

Abstract
Enterococci have emerged as one of the leading causes of nosocomial bloodstream, surgical site, and urinary tract infections. More recently, enterococci have been associated with biofilms, which are bacterial communities attached to a surface and encased in an extracellular polymeric matrix. The enterococcal cell surface-associated protein, Esp, enhances biofilm formation by Enterococcus faecalis in a glucose-dependent manner. Mature Esp consists of a nonrepeat N-terminal domain and a central region made up of two types of tandem repeats followed by a C-terminal membrane-spanning and anchor domain. This study was undertaken to localize the specific domain(s) of Esp that plays a role in Esp-mediated biofilm enhancement. To achieve this objective, we constructed in-frame deletion mutants expressing truncated forms of Esp in an isogenic background. By comparing strains expressing the mutant forms of Esp to those expressing wild-type Esp, we found that the strain expressing Esp lacking the N-terminal domain formed biofilms that were quantitatively less in biovolume than the strain expressing wild-type Esp. Furthermore, an E. faecalis strain expressing only the N-terminal domain of Esp fused to a heterologous protein anchor formed biofilms that were quantitatively similar to those formed by a strain expressing full-length Esp. This suggested that the minimal region contributing to Esp-mediated biofilm enhancement in E. faecalis was confined to the nonrepeat N-terminal domain. Expression of full-length E. faecalis Esp in heterologous host systems of esp-deficient Lactococcus lactis and Enterococcus faecium did not enhance biofilm formation as was observed for E. faecalis. These results suggest that Esp may require interaction with an additional E. faecalis-specific factor(s) to result in biofilm enhancement.
Enterococci have frequently been associated with bacteremia and endocarditis and with infections of the urinary tract, abdominal cavity, pelvis, and soft tissues, especially in the nosocomial setting (13). Although primarily categorized as members of the commensal human microbial flora, enterococci now rank among the leading causes of nosocomial infections (36), with two species, Enterococcus faecalis and Enterococcus faecium, remaining clinically the most significant species by virtue of their association with infection as well as antibiotic resistance dissemination (1, 2, 5, 35).
Biofilms are bacterial communities, single- or multispecies, attached to a surface and surrounded by a self-produced polymeric matrix (11, 23, 27, 50). Biofilms are clinically significant since they exhibit increased resistance to several antimicrobials, thereby making treatment of biofilm-related infections a therapeutic challenge (11). Several chronic infections such as bacterial endocarditis, infectious kidney stones, and lung infections in cystic fibrosis have been attributed to bacterial biofilms (28).
Several E. faecalis factors involved in biofilm formation have been identified, including the cell surface-localized enterococcal surface protein Esp (46, 47), the sugar-binding transcriptional regulator BopD (19), the quorum-sensing locus fsr (16, 32), and the secreted metalloprotease GelE (16, 21, 32). The gene bopD, which putatively encodes a sugar-binding transcriptional regulator, is a member of the bop (biofilm on plastic) locus comprised of four genes, bopA, bopB, bopC, and bopD (19). Seventeen two-component systems and one orphan response regulator have been identified in the genome of E. faecalis strain V583 (31, 38). A study aimed at systematically inactivating all two-component systems present in V583 helped identify the fsr system (34, 44) as the sole two-component system affecting biofilm formation when inactivated (16). It was further found that the fsr system may regulate the process of biofilm formation through the production of a secreted metalloprotease, gelatinase (16, 32).
The esp gene was first detected in a highly virulent gentamicin-resistant clinical isolate, MMH594, that caused a hospital ward outbreak in the mid-1980s (20, 41). It was later found to exist on a large 153-kb pathogenicity island in the same isolate (40). A significant association (P < 0.0001) between the presence of the esp gene and the ability of E. faecalis to form biofilms in vitro was demonstrated earlier (47). In a recent study, although esp was found to be dispensable for biofilm formation by E. faecalis, the presence of esp was associated with an increased ability to form biofilms (24). Our earlier study (46) showed that expression of Esp at the cell surface can significantly enhance biofilm formation by E. faecalis. However, this Esp-mediated effect was only observed in the presence of 0.5% (wt/vol) or greater amounts of glucose for the isolates tested (46).
Esp is a large protein of deduced molecular mass of ∼202 kDa and is cell surface localized. The Esp protein consists of an extensive array of highly conserved repeat blocks designated as A and C repeats, as depicted in Fig. 1A. Owing to homologous recombination between the identical tandem repeat units of esp, enterococcal strains with various numbers of A and C repeats have been isolated. The A repeats located downstream of the N-terminal domain consist of one to three 84-residue repeats specified by nearly identical 252-bp sequences. The C repeats consist of three to nine 82-amino-acid reiterations that are encoded by tandem repeating units of 246 nucleotides. The C repeats are flanked by sequences sharing 50% identity, termed B repeats (69 amino acids). The nonrepeat N-terminal domain is comprised of 694 amino acids and is located downstream of a 49-residue transport signal sequence (41).
FIG. 1.

Construction of N-terminal and A-repeat deletion mutants of Esp. (A) Strategy used to generate pNdel and pAdel plasmid constructs from plasmid pEsp219 (pAT28 harboring the entire esp gene from E. faecalis strain EV219). EspN1 and EspN2a (filled arrows) and EspA1(K) and EspA2(K) (line arrows) primer pairs, designed with KpnI recognition sequence, were used to perform an inverse PCR on the plasmid template pEsp219. The amplified products were restricted with KpnI and self-ligated to generate plasmid constructs pNdel and pAdel, respectively. FA2-2 was transformed with pNdel and pAdel to generate the N-terminal deletion mutant FA2-2(pNdel) and the A-repeat deletion mutant FA2-2(pAdel), respectively. P, promoter region; S, signal sequence; N, N-terminal domain; R, repeat region (consisting of the A and C repeats); and C, C-terminal domain. (B) PCR was performed on the genomic DNA extracted from strains FA2-2(pAT28) (lane 1), FA2-2(pEsp219) (lane 2), FA2-2(pNdel) (lane 3), and FA2-2(pAdel) (lane 4) using oligonucleotide primers VS1 and Esp104. A 1-kb ladder (New England Biolabs, Beverly, MA) was used for size reference (lane M). As expected, amplified products of 5.6 kb, 3.6 kb, and 4.9 kb were observed with FA2-2(pEsp219), FA2-2(pNdel), and FA2-2(pAdel) DNA, respectively. No amplification was observed with the negative control FA2-2(pAT28) DNA.
