

Abstract
A large variety of lactic acid bacteria (LAB) can utilize citrate under fermentative conditions. Although much information concerning the metabolic pathways leading to citrate utilization by LAB has been gathered, the mechanisms regulating these pathways are obscure. In Weissella paramesenteroides (formerly called Leuconostoc paramesenteroides), transcription of the citMDEFCGRP citrate operon and the upstream divergent gene citI is induced by the presence of citrate in the medium. Although genetic experiments have suggested that CitI is a transcriptional activator whose activity can be modulated in response to citrate availability, specific details of the interaction between CitI and DNA remained unknown. In this study, we show that CitI recognizes two A+T-rich operator sites located between citI and citM and that the DNA-binding affinity of CitI is increased by citrate. Subsequently, this citrate signal propagation leads to the activation of the cit operon through an enhanced recruitment of RNA polymerase to its promoters. Our results indicate that the control of CitI by the cellular pools of citrate provides a mechanism for sensing the availability of citrate and adjusting the expression of the cit operon accordingly. In addition, this is the first reported example of a transcription factor directly functioning as a citrate-activated switch allowing the cell to optimize the generation of metabolic energy.
Variability and adaptability are crucial characteristics of organisms possessing the ability to survive and prosper under a wide variety of environmental conditions. In order for bacteria to effectively compete and survive, they have to sense and respond to changes in the availability of specific nutrients. Citrate is an abundant nutrient in nature since it is a natural constituent of all living cells. It is therefore not surprising that a large variety of bacterial species can utilize citrate under aerobic or anaerobic conditions. Although the citrate degradation pathways in bacteria are well established, less information is available on the mechanisms for sensing the availability of citrate and controlling its utilization. These mechanisms were studied mainly in Klebsiella pneumoniae and Bacillus subtilis. In these bacteria, two-component systems regulate the expression of citrate utilization genes. In particular, the CitA/B two-component system of K. pneumoniae has been extensively studied (4-6, 16). This system regulates the expression of two operons, citCDEFG and CitS-oadGAB-citAB, which are involved in the transport and metabolism of citrate. Expression of these operons is induced under anoxic conditions in the presence of citrate and is subject to catabolic repression (6, 16). In Bacillus subtilis, the CitS/T two-component system (a member of the CitA/B family) is involved in the regulation of the expression of genes encoding citrate transporters required for the utilization of citrate in aerobic conditions (22, 23).
Citrate metabolism has been extensively studied in lactic acid bacteria (LAB) because it significantly contributes to growth (12) and resistance to acidic stress (10, 13, 15). Understanding the mechanisms by which these organisms perceive the presence of citrate and using this information to activate its transport and degradation are important not only from a basic research point of view but also because citrate fermentation is relevant for the production of cheese and wine (11). Despite that, no regulator involved in the control of the expression of citrate utilization genes has been characterized in LAB. The dairy Weissella paramesenteroides J1 carries the cit operon, which includes the genes encoding the oxaloacetate decarboxylase (citM), the citrate lyase active complex (citCDEFG), and the citrate permease (citP) (14) (Fig. 1A), which are key proteins involved in the citrate fermentation pathway. Located upstream of the cit operon is the citI gene with divergent polarity. CitI, the citI gene product, belongs to the SorC/DeoR family, which includes more than 50 transcriptional regulators. Among them, only DeoR has been purified and has had its interactions with its operators investigated (24). In a previous study, we demonstrated that the transcription of the citI gene and the cit operon is induced when the J1 strain is grown in the presence of citrate (14). Overexpression of CitI in Escherichia coli resulted in trans activation of the Pcit promoter, suggesting that CitI is a transcriptional regulator (14). Nevertheless, the molecular mechanism by which the transcription of the W. paramesenteroides J1 operon is induced by citrate remains unsolved. Here we show that CitI, in the presence of citrate, stimulates its own transcription, leading to increased expression of all the genes required for citrate metabolism. Our results provide evidence for a novel mechanism for the control of citrate degradation, in which the CitI regulator couples the availability of citrate in the cells with the expression of genes involved in citrate fermentation.
FIG. 1.

Binding of CitI to the citI-citM intergenic region. (A) Schematic representation of the 8.8-kb cit operon from W. paramesenteroides J1, including the genes involved in citrate fermentation and the divergent citI gene. The Pcit and PcitI promoters driving the expressions of the cit operon and of the citI gene, respectively, are indicated with arrows. The black bars indicate the locations of the 344-bp DNA probe (amplicon A), the 65-bp probe (amplicon B), and the 78-bp probe (amplicon C) used in the gel shift experiments performed in this work. (B) Autoradiogram of gel shift assays performed by use of amplicon A and increasing concentrations of CitI. Arrows indicate the positions of the retarded complexes CI and CII and free DNA. (C) The radioactivity present in the complexes shown in panel B was quantified, and the percentages of bound DNA were plotted as functions of CitI concentration. The values given in the plot correspond to means from three independent experiments.
MATERIALS AND METHODS
E. coli strains and growth conditions.
BL21(DE3) (Stratagene) and DH5α (laboratory stock) strains were grown at 37°C with shaking in LB medium (19). When required, ampicillin at 100 μg ml−1 and kanamycin at 30 μg ml−1 were added to the medium.
DNA analysis and manipulation.
