 871–874 (1988). [DOI] [PubMed] [Google Scholar]
- 223.Ogasawara Y. et al. The intussusception antireflux valve is ineffective for preventing cholangitis in biliary atresia: a prospective study. J. Pediatr. Surg 38, 1826–1829 (2003). [DOI] [PubMed] [Google Scholar]
- 224.Li Y. et al. Medium-term outcome of laparoscopic Kasai portoenterostomy for biliary atresia with 49 cases. J. Pediatr. Gastroenterol. Nutr 66, 857–860 (2018). [DOI] [PubMed] [Google Scholar]
- 225.Ji Y. et al. The short-term outcome of modified laparoscopic Kasai portoenterostomy for biliary atresia. Surg. Endosc 35, 1429–1434 (2021). [DOI] [PubMed] [Google Scholar]
- 226.Zhang M. et al. Robotic-assisted Kasai portoenterostomy for biliary atresia. Surg. Endosc 37, 3540–3547 (2023). [DOI] [PubMed] [Google Scholar]
- 227.Phelps HM et al. Enhancing recovery after kasai portoenterostomy with epidural analgesia. J. Surg. Res. 243, 354–362 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Sun X. et al. A prospective study comparing laparoscopic and conventional Kasai portoenterostomy in children with biliary atresia. J. Pediatr. Surg 51, 374–378 (2016). [DOI] [PubMed] [Google Scholar]
- 229.Tsukui T. et al. Biochemical evaluation of laparoscopic portoenterostomy for treating biliary atresia and redo for failed portoenterostomy. J. Laparoendosc. Adv. Surg. Tech. A 32, 1212–1219 (2022). [DOI] [PubMed] [Google Scholar]
- 230.Chan KWE et al. Ten-year native liver survival rate after laparoscopic and open Kasai portoenterostomy for biliary atresia. J. Laparoendosc. Adv. Surg. Tech. A 29, 121–125 (2019). [DOI] [PubMed] [Google Scholar]
- 231.de Vries W. et al. Biliary atresia in the Netherlands: outcome of patients diagnosed between 1987 and 2008. J. Pediatr 160, 638–644.e2 (2012). [DOI] [PubMed] [Google Scholar]
- 232.Madadi-Sanjani O. et al. Centralization of biliary atresia: has germany learned its lessons. Eur. J. Pediatr. Surg 32, 233–239 (2022). [DOI] [PubMed] [Google Scholar]
- 233.Al-Hussaini A. et al. The epidemiology and outcome of biliary atresia: Saudi Arabian National Study (2000-2018). Front. Pediatr 10, 921948 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Schreiber RA, Barker CC, Roberts EA, Martin SR & Canadian Pediatric Hepatology Research, G. Biliary atresia in Canada: the effect of centre caseload experience on outcome. J. Pediatr. Gastroenterol. Nutr 51, 61–65 (2010). [DOI] [PubMed] [Google Scholar]
- 235.Shneider BL et al. Total serum bilirubin within 3 months of hepatoportoenterostomy predicts short-term outcomes in biliary atresia. J. Pediatr 170, 211–217.e2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Superina R. et al. The anatomic pattern of biliary atresia identified at time of Kasai hepatoportoenterostomy and early postoperative clearance of jaundice are significant predictors of transplant-free survival. Ann. Surg 254, 577–585 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kelley-Quon LI et al. The need for early Kasai portoenterostomy: a Western Pediatric Surgery Research Consortium study. Pediatr. Surg. Int 38, 193–199 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Davenport M. et al. The outcome of a centralization program in biliary atresia: 20 years and beyond. Ann. Surg 10.1097/SLA.0000000000006273 (2024). [DOI] [PubMed] [Google Scholar]; Definitive evidence that centralization of surgery leads to improved outcomes.