Due to the presence of multiple domains in the structure of Esp and the identification of Esp as an enhancer of biofilm formation by E. faecalis, it was of interest to determine which element(s) of Esp structure is involved in this process. In the work described here, mutant forms of Esp lacking specific domains were expressed in an isogenic background, and the strains were subsequently assessed for their ability to form biofilms. Employing this approach, we identified the N-terminal domain of Esp as the functional domain sufficient for Esp-mediated biofilm enhancement by E. faecalis.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
E. faecalis strain EV219 is a bacteremia isolate and strong biofilm former and is positive for Esp (our unpublished results). FA2-2 is a plasmid-free, esp-negative E. faecalis strain (4). E. faecium isolate 617 (kindly provided by Mark M. Huycke) and Lactoccus lactis NZ9700 (8) were used as heterologous hosts for Esp expression. Escherichia coli XL1-Blue was obtained from Stratagene (La Jolla, CA) and used as a host for plasmid purifications. Enterococcal strains were cultured in brain heart infusion, or trypticase soy broth (TSB) supplemented with 0.75% glucose. Luria-Bertani broth was used for cultivation of E. coli strains. Antibiotics (Sigma, St. Louis, MO) used for selection of E. faecalis strains included kanamycin (500 μg/ml) for EV219 and rifampin (25 μg/ml) plus fusidic acid (10 μg/ml) for FA2-2. The shuttle vector pAT28 (48) was used as a vector to generate various constructs of Esp. Spectinomycin at 500 μg/ml for E. faecalis and 150 μg/ml for E. coli was used for the selection of strains containing pAT28-based constructs. Custom oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA). Restriction and modifying enzymes were purchased from New England Biolabs Inc. (Beverly, MA). Plasmids were introduced into electrocompetent E. coli or E. faecalis cells using a Gene Pulser unit (Bio-Rad Laboratories, Hercules, CA). Sequence information was obtained using an ABI 3730 capillary sequencer at the Oklahoma Medical Research Foundation (Oklahoma City, OK). All PCR amplifications for cloning purposes were performed using high fidelity Takara LA Taq polymerase (TaKaRa Biomedicals, Shiga, Japan).
Generation of Esp deletion mutants.
The esp gene, together with the 160-bp region upstream of the putative start codon known to be sufficient for the expression of esp, was amplified from the E. faecalis strain EV219 using primers Esp103 and Esp104 containing the recognition sequences for SacI and BamHI, respectively. The amplified product (5.84 kb) was restricted with SacI and BamHI and ligated to SacI/BamHI-restricted shuttle vector pAT28 (48). The generated construct was labeled pEsp219. An inverse PCR (49) approach was used to generate a plasmid construct expressing Esp lacking the nonrepeat N-terminal region. Plasmid pESP219 was used as a template with outward facing primers EspN1 and EspN2a (containing the recognition sequence for the endonuclease KpnI) for the PCR amplification. Due to the possibility of hybridization to complementary sequences within each of the three A repeats, primer EspN2a was designed 54 bp 5′ to the region encoding the A repeats. Primer EspN1 was designed to hybridize at the 3′ end of the predicted signal sequence. The amplified product (10.5 kb) was restricted with KpnI and purified from a 0.8% low-melting-point agarose gel using Wizard Preps DNA purification system (Promega, Madison, WI). The linear, KpnI-restricted fragment was circularized by self-ligation using T4 DNA ligase (Promega, Madison, WI). An aliquot of the ligation mix was used to transform E. coli XL1-Blue, and plasmid DNA was prepared from a single transformant. The plasmid thus generated, pNdel, was sequenced across the KpnI junction site using primer VS1 to ensure an in-frame ligation. Wild-type, Esp-negative E. faecalis strain FA2-2 was transformed with pNdel to generate FA2-2(pNdel) expressing mutant Esp lacking the N-terminal region. To generate a mutant lacking the A-repeat region of Esp, an approach similar to the one above was used. Divergent primers EspA1(K) and EspA2(K) were used for an inverse PCR to amplify an 11.84-kb region from pEsp219. Primer Esp47 was used to verify the nucleotide sequence across the KpnI site of the resulting plasmid, pAdel, to ensure an in-frame ligation. E. faecalis strain FA2-2 was transformed with pAdel to generate FA2-2(pAdel) expressing mutant Esp lacking the A-repeat region.
Construction of Esp N terminus and protein F chimera.
The part of the esp gene encoding the signal sequence and the entire N-terminal domain (GenBank accession number AF034779; corresponding to nucleotide positions 82 to 228 and 229 to 2314, respectively), together with a 160-bp region upstream of the putative translational start site, was amplified from EV219 using primers Esp103 and Esp202R containing the recognition sequences for SacI and XhoI, respectively. The amplified product was restricted with XhoI and subsequently purified from a 0.8% low-melting-point agarose gel. Nucleotide sequence information available for the Streptococcus pyogenes prtF gene and flanking region (GenBank accession number L10919) was used to design primers PrtF-L1 (corresponding to nucleotide positions 845 to 868) and PrtF-R1 (corresponding to nucleotide positions 2405 to 2428) with recognition sequences for XhoI and XbaI, respectively. Primers PrtF-L1 and PrtF-R1 were used to PCR amplify a 1.5-kb region corresponding to a part of the prtF gene from the plasmid pPTF8 (kindly provided by Michael G. Caparon). The amplification product was then restricted with XhoI and purified from a low-melting-point agarose gel. The XhoI-restricted prtF and Esp N-terminal fragments were ligated using T4 DNA ligase at 16°C. One microliter of the ligation mix was used for PCR amplification with primers Esp103 and PrtF-R1 to enrich for the ligated EspN/PrtF1 product. The amplified product was restricted with SacI and XbaI, purified from a low-melting-point agarose gel, and subsequently ligated to SacI/XbaI-restricted pAT28. To confirm an in-frame ligation, nucleotide sequence information was obtained from the generated construct, pPFEN, across the XhoI junction site using primer Esp16.
An 8.6-kb region was amplified from pPFEN using divergent primers EspN1 and EspN2a (containing the recognition sequence for KpnI). The amplified product was restricted with KpnI, purified from a 0.8% low-melting-point agarose gel, and then self-ligated to generate the plasmid pPF1. The plasmid construct, pPF1, thus contained the prtF region from plasmid pPFEN but lacked the region encoding the N-terminal domain of Esp. FA2-2 was transformed with plasmids pPFEN and pPF1 to generate the strains FA2-2(pPFEN) and FA2-2(pPF1), respectively.
Biofilm assay.