Plasmid DNA preparations for cloning and sequencing experiments, treatment of DNA with restriction enzymes, and ligation with T4 DNA ligase, as well as transformations of E. coli, were performed as described by Sambrook et al. (19). In all cases, the authenticity of the DNA inserts generated by PCR was confirmed by sequencing determined with automated DNA sequencing instrumentation (ABI PRISM; Perkin-Elmer) at the Centro de Investigaciones Biológicas. The oligonucleotides used in this work are listed in Table 1.
TABLE 1.
Primers used in this study
| Primera | Sequenceb | Coordinate setc |
|---|---|---|
| F(−1285m) | 5′-GTGCAGAATTCTCGTCATCGACGGTGGATAC-3′ | 457-485 |
| F(−253) | 5′-TCTTCGGCAATTTTAGC-3′ | 1489-1505 |
| F(−199m) | 5′-GTGAGAATTCTAATATCATCATACACC-3′ | 1543-1569 |
| F(−199) | 5′-GTGAATGTTGTAATATCATCATACACC-3′ | 1543-1569 |
| F(−169) | 5′-TTAAAAATTTGAACTAGTATGTAAA-3′ | 1573-1597 |
| F(−101) | 5′-ATCAATTGAAAAAGCGTAAGTGC-3′ | 1619-1641 |
| F(−78m) | 5′-GCGAGAATTCAACTGGAAATTCGCAG-3′ | 1664-1689 |
| R(−207m) | 5′-GGATATGCACATATGGATGAAGAACAATTAGC-3′ | 1535-1504 |
| R(−135) | 5′-ATATTTATTTTTTACATACTAGTTC-3′ | 1607-1583 |
| R(−46) | 5′-AAAAAAACTGCGAATTTCCAGTTTAAATCTCG-3′ | 1696-1665 |
| R90 | 5′-TGGGATTTGTGCACCTT-3′ | 1832-1816 |
| R91m | 5′-GTGGGATCCGTGCACCTTCAAAA-3′ | 1833-1811 |
The numbers in the primer names indicate the 5′ positions of the primers relative to the transcription start site (+1) of the cit operon (Fig. 2). F, forward primer; R, reverse primer (denotes complementarity to indicated coordinates); m, mutagenic primer.
Restriction sites are underlined.
Coordinates in pCITJ1 cit region (EMBL nucleotide databank accession number AJ132782).
Construction and analysis of E. coli strains carrying plasmids with Pcit-lacZ transcriptional fusions.
To analyze in vivo the influence of CitI operators in the transcriptional activation of Pcit, plasmids pJMM13 and pJMM14 were constructed. Plasmid pJMM13 harbors a pCITJ1 insert expanding from the transcriptional start site (+1) of citI to the +83 nucleotide of the cit transcript. A 291-bp amplicon was generated by PCR by use of oligonucleotides F(−199m) and R91m, containing EcoRI and BamHI restriction sites, respectively. The amplicon was located upstream of the ribosomal binding site of lacZ by digestion with EcoRI and BamHI and cloning into the same restriction sites of the ColE1 derivative pJM116 (14), generating pMM13. Plasmid pJMM14 harbors the same transcriptional Pcit-lacZ fusion as pJMM13, but lacks the O1 and O2 CitI operators, since its pCITJ1 insert extends from the −69 to the +83 nucleotides of the cit transcript. A 170-bp amplicon was generated by PCR by use of the oligonucleotides F(−78m) and R91m, digested with EcoRI and BamHI, and cloned in the same sites of the pJM116 vector. The constructs were independently established in E. coli DH5α containing the pSUI plasmid (14). pSUI carries the citI gene under the control of the lacZ promoter and bears the P15a replicon compatible with the pJMMxx plasmids.
To determine the β-galactosidase activity encoded by the Pcit-lacZ fusions, three independent cultures of each E. coli strain were grown overnight with aeration at 37°C in medium supplemented with ampicillin and kanamycin, diluted 1:100 in 10 ml of fresh medium, and grown to an absorbance at 600 nm (A600) of 0.2. Then, 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added to induce citI expression from pSUI, and the culture was incubated for a further 60 min. The final absorbance was measured after 10 min in an ice bath to stop the cell growth, and aliquots of 1 ml were harvested. Pellets were frozen at −20°C until they were used. β-Galactosidase activity was measured by the method of Miller (17). Specific activities were expressed in Miller units · A600 of the cultures−1. The average values and standard deviations of three different experiments are described in Results.
Expression and purification of recombinant CitI.
The coding region of citI and its transcriptional terminator were amplified by PCR by use of the primers R1F(−207) and (−1285) containing, respectively, an NdeI site at the translation start site and an EcoRI site at the end of the amplified coding region. After digestion with NdeI and EcoRI, the citI coding region was cloned between the same sites on the expression vector pET22b (Novagen) to yield pMI22. This plasmid, directing synthesis of the CitI protein without a histidine tag, was transformed into BL21(DE3) cells.
