Ana Tereza R Vasconcelos
Ana Tereza R Vasconcelos
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
1,
Henrique B Ferreira
Henrique B Ferreira
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Cristiano V Bizarro
Cristiano V Bizarro
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Sandro L Bonatto
Sandro L Bonatto
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
3,
Marcos O Carvalho
Marcos O Carvalho
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Paulo M Pinto
Paulo M Pinto
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Darcy F Almeida
Darcy F Almeida
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
4,
Luiz G P Almeida
Luiz G P Almeida
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
1,
Rosana Almeida
Rosana Almeida
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
5,
Leonardo Alves-Filho
Leonardo Alves-Filho
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Enedina N Assunção
Enedina N Assunção
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
6,
Vasco A C Azevedo
Vasco A C Azevedo
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
7,
Maurício R Bogo
Maurício R Bogo
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
3,
Marcelo M Brigido
Marcelo M Brigido
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
8,
Marcelo Brocchi
Marcelo Brocchi
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
5,9,
Helio A Burity
Helio A Burity
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
10,
Anamaria A Camargo
Anamaria A Camargo
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
11,
Sandro S Camargo
Sandro S Camargo
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
12,
Marta S Carepo
Marta S Carepo
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
13,
Dirce M Carraro
Dirce M Carraro
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
11,
Júlio C de Mattos Cascardo
Júlio C de Mattos Cascardo
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
14,
Luiza A Castro
Luiza A Castro
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Gisele Cavalcanti
Gisele Cavalcanti
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
1,
Gustavo Chemale
Gustavo Chemale
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Rosane G Collevatti
Rosane G Collevatti
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
15,
Cristina W Cunha
Cristina W Cunha
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
16,
Bruno Dallagiovanna
Bruno Dallagiovanna
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
17,
Bibiana P Dambrós
Bibiana P Dambrós
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
18,
Odir A Dellagostin
Odir A Dellagostin
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
16,
Clarissa Falcão
Clarissa Falcão
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
15,
Fabiana Fantinatti-Garboggini
Fabiana Fantinatti-Garboggini
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
9,
Maria S S Felipe
Maria S S Felipe
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
8,
Laurimar Fiorentin
Laurimar Fiorentin
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
19,
Gloria R Franco
Gloria R Franco
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
7,
Nara S A Freitas
Nara S A Freitas
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
20,
Diego Frías
Diego Frías
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
14,
Thalles B Grangeiro
Thalles B Grangeiro
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
21,
Edmundo C Grisard
Edmundo C Grisard
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
18,
Claudia T Guimarães
Claudia T Guimarães
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
22,
Mariangela Hungria
Mariangela Hungria
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
23,
Sílvia N Jardim
Sílvia N Jardim
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
22,
Marco A Krieger
Marco A Krieger
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
17,
Jomar P Laurino
Jomar P Laurino
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
3,
Lucymara F A Lima
Lucymara F A Lima
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
24,
Maryellen I Lopes
Maryellen I Lopes
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
25,
Élgion L S Loreto
Élgion L S Loreto
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
26,
Humberto M F Madeira
Humberto M F Madeira
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
27,
Gilson P Manfio
Gilson P Manfio
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
9,
Andrea Q Maranhão
Andrea Q Maranhão
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
8,
Christyanne T Martinkovics
Christyanne T Martinkovics
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Sílvia R B Medeiros
Sílvia R B Medeiros
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
24,
Miguel A M Moreira
Miguel A M Moreira
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
28,
Márcia Neiva
Márcia Neiva
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
6,
Cicero E Ramalho-Neto
Cicero E Ramalho-Neto
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
29,
Marisa F Nicolás
Marisa F Nicolás
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
23,
Sergio C Oliveira
Sergio C Oliveira
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
7,
Roger F C Paixão
Roger F C Paixão
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
1,
Fábio O Pedrosa
Fábio O Pedrosa
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
30,
Sérgio D J Pena
Sérgio D J Pena
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
