

Abstract
Mitochondria play central roles in cellular metabolism and apoptosis and are a major source of reactive oxygen species (ROS). We investigated the role of ROS and mitochondria in radiation-induced apoptosis in multiple myeloma cells. Two distinct levels of ROS were generated following irradiation: a small increase observed early, and a pronounced late increase, associated with depletion of reduced glutathione (GSH) and collapse of mitochondrial membrane potential (Δψm). Exogenous ROS and caspase-3 induced Δψm drop and cytochrome c release from mitochondria, which could be prevented by molecular (dominant-negative caspase-9) and pharmacologic (zVAD-fmk) caspase inhibitors and overexpression of Bcl-2. Exogenous ROS also induced mitochondrial permeability transition (PT) pore opening and cytochrome c release in isolated mitochondria, which could be blocked by inhibition of PT with cyclosporin A. These results indicate that the late ROS production is associated with increased PT pore opening and decreased Δψm, and GSH, events associated with caspase activation and cytochrome c release.
Keywords: apoptosis, reactive oxygen species, mitochondria, Bcl-2, caspases, cytochrome c, glutathione, ionizing radiation
Abbreviations: cyt c; cytochrome c; CM-H2 CFDA; 2′, 7′-dichlorodihydrofluorescein diacetate; DEVD, acetyl-asp-glu-val-asp; DHE, dihydroethidium; fmk, fluoromethyl ketone; GSH, reduced glutathione; GSSG, oxidized glutathione; H2O2, hydrogen peroxide; IR, ionizing radiation; mAb, monoclonal antibody; PI, propidium iodide; PBS, phosphate-buffered saline; PT, mitochondrial permeability transition; ROS, reactive oxygen species; YVAD, acetyl-Tyr-Val-Ala-Asp; zVAD, Benzyloxycarbonyl-Val-Ala-Asp; Δψm; mitochondrial membrane potential
Introduction
Mitochondria play a central role in regulating apoptosis.1 During the initial phases of apoptosis, a wide variety of cellular signaling pathways are activated by cell death stimuli. These signals could consist of perturbations of redox and energy metabolism, ceramide generation, Ca2+ mobilization, or modulation of expression levels of Bcl-2 family members. Mitochondria sense the catastrophic cellular changes and irreversibly commit cells to apoptosis by releasing death factors into the cytosol, such as cytochrome c (cyt c),1 Smac 2/DIABLO,2 AIF,3 EndoG,4 and Omi/HatrA2.5 The best characterized of these factors is cyt c, which plays a dual role in triggering apoptosis and maintaining the oxidative phosphorylation of mitochondria. Cyt c is a soluble protein located in the mitochondrial intermembrane space, which functions as an electron carrier of the mitochondrial respiration chain between complexes III and IV. Once released into the cytosol, cyt c binds to Apaf-1 to form a complex with caspase-9.6,7 This large complex, termed the apoptosome, which may contain as many as 20 additional proteins, initiates the activation of downstream caspases, which cleave cellular substrates at a specific tetra-peptide sequence on the carboxyl side of aspartate residues.8
Bcl-2 is a proto-oncoprotein that functions at multiple levels to prevent morphological changes of apoptosis induced by diverse death stimuli. It has been suggested that Bcl-2 could prevent apoptosis by regulating the cellular redox potential. Thus, Bcl-2 could act as an antioxidant by increasing the GSH pool or by redistributing GSH to various cellular compartments9 and prevent ROS production, GSH depletion, and cellular damage caused by lipid peroxidation.9–12 However, other reports indicate that Bcl-2 could also function as a pro-oxidant.13 The mechanism of how Bcl-2 regulates cellular redox potential and mitochondrial cyt c release are not clear. Bcl-2 and Bcl-XL regulate the homeostasis of mitochondria by blocking cyt c release from mitochondria and thereby preventing caspase activation.14–16 It was suggested that Bcl-2 could be a general regulator of the mitochondrial homeostasis rather than simply a regulator of apoptosis.16,17 Alternatively, Bcl-2 could regulate the mitochondrial permeability transition (PT) pore opening by opposing the effect of Bax, a component of the PT pore.18 The PT pore opening leading to collapse of mitochondrial inner transmembrane potential (Δψm) was thought to be the irreversible step during the postmitochondrial execution phase of apoptosis.18,19 However, the relationships between the antioxidant properties of Bcl-2 and its protective role in preventing cyt c release and apoptosis have not been well established.