The expression of citI was induced in BL21(DE3)/pMI22 cells by the addition of 1 mM IPTG when cultures reached A600 values of 0.5. Cultures were further incubated for 5 h at 37°C. Cells were harvested by centrifugation and resuspended in ice-cold lysis buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5% glycerol, 1 mM phenylmethylsulfonyl fluoride, and 4 mg/ml lysozyme. Crude extracts were prepared after incubation during 30 min at room temperature by passing cells four times through a French pressure cell at 12,000 lb/in2. Expressed CitI protein was found to be in inclusion bodies, which were recovered by centrifugation at 10,600 × g for 15 min at 4°C. Protein pellets were resuspended and resedimented, first in a buffer containing 50 mM Tris-HCl (pH 6.2), 10 mM EDTA, 2 M urea, and 1% Triton X-100 and second in water. The CitI inclusion bodies were resuspended in 20 mM Tris-HCl pH (6.2) and 8 M urea at a final protein concentration of 0.5 μg ml−1. At this stage, the protein preparation consisted of >90% CitI, as judged by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. CitI was refolded by successive dialyses in a buffer consisting of 20 mM Tris-HCl (pH 6.2), 500 mM NaCl, and 10% glycerol and containing decreasing concentrations of urea ranging from 4 M to 1 M. Insoluble material was removed by centrifugation at 10,600 × g for 15 min at 4°C between each step of dialysis. Finally, urea was eliminated by dialysis overnight in the same buffer, and samples were frozen at −80°C.
DNA labeling.
Eighty picomoles of oligonucleotides employed to amplify DNA probes for band shifts or footprinting experiments was phosphorylated for 30 min at 37°C with 250 μCi of [γ-32P]ATP (DuPont/NEN) by use of a T4 polynucleotide kinase (U.S. Biochemical Corp.). Free [γ-32P]ATP was removed by filtration on a Microspin G25 column (Amersham Biosciences).
Electrophoretic mobility shift assays.
For these experiments, the DNAs used were the A, B, and C amplicons (Fig. 1A). The amplicons were generated by PCR by use of the following 32P-5′-end-labeled oligonucleotides: F(−253) and R90 for amplicon A, F(−199) and R(−135) for amplicon B, and F(−101) and R(−46) for amplicon C. The amplicons were purified from a 5% polyacrylamide gel prior to their use for binding reactions, which were performed in a 20-μl final volume of 20 mM Tris-HCl (pH 6.2), 250 mM NaCl, and 10% glycerol. Labeled DNA fragment (0.5 to 1.0 nM) and proteins (0.125 to 1.25 μM) were incubated at 0°C for 30 min. The effect of citrate was investigated by adding 15 mM citrate to the binding mixtures. The reactions were loaded onto a 5% nondenaturing polyacrylamide gel. After the completion of electrophoresis, the gels were dried and autoradiographed, and the bands were quantified by use of a GS800 phosphorimager (Fujifilm).
The template DNA used to identify ternary complexes on the Pcit promoter was amplicon D (see Fig. 5), generated by PCR by use of the 32P-5′-end-labeled oligonucleotides F(−169) and R90. In these experiments, 0.04 pmol of probe DNA was incubated either in the presence or in the absence of 1.0 μM CitI as described above. After binding, 80 nM E. coli RNA polymerase (RNAP) σ70 holoenzyme (Epicenter Technologies) was added and further incubated at 22°C for 30 min to allow the formation of stable complexes. Finally, samples were incubated for 3 min with 1% heparin as an unspecific competitor prior to being loaded on the polyacrylamide gels. Citrate was added, when required, to final concentrations of 15 mM.
FIG. 5.

Citrate increases interaction of RNAP with Pcit in the presence of citrate. (A) Schematic representation of the 260-bp template (amplicon D) used to identify RNAP-DNA-CitI ternary complexes. To simplify the analysis, assays were performed with a DNA fragment including the Pcit promoter (arrow) and O2 (empty rectangle). (B) Electrophoretic mobility shift assays. The experiments were performed with 8 nM of RNAP in the presence (left panel) or absence (right panel) of citrate. In the lanes indicated, CitI was included (+) in the binding reactions. The positions of the different retarded complexes are shown between the panels.
DNase I footprinting.
The DNA probe was amplicon A, γ-32P labeled in either the plus or minus strand as indicated above. The one-strand-labeled DNA fragment (10,000 cpm) was incubated as for the gel retardation experiments with the concentration of CitI necessary to saturate both operator sites. After binding, 1 U of RQ1 DNase I (Promega) diluted in 80 μl of DNase buffer (400 mM Tris [pH 8.0], 100 mM MgSO4, 10 mM CaCl2) was added and incubated for 2 min at room temperature. The reaction was stopped by addition of 90 μl of buffer (20 mM EDTA [pH 8], 200 mM NaCl, 100 μg ml−1 tRNA). The samples were extracted with equal volumes of phenol-chloroform, precipitated with 3 volumes of ethanol, and resuspended in formamide loading buffer (80% deionized formamide, 10 mM NaOH, 1 mM EDTA, 0.1% bromophenol blue, 0.1% xylene cyanol). After heating for 5 min at 94°C, the samples were applied to a denaturing 6% polyacrylamide gel containing 8 M urea. Sanger sequencing reactions of DNA from M13mp18 with the −40 primer from a Sequenase kit (U. S. Biochemical Corp.) were used to generate molecular weight markers.
Hydroxyl radical footprinting.