The biofilm assay was performed using flat-bottom polystyrene microtiter plates (Corning Inc., Corning, NY) as described earlier (46, 47). Bacterial strains were grown at 37°C for 16 h in TSB medium containing 0.75% glucose, following which the bacterial cells were harvested at 6,000 × g for 10 min, and the cell pellet was resuspended in 5 ml of fresh medium. The optical densities of the bacterial suspensions were normalized using a UV-1201 spectrophotometer (Shimadzu, Kyoto, Japan). Bacterial cultures were diluted 1:40 in fresh TSB medium supplemented with 0.75% glucose and suitable antibiotics. From the diluted culture, 200 μl was dispensed into each of the 12 wells in a single row of a sterile 96-well flat-bottom microtiter plate (Corning Inc., Corning, NY). Following incubation at 37°C for 24 h, the planktonic cells were aspirated, and the wells were washed thrice with sterile phosphate-buffered saline (PBS). The plates were inverted, and the wells were allowed to dry for 1 h at room temperature. For biofilm quantification, 200 μl of 0.2% aqueous crystal violet solution was added to each well, and the plate was allowed to stand for 15 min at room temperature. The wells were subsequently washed thrice with sterile PBS to wash off excess crystal violet. Crystal violet bound to the biofilm was extracted using 200 μl of an 80:20 (vol/vol) mixture of ethyl alcohol and acetone, and the absorbance was measured at 595 nm using an ELX-800 (Bio-Tek Instruments Inc., Winooski, VT) automatic microplate reader. As a control, background crystal violet binding was measured for wells exposed only to the medium with no bacteria, and the background optical density (OD) was subtracted from the OD values of test wells. All biofilm assays were performed thrice independently, with 12 replicates for each strain per assay. Statistical significance between the mean OD at 595 nm (OD595) values was tested using a Student's t test.
Cell surface hydrophobicity determination.
Bacterial cell surface hydrophobicity was measured as previously described (37). Briefly, bacterial strains were grown overnight at 37°C in TSB containing 0.75% glucose. The cultures were subsequently diluted 1:50 in 5 ml of fresh medium. The subcultures were incubated further at 37°C for 4 h. Log-phase bacteria were harvested by centrifugation, washed twice with PUM buffer (22.2 g/liter potassium phosphate trihydrate, 7.26 g/liter monobasic potassium phosphate, 1.8 g/liter urea, 0.2 g/liter magnesium sulfate heptahydrate, pH 7.1), and resuspended in 5 ml of PUM buffer. Two hundred microliters of n-hexadecane was added to a bacterial cell suspension normalized to an OD of 1.0 at 400 nm. The mixtures were incubated at 30°C for 10 min, subsequently vortexed vigorously for 2 min, and then allowed to stand for 15 min at room temperature to ensure complete separation of the organic and aqueous phases. The absorbance of the aqueous layer was measured at 400 nm. The percent cell surface hydrophobicity, which is a measure of the percent of bacterial cells partitioning into the organic phase, was calculated using the following formula: [1 − (final OD400 /initial OD400)] × 100. The assay was performed in triplicate. Statistical significance between the percent cell surface hydrophobicities of the assayed strains was tested using a Student's t test.
CLSM.
Confocal laser scanning microscopy (CLSM) was performed as described earlier (46). Bacterial biofilms were grown in six-well polystyrene tissue culture plates (Corning Inc., Corning, NY) for 24 h in TSB supplemented with 0.75% glucose and antibiotics. For imaging, the cell culture supernatant and planktonic cells were removed by gentle vacuum suction, and the biofilms washed twice with sterile PBS. The biofilms were stained with 100 μl of 1% aqueous acridine orange solution for 10 min and subsequently washed twice with sterile PBS. The biofilms were kept hydrated in 2 ml of PBS during imaging. Confocal images were collected using a Leica TCS NT microscope at a magnification of ×63. An argon laser (488 nm) was used for sample excitation, and the captured images were viewed using the LCS Lite software (ftp://ftp.llt.de/pub/softlib/LCSLite/). The acquired image stacks were quantified using the COMSTAT program (18). Three biofilm parameters were measured using this program: biovolume, average thickness, and maximum thickness. Biovolume is defined as the ratio of the product of the number of biomass pixels and the voxel size [(pixel size)X × (pixel size)Y × (pixel size)Z] to the substratum area and is expressed in μm3/μm2. The average and maximum thicknesses are expressed in microns (μm). In each experiment, these parameters were computed for 10 different image fields per bacterial strain. The experiment was performed twice independently. Statistical significance between the parameters was tested using a Student's t test.
Generation of antibodies to the A-repeat region of Esp.
A 775-bp region corresponding to nucleotides 2311 to 3085 of the esp gene was amplified by PCR using primers 203-F and 204-R with recognition sequences for BamHI and XhoI, respectively (Table 1). The amplified product was restricted with BamHI and XhoI and ligated into BamHI/XhoI-cut expression vector pET29b(+) (Novagen, Madison, WI) containing a C-terminal oligohistidine (His-tag) domain. The A-repeat region was then purified from the expression host E. coli BL21(DE3) using a method described previously for the purification of the N-terminal region of Esp (41). The fusion protein was used as an immunogen to raise polyclonal antibodies in rabbits (Proteintech Group, Inc., Chicago, IL).
TABLE 1.
Oligonucleotides used in this study
| Primer name | Sequencea |
|---|---|
| Esp103 | GAGAGAGCTCGGGATGTTCCAGTGACCCC |
| Esp104 | GAGAGGATCCGAGGAAGAGACTTCTTCCTCTTGT |
| EspN1 | GAGAGGTACCGCATTAACTATTCCAGTTGC |
| EspN2a | GAGAGGTACCTTACCAAGATGGTTCTGTAG |
| EspA1(K) | GAGAGGTACCATTTTTACTTACAGTTACTGC |
| EspA2(K) | GAGAGGTACCTTGGATGATAAATCTGATGCTGAC |
| Esp202R | GAGACTCGAGACTTACAGTTACTGCTAAAT |
| VS1 | GCATAAAAAAATTAAGTGTGGGTGTTGCATCA |
| Esp47 | CCAAGTATACTTAGCATCTTTTGG |
| PrtF-L1 | GAGACTCGAGGATTATGCTCACACTACTAAACTA |
| PrtF-R1 | GAGATCTAGATATTTCTTATGGAACACTTTTACG |
| Esp16 | CAGCTAAAGGTGTAGGAGAATCGGAGCCGAT |
| 203-F | GAGAGGATCCTATTTATGAAAATCCAGGAG |
| 204-R | GAGACTCGAGTTTGTCAGCATCAGATTTAT |
Underlining indicates a recognition site for a restriction endonuclease not present in the template sequence.
ELISA.