Mitochondria are the center for intracellular free radical oxygen production in the process of oxidative phosphorylation. During this process, 0.4–4% of the oxygen consumed in the cell is reduced to generate superoxide anions, O2−· which could be converted to hydrogen peroxide (H2O2) by mitochondrial superoxide dismutase 2 (SOD2). H2O2 is then converted into H2O by mitochondrial GSH peroxidase at the expense of GSH or it participates in the Fenton reaction, giving rise to other ROS species, such as hydroxyl radicals (OH·), the most damaging species of ROS. Reduced levels of cellular GSH can lead to increased steady-state concentrations of peroxides without an increase in their rate of ROS production, thus causing cellular oxidative stress.12 ROS have been implicated as second messengers in apoptosis,20 although other reports challenge this view.21 Nevertheless, mitochondrial ROS play a clear role in cellular cytotoxicity caused by TNF, Fas, p53, and viruses.22–24 Moreover, expression of SOD2, an antioxidant gene, was able to protect these cells from TNF-induced apoptosis.25 Mice lacking SOD2 show a profound increase in oxidative damage-related diseases and malfunction of mitochondria owing to oxidative damage.26 Although almost half of cancer patients are treated with ionizing radiation (IR), the mechanisms of IR-induced apoptosis with regard to its impact on mitochondria and ROS regulation are not known.
Recently, we reported that genotoxic stress and interferon-induced apoptosis in multiple myeloma cells are characterized by two distinct stages of cyt c release and a positive feedback loop linking caspase activation to cyt c release and mitochondrial dysfunction.27,28 This study further addresses the role played by ROS production in this model, by examining its effect on mitochondrial functions, release of apoptogenic factors such as cyt c, and caspase activation. We show that two distinct levels of ROS are generated following irradiation, with a pronounced, late ROS increase associated with reduced GSH levels, collapse of Δψm, and cyt c depletion from mitochondria, events prevented by molecular and pharmacological caspase inhibitors and Bcl-2.
Results
IR induces apoptosis blocked by ectopic bcl-2 expression
We have previously characterized IR-induced apoptosis in IM-9 cells.27 This study has examined apoptosis in this multiple myeloma cell line and a derivative cell population stably expressing ectopic Bcl-2. Flow cytometric determination of the DNA content of control and irradiated propidium iodide (PI)-stained IM-9 and IM-9/Bcl-2 cells indicated that a sub-G1 cell population appeared at 24 h following IR (Figure 1a), an indication of the DNA loss following its fragmentation to a typical 123-bp DNA ladder.29 Overexpression of Bcl-2 prevented the appearance of this sub-G1 population. The nuclease responsible for this DNA fragmentation in human cells has been identified as the DNA fragmentation factor (DFF) 40, which is activated following caspase-3-mediated cleavage of its inhibitor DFF45.4 Immunoblot analyses indicate, indeed, that DFF45 was proteolytically cleaved to its 35-kDa derivative as early as 4 h after IR (Figure 1b). This cleavage was effectively inhibited by both the broad caspase inhibitor zVAD-fmk and Bcl-2 (Figure 1b). These findings are consistent with our previous report indicating that IR induces activation of caspase-9 and caspase-3 as early as 2 h after IR and that Bcl-2 prevents activation of these caspases in IM-9 cells.
Figure 1.

IR induces apoptosis, which is inhibited by Bcl-2. (a) Flow cytometric assays for DNA content were performed in parental IM-9 and derivative cells expressing ectopic Bcl-2 (Clone 1-1), as described in the ‘Materials and methods’ section. The sub G1 DNA content is indicative of apoptotic cells. (b) IM-9 and IM-9/Bcl-2 cells were irradiated in the presence or absence of the pancaspase inhibitor zVAD-fmk and prepared for immunoblotting for DFF45 as described in the ‘Materials and methods’ section. The DFF45 proteolytic cleavage was examined following irradiation (10 Gy) in the absence or presence of the pharmacologic caspase inhibitor zVAD-fmk and Bcl-2 overexpression. DFF-Δdesignates the truncated form of DFF45
ROS are produced in distinct stages during IR-induced apoptosis
IR produces hydroxyl radicals (OH·) in aqueous solutions by direct radiolytic attack on H2O. These hydroxyl radicals are capable of causing direct oxidative damage to cellular molecules, which result in DNA lesions, protein malfunctions, and lipid membrane peroxidation.10,30 Cells may respond to such damage by growth arrest, cell senescence, DNA repair or, if the damage is too severe to be repaired, undergo apoptosis.31
We examined the production of the two most abundant ROS following irradiation, O2−· and H2O2, using oxidation-sensitive fluorescent probes and flow cytometry.10,11,24 Examining the formation of O2−· anions by dihydroethidium (DHE) staining of IM-9 cells revealed a small early, as well as a significant (three-fold) later increase in superoxide anion levels (Figure 2a). We next determined the levels of H2O2, to which O2−· is converted by SOD, using CM-H2 DCFDA staining.10,11,24 Interestingly, we found that H2O2, similarly to O2−·, was also generated in two distinct phases following IR. First, there was a small but consistent increase, which appeared early and persisted for at least 8 h following irradiation (Figure 2b). In contrast, a more substantial two-fold increase in H2O2 levels was observed at 16 h (Figure 2b), at a time we have previously reported to correspond to a late-stage, massive, mitochondrial cyt c release.27 Most cells displayed a readily detectable increase in H2O2 levels compared to untreated cells by 24 h following IR, with only a few cells remaining in which this increase in H2O2 did not take place. These ROS changes were determined only in the viable cell population (PI-negative and eliminating additional apoptotic cells by gating on a forward-scatter versus side-scatter dot plot), in order to exclude the possible contribution of ROS from necrotic cells (Figure 2c). As expected, exogenous H2O2 (0.1 mM), a well-known membrane permeable oxidant,10 induced a significant accumulation of cellular ROS in IM-9 cells following 5 min of treatment (data not shown). Bcl-2 did not prevent the small, early increase in the levels of O2−· and H2O2, but efficiently blocked the later stage of ROS production (Figure 2a–c). These analyses indicate the presence of constitutive levels of intracellular ROS in exponentially growing cells, which are further increased immediately following IR. A more pronounced increase, contemporaneous with mitochondrial dysfunction, is observed at 16 and 24 h following treatment.