7,
Maristela Pereira
Maristela Pereira
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
31,
Lilian Pereira-Ferrari
Lilian Pereira-Ferrari
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
27,
Itamar Piffer
Itamar Piffer
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
19,
Luciano S Pinto
Luciano S Pinto
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
21,
Deise P Potrich
Deise P Potrich
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Anna C M Salim
Anna C M Salim
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
11,
Fabrício R Santos
Fabrício R Santos
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
7,
Renata Schmitt
Renata Schmitt
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
25,
Maria P C Schneider
Maria P C Schneider
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
13,
Augusto Schrank
Augusto Schrank
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Irene S Schrank
Irene S Schrank
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Adriana F Schuck
Adriana F Schuck
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Hector N Seuanez
Hector N Seuanez
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
28,
Denise W Silva
Denise W Silva
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
29,
Rosane Silva
Rosane Silva
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
4,
Sérgio C Silva
Sérgio C Silva
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Célia M A Soares
Célia M A Soares
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
31,
Kelly R L Souza
Kelly R L Souza
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
28,
Rangel C Souza
Rangel C Souza
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
1,
Charley C Staats
Charley C Staats
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Maria B R Steffens
Maria B R Steffens
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
30,
Santuza M R Teixeira
Santuza M R Teixeira
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
7,
Turan P Urmenyi
Turan P Urmenyi
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
4,
Marilene H Vainstein
Marilene H Vainstein
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,
Luciana W Zuccherato
Luciana W Zuccherato
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
7,
Andrew J G Simpson
Andrew J G Simpson
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
32,
Arnaldo Zaha
Arnaldo Zaha
LNCC/MCT, Petrópolis, RJ,1 Centro de Biotecnologia, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,2 Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS,3 Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,4 Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP,5 Universidade Federal do Amazonas, Manaus, AM,6 Universidade Federal de Minas Gerais, Belo Horizonte, MG,7 Universidade de Brasília, Brasília, DF,8 Universidade Estadual de Campinas, Campinas, SP,9 EMBRAPA/Empresa Pernambucana de Pesquisa Agropecuária, IPA, Recife, PE,10 Ludwig Institute for Cancer Research, São Paulo, SP,11 Instituto de Informática, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS,12 Universidade Federal do Pará, Belém, PA,13 Universidade Estadual de Santa Cruz, Ilhéus, BA,14 Universidade Católica de Brasília, Brasília, DF,15 Universidade Federal de Pelotas, Pelotas, RS,16 Instituto de Biologia Molecular do Paraná, Curitiba, PR,17 Universidade Federal de Santa Catarina, Florianópolis, SC,18 CNPSA/EMBRAPA, Concórdia, SC,19 Universidade Federal Rural de Pernambuco, Recife, PE,20 Universidade Federal do Ceará, Fortaleza, CE,21 EMBRAPA Milho e Sorgo, Sete Lagoas, MG,22 EMBRAPA Soja, Londrina, PR,23 Universidade Federal do Rio Grande do Norte, Natal, RN,24 Instituto Nacional de Pesquisas da Amazônia, Manaus, AM,25 Universidade Federal de Santa Maria, Santa Maria, RS,26 Pontifícia Universidade Católica do Paraná, São José dos Pinhais, PR,27 Instituto Nacional de Cāncer, Rio de Janeiro, RJ,28 Universidade Federal de Alagoas, Maceió, AL,29 Universidade Federal do Paraná, Curitiba, PR,30 Universidade Federal de Goiás, Goiānia, GO, Brazil,31 Ludwig Institute for Cancer Research, New York, New York32
2,*






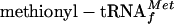 . The physiological consequences, if any, to bacterial growth behavior remain to be studied. Interestingly, disruption of the fmt gene severely impairs growth of Escherichia coli (23). This could be related to the higher binding affinity of IF-2 to
. The physiological consequences, if any, to bacterial growth behavior remain to be studied. Interestingly, disruption of the fmt gene severely impairs growth of Escherichia coli (23). This could be related to the higher binding affinity of IF-2 to 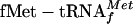 than to
than to 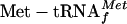 (24). However, a fmt-deficient strain of Pseudomonas aeruginosa can carry out formylation-independent initiation of protein synthesis, conceivably because IF-2 has a dual substrate specificity (38, 53).
(24). However, a fmt-deficient strain of Pseudomonas aeruginosa can carry out formylation-independent initiation of protein synthesis, conceivably because IF-2 has a dual substrate specificity (38, 53).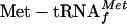 formyltransferase severely impairs growth of Escherichia coli. J. Bacteriol. 174:4294-4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
formyltransferase severely impairs growth of Escherichia coli. J. Bacteriol. 174:4294-4301. [DOI] [PMC free article] [PubMed] [Google Scholar]