An enzyme linked immunosorbent assay (ELISA) was performed as described previously (46). Bacterial cultures were grown overnight in TSB supplemented with 0.75% glucose and appropriate antibiotics. Cells were harvested at 6,000 × g for 10 min, and the cell pellet was washed once with sterile PBS. Following the wash, the cells were resuspended in 5 ml of carbonate-bicarbonate buffer (1.59 g/liter sodium carbonate and 2.92 g/liter sodium bicarbonate, pH 9.6). The optical densities of bacterial suspensions were measured at 600 nm, and cell densities were normalized by appropriately diluting the cultures in sodium carbonate-bicarbonate buffer. The normalized cultures (100 μl) were added to the wells of a 96-well Immulon2 HB plate (Dynex Technologies Inc., Chantilly, VA). The plate was incubated at 4°C for 16 h, following which the wells were washed three times using PBS supplemented with 0.05% Tween-20 to reduce nonspecific binding. A 4% (wt/vol) suspension of nonfat dry milk (Bio-Rad Laboratories, Hercules, CA) in PBS was added to the wells as a blocking agent, and the plate was incubated at 37°C for 2 h. Rabbit polyclonal antibody (specific to either a part of the N-terminal domain of Esp, the A-repeat region of Esp, or the entire Sfb-I protein, depending on the experiment performed) diluted 1:500 was added to each well. The plate was incubated at room temperature for 4 h. Following the incubation, the wells were washed thrice with sterile PBS containing Tween-20. Goat anti-rabbit immunoglobulin G conjugated to alkaline phosphatase (Sigma-Aldrich Co., St. Louis, MO) was used as the secondary antibody, and the plate was incubated at 37°C for 2 h. A 0.1% solution of p-nitrophenylphosphate was added as a substrate for colorimetric detection, and absorbance was measured at 405 nm after 30 min incubation at 37°C.
Expression of Esp in heterologous host systems.
E. faecium strain 617 and L. lactis strain NZ9700 were transformed with pEsp219 to obtain 617(pEsp219) and NZ9700(pEsp219), respectively. The cell surface expression of Esp was confirmed in the transformants by ELISA using antibodies to an internal region of the N-terminal domain of Esp, as described above.
RESULTS
Generation of isogenic mutants lacking the N-terminal or A-repeat region of Esp.
The E. faecalis surface protein, Esp, consists of a large 694-residue N-terminal region and an array of highly conserved tandem repeat units designated A repeats and C repeats (41) (Fig. 1A). In order to assess the contribution of these different domains to the process of biofilm formation, isogenic mutants lacking the N terminus or the A-repeat region of Esp were first generated by transforming wild-type Esp-negative E. faecalis strain FA2-2 with pNdel or pAdel, respectively. Repeated attempts to generate a C-repeat deletion mutant of Esp that could be expressed in FA2-2 failed. Plasmids pNdel and pAdel were constructed using an inverse PCR approach from the plasmid pEsp219 as outlined in Materials and Methods. The strategy used for constructing plasmids pNdel and pAdel is depicted in Fig. 1A. The plasmid pEsp219 encoded the entire Esp protein; pNdel encoded the signal sequence, 18 amino-acids of the N-terminal region, intact A-, B-, and C-repeat regions, and the C-terminal anchor region of Esp; pAdel encoded the signal sequence, the entire N-terminal domain, three amino acids of the A-repeat region, intact B- and C-repeat regions, and the C-terminal region of Esp. PCR with primers VS1 and Esp104 amplified an expected 3.6-kb product from pNdel, confirming the loss of the major part of the N-terminal region. Similarly, as expected, a 4.9-kb region was amplified from pAdel (Fig. 1B).
The cell surface expression of Esp in these mutants was confirmed by ELISA as well as by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blot analysis (data not shown). Antibodies specific to an internal portion of the N-terminal region (corresponding to nucleotides 441 to 2300 of esp) reacted with FA2-2(pEsp219) and FA2-2(pAdel), whereas only antibodies specific to the A-repeat region reacted with FA2-2(pNdel). Cell wall-associated proteins were isolated from FA2-2(pEsp219), FA2-2(pNdel), and FA2-2(pAdel) using lysozyme digestion and were electrophoresed by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis prior to staining with Coomassie brilliant blue R-250 to visualize protein. Proteins of the expected size corresponding to the full-length Esp, Esp lacking the N-terminal domain, and Esp lacking the A-repeat region were detected. Western blot analysis further confirmed the reactivity of the full-length Esp and the mutant Esp proteins with specific antibodies.
All the strains were also compared for growth rates. No significant difference was observed between the strain expressing wild-type Esp and those expressing mutant forms of Esp (data not shown).
Analysis of biofilm formation in the deletion mutants.
Since previous studies have shown that Esp significantly enhances biofilm formation by E. faecalis, it was of interest to determine which elements of Esp structure are essential for this process. Crystal violet binding was used to quantify biofilm formation by Esp-negative vector control FA2-2(pAT28), Esp-positive FA2-2(pEsp219), N-terminal deletion mutant FA2-2(pNdel), and A-repeat deletion mutant FA2-2(pAdel). As shown in Fig. 2, no significant difference between the abilities of FA2-2(pEsp219) and FA2-2(pAdel) to form biofilms was observed (P = 0.169); however, the N-terminal deletion mutant, FA2-2(pNdel), was significantly attenuated in this process compared to FA2-2(pEsp219) and FA2-2(pAdel) (P = 0.0007 and 0.00009, respectively). Moreover, no statistically significant difference was observed between the abilities of FA2-2(pAT28) and FA2-2(pNdel) to form biofilms (P = 0.112).
FIG. 2.

Biofilm formation by Esp-negative FA2-2(pAT28), Esp-positive FA2-2(pEsp219), Esp N-terminal deletion mutant FA2-2(pNdel), and Esp A-repeat deletion mutant FA2-2(pAdel) grown in TSB supplemented with 0.75% glucose. Biofilm formation was quantified by crystal violet staining followed by extraction of bound crystal violet into an ethanol-acetone mixture. The y axis represents the optical densities of dissolved crystal violet measured at 595 nm. The error bars represent means ± standard errors.
CLSM and COMSTAT analyses.
We have previously shown that crystal violet staining does not accurately reflect biofilm volume (46), and, hence, the biofilms formed by FA2-2(pAT28), FA2-2(pEsp219), FA2-2(pNdel), and FA2-2(pAdel) were also characterized using CLSM and COMSTAT analysis of the acquired images. As shown in Fig. 3A, there was a striking difference observed between the biofilms formed by the Esp-positive FA2-2(pEsp219) and the A-repeat deletion mutant FA2-2(pAdel) compared to the N-terminal deletion mutant FA2-2(pNdel). The biofilms formed by FA2-2(pNdel) lacked the dense architectural network of the FA2-2(pEsp219) and FA2-2(pAdel) biofilms and appeared more like those formed by Esp-negative FA2-2(pAT28). COMSTAT analysis further confirmed this observation. As shown in Table 2, the biofilm volume and the average and maximum thicknesses of FA2-2(pNdel) biofilms were significantly less that those of FA2-2(pEsp219) (P ≤ 0.05) and were similar to those of Esp-negative FA2-2(pAT28) (P > 0.05). There were no significant differences observed between FA2-2(pEsp219) and FA2-2(pAdel) biofilms with respect to the biovolume, average thickness, and maximum thickness.
FIG. 3.