Figure 2.

IR induces distinct phases of ROS production during apoptosis. Control or irradiated (10 Gy) IM-9 or IM-9/Bcl-2 cells were harvested at the indicated times and washed with serum-free medium, then incubated with 5 mM DHE (a) or 1 mM DCFDA (b, c) in PBS at 37°C for 10 min. The cellular fluorescence levels (Fl-1, emission wavelength 620 or 530 nm) were analyzed by flow cytometry, after the cell debris and PI-positive population were gated out. P: percentage of PI-negative cells; x: relative fluorescence. ROS levels, determined by DHE (a) or H2DFCDA (b) staining, are presented as relative fluorescence of DCFDA or DHE. The dot plots shown (c) represent only the viable cell population and are presented gated as relative fluorescence of DCFDA. The mean value of Fl-1 fluorescence was normalized against untreated cells. Data represent mean values from duplicate values from three separate experiments
Late increase of ROS and the reduction of Δψm is inhibited by zVAD-fmk
To determine the molecular determinants of ROS in apoptosis, we next examined whether activated caspases were required for ROS production. Caspase activation was measured following IR in the absence or presence of zVAD-fmk, or of the caspase-1 inhibitor, Ac-YVAD-cho. zVAD-fmk effectively prevented DEVD cleavage activity (Figure 3a) and apoptosis.
Figure 3.
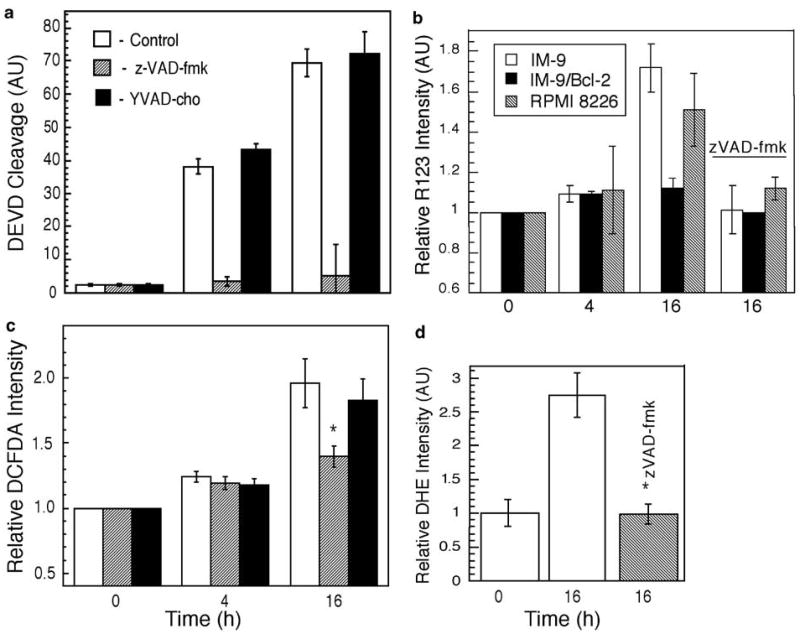
zVAD-fmk blocks DEVD cleavage activity and late ROS increase. (a) IM-9 cells were lysed at the indicated times following irradiation and assayed for caspase-3 activity toward the peptide substrate Ac-DEVD-p-nitroanilide, which was normalized to that of control cells. (b) The effect of IR on Δψm was examined in IM-9 and RPMI-8226 cells, irradiated in the absence or presence of zVAD-fmk. Δψm was measured using R123 as described in the ‘Materials and methods’ section. Data were normalized against untreated cells and represent the mean values of three separate experiments. Cells were stained with 1 μM CM-H2DCFDA (c) or 5 mM DHE (d) and cellular fluorescence levels were analyzed by flow cytometry, similar to Figure 2. The data shown represent the mean values of two duplicate experiments. The values shown are for control, zVAD, and YVAD-treated cells as represented by empty, shaded, and filled symbols, respectively. *P<0.05, as determined by t-test
We have reported that the Δψm reduction could be a consequence of cyt c release and that it is inhibitable by zVAD-fmk and ectopic expression of Bcl-2 in IM-9 cells.27 To further examine the generality of these findings, we determined Δψm in irradiated IM-9, IM-9/Bcl-2, and RPMI 8226 cells in the absence and presence of zVAD-fmk. We found that, similarly to IM-9, IR induced ψm reduction in IR-sensitive RPMI-8226 multiple myeloma cells 16 h after IR, in a zVAD-fmk inhibitable manner (Figure 3b).