Hydroxyl radical footprintings were performed as described previously by Tullius et al. (21). The DNA probe was generated by PCR and by use of oligonucleotides F(−199) and R(−46) and purified as described for the electrophoretic mobility shift assays. Before amplification, either F(−199) or R(−46) was 32P 5′ end labeled, and the resulting amplicons were used to protect the plus and minus strands, respectively. Binding reactions were performed in a volume of 200 μl, as was used for the gel retardation experiments, but, after binding, samples were dialyzed in 20 mM Tris-HCl (pH 6.2) and 250 mM NaCl to eliminate glycerol. Dialyzed samples were treated with 36 μl of 20 mM sodium ascorbate, 0.6% H2O2, 2 mM (NH4)2Fe(SO4)2 · 6H2O, and 4 mM EDTA for 10 min at room temperature. Reactions were terminated by addition of a 59-μl volume of stop solution (0.1 M thiourea, 20 mM EDTA, pH 8.0). Finally, CitI-bound complexes were isolated from 5% acrylamide gels and eluted by diffusion, and DNA fragments were resolved on a 6% polyacrylamide-8 M urea gel. Maxam and Gilbert sequencing reactions of the same templates were used as molecular weight standards.
In vitro transcription.
Transcription reactions were performed at 37°C for 15 min in a volume of 50 μl containing 40 mM Tris-HCl (pH 8), 10 mM MgCl2, 200 mM NaCl, 0.1 mM EDTA, 8% glycerol, 200 μM GTP, 200 μM ATP, 50 μM UTP, 50 μM CTP, 0.5 μM [α-32P]UTP (400 Ci mmol−1; 10 μCi μl−1), 1 nM template DNA, and 0.2 U RNA polymerase σ70 holoenzyme. After the binding of 1.25 μM CitI to the DNA template for 30 min at 22°C in a 40-μl volume under the described conditions, transcription was started by the addition of 10 μl of nucleoside triphosphate mix containing the RNA polymerase. Reactions were terminated by additions of equal volumes of phenol, and samples were precipitated by the addition of 10 μg tRNA, NaAc (pH 7), and 3 volumes of ethanol. Pellets were resuspended in RNA loading buffer (80% deionized formamide, 1× Tris-borate-EDTA, 0.025% bromophenol blue, 0.025% xylene cyanol) and analyzed on an 8% polyacrylamide-urea gel followed by autoradiography. Transcription products were quantified with a phosphorimager. Citrate and other compounds tested were added to binding reactions at final concentrations of 15 mM. To generate the 595-bp linear template for transcription, pJMM12 (14) was digested with HindIII and PstI (coordinates 1413 to 2007 in EMBL nucleotide database accession number AJ132782), which cut the plasmid 111 bp and 207 bp, respectively, downstream of the citI and citM translational start codons. The molecular weight standard was the A+G Maxam and Gilbert sequencing reaction of a 344-bp amplicon generated by PCR by use of the oligonucleotides 5′-32P-labeled F(−253) and R90.
Analysis of CitI binding to its operators.
The analysis of the cooperative interaction of CitI to O1 and O2 operators is based on a two-site system as described by Carlson and Little (7), in which the cooperativity parameter ω is defined as follows:
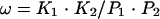 |
(1) |
where K1 and K2 are the apparent equilibrium dissociation rate constants (KDs) for independent O1 and O2 operators, and P1 and P2 are the KDs for the linked O1 and O2 operators.
To determine the KDs, the complex formation between CitI and DNA was measured as a function of protein concentration, and the saturation curves are depicted in Fig. 1C (see also Fig. 3). The data obtained from the electrophoretic mobility shift assay were fitted by a direct nonlinear least-squares regression program to the following equilibrium binding equation:
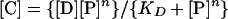 |
(2) |
where [C], [D], and [P] represent concentrations of formed complex, DNA, and free protein, respectively, and n is the Hill coefficient.
FIG. 3.

Citrate effect on CitI affinity to DNA. The top panels show gel shift assays performed by use of CitI and end-labeled amplicon B and amplicon C including either O1 or O2, respectively. Complexes CitI-O1 and CitI-O2 are indicated with arrows. The bottom panel shows the quantification of radioactivity present in the complexes obtained in each experiment shown in the upper panels and a plot of the percentages of bound DNA as functions of CitI concentration. The values given in the plot correspond to means of three independent experiments.
To determine the n values, equation 2 was transformed into a linear form, yielding equation 3:
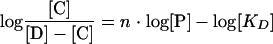 |
(3) |
The n values were derived from the slopes of the Hill plots: log{[C]/([D] − [C])} versus log[P].
K1 and K2 were calculated from equation 4 below and data shown in Fig. 1, and P1 and P2 were calculated from equation 5 and equation 6 below, respectively, and from data shown elsewhere (see Fig. 3). The KDs were calculated at each protein concentration tested, and the average values and standard deviations are given in Results.
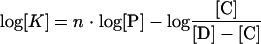 |
(4) |
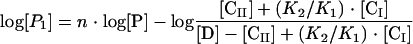 |
(5) |
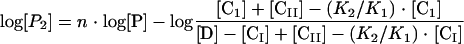 |
(6) |
In the above equations, [CI] and [CII] represent, respectively, concentrations of formed complex I (in which CitI is bound to either O1 or O2) and complex II (in which CitI is bound to both operators).
RESULTS
CitI binds to two operators located in the citI-citM intergenic region.