Confocal laser scanning micrographs of Esp-negative FA2-2(pAT28), Esp-positive FA2-2(pEsp219), Esp N-terminal deletion mutant FA2-2(pNdel), and Esp A-repeat deletion mutant FA2-2(pAdel) grown in TSB containing 0.75% glucose. The 24-h biofilms were stained with acridine orange and visualized in situ by CLSM. Representative micrographs for each of the analyzed strains are shown: FA2-2(pAT28) (A), FA2-2(pEsp219) (B), FA2-2(pNdel) (C), and FA2-2(pAdel) (D).
TABLE 2.
Quantitative COMSTAT analyses of CLSM image stacks from biofilms of FA2-2 deletion mutants
| Biofilm parameter | Value for strain:
|
|||
|---|---|---|---|---|
| FA2-2(pAT28) | FA2-2(pEsp219) | FA2-2(pNdel) | FA2-2(pAdel) | |
| Biovolumea | 3.647 ± 0.069 | 13.196 ± 2.143 | 3.203 ± 1.379 | 10.72 ± 0.4 |
| Average thickness (μm) | 3.799 ± 0.3 | 16.437 ± 0.951 | 3.089 ± 1.291 | 15.489 ± 0.525 |
| Maximum thickness (μm) | 9 ± 2.1 | 23.775 ± 0.375 | 6.9 ± 0.45 | 25.125 ± 0.525 |
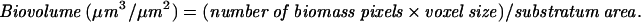
Cell surface hydrophobicity determination.
Bacterial cell surface hydrophobicity has been thought to influence the primary adhesion process in biofilm formation (10). We therefore ascertained cell surface hydrophobicities of FA2-2 expressing wild-type Esp, Esp lacking the N-terminal region, and Esp lacking the A-repeat region. The lack of the A-repeat region in FA2-2(pAdel) did not significantly alter the bacterial cell surface hydrophobicity relative to FA2-2(pEsp219). Although deleting the N-terminal region of Esp in FA2-2(pNdel) resulted in a significant reduction in the cell surface hydrophobicity compared to Esp-positive FA2-2(pEsp219) (P = 0.01), the strain was significantly more hydrophobic than the isogenic Esp-negative vector control, FA2-2(pAT28) (P = 0.02) (Fig. 4). However, both FA2-2(pAT28) and FA2-2(pNdel) were indistinguishable in their abilities to form biofilms as analyzed by crystal violet staining (Fig. 2) as well as by COMSTAT analysis of the CLSM acquired biofilm images (Table 2).
FIG. 4.

Cell surface hydrophobicities of Esp-negative FA2-2(pAT28), Esp-positive FA2-2(pEsp219), Esp N-terminal deletion mutant FA2-2(pNdel), and Esp A-repeat deletion mutant FA2-2(pAdel) grown in TSB containing 0.75% glucose. The bacteria were cultured in TSB containing 0.75% glucose, and the respective cell surface hydrophobicities were measured as described in Materials and Methods. The error bars represent means ± standard errors. Deletion of the N-terminal region of Esp resulted in a significant decrease in the cell surface hydrophobicity, whereas no effect was observed on deleting the A-repeat region. Nevertheless, the cell surface hydrophobicity of FA2-2(pNdel) was significantly greater than FA2-2(pAT28).
Generation of chimeric Esp N terminus and PrtF protein and analysis of biofilm formation by E. faecalis expressing the chimeric protein.
In order to confirm that the attenuation in the ability of the N-terminal deletion mutant FA2-2(pNdel) to form biofilms was not due to a disruption in the global conformation of the Esp protein, the N-terminal region of Esp was anchored to the cell surface of E. faecalis using a heterologous protein anchor, and the strain expressing the chimeric protein was then assessed for biofilm formation. Consistent with other surface proteins of gram-positive bacteria, PrtF, a fibronectin-binding protein identified in S. pyogenes (17) possesses the conserved LPXTG motif (39) in its C-terminal region. To generate a chimeric Esp N terminus and PrtF surface protein, we utilized the LPXTG motif of PrtF to anchor the N-terminal domain of Esp to the bacterial cell wall. Essentially, the part of the esp gene encoding the entire nonrepeat N terminus of mature Esp (referred to as espN and EspN hereafter) was ligated in-frame to a part of the prtF gene corresponding to a portion of the N-terminal domain, upstream fibronectin binding domain, the RD2 domain, and the C-terminal domain of PrtF (referred to as prtF1 and PrtF1 hereafter) as shown in Fig. 5. The espN/prtF1 fragment was then ligated to plasmid pAT28 to generate pPFEN as described in Materials and Methods. Plasmid pPF1 generated from pPFEN served as a control expressing PrtF1 but not EspN.
FIG. 5.

Schematic of the strategy used to generate a plasmid construct expressing a chimeric Esp N terminus and PrtF protein. The N-terminal region of esp with its signal sequence and putative promoter was amplified using primers Esp103 and Esp202R and ligated in-frame to the PrtF-L1 and PrtF-R1 amplified region of prtF. The ligated product was then cloned into the SacI/XbaI-restricted shuttle vector pAT28 to generate a plasmid pPFEN. P, promoter region; S, signal sequence; N, N-terminal domain; R, repeat region; C, C-terminal domain; and F, upstream fibronectin binding domain and RD2 domains.
The expression of the fusion EspN/PrtF1 protein in FA2-2(pPFEN) as well as PrtF1 alone in the control strain FA2-2(pPF1) was confirmed by ELISA employing antibodies to the S. pyogenes protein, SfbI, which is an allelic variant of PrtF (25). Thus, antibodies to Sfb-I (kindly provided by Gursharan S. Chhatwal) cross-react extensively with PrtF. FA2-2 expressing the EspN/PrtF1 fusion protein, FA2-2(pPFEN), was then assessed for its ability to form biofilms by crystal violet staining. As shown in Fig. 6, there was no statistically significant difference between the abilities of Esp-positive FA2-2(pEsp219) and EspN/PrtF1-expressing FA2-2(pPFEN) (P = 0.77) to form biofilms. Also, no significant difference was observed between the abilities of Esp-negative FA2-2(pAT28) and PrtF1-expressing control FA2-2(pPF1) (P = 0.88) to form biofilms, confirming that the enhanced biofilm forming ability of FA2-2(pPFEN) was specifically due to EspN and not due to PrtF1. The abilities of FA2-2(pPFEN) and FA2-2(pEsp219) to form biofilms were thus indistinguishable, implying that Esp mediates biofilm formation through its N-terminal domain.
FIG. 6.

Biofilm formation by Esp-negative FA2-2(pAT28), Esp-positive FA2-2(pEsp219), Esp N-terminal deletion mutant FA2-2(pNdel), PrtF1-expressing control strain FA2-2(pPF1), and EspN/PrtF1-expressing FA2-2(pPFEN) strain grown in TSB supplemented with 0.75% glucose. Biofilm formation was quantified by crystal violet staining. The y axis represents the optical densities of dissolved crystal violet measured at 595 nm. The error bars represent means ± standard errors.