Since the late-stage ROS levels temporally followed caspase activation, we next examined the effect of caspase inhibitors on ROS production. Interestingly, zVAD-fmk prevented the late increase of O2−· and H2O2 generation resulting from mitochondrial cyt c depletion, as assayed by CM-H2DCFDA or DHE staining and flow cytometry (Figure 3c, d). In contrast, YVAD-cho, which did not have any effect on caspase activity, could not prevent H2O2 elevation. zVAD-fmk inhibited the increase of ROS even when it was added at 8 h after IR, at the time when caspases were activated but when mitochondrial cyt c levels remained unchanged (data not shown). The inhibition of ROS was not because of the effect of zVAD-fmk itself, since zVAD-fmk had no effect on ROS levels assayed at an early time (4 h) in parental or Bcl-2 overexpressing IM-9 cells. These results are consistent with the observations that IR does not induce caspase activation in cells ectopically expressing Bcl-2 at 24 h. These data further reinforce the notion that the late, but not the early, increase of ROS is associated with caspase activity.
Blocking caspase-9 activation prevents cell death and increased ROS production
We have previously shown that caspase-9 and caspase-3 are activated following the initial, low-level cyt c release into the cytosol.27 Activation of the initiator caspase-9 by cyt c is considered to be a critical event for the activation of the caspase cascade, which may then further amplify cyt c release, thus leading to ROS production and mitochondrial dysfunction. To test this hypothesis directly, we stably overexpressed a dominant-negative (DN) caspase-9 in IM-9 cells and examined its effects on cell death and ROS production. IR-induced apoptosis, as assayed by examining phosphatidyl-serine exposure on the plasma membrane, was significantly inhibited in caspase-9-DN-expressing cells as compared to that observed in parental IM-9 cells 24 h after IR (Figure 4a). Furthermore, the late-phase increase of O2−· levels was completely inhibited, as assayed by DHE staining (Figure 4b). Caspase-9 protein levels were approximately sixfold higher in a representative clone, B-4, stably expressing caspase-9-DN, as compared to those in parental IM-9 cells (Figure 4c). The effects of caspase-9-DN in abrogating IR-induced ROS were comparable with that of Bcl-2. These results strongly suggest a feedback control of O2−· production by a caspase-9-mediated caspase cascade in irradiated IM-9 cells.
Figure 4.

Caspase-9-DN inhibits IR-induced apoptosis and late increase in ROS. (a) IM-9 cells were transfected and selected for expression of caspase-9-DN. Clone B-4, which abundantly expressed caspase-9 and parental IM-9 cells were then used to examine phosphatidylserine exposure, an early marker of apoptosis, by annexin V staining and flow cytometry. (b) Parental IM-9 or derivative cells ectopically expressing Bcl-2 or caspase-9-DN were subjected to DHE staining and flow cytometry at 24 h following irradiation, as described above. The data shown are mean values of three experiments with duplicate samples and are representative of three separate experiments. (c) Level of expression of caspase-9 as determined by immunoblotting with anti-caspase-9, and as a control, anti-β-actin primary antibodies. DN, dominant-negative
Radiation-induced depletion of reduced GSH is inhibited by Bcl-2 and zVAD-fmk
Reduced glutathione (GSH) is a major antioxidant and protects cells from oxidative damage by scavenging peroxides in the cytosol and mitochondria.12 It has been reported that oxidative stress could lead to GSH consumption, which is also a major oxidant signal for apoptosis.12, 32 We therefore examined the intracellular levels of reduced (GSH) and oxidized glutathione (GSSG) in parental and Bcl-2 overexpressing IM-9 cells following IR. By using HPLC to determine the intracellular GSH and GSSG levels, we found that IR caused a 2.2-fold decrease in the levels of GSH as early as 2 h, with almost all GSH being exhausted by 24 h (Table 1). In contrast, the levels of GSSG remained unchanged during this time, indicating that the depletion of GSH is specific. Therefore, the ratio of GSH to GSSG decreased following IR. In parallel experiments, we examined GSH levels in Bcl-2 overexpressing cells. GSH levels in untreated IM-9/Bcl-2 cells were equivalent to those found in parental IM-9 cells. The GSH levels were only slightly decreased at 2 h after IR and were maintained at high levels at 24 h (80–85% of control). These data suggest that depletion of GSH following IR may be because of the increase of ROS, an event associated with caspase activation, and which can be prevented by Bcl-2 overexpression.
Table 1.