- 239.LeeVan E, Matsuoka L, Cao S, Groshen S & Alexopoulos S Biliary-enteric drainage vs primary liver transplant as initial treatment for children with biliary atresia. JAMA Surg. 154, 26–32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Yoeli D. et al. Primary vs. salvage liver transplantation for biliary atresia: a retrospective cohort study. J. Pediatr. Surg 57, 407–413 (2022). [DOI] [PubMed] [Google Scholar]
- 241.Davenport M & Superina R Primary liver transplant in biliary atresia: the case for and against. J. Pediatr. Surg 10.1016/j.jpedsurg.2024.03.005 (2024). [DOI] [PubMed] [Google Scholar]
- 242.Hukkinen M, Pihlajoki M & Pakarinen MP Predicting native liver injury and survival in biliary atresia. Semin. Pediatr. Surg 29, 150943 (2020). [DOI] [PubMed] [Google Scholar]
- 243.Davenport M & Grieve A Maximizing Kasai portoenterostomy in the treatment of biliary atresia: medical and surgical options. S Afr. Med. J 102, 865–867 (2012). [DOI] [PubMed] [Google Scholar]
- 244.Sun S. et al. Low gamma-glutamyl transpeptidase levels at presentation are associated with severity of liver illness and poor outcome in biliary atresia. Front. Pediatr 10, 956732 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Chi S. et al. Dynamic analysis of serum MMP-7 and its relationship with disease progression in biliary atresia: a multicenter prospective study. Hepatol. Int 16, 954–963 (2022). [DOI] [PubMed] [Google Scholar]
- 246.Godbole N. et al. Liver secretin receptor predicts portoenterostomy outcomes and liver injury in biliary atresia. Sci. Rep 12, 7233 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Godbole N. et al. Prognostic and pathophysiologic significance of IL-8 (CXCL8) in biliary atresia. J. Clin. Med 10, 2705 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Nyholm I. et al. Serum FGF19 predicts outcomes of Kasai portoenterostomy in biliary atresia. Hepatology 77, 1263–1273 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Yoshii D. et al. Ki67 expression at Kasai portoenterostomy as a prognostic factor in patients with biliary atresia. BJS Open. 4, 873–883 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Lampela H. et al. National centralization of biliary atresia care to an assigned multidisciplinary team provides high-quality outcomes. Scand. J. Gastroenterol 47, 99–107 (2012). [DOI] [PubMed] [Google Scholar]
- 251.Tu CG, Khurana S, Couper R & Ford AW Kasai hepatoportoenterostomy in South Australia: a case for ‘centralized decentralization’. Anz. J. Surg 85, 865–868 (2015). [DOI] [PubMed] [Google Scholar]
- 252.Baek SH et al. The epidemiology and etiology of cholangitis after Kasai portoenterostomy in patients with biliary atresia. J. Pediatr. Gastroenterol. Nutr 70, 171–177 (2020). [DOI] [PubMed] [Google Scholar]
- 253.Wang P. et al. Severity assessment to guide empiric antibiotic therapy for cholangitis in children after Kasai portoenterostomy: a multicenter prospective randomized control trial in China. Int. J. Surg 109, 4009–4017 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Alatas FS, Lazarus G, Junaidi MC & Oswari H Prophylactic antibiotics to prevent cholangitis in children with biliary atresia after Kasai portoenterostomy: a meta-analysis. J. Pediatr. Gastroenterol. Nutr 77, 648–654 (2023). [DOI] [PubMed] [Google Scholar]
- 255.Willot S. et al. Effect of ursodeoxycholic acid on liver function in children after successful surgery for biliary atresia. Pediatrics 122, e1236–e1241 (2008). [DOI] [PubMed] [Google Scholar]
- 256.Davenport M, Parsons C, Tizzard S & Hadzic N Steroids in biliary atresia: single surgeon, single centre, prospective study. J. Hepatol 59, 1054–1058 (2013). [DOI] [PubMed] [Google Scholar]
- 257. Lu X. et al. Effect of adjuvant steroid therapy in type 3 biliary atresia: a single-center, open-label, randomized controlled trial. Ann. Surg 277, e1200–e1207 (2023). Latest finding that postoperative adjuvant steroid therapy improves bile drainage and native liver survival.