Expression of Esp in heterologous systems.
In order to investigate the effect of Esp expression in a heterologous species and genus, respectively, Esp was expressed in Esp-negative E. faecium strain 617 (kindly provided by Mark M. Huycke) and Esp-negative L. lactis strain NZ9700 (8). Compared to the isogenic Esp-negative controls, strains 617 and NZ9700, there was no statistically significant difference in the abilities of 617(pEsp219) and NZ9700(pEsp219) to form biofilms (P > 0.05) at either of the glucose concentrations tested, as assessed by crystal violet staining (Fig. 7). Since strain NZ9700 was routinely grown in medium supplemented with 0.5% glucose, the effect of Esp on the ability of this strain to form biofilms was tested only in the presence of 0.5%, 0.75%, and 1% glucose. Thus, the expression of Esp at the cell surface of both E. faecium as well as L. lactis did not enhance their abilities to form biofilms as was observed in E. faecalis (Fig. 2 and 3), even though Esp was expressed at similar levels in all the strains as assessed by ELISA (data not shown).
FIG. 7.

Effect of Esp on biofilm formation when expressed in heterologous hosts. E. faecium strain 617 representing heterologous species (A) and L. lactis strain NZ9700 representing a heterologous genus (B) were transformed with pEsp219 to generate Esp-positive strains NZ9700(pEsp219) and 617(pEsp219). The expression of Esp at the cell surface was confirmed by ELISA. Esp did not enhance biofilm formation by strains 617 and NZ9700 under different glucose conditions tested.
DISCUSSION
Biofilms, which have been defined as bacterial communities attached to a surface and encased in a sticky extracellular matrix, have gained considerable clinical importance because of their ability to resist antibiotics (11). The first step in biofilm formation is attachment of the bacterial cells to a surface, which may either be inanimate, such as an implanted medical device, or host tissue such as tooth enamel, heart valves, lung, etc. The subsequent steps of biofilm development include the production of extracellular matrix and finally growth and maturation (9).
Enterococci have been frequently associated with biofilms formed on various indwelling medical devices such as prosthetic heart valves, intrauterine devices, artificial hip prostheses, urinary catheters, and central venous catheters (10). Novel factors involved in the process of enterococcal biofilm formation that are regulatory (Fsr and BopD) (19, 32), cell-wall anchored (Esp) (46, 47), or secreted (GelE) (16, 21) have been identified. It is now well appreciated that biofilm development and maturation are the result of a complex interplay of a number of factors. However, the regulatory network as well as the precise role that each of the identified factors plays in the process of biofilm formation remains to be elucidated.
The vast majority of gram-positive cell wall-anchored proteins perform a multitude of functions that are crucial for the survival or persistence of these microorganisms (26). These include functions such as binding to extracellular matrix proteins like fibronectin, collagen (17, 30, 42), immunoglobulin (12), and some surface proteins, such as β-N-acetyl glucosaminidase from Streptococcus pneumoniae, even possess enzymatic activities (3). A few of these surface proteins have also been found to contribute to biofilm formation. Surface proteins that bind to the extracellular matrix components of the host, collectively called microbial surface components recognizing adhesive matrix molecules (29), mediate bacterial attachment, which is the first step in biofilm formation, further leading to chronic infections (15). The Centers for Disease Control and Prevention estimates that 65% of bacterial infections in humans are associated with biofilms (33). Another surface protein, biofilm-associated protein (Bap), which shares global similarity with Esp, has been implicated in biofilm formation by Staphylococcus aureus isolated from chronic mastitis infections (6). Interestingly, Bap was recently found to interfere with adherence mediated by microbial surface components recognizing adhesive matrix molecules to host tissues of S. aureus (7). Surface proteins can thus play differential, but crucial, roles in biofilm formation depending on the environmental conditions that the bacteria encounter.
The domain organization of many of these surface proteins found in gram-positive bacteria is similar: presence of a carboxyl terminus that spans the cell wall and anchors the protein to the cell wall, a central repeat domain that may confer an extended structure to the protein core, and a nonrepeat amino terminus. In agreement with this theme of domain organization of gram-positive surface proteins, Esp consists of four major structural domains: a nonrepeat N-terminal region, more centrally located A repeats and C repeats, and a C-terminal anchor region (Fig. 1A) (41). Owing to the possibility of the different domains of Esp playing variable roles in the process of biofilm formation, this study was undertaken to understand the structure-function correlation of Esp in the process of biofilm formation.
Using a deletion analysis approach, we have demonstrated that relative to the wild-type Esp-positive strain, the isogenic mutant lacking the N-terminal region of Esp was significantly attenuated in its ability to form biofilms. Moreover the N-terminal deletion mutant formed biofilms that are structurally (Fig. 3) as well as quantitatively (Fig. 2 and Table 2) similar to those formed by the strain lacking Esp. A potential confounding effect of deleting an entire domain from a protein is the possible consequential global misfolding of the protein. Therefore, we also used an in-frame gene fusion strategy to construct a hybrid Esp N terminus and protein F construct that was generated by an in-frame ligation of the N terminus of Esp to the C-terminal region of S. pyogenes surface protein PrtF. Using this construct, we were able to show that the N-terminal domain of Esp by itself was sufficient to confer on the transformed cells an ability to form biofilms that was quantitatively similar to that of the cells expressing the entire Esp protein. The biofilm formed by the A-repeat deletion mutant, however, was similar to that of the wild-type Esp-positive strain. Deleting the 252-amino-acid A-repeat region from Esp did not alter the ability of the mutant strain to form biofilms. Thus, using two approaches, we show here that the nonrepeat N-terminal domain of Esp is involved in Esp-mediated biofilm enhancement in E. faecalis.
Studies in E. coli with certain LacZ fusion proteins have shown that these proteins can assume conformations that can interfere with the protein secretion machinery of the organism, sometimes even proving toxic to the cell (43, 45). Since repeated attempts to express Esp lacking the C-repeat region failed in two different E. faecalis strains, FA2-2 (4) and OG1RF (14), it is possible that expression of such a mutant form of Esp had a similar effect in E. faecalis. Thus, the role of the C-repeat region in the process of biofilm formation remains to be ascertained. Whether the C repeats serve merely to anchor the N-terminal region of Esp in the correct orientation or whether they possess any additional function remains unclear.
A considerable amount of sequence heterogeneity has been observed in the region encoding the nonrepeat N-terminal domain of Esp in E. faecium (22). The existence of sequence variation in the N-terminal domain of Esp from E. faecalis isolates has not yet been investigated. However, nucleotide sequences corresponding to the region encoding the N-terminal domain of Esp derived from EV219 (a strong biofilm former and a strain used in this study) and MMH594 (a weak biofilm former and the prototype strain from which Esp was first detected and characterized) strains were nearly identical (data not shown). Nevertheless, a more comprehensive sequence analysis of Esp from multiple isolates would be required to eliminate the possibility of N-terminal sequence variation accounting for differential biofilm phenotypes among nonisogenic E. faecalis isolates.