Our previous results suggested that CitI could directly activate transcription from both its own promoter (PcitI) and the Pcit operon promoter (Fig. 1A). To test the DNA-binding ability of CitI, the protein was purified as described in Materials and Methods and tested in gel mobility shift assays. To this end, the γ-32P-labeled 344-bp amplicon A (Fig. 1A), including the citI-citM intergenic region, was incubated with increasing concentrations of purified CitI. Two CitI-DNA complexes, designated CI and CII, were identified (Fig. 1B), and the binding specificity was confirmed by determining that CitI did not shift an unrelated 367 bp belonging to the B. subtilis des promoter (1) (results not shown). Only the high-mobility complex I was detectable at low concentrations of CitI. A quantification of the levels of bound DNA in each complex is shown in Fig. 1C. A Gaussian response of formation of CI to different protein concentrations was observed, reaching a maximum of 40% of total DNA bound at a CitI concentration of 750 nM, whereas a sigmoid curve of binding of CitI in CII was detected. In addition, the decrease of CI levels was accompanied by an increase of formation of CII, which reached levels containing 93% of total DNA. These binding patterns suggested the existence of two operators for CitI in the citI-citM intergenic region.
To identify the CitI binding sites in the citI-citM intergenic region, DNase I footprinting assays were performed. To favor the formation of CII, 1.25 μM CitI was used in the DNA-binding reaction. As indicated in Fig. 2B (also see Fig. S1 in the supplemental material), on the plus strand, the binding of CitI resulted in protected sites which extend from −178 to −82 positions relative to the cit operon transcriptional start site. On the minus strand, protected sites were included in a region between complementary nucleotides from positions −177 to −80. Interestingly, the footprints appeared to be rather complex, as several hypersensitive sites were found on both strands, indicating local deformations in the DNA helix after protein binding. Due to the extended region protected by CitI, the low resolution of the DNase I footprints did not allow the identification of the sequences recognized by this protein. Thus, to obtain a high-resolution profile of the contacts between CitI and its target DNA, we resorted to hydroxyl radical footprinting analysis. CitI (1.25 μM) was used in the DNA-binding reaction, and, after the Fenton reaction, CII complex was purified and the footprinting pattern identified (Fig. 2) as described in Materials and Methods. Figure 3 shows typical results of the OH cleavage in the absence and presence of CitI for both coding and noncoding strands. The CitI binding generated two protected regions rich in A/T nucleotides. Inspection of the nucleotide sequence in these regions revealed the presence of two almost identical motifs exhibiting dyad symmetry (designated O1 and O2), which could correspond to the operators recognized by CitI (Fig. 2B).
FIG. 2.

Protection of the DNA sequence containing PcitI and Pcit promoters by CitI. (A) Hydroxyl radical footprinting of CitI binding. A 150-bp DNA probe including the DNase I protected region (see Fig. S1 in the supplemental material) was γ-32P labeled in either the plus or the minus strand (see details in Materials and Methods). The assays were performed in the presence (+) or absence (−) of CitI. The footprintings of the plus (left panel) and minus (right panel) strands are depicted, and they correspond to CitI-DNA complex II. Regions protected from the cleaving agent by CitI on the plus strand (A, B, C, and D) and on the minus strand (1 to 4) are indicated. Footprintings are flanked by the corresponding G+A sequence ladders (AG) generated with the same DNA probe (see details in Materials and Methods). (B) Schematic representation of DNA regions protected by CitI. Closed circles and gray bars indicate protections by CitI obtained, respectively, in the hydroxyl radical (A) and DNase I (see Fig. S1 in the supplemental material) footprintings. The inverted repeats identified in the CitI binding sites are shown with arrows. The operator sites identified, named O1 and O2, are enclosed in ovals. The transcriptional start sites (+1) identified in W. paramesenteroides (14) and the putative −10 and −35 boxes of the Pcit and PcitI promoters are depicted.
To confirm the existence of two independent operators for CitI, gel mobility shift assays (Fig. 3) were performed with either the 65-bp amplicon B or the 78-bp amplicon C (Fig. 2A) including, respectively, O1 or O2. Only one complex was detected with each DNA fragment (Fig. 3). Complex formation between CitI and each DNA was quantified as a function of protein concentration, and the curves are depicted in Fig. 3. The data were fitted to an equilibrium binding equation, and the apparent KD and the Hill coefficient for each operator were calculated (see the details in Materials and Methods). Intrinsic KD values of 1.43 ± 0.29 μM (K1) and 0.36 ± 0.07 μM (K2) for O1 and O2, respectively, were obtained. These results show a fourfold-higher affinity of CitI for the operator O2, which is located closer to the Pcit promoter. In addition, Hill coefficients of 3.63 ± 0.07 for both operators indicated a stoichiometry of 4:1 for the protein-DNA complexes.
To analyze a possible cooperative binding of CitI to the two operators, the data depicted in Fig. 2C were used for calculation of the apparent KD values for each operator when they were adjacent in amplicon A (see the details in Materials and Methods). The obtained values, i.e., 0.319 ± 0.06 μM (P1) and 0.194 ± 0.05 μM (P2) for O1 and O2, respectively, together with the above K1 and K2 values, allowed us to determine a cooperativity parameter (ω) of 8.08 (see the details in Materials and Methods), assuming a tetrameric active form for CitI, by use of equation 1.
Reported cooperativity parameter values can range from 1, for noncooperative systems, to values close to 2,000, as described for the binding of the HK022 CI repressor (7), which is a system with one of the highest degrees of cooperativity known for regulatory proteins. Most current estimates of ω are in the range of 50 to 300, although it is not entirely clear whether these values depend markedly on the experimental conditions. Thus, our results indicate the existence of a low but nevertheless significant cooperativity for CitI binding to its operators O1 and O2.