- 258.Davenport M. et al. Randomized, double-blind, placebo-controlled trial of corticosteroids after Kasai portoenterostomy for biliary atresia. Hepatology 46, 1821–1827 (2007). [DOI] [PubMed] [Google Scholar]
- 259.Bezerra JA et al. Use of corticosteroids after hepatoportoenterostomy for bile drainage in infants with biliary atresia: the START randomized clinical trial. JAMA 311, 1750–1759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Pandurangi S. et al. Customized postoperative therapy improves bile drainage in biliary atresia: a single center preliminary report. J. Pediatr. Surg 58, 1483–1488 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Wong ZH & Davenport M What happens after Kasai for biliary atresia? A European Multicenter Survey. Eur. J. Pediatr. Surg 29, 1–6 (2019). [DOI] [PubMed] [Google Scholar]
- 262.Tyraskis A & Davenport M Steroids after the Kasai procedure for biliary atresia: the effect of age at Kasai portoenterostomy. Pediatr. Surg. Int 32, 193–200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Alonso EM et al. Impact of steroid therapy on early growth in infants with biliary atresia: the multicenter Steroids in Biliary Atresia Randomized Trial. J. Pediatr 202, 179–185.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Mack CL et al. A phase I/IIa trial of intravenous immunoglobulin following portoenterostomy in biliary atresia. J. Pediatr. Gastroenterol. Nutr 68, 495–501 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Fischler B, Casswall TH, Malmborg P & Nemeth A Ganciclovir treatment in infants with cytomegalovirus infection and cholestasis. J. Pediatr. Gastroenterol. Nutr 34, 154–157 (2002). [DOI] [PubMed] [Google Scholar]
- 266.Shah I & Bhatnagar S Biliary atresia with cytomegalovirus infection and its response to ganciclovir. Trop. Gastroenterol 35, 56–58 (2014). [DOI] [PubMed] [Google Scholar]
- 267.Parolini F, Hadzic N & Davenport M Adjuvant therapy of cytomegalovirus IgM+ve associated biliary atresia: Prima facie evidence of effect. J. Pediatr. Surg 54, 1941–1945 (2019). [DOI] [PubMed] [Google Scholar]
- 268.US National Library of Medicine. ClinicalTrials.gov clinicaltrials.gov/study/NCT04524390 (2024). [DOI] [PubMed]
- 269.US National Library of Medicine. ClinicalTrials.gov clinicaltrials.gov/study/NCT04336722 (2024). [DOI] [PubMed]
- 270.Jeyaraj R, Maher ER & Kelly D Paediatric research sets new standards for therapy in paediatric and adult cholestasis. Lancet Child. Adolesc. Health 8, 75–84 (2024). [DOI] [PubMed] [Google Scholar]
- 271.US National Library of Medicine. ClinicalTrials.gov clinicaltrials.gov/study/NCT05321524 (2023). [DOI] [PubMed]
- 272.Fuchs CD & Trauner M Role of bile acids and their receptors in gastrointestinal and hepatic pathophysiology. Nat. Rev. Gastroenterol. Hepatol 19, 432–450 (2022). [DOI] [PubMed] [Google Scholar]
- 273.Gonzales E. et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. Lancet 398, 1581–1592 (2021). [DOI] [PubMed] [Google Scholar]
- 274.Baumann U. et al. Effects of odevixibat on pruritus and bile acids in children with cholestatic liver disease: phase 2 study. Clin. Res. Hepatol. Gastroenterol 45, 101751 (2021). [DOI] [PubMed] [Google Scholar]
- 275. Tam PKH et al. Regenerative medicine: postnatal approaches. Lancet Child. Adolesc. Health 6, 654–666 (2022). State-of-the art review of the potential applications of regenerative medicine to paediatric disease including biliary atresia.
- 276.Afonso MB, Marques V, van Mil SWC & Rodrigues CMP Human liver organoids: from generation to applications. Hepatology 79, 1432–1451 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Sharma S. et al. Bone marrow mononuclear stem cell infusion improves biochemical parameters and scintigraphy in infants with biliary atresia. Pediatr. Surg. Int 27, 81–89 (2011). [DOI] [PubMed] [Google Scholar]
- 278.Nguyen TL et al. Autologous bone marrow mononuclear cell infusion for liver cirrhosis after the Kasai operation in children with biliary atresia. Stem Cell Res. Ther 13, 108 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Holterman A. et al. Granulocyte-colony stimulating factor GCSF mobilizes hematopoietic stem cells in Kasai patients with biliary atresia in a phase 1 study and improves short term outcome. J. Pediatr. Surg 56, 1179–1185 (2021). [DOI] [PubMed] [Google Scholar]