Whereas Esp expression in E. faecalis significantly enhances its ability to form biofilms, expression in a heterologous species, E. faecium (Fig. 7A), as well as in a heterologous genus, L. lactis (Fig. 7B), had no effect on the ability of these strains to form biofilms in the presence of an increasing amount of glucose, as was observed with E. faecalis (Fig. 2). This observation suggests the presence of at least one other factor in E. faecalis that may act synergistically with the N-terminal domain of Esp to enhance biofilm formation in this organism.
Acknowledgments
This work was supported, in part, by Public Health Services grant AI 059673 (N.S.) from the National Institutes of Health.
We thank Michael G. Caparon for providing us with the pPTF8 plasmid construct and Arne Heydorn for the COMSTAT program. We are indebted to Jim Henthorn for assistance with confocal laser scanning microscopy. We also thank Gursharan S. Chhatwal for providing us the anti-SfbI antibodies and Mark M. Huycke for the E. faecium strain 617. We are grateful to Michael S. Gilmore and Phillip S. Coburn for helpful discussions and for critical reading of the manuscript.
REFERENCES
- 1.Chavers, L. S., S. A. Moser, W. H. Benjamin, S. E. Banks, J. R. Steinhauer, A. M. Smith, C. N. Johnson, E. Funkhouser, L. P. Chavers, A. M. Stamm, and K. B. Waites. 2003. Vancomycin-resistant enterococci: 15 years and counting. J. Hosp. Infect. 53:159-171. [DOI] [PubMed] [Google Scholar]
- 2.Clark, N. M., E. Hershberger, M. J. Zervosc, and J. P. Lynch, 3rd. 2003. Antimicrobial resistance among gram-positive organisms in the intensive care unit. Curr. Opin. Crit. Care 9:403-412. [DOI] [PubMed] [Google Scholar]
- 3.Clarke, V. A., N. Platt, and T. D. Butters. 1995. Cloning and expression of the beta-N-acetylglucosaminidase gene from Streptococcus pneumoniae. Generation of truncated enzymes with modified aglycon specificity. J. Biol. Chem. 270:8805-8814. [DOI] [PubMed] [Google Scholar]
- 4.Clewell, D. B., P. K. Tomich, M. C. Gawron-Burke, A. E. Franke, Y. Yagi, and F. Y. An. 1982. Mapping of Streptococcus faecalis plasmids pAD1 and pAD2 and studies relating to transposition of Tn917. J. Bacteriol. 152:1220-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crossley, K. 2001. Long-term care facilities as sources of antibiotic-resistant nosocomial pathogens. Curr. Opin. Infect. Dis. 14:455-459. [DOI] [PubMed] [Google Scholar]
- 6.Cucarella, C., C. Solano, J. Valle, B. Amorena, I. Lasa, and J. R. Penades. 2001. Bap, a Staphylococcus aureus surface protein involved in biofilm formation. J. Bacteriol. 183:2888-2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cucarella, C., M. A. Tormo, E. Knecht, B. Amorena, I. Lasa, T. J. Foster, and J. R. Penades. 2002. Expression of the biofilm-associated protein interferes with host protein receptors of Staphylococcus aureus and alters the infective process. Infect. Immun. 70:3180-3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Ruyter, P. G., O. P. Kuipers, M. M. Beerthuyzen, I. van Alen-Boerrigter, and W. M. de Vos. 1996. Functional analysis of promoters in the nisin gene cluster of Lactococcus lactis. J. Bacteriol. 178:3434-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donlan, R. M. 2001. Biofilm formation: a clinically relevant microbiological process. Clin. Infect. Dis. 33:1387-1392. [DOI] [PubMed] [Google Scholar]
- 10.Donlan, R. M. 2002. Biofilms: microbial life on surfaces. Emerg. Infect. Dis. 8:881-890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donlan, R. M., and J. W. Costerton. 2002. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 15:167-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forsgren, A., and J. Sjoquist. 1966. “Protein A” from S. aureus. I. Pseudo-immune reaction with human gamma-globulin. J. Immunol. 97:822-827. [PubMed] [Google Scholar]
- 13.Gilmore, M. S. (ed.). 2002. The enterococci: pathogenesis, molecular biology, and antibiotic resistance. ASM Press, Washington, D.C.
- 14.Gold, O. G., H. V. Jordan, and J. van Houte. 1975. The prevalence of enterococci in the human mouth and their pathogenicity in animal models. Arch. Oral. Biol. 20:473-477. [DOI] [PubMed] [Google Scholar]
- 15.Gotz, F. 2002. Staphylococcus and biofilms. Mol. Microbiol. 43:1367-1378. [DOI] [PubMed] [Google Scholar]
- 16.Hancock, L. E., and M. Perego. 2004. The Enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J. Bacteriol. 186:5629-5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanski, E., and M. Caparon. 1992. Protein F, a fibronectin-binding protein, is an adhesin of the group A streptococcus Streptococcus pyogenes. Proc. Natl. Acad. Sci. USA 89:6172-6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heydorn, A., A. T. Nielsen, M. Hentzer, C. Sternberg, M. Givskov, B. K. Ersboll, and S. Molin. 2000. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146:2395-2407. [DOI] [PubMed] [Google Scholar]
- 19.Hufnagel, M., S. Koch, R. Creti, L. Baldassarri, and J. Huebner. 2004. A putative sugar-binding transcriptional regulator in a novel gene locus in Enterococcus faecalis contributes to production of biofilm and prolonged bacteremia in mice. J. Infect. Dis. 189:420-430. [DOI] [PubMed] [Google Scholar]
- 20.Huycke, M. M., C. A. Spiegel, and M. S. Gilmore. 1991. Bacteremia caused by hemolytic, high-level gentamicin-resistant Enterococcus faecalis. Antimicrob. Agents Chemother. 35:1626-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kristich, C. J., Y. H. Li, D. G. Cvitkovitch, and G. M. Dunny. 2004. Esp-independent biofilm formation by Enterococcus faecalis. J. Bacteriol. 186:154-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leavis, H., J. Top, N. Shankar, K. Borgen, M. Bonten, J. van Embden, and R. J. Willems. 2004. A novel putative enterococcal pathogenicity island linked to the esp virulence gene of Enterococcus faecium and associated with epidemicity. J. Bacteriol. 186:672-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis, K. 2001. Riddle of biofilm resistance. Antimicrob. Agents Chemother. 45:999-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohamed, J. A., W. Huang, S. R. Nallapareddy, F. Teng, and B. E. Murray. 2004. Influence of origin of isolates, especially endocarditis isolates, and various genes on biofilm formation by Enterococcus faecalis. Infect. Immun. 72:3658-3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molinari, G., and G. S. Chhatwal. 1999. Role played by the fibronectin-binding protein SfbI (protein F1) of Streptococcus pyogenes in bacterial internalization by epithelial cells. J. Infect. Dis. 179:1049-1050. [DOI] [PubMed] [Google Scholar]