CitI stimulates transcription from cit promoters.
In W. paramesenteroides, the presence of citrate in the growth medium results in increased levels of mRNAs synthesized from PcitI and Pcit promoters, indicating that this compound or some of its metabolites could be implicated in transcriptional stimulation (14). In addition, previous studies performed in E. coli revealed activation of Pcit when CitI was overproduced in trans (14). Moreover, this host comparison of the trans complementation effect of CitI on the β-galactosidase levels encoded by Pcit-lacZ fusions, carrying or not carrying the O1 and O2 operators (see details in Materials and Methods), revealed a decrease of activity from 24,987 ± 271 U A600−1 to 170 ± 20 U A600−1, indicating that the operators are indeed required for CitI transcriptional activation. To directly test whether CitI is a transcriptional activator and whether citrate is the effector of this regulatory mechanism, we performed in vitro runoff transcription assays with the E. coli σ70 holoenzyme RNAP, using as the template amplicon A (Fig. 4A), which contains the divergent PcitI and Pcit promoters, in the presence of purified CitI and citrate. The expected runoff transcripts of 141 nucleotides (nt) and 266 nt, respectively, were detected (Fig. 4B), indicating that E. coli and W. paramesenteroides RNAPs initiate transcription from PcitI and Pcit at the same nucleotides (see Fig. 2B). The E. coli RNAP was able to drive efficient transcription from both promoters in the presence of CitI (Fig. 4B), and the levels of transcription from the Pcit promoter were approximately 70-fold higher than those from PcitI. When citrate was added to the reaction mixtures, the levels of runoff transcripts from both promoters showed increases of 2.5-fold in the presence of CitI (Fig. 4B). As expected, the citrate did not affect RNA synthesis in the absence of the regulator (data not shown). The effect of citrate is highly specific, since its degradation products (lactate, pyruvate, and acetate) and malate (a citrate analogue) were not able to enhance the induction mediated by CitI on transcription from Pcit (Fig. 4C). In addition, the KD values inferred from band shift experiments performed with amplicon A in the presence of citrate (results not shown) revealed that this effector increases by 3.5- and 6-fold the CitI affinity for O1 and for O2, respectively.
FIG. 4.

In vitro transcription from cit promoters. (A) The 595-bp linear DNA template used in the transcription in vitro assays, including the complete citI-citM intergenic region, is depicted. The expected lengths (in nucleotides) of the runoff transcripts from PcitI and Pcit promoters are indicated. The empty rectangles represent O1 and O2 operators. (B) Effect of CitI and citrate on transcription. The runoff transcripts obtained from PcitI (left panel) and from Pcit (right panel) synthesized by the E. coli RNAP in the presence (+) and absence (−) of CitI and citrate are depicted. The positions and lengths of the transcripts generated are indicated. (C) Effect of different citrate-related compounds on CitI activation of transcription. The 266-nt runoff transcripts synthesized from the Pcit promoter in the presence of CitI and the indicated compounds are shown. The in vitro transcriptions shown in panels B and C are flanked by G+A sequence ladders (AG) generated with an unrelated DNA fragment (see details in Materials and Methods).
Binding of RNAP to Pcit is enhanced by CitI.
The above results indicate that a citrate-CitI complex stimulates transcription from cit promoters. To determine whether CitI also increases interactions of RNAP with these promoters and whether citrate enhances such binding, we assayed the formation of ternary complexes with the strong Pcit promoter by use of amplicon D, which carries the promoter and the O2 operator (Fig. 5A), in the absence or in the presence of citrate. As shown in Fig. 5B, RNAP was able to interact with the Pcit promoter to give a stable complex (lanes 2 and 4), and the addition of CitI led to the formation of a ternary complex with lower mobility whose intensity corresponded to that of 20% of the DNA bound to RNAP (lane 5). However, in the presence of citrate, CitI was also able to further stimulate RNAP binding to Pcit and resulted in increases of ternary complex levels up to 78% of DNA bound to RNAP (Fig. 5B, lane 3), even when the presence of citrate seems to affect the stability of the DNA-RNAP complex (Fig. 5B, compare lanes 2 and 4). Moreover, in these experiments, the levels of CitI-DNA complex increased from 7% in the absence of citrate to 24% in its presence (Fig. 5B, lanes 5 and 3). This result was apparently due to the citrate-dependent increase of CitI binding to DNA, since in the presence of the effector a twofold-higher rate of DNA-CitI complex formation was observed in band shift experiments when binding conditions the same as those for ternary complex analysis were used (see Fig. S2 in the supplemental material).
All of these results support the results presented above, in which a stimulatory effect of citrate on transcriptional activation mediated by CitI was observed (Fig. 4B).