- 280.Sampaziotis F. et al. Cholangiocyte organoids can repair bile ducts after transplantation in the human liver. Science 371, 839–846 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Cross-Najafi AA et al. The long road to develop custom-built livers: current status of 3D liver bioprinting. Transplantation 108, 357–368 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Sundaram SS, Mack CL, Feldman AG & Sokol RJ Biliary atresia: indications and timing of liver transplantation and optimization of pretransplant care. Liver Transpl. 23, 96–109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Jain V. et al. Prognostic markers at adolescence in patients requiring liver transplantation for biliary atresia in adulthood. J. Hepatol 71, 71–77 (2019). [DOI] [PubMed] [Google Scholar]
- 284.Howard ER et al. Survival patterns in biliary atresia and comparison of quality of life of long-term survivors in Japan and England. J. Pediatr. Surg 36, 892–897 (2001). [DOI] [PubMed] [Google Scholar]
- 285.Kerola A. et al. Molecular signature of active fibrogenesis prevails in biliary atresia after successful portoenterostomy. Surgery 162, 548–556 (2017). [DOI] [PubMed] [Google Scholar]
- 286.Kyronlahti A. et al. Evolving up-regulation of biliary fibrosis-related extracellular matrix molecules after successful portoenterostomy. Hepatol. Commun 5, 1036–1050 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Sato K. et al. Ductular reaction in liver diseases: pathological mechanisms and translational significances. Hepatology 69, 420–430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Nyholm I. et al. Deep learning quantification reveals a fundamental prognostic role for ductular reaction in biliary atresia. Hepatol. Commun 7, e0333 (2023). This paper demonstrates the central prognostic importance of ductular reaction both at the time of KPE and during the postoperative follow-up using a large biopsy material and a neural network model.
- 289.Harpavat S. et al. Serum bile acids as a prognostic biomarker in biliary atresia following Kasai portoenterostomy. Hepatology 77, 862–873 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Venkat V. et al. Modeling outcomes in children with biliary atresia with native liver after 2 years of age. Hepatol. Commun 4, 1824–1834 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Witt M. et al. Prognosis of biliary atresia after 2-year survival with native liver: a nationwide cohort analysis. J. Pediatr. Gastroenterol. Nutr 67, 689–694 (2018). [DOI] [PubMed] [Google Scholar]
- 292.Hukkinen M. et al. Noninvasive evaluation of liver fibrosis and portal hypertension after successful portoenterostomy for biliary atresia. Hepatol. Commun 3, 382–391 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Lykavieris P. et al. Outcome in adulthood of biliary atresia: a study of 63 patients who survived for over 20 years with their native liver. Hepatology 41, 366–371 (2005). One of the first studies to investigate adulthood outcomes in patients surviving with their native liver.
- 294.de Vries W. et al. Twenty-year transplant-free survival rate among patients with biliary atresia. Clin. Gastroenterol. Hepatol 9, 1086–1091 (2011). [DOI] [PubMed] [Google Scholar]
- 295.Yoon HJ et al. Hepatic tumours in children with biliary atresia: single-centre experience in 13 cases and review of the literature. Clin. Radiol 69, e113–e119 (2014). [DOI] [PubMed] [Google Scholar]
- 296.Hadzic N. et al. Hepatocellular carcinoma in biliary atresia: King’s College Hospital experience. J. Pediatr 159, 617–622.e1 (2011). [DOI] [PubMed] [Google Scholar]
- 297.Joshi D. et al. The management of childhood liver diseases in adulthood. J. Hepatol 66, 631–644 (2017). [DOI] [PubMed] [Google Scholar]
- 298. Bass LM et al. Risk of variceal hemorrhage and pretransplant mortality in children with biliary atresia. Hepatology 76, 712–726 (2022). This study addresses the risk of variceal bleeding and associated mortality in two large cohorts of paediatric patients surviving with their native liver.
- 299.Lampela H, Hukkinen M, Kosola S, Jahnukainen T & Pakarinen MP Poor performance of noninvasive predictors of esophageal varices during primary prophylaxis surveillance in biliary atresia. J. Pediatr. Surg 55, 2662–2667 (2020). [DOI] [PubMed] [Google Scholar]
- 300. Ng VL et al. Medical status of 219 children with biliary atresia surviving long-term with their native livers: results from a North American multicenter consortium. J. Pediatr. 165, 539–546.e2 (2014). A benchmark study for long-term health outcomes in children with biliary atresia surviving with their native liver.