Radiation-induced GSH depletion is inhibited by z-VAD-fmk, overexpression of Bcl-2 and DN caspase-9
|
IM-9 |
IM-9/Bcl-2 |
IM-9/Casp9-DN |
||||
|---|---|---|---|---|---|---|
| Time (h) | GSH | GSSG | GSH | GSSG | GSH* | GSSG* |
| 0 | 8.35±2.55 | 0.87±0.18 | 8.72±1.55 | 0.96±0.13 | 9.8 | 0.81 |
| 2 | 3.77±0.57 | 0.78±0.13 | 6.84±0.42 | 0.85±0.15 | 6.53 | 0.84 |
| 6 | 4.30±1.49 | 0.73±0.13 | 7.40±1.45 | 0.90±0.18 | 6.32 | 0.72 |
| 24 | 1.78±0.42 | 0.73±0.18 | 7.45±0.57 | 0.64±0.13 | 5.9 | 0.79 |
| 24+zVAD | 6.10±1.92 | 0.87±0.21 | ND | ND | ND | ND |
Cells were irradiated (10 Gy) and incubated for the durations indicated. Cells were then harvested and GSH and GSSG (nM/mg protein) were assayed by HPLC. Data represent mean values from three separate experiments, unless indicated. *Mean values of two separate experiments.
Since zVAD-fmk may block the late ROS production through inhibition of caspase activity, we further examined the effect of caspases on GSH levels. Caspase inhibition by zVAD-fmk prevented the reduction of GSH levels at 24 h after IR, indicating that caspases were important for the late GSH depletion. These results indicate that the GSH oxidation is an early event following irradiation, which intensifies by 24 h, and that both Bcl-2 and zVAD-fmk prevent the late GSH depletion without affecting the initial decrease of GSH levels. Consistent with the above data, the GSH levels at 24 h following IR were unaffected in IM-9/caspase-9-DN cells (Table 1), further supporting the notion that caspase activation could result in ROS burst and an associated GSH exhaustion. The depletion of GSH was not because of the increase of peroxidase activity, which was unchanged following irradiation in both parental and ectopically expressing Bcl-2 cells (Chen and Almasan, unpublished). Buthionine-l-sulfoximine (BSO; 100 μM), a potent inhibitor of GSH synthesis, completely depleted GSH, but without affecting intracellular ROS levels and cell viability (data not shown). These data suggest that GSH depletion is not responsible for apoptosis induction by IR in IM-9 cells. Rather, GSH depletion may be a downstream event associated with the increase of ROS, most likely mediated by caspase activation.
ROS induce apoptosis, Δψm, and PT pore opening
To determine whether ROS were responsible for cell death, we examined the effect of two well-characterized antioxidants, pyrrolidine-dithiocarbamate (PDTC; 50 mM) and N-acetyl-l-cysteine (NAC; 10 mM), known to be capable of preventing the production of ROS and hence apoptosis.10,11 Both of these reducing agents significantly prevented IR-induced apoptosis (Figure 5a).
Figure 5.
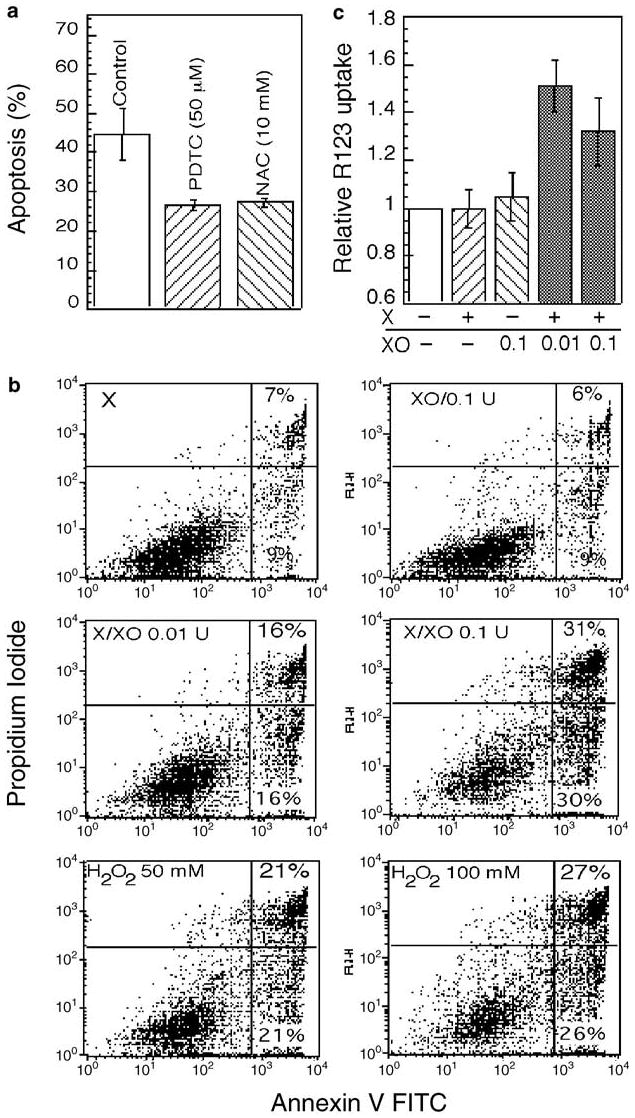
Effect of anti- and pro-oxidants on apoptosis and Δψm. (a) Morphological changes characteristic of apoptosis were determined by DAPI staining in cells pretreated with the antioxidants PDTC (50 μM) and NAC (10 mM) for 1 h before irradiaton (10 Gy). Apoptosis was then examined at 28 h after IR. The data represent mean values from three separate experiments. (b, c) The effect of xanthine (X) and xanthine oxidase (XO) on apoptosis and Δψm was examined in cells treated with X and XO, alone or in combination, or H2O2 for 30 min at 30°C and then subjected to R123 staining (c) or incubated in complete medium for 24 h before annexin V staining and flow cytometry (b)
To mimic the effects of ROS production in mitochondria, we used the reaction of xanthine (X) with xanthine oxidase (XO) to generate O2−·.33 The combination of X (1 mM) with different concentrations of XO (0.01 or 0.1 units) induced apoptosis in a dose-dependent manner, as determined by annexin V staining (Figure 5b). In contrast, X or XO alone had no effect compared to treatment with vehicle alone. H2O2, a known oxidant, also induced annexin V staining when used at concentrations of 50 and 100 mM. ROS generated by the X/XO system appeared to directly impact on mitochondrial PT, since 30 min of this treatment induced a reduction in Δψm, as determined by R123 staining and flow cytometry (Figure 5c).