DISCUSSION
The metabolism of citrate in bacteria is an adaptive response that requires important changes in the gene expression pattern. Studies of this adaptive response in K. pneumoniae (4-6, 16), B. subtilis (22, 23), and W. paramesenteroides (14 and this work) have shown that the expression of the proteins required for the transport and degradation of citrate is regulated at the transcriptional level. To date, K. pneumoniae CitA, B. subtilis CitT, and W. paramesenteroides CitI have been identified as transcription factors specifically involved in citrate metabolism. However, the mechanisms by which these transcription factors are activated differ greatly. Whereas CitA and CitT are activated by a cognate sensor kinase-mediated phosphorylation, the in vitro transcriptional analysis performed in this work demonstrates that CitI is an activator directly controlled by citrate. Moreover, electrophoretic mobility shift assays revealed that CitI is able to bind cooperatively to the operators O1 and O2 located between the PcitI and Pcit promoters and that citrate increases the affinity of CitI for these and enhances the formation of the Pcit-RNAP-CitI ternary complex. Hydroxyl radical footprintings allowed us to define the DNA sequences of these operators. Each binding site consists of one AT-rich motif exhibiting dyad symmetry. The O2 operator TTTTAAA-AA-TTTAAAA, with perfect matched inverted repeats (centered at position −92 with respect to the cit operon transcriptional start site), corresponds to a high-affinity site, whereas the O1 operator TTTTAAA-AA-TTTGAAC (centered at position −164 with respect to the same transcriptional start site), corresponds to the site recognized with less affinity. In addition, the presence of additional A+T-rich sequences between O1 and O2, with some similarity to these operators, can be observed (Fig. 2B). Therefore, it is possible that additional weak CitI operators located between O1 and O2 exist and, due to the low affinity of CitI for them, they could not be detected by the footprinting analysis of the DNA-CitI complex.
The results presented in this work suggest a scenario for the transcriptional activation of the Pcit and PcitI promoters in W. paramesenteroides. In the absence of citrate, CitI would bind to the operator sites, stimulating the RNAP to form complexes on both promoters. This would result in low levels of expression of the citrate fermentation enzymes as well as of citrate permease P, which catalyzes the uptake of citrate when this compound becomes available in the environment. Once transported inside the cell, citrate would bind to CitI, enhancing the regulator affinity for its DNA operators and resulting in increased RNAP recruitment at Pcit and PcitI. This would result in transcriptional activation from both promoters, as demonstrated here by in vitro transcription experiments. The activation of Pcit results in coordinated synthesis of the citrate fermentation enzymes and the breakdown of citrate. It should be noted that in vivo, the levels of citI and cit mRNAs were about 2.5- and 20-fold higher, respectively, in cultures grown in the presence of citrate. Thus, our model predicts that a small increase in CitI transcription could account for the large increase of the cit mRNA detected in W. paramesenteroides growing in a citrate-supplemented medium. However, our present in vitro experiments showed only threefold induction by citrate of cit RNA synthesis. Thus, it is possible that in the in vivo intracellular environment, additional factors, such as DNA bending (9, 18), might be acting on this system, and it has been demonstrated that histone-like proteins, such as HU, are required for the efficient activation or repression of promoters (2, 8). Further supporting this possibility, the intergenic region containing the citrate promoters has an unusually high AT content (70%), and consequently the DNA in this region could have an intrinsic propensity to bend (20). The contribution of these changes in the DNA bending and the participation of host factors to the transcriptional stimulation remain to be investigated.
CitI is highly conserved in several lactic acid bacteria (Fig. 6B). Moreover, in all the organisms analyzed, the citI gene was found associated with gene clusters involved in the citrate fermentation pathway (Fig. 6B). Similar genetic organizations were found in W. paramesenteroides, Leuconostoc mesenteroides, Oenococcus oeni, and Lactobacillus plantarum, where citI is placed in opposite orientation with respect to the cit operon (Fig. 6B). In these bacteria, two consensus CitI binding operators could be found in the intergenic region between citI and citM (Fig. 6C), suggesting a similar mechanism of transcriptional activation by CitI in conjunction with citrate. Supporting this hypothesis, the induction of expression from the Pcit and PcitI promoters in the presence of citrate has been described for L. mesenteroides (3). These observations indicate that the regulation mechanism detected in W. paramesenteroides is probably conserved in other LAB. Thus, CitI seems to play a pivotal role in citrate sensing and in the transcriptional stimulation of the operons involved in the transport and metabolism of citrate in LAB. However, the different genetic organization found in Lactococcus lactis (Fig. 6A) could reflect a different regulatory model. In accordance with this hypothesis, the induction of the cit operon in this bacterium occurs at low pH independently of the presence of citrate (15). In addition, only operator O1 is present in the intergenic region between citI and citC. In this region, only one promoter driving the expression of citI has been described (15), and the role of CitI in this microorganism is under investigation.
FIG. 6.

The cit clusters and putative CitI operators are present in different lactic acid bacteria. (A) Schematic representation of citrate utilization clusters in W. paramesenteroides, L. mesenteroides, Lactobacillus plantarum, Oenococcus oeni, and Lactococcus lactis. Arrows indicate the Pcit and PcitI promoters. The open squares represent the putative CitI operators proposed for the different organisms. (B) Nucleotide sequences of the putative CitI binding boxes identified in the organisms listed above. The consensus sequences proposed for O1 and O2 are indicated in boldface. The numbers of nucleotides between O2 and translation start codons (ATG) and between operators are indicated.
Supplementary Material
Acknowledgments
We thank M. A. Corrales and M. Arevalo for technical assistance. We thank Steve Elson for the critical reading of the manuscript.
This work was supported by the European Union grant QLKI-2002-02388, Consejo Superior de Investigaciones Científica (CSIC, Spain), and Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, Argentina) exchange program grant 2003AR0001.