- 301.Sadiq J. et al. Long-term clinical and socioeconomic outcomes of children with biliary atresia. JGH Open. 7, 841–847 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Bijl EJ, Bharwani KD, Houwen RH & de Man RA The long-term outcome of the Kasai operation in patients with biliary atresia: a systematic review. Neth. J. Med 71, 170–173 (2013). [PubMed] [Google Scholar]
- 303.Duche M. et al. Experience with endoscopic management of high-risk gastroesophageal varices, with and without bleeding, in children with biliary atresia. Gastroenterology 145, 801–807 (2013). [DOI] [PubMed] [Google Scholar]
- 304.Samyn M. Transitional care of biliary atresia. Semin. Pediatr. Surg 29, 150948 (2020). [DOI] [PubMed] [Google Scholar]
- 305.Karrer FM, Wallace BJ & Estrada AE Late complications of biliary atresia: hepatopulmonary syndrome and portopulmonary hypertension. Pediatr. Surg. Int 33, 1335–1340 (2017). [DOI] [PubMed] [Google Scholar]
- 306.Matcovici M, Stoica I, Smith K & Davenport M What makes a “successful” Kasai portoenterostomy “unsuccessful”? J. Pediatr. Gastroenterol. Nutr 76, 66–71 (2023). [DOI] [PubMed] [Google Scholar]
- 307.Ginstrom DA, Hukkinen M, Kivisaari R & Pakarinen MP Biliary atresia-associated cholangitis: the central role and effective management of bile lakes. J. Pediatr. Gastroenterol. Nutr 68, 488–494 (2019). [DOI] [PubMed] [Google Scholar]
- 308.Calinescu AM et al. Cholangitis definition and treatment after Kasai hepatoportoenterostomy for biliary atresia: a Delphi process and international expert panel. J. Clin. Med 11, 494 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Ruuska S. et al. Impaired bone health in children with biliary atresia. J. Pediatr. Gastroenterol. Nutr 71, 707–712 (2020). [DOI] [PubMed] [Google Scholar]
- 310.Ng VL et al. Neurodevelopmental outcome of young children with biliary atresia and native liver: results from the ChiLDReN study. J. Pediatr 196, e133 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Squires JE et al. Neurodevelopmental outcomes in preschool and school aged children with biliary atresia and their native liver. J. Pediatr. Gastroenterol. Nutr 70, 79–86 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Ruuska S. et al. Neurocognitive and motor functions in biliary atresia patients: a cross-sectional, prospective national cohort study. J. Pediatr. Gastroenterol. Nutr 73, 491–498 (2021). [DOI] [PubMed] [Google Scholar]
- 313.Rodijk LH et al. Long-term neurodevelopmental outcomes in children with biliary atresia. J. Pediatr 217, 118–124.e3 (2020). [DOI] [PubMed] [Google Scholar]
- 314.Sundaram SS et al. Health related quality of life in patients with biliary atresia surviving with their native liver. J. Pediatr 163, 1052–1057.e2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.de Vries W. et al. Overall quality of life in adult biliary atresia survivors with or without liver transplantation: results from a national cohort. Eur. J. Pediatr. Surg 26, e1 (2016). [DOI] [PubMed] [Google Scholar]
- 316.Wong CWY, Chung PHY, Tam PKH & Wong KKY Long-term results and quality of life assessment in biliary atresia patients: a 35-year experience in a tertiary hospital. J. Pediatr. Gastroenterol. Nutr 66, 570–574 (2018). [DOI] [PubMed] [Google Scholar]
- 317.Hukkinen M, Ruuska S, Pihlajoki M, Kyronlahti A & Pakarinen MP Long-term outcomes of biliary atresia patients surviving with their native livers. Best. Pract. Res. Clin. Gastroenterol 56-57, 101764 (2022). [DOI] [PubMed] [Google Scholar]
- 318.Cui MM et al. Contribution of ADD3 and the HLA genes to biliary atresia risk in Chinese. Int. J. Mol. Sci 24, 14719 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Zeng S. et al. Association between single nucleotide polymorphisms in the ADD3 gene and susceptibility to biliary atresia. PLoS ONE 9, e107977 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Kaewkiattiyot S, Honsawek S, Vejchapipat P, Chongsrisawat V & Poovorawan Y Association of X-prolyl aminopeptidase 1 rs17095355 polymorphism with biliary atresia in Thai children. Hepatol. Res 41, 1249–1252 (2011). [DOI] [PubMed] [Google Scholar]
- 321.La Pergola E, Zen Y & Davenport M Developmental histology of the portal plate in biliary atresia: observations and implications. Pediatr. Surg. Int 37, 715–721 (2021). [DOI] [PubMed] [Google Scholar]