To examine the effect of ROS on mitochondria directly, we carried out similar assays with isolated mitochondria. ROS generated by the X/XO donor system could induce PT pore opening in mitochondria, as evidenced by the changes in light scatter properties determined by spectrofluorometry (Figure 6a). Moreover, we found that a similar treatment of mitochondria with exogenous ROS induced cyt c release from mitochondria (Figure 6b). Cyt c release was completely inhibited by cyclosporin A (CSA), indicating that PT was critical for cyt c release. The effect of cyclosporin A was specific as it also led, as expected, to the abrogation of the PT decrease caused by ROS (Figure 6a).
Figure 6.
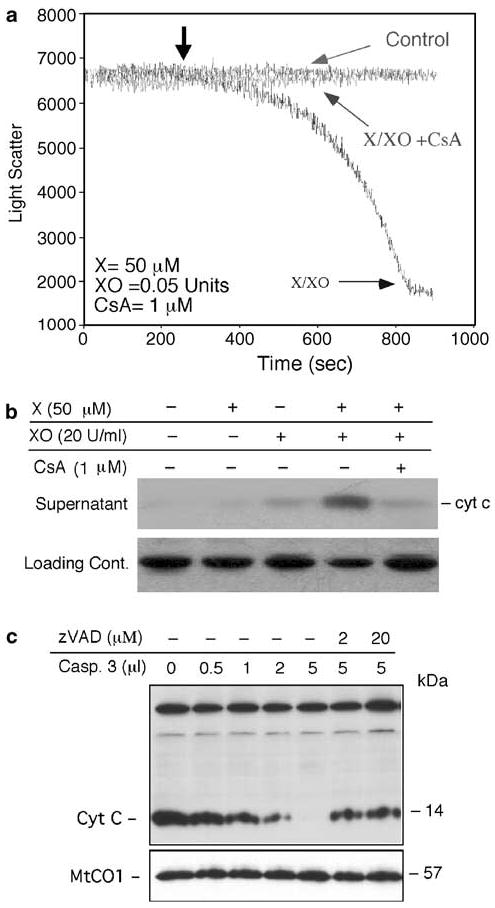
Effect of ROS and caspase-3 on isolated mitochondria. (a) The effect of ROS generation from the reaction of X with XO on PT pore opening was examined in isolated mitochondria from mouse liver, as described in the ‘Materials and Methods’ section. XO was added at the indicated times with X being added as designated by the arrow. Control and CsA represent mitochondria alone or those treated with cyclosporin A (1 μM) in addition to X and XO. (b) The release of cyt c from X/XO-treated mouse liver mitochondria was determined by immunoblotting with an anti-cyt c antibody. Loading control represents a nonspecific antibody-reactive band. (c) P10 mitochondrial fractions from IM-9 cells were incubated with different concentrations of recombinant caspase-3 in the absence, or presence, of the pancaspase inhibitor zVAD-fmk at 30°C for 2.5 h. The reaction mixtures were then pelleted and examined for cyt c levels by immunoblotting with anti-cyt c mAb
To further explore the mechanisms by which zVAD-fmk inhibits cyt c release, and thereby the late increase of ROS production in IM-9 cells, we employed a cell-free system in which isolated mitochondria from untreated IM-9 cells were treated with recombinant caspase-3. Caspase-3 induced depletion of cyt c from mitochondria in a concentration-dependent manner (Figure 6c). In contrast, the levels of another mitochondrial protein, MtCO1, were unchanged, indicating the specificity of cyt c release. zVAD-fmk inhibited the caspase-3-induced cyt c release. These data indicate that exogenous ROS could directly induce apoptosis and mitochondrial functional changes, such as the increased PT pore opening and cyt c release in isolated IM-9 and mouse liver mitochondria. ROS has a profound effect on mitochondria by inducing the opening of the PT pore and loss of Δψm, events associated with cyt c release and caspase activation.