The work at the CIB was performed under the auspices of the CSIC. The work at the IBR was performed under the auspices of the CONICET, Fundación Antochas, ANPCyT (PICT2000). D.D.M. is career investigator of CONICET and an international research scholar at the Howard Hughes Medical Institute.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Aguilar, P. S., A. M. Hernandez-Arriaga, L. E. Cybulski, A. C. Erazo, and D. de Mendoza. 2001. Molecular basis of thermosensing: a two-component signal transduction thermometer in Bacillus subtilis. EMBO J. 20:1681-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aki, T., H. E. Choy, and S. Adhya. 1996. Histone-like protein HU as a specific transcriptional regulator: co-factor role in repression of gal transcription by GAL repressor. Genes Cells 1:179-188. [DOI] [PubMed] [Google Scholar]
- 3.Bekal, S., C. Diviés, and H. Prévost. 1999. Genetic organization of the citCDEF locus and identification of mae and clyR genes from Leuconostoc mesenteroides. J. Bacteriol. 181:4411-4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bott, M., and P. Dimroth. 1994. Klebsiella pneumoniae genes for citrate lyase and citrate lyase ligase: localization, sequencing, and expression. Mol. Microbiol. 14:347-356. [DOI] [PubMed] [Google Scholar]
- 5.Bott, M., M. Meyer, and P. Dimroth. 1995. Regulation of anaerobic citrate metabolism in Klebsiella pneumoniae. Mol. Microbiol. 18:533-546. [DOI] [PubMed] [Google Scholar]
- 6.Bott, M. 1997. Anaerobic citrate metabolism and its regulation in enterobacteria. Arch. Microbiol. 167:78-88. [PubMed] [Google Scholar]
- 7.Carlson, N. G., and J. W. Little. 1993. Highly cooperative DNA binding by the coliphage HK022 repressor. J. Mol. Biol. 230:1108-1130. [DOI] [PubMed] [Google Scholar]
- 8.Carmona, M., and B. Magasanik. 1996. Activation of transcription at sigma 54-dependent promoters on linear templates requires intrinsic or induced bending of the DNA. J. Mol. Biol. 261:348-356. [DOI] [PubMed] [Google Scholar]
- 9.Choy, H. E., and S. Adhya. 1992. Control of gal transcription through DNA looping: inhibition of the initial transcribing complex. Proc. Natl. Acad. Sci. USA 89:11264-11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Quintans, N., C. Magni, D. de Mendoza, and P. Lopez. 1998. The citrate transport system of Lactococcus lactis subsp. lactis biovar diacetylactis is induced by acid stress. Appl. Environ. Microbiol. 64:850-857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hugenholtz, J. 1993. Citrate metabolism in lactic acid bacteria. FEMS Microbiol. Rev. 12:165-178. [Google Scholar]
- 12.Lolkema, J. S., B. Poolman, and W. N. Konings. 1995. Role of scalar protons in metabolic energy generation in lactic acid bacteria. J. Bioenerg. Biomembr. 27:467-473. [DOI] [PubMed] [Google Scholar]
- 13.Magni, C., D. de Mendoza, W. N. Konings, and J. S. Lolkema. 1999. Mechanism of citrate metabolism in Lactococcus lactis: resistance against lactate toxicity at low pH. J. Bacteriol. 181:1451-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin, M., C. Magni, P. Lopez, and D. de Mendoza. 2000. Transcriptional control of the citrate-inducible citMCDEFGRP operon encoding genes involved in citrate fermentation in Leuconostoc paramesenteroides. J. Bacteriol. 182:3904-3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin, M. G., P. D. Sender, S. Peiru, D. de Mendoza, and C. Magni. 2004. Acid-inducible transcription of the operon encoding the citrate lyase complex of Lactococcus lactis biovar diacetylactis CRL264. J. Bacteriol. 186:5649-5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer, M., P. Dimroth, and M. Bott. 2001. Catabolite repression of the citrate fermentation genes in Klebsiella pneumoniae: evidence for involvement of the cyclic AMP receptor protein. J. Bacteriol. 183:5248-5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 18.Munteanu, M. G., K. Vlahovicek, S. Parthasaraty, I. Simon, and S. Pongor. 1998. Rod models of DNA: sequence-dependent anisotropic elastic modelling of local bending phenomena. Trends Biochem. Sci. 23:341-346. [DOI] [PubMed] [Google Scholar]
- 19.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 20.Shpigelman, E. S., E. N. Trifonov, and A. Bolshoy. 1993. CURVATURE: software for the analysis of curved DNA. Comput. Appl. Biosci. 9:435-440. [DOI] [PubMed] [Google Scholar]
- 21.Tullius, T. D., B. A. Dombrosky, M. E. A. Churchill, and L. Kam. 1987. Hydroxyl radical footprinting: a high resolution method for mapping protein-DNA contacts. Methods Enzymol. 155:537-558. [DOI] [PubMed] [Google Scholar]
- 22.Warner, J. B., B. P. Krom, C. Magni, W. N. Konings, and J. S. Lolkema. 2000. Catabolite repression and induction of the Mg2+-citrate transporter CitM of Bacillus subtilis. J. Bacteriol. 182:6099-6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto, H., M. Murata, and J. Sekiguchi. 2000. The CitST two-component system regulates the expression of the Mg-citrate transporter in Bacillus subtilis. Mol. Microbiol. 37:898-912. [DOI] [PubMed] [Google Scholar]
- 24.Zeng, X., H. H. Saxild, and R. L. Switzer. 2000. Purification and characterization of the DeoR repressor of Bacillus subtilis. J. Bacteriol. 182:1916-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.