- 26.Navarre, W. W., and O. Schneewind. 1999. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 63:174-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Toole, G., H. B. Kaplan, and R. Kolter. 2000. Biofilm formation as microbial development. Annu. Rev. Microbiol. 54:49-79. [DOI] [PubMed] [Google Scholar]
- 28.Parsek, M. R., and P. K. Singh. 2003. Bacterial biofilms: an emerging link to disease pathogenesis. Annu. Rev. Microbiol. 57:677-701. [DOI] [PubMed] [Google Scholar]
- 29.Patti, J. M., B. L. Allen, M. J. McGavin, and M. Hook. 1994. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 48:585-617. [DOI] [PubMed] [Google Scholar]
- 30.Patti, J. M., H. Jonsson, B. Guss, L. M. Switalski, K. Wiberg, M. Lindberg, and M. Hook. 1992. Molecular characterization and expression of a gene encoding a Staphylococcus aureus collagen adhesin. J. Biol. Chem. 267:4766-4772. [PubMed] [Google Scholar]
- 31.Paulsen, I. T., L. Banerjei, G. S. Myers, K. E. Nelson, R. Seshadri, T. D. Read, D. E. Fouts, J. A. Eisen, S. R. Gill, J. F. Heidelberg, H. Tettelin, R. J. Dodson, L. Umayam, L. Brinkac, M. Beanan, S. Daugherty, R. T. DeBoy, S. Durkin, J. Kolonay, R. Madupu, W. Nelson, J. Vamathevan, B. Tran, J. Upton, T. Hansen, J. Shetty, H. Khouri, T. Utterback, D. Radune, K. A. Ketchum, B. A. Dougherty, and C. M. Fraser. 2003. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:2071-2074. [DOI] [PubMed] [Google Scholar]
- 32.Pillai, S. K., G. Sakoulas, G. M. Eliopoulos, R. C. Moellering, Jr., B. E. Murray, and R. T. Inouye. 2004. Effects of glucose on fsr-mediated biofilm formation in Enterococcus faecalis. J. Infect. Dis. 190:967-970. [DOI] [PubMed] [Google Scholar]
- 33.Potera, C. 1999. Forging a link between biofilms and disease. Science 283:1837:1839. [DOI] [PubMed] [Google Scholar]
- 34.Qin, X., K. V. Singh, G. M. Weinstock, and B. E. Murray. 2000. Effects of Enterococcus faecalis fsr genes on production of gelatinase and a serine protease and virulence. Infect. Immun. 68:2579-2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rapp, R. P. 2000. Overview of resistant gram-positive pathogens in the surgical patient. Surg. Infect. 1:39-47. [DOI] [PubMed] [Google Scholar]
- 36.Richards, M. J., J. R. Edwards, D. H. Culver, and R. P. Gaynes. 2000. Nosocomial infections in combined medical-surgical intensive care units in the United States. Infect. Control Hosp. Epidemiol. 21:510-515. [DOI] [PubMed] [Google Scholar]
- 37.Rosenberg, M., A. Perry, E. A. Bayer, D. L. Gutnick, E. Rosenberg, and I. Ofek. 1981. Adherence of Acinetobacter calcoaceticus RAG-1 to human epithelial cells and to hexadecane. Infect. Immun. 33:29-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sahm, D. F., J. Kissinger, M. S. Gilmore, P. R. Murray, R. Mulder, J. Solliday, and B. Clarke. 1989. In vitro susceptibility studies of vancomycin-resistant Enterococcus faecalis. Antimicrob. Agents Chemother. 33:1588-1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneewind, O., D. Mihaylova-Petkov, and P. Model. 1993. Cell wall sorting signals in surface proteins of gram-positive bacteria. EMBO J. 12:4803-4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shankar, N., A. S. Baghdayan, and M. S. Gilmore. 2002. Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature 417:746-750. [DOI] [PubMed] [Google Scholar]
- 41.Shankar, V., A. S. Baghdayan, M. M. Huycke, G. Lindahl, and M. S. Gilmore. 1999. Infection-derived Enterococcus faecalis strains are enriched in esp, a gene encoding a novel surface protein. Infect. Immun. 67:193-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Signas, C., G. Raucci, K. Jonsson, P. E. Lindgren, G. M. Anantharamaiah, M. Hook, and M. Lindberg. 1989. Nucleotide sequence of the gene for a fibronectin-binding protein from Staphylococcus aureus: use of this peptide sequence in the synthesis of biologically active peptides. Proc. Natl. Acad. Sci. USA 86:699-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silhavy, T. J., H. A. Shuman, J. Beckwith, and M. Schwartz. 1977. Use of gene fusions to study outer membrane protein localization in Escherichia coli. Proc. Natl. Acad. Sci. USA 74:5411-5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh, K. V., X. Qin, G. M. Weinstock, and B. E. Murray. 1998. Generation and testing of mutants of Enterococcus faecalis in a mouse peritonitis model. J. Infect. Dis. 178:1416-1420. [DOI] [PubMed] [Google Scholar]
- 45.Snyder, W. B., and T. J. Silhavy. 1995. Beta-galactosidase is inactivated by intermolecular disulfide bonds and is toxic when secreted to the periplasm of Escherichia coli. J. Bacteriol. 177:953-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tendolkar, P. M., A. S. Baghdayan, M. S. Gilmore, and N. Shankar. 2004. Enterococcal surface protein, Esp, enhances biofilm formation by Enterococcus faecalis. Infect. Immun. 72:6032-6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toledo-Arana, A., J. Valle, C. Solano, M. J. Arrizubieta, C. Cucarella, M. Lamata, B. Amorena, J. Leiva, J. R. Penades, and I. Lasa. 2001. The enterococcal surface protein, Esp, is involved in Enterococcus faecalis biofilm formation. Appl. Environ. Microbiol. 67:4538-4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trieu-Cuot, P., C. Carlier, C. Poyart-Salmeron, and P. Courvalin. 1990. A pair of mobilizable shuttle vectors conferring resistance to spectinomycin for molecular cloning in Escherichia coli and in gram-positive bacteria. Nucleic Acids Res. 18:4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Triglia, T., M. G. Peterson, and D. J. Kemp. 1988. A procedure for in vitro amplification of DNA segments that lie outside the boundaries of known sequences. Nucleic Acids Res. 16:8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whitchurch, C. B., T. Tolker-Nielsen, P. C. Ragas, and J. S. Mattick. 2002. Extracellular DNA required for bacterial biofilm formation. Science 295:1487. [DOI] [PubMed] [Google Scholar]