Discussion
This study has found that the late increase of ROS production coincides with cyt c release into the cytosol, both events likely to be mediated by caspase-induced feedback amplification. We have previously identified and characterized two distinct stages of cyt c release into the cytosol during genotoxic stress-induced apoptosis.27 A two-step process for cyt c release has been recently expanded and connected to the loosely and tightly bound pools of cyt c. The latter is attached to the inner mitochondrial membrane by its association with cardiolipin, an interaction that has to be disrupted to generate a soluble pool of this protein.34 A link between ROS production and mitochondrial cyt c depletion during genotoxic stress-induced apoptosis is supported by data from this and our previous study.27 This late cellular ROS increase, associated with mitochondrial cyt c depletion, is inhibited not only by Bcl-2 but also by zVAD-fmk and caspase-9-DN, thus implicating caspases in a positive feedback control of mitochondrial functions, including ROS production. Recombinant caspase-3 induces cyt c release (Figure 6), as well as ROS production and PT pore opening in isolated mitochondria.35 These data, taken together with our previous report,27 suggest that mitochondrial functions, such as reductions of Δψm, ATP, and GSH levels that occur during the late stages of apoptosis, are dependent on caspase activation and cyt c release. These events are prevented by molecular and pharmacological caspase inhibition and by Bcl-2 overexpression.
The regulation of ROS and its role in apoptosis have not been resolved. It has been generally believed that cyt c release from mitochondria could lead to the production of cellular ROS.36,37 Our results, suggesting caspase participation in the late increase of ROS production during apoptosis, reconcile a large body of recent contradictory reports on ROS regulation during apoptosis in different systems.38,39 Kroemer and colleagues40 first connected a protease activation pathway to the mitochondrial phase of apoptosis regulation. Two phases of ROS were also reported in a model of apoptosis induced by glutamate in HT22 cells, with zVAD-fmk also inhibiting ROS production.38 In contrast, a recent report suggests that cyt c release from mitochondria is an ‘all or nothing’ event.41 Our previous results implicating a feedback amplification of cyt c release by caspases27 are not contradictory but rather complementary to this observation. It is possible that only a small proportion of mitochondria within any individual cell open the PT pore following irradiation, resulting in release of cyt c from the affected mitochondria. This small amount of cyt c may be sufficient to activate caspases, which in turn may cause a feedback amplification of cyt c release by inducing ROS production and PT pore opening in neighboring mitochondria. If this is the case, caspases could represent an amplifying signal responsible for the overall cellular mitochondrial dysfunction. It is not known whether other molecules, such as BH3-domain only or multidomain Bcl-2 family proteins,42 are also involved in this amplification process. Interestingly, Bid does not seem to be activated in irradiated IM-9 cells.27 Determining ROS production in individual mitochondria and establishing whether ROS per se is responsible for the PT pore opening and cyt c release in vivo, remain among the technical challenges ahead.
The relationship between ROS and mitochondrial functional readouts, such as cyt c release and Δψm reduction, and other related apoptotic events are not clear. Our examination of mitochondrial apoptotic determinants following IR points to their activation downstream of the early stage, limited cyt c release. Indeed, treatment of isolated mitochondria with caspase-3 directly induces PT pore opening and massive cyt c release, events inhibited by Bcl-xL, cyclosporin A, and z-VAD-fmk.35 It is possible that caspase-3 could directly target the PT pore to induce the massive cyt c release and subsequent apoptosis. Recently, it has been shown that caspase-2 may also cause release of cyt c from mitochondria.43–45 These data are consistent with reports indicating that cyt c could be released either before or after PT pore opening, depending on the cell system and the death stimuli.42 A number of apoptotic stimuli, including ROS, impact on PT pore opening to induce cyt c release and apoptosis. Our data indicate that ROS generated by the X/XO reaction could directly affect Δψm through induction of PT, as suggested.33 PT pore opening could then lead to more cyt c release and a further increase in ROS levels.27,46 It is possible that there is a self-amplifying loop that depends on ROS, cyt c release, caspase activation, and PT.
Understanding the complex functional relationships between the biochemical and metabolic hallmarks for apoptosis, such as cyt c release, caspase activation, ROS production, PT, and GSH depletion, and their regulation by Bcl-2 are important for advancing our knowledge of apoptosis. The redox potential and cellular ROS are known to be influenced by a number of endogenous and exogenous processes and under the control of several radical producing and scavenging enzymes.47 GSH is the major intracellular free radical scavenger and reacts rapidly and nonenzymatically with different types of free radicals during normal metabolic processes and during oxidative stress. One of the consequences of the late ROS increase could be the depletion of GSH, both events being associated with secondary necrosis, a phenomenon known to be associated with the late stages of apoptosis. Sequential cellular changes are characterized by GSH depletion followed by increased ROS production, with these cells manifesting a near-to-simultaneous Δψm dissipation and GSH depletion early during the apoptotic process.32,46 In agreement with these reports, we observed that the GSH levels decreased following IR, becoming depleted 24 h later in IR-sensitive cells, at the time at which PT pore opening and ROS were induced. GSH depletion was inhibited by zVAD-fmk, caspase-9-DN, and Bcl-2. Moreover, BSO could completely deplete GSH without any significant impact on intracellular ROS and cell viability. Taken together, these data suggest that the late GSH depletion is the consequence, at least in part, of caspase-mediated mitochondrial dysfunction and ROS production, although the mechanisms of GSH depletion and the cause of the early GSH consumption remain to be determined.
At least two major functions are attributed to the protective role of Bcl-2 in apoptosis, its regulation of the cellular redox potential and cyt c release from mitochondria. We find that Bcl-2 prevents the late GSH depletion and increased ROS production. Bcl-2 itself is known to be able to increase the intracellular levels of GSH or mobilization in some systems,12 although generalization of these findings may be difficult. However, the mechanism by which Bcl-2 acts to maintain cellular GSH levels is not clear. Bcl-2 overexpression appears not to impact on the initial GSH loss in our system, but rather prevents further loss of GSH at 24 h. In cells expressing high levels of ectopic Bcl-2, in which cyt c release is blocked, there was no significant late-phase change in cellular ROS and GSH levels. Our data strongly indicate that Bcl-2 prevents GSH depletion by blocking cyt c depletion mediated by caspases and the subsequent ROS increase, thus reconciling its dual role in regulating cyt c release and redox potential. This further reinforces the notion that the functions of Bcl-2 in regulating the redox potential, ROS production, and cyt c release from mitochondria are inter-related, and the suppression of cyt c release could be the primary function of Bcl-2. Thus, our results are consistent with a large body of evidence indicating that Bcl-2 prevents caspase activation, cyt c release, GSH depletion, and the associated mitochondrial dysfunction. We extend these observations further to a critical role for Bcl-2 in preventing the late-stage increase in cellular ROS and decrease in GSH levels, likely to be achieved by blocking the caspase-mediated amplification of cyt c release.
In summary, we provide evidence for two distinct phases of ROS generated following irradiation. A pronounced late ROS increase is associated with increased PT pore opening and reduced GSH, and Δψm, all events that can be prevented by molecular and pharmacologic caspase inhibitors and Bcl-2. Exogenous ROS induce PT pore opening and cyt c release directly in isolated mitochondria, indicating that the late ROS production is associated with critical mitochondrial functions, likely to be tightly linked to caspase activation and cyt c release.
Materials and Methods
Cell culture and treatments
IM-9 and RPMI-8226 multiple myeloma cells were obtained from the American Type Culture Collection and cultured at 37°C in a 5% CO2 incubator in RPMI 1640 medium, supplemented with 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin, 100 mg/ml streptomycin, and 2 mM l-glutamine (InVitrogen, Inc.). Exponentially growing cells were subject to γ-irradiation31 or incubated with the indicated amount of xanthine oxidase (XO) in the presence of 1 mM xanthine (X) in phosphate-buffered saline (PBS) and assayed for ROS. The generation of ROS by the X/XO system, as revealed by the luminol assay, showed a burst of ROS immediately after mixing X with XO.
DN caspase-9 and pSFFV-neo FLAG.Bcl-2 (gifts from A Alnemri6 and G Nunez48) were used for stable transfection of IM-9 cells. Individual clones stably expressing different levels of ectopic Bcl-2 and caspase-9-DN were isolated by limited dilution in neomycin (0.7 mg/ml)-containing medium following transfection (1 × 106 cells/ml) using 2 μg DNA and 4 μg DMRIE-C reagent in OPTI-MEM I (Invitrogen, Inc.).
Caspase-3-like protease assay
Cells (3 × 105) were washed with PBS, lysed on ice in 25 mM HEPES (pH 7.4), 0.5% NP-40, 2 mM EDTA, and 1 mM DTT in the presence of a protease inhibitor cocktail. The lysates were centrifuged at 13,000 rpm for 12 min at 4°C and assayed for protease activity using 40 μg of protein and 100 nM of acetyl-Asp-Glu-Val-Asp-p-nitroanilide (Ac-DEVD-p-nitroanilide) substrate.27,31 Samples were incubated at 30°C for 60 min, after which the enzyme-catalyzed release of p-nitroanilide was monitored by measuring optical density at a wavelength of 405 nm using a microplate reader. In some experiments, the cell lysates were first incubated with the caspase inhibitors benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (zVAD-fmk) or acetyl-Tyr-Val-Ala-Asp-cho (YVAD) on ice for 30 min. Caspase peptide inhibitors or substrates were from CalBiochem; all other chemicals were from Sigma Chemicals, unless otherwise specified.
Immunoblot analysis
Cells were washed in PBS and lysed in buffer A containing 150 mM NaCl, 25 mM HEPES pH 7.4, 1% (v/v) NP-40, 0.25% (w/v) sodium deoxycholate, 1 mM EGTA, EDTA, and PMSF, 10 mg/ml aprotinin, leupeptin, and pepstatin. Cellular proteins from total cell lysates (10 μg/sample) were separated by SDS-PAGE and transferred onto a nitrocellulose membrane (Schleicher and Schull). The membranes were probed using immunoblot analyses with mAbs specific to the human caspase-9, DFF45 (Santa Cruz), cyt c (Pharmingen), or Cyt Oxidase I (MTCO1) (Molecular Probes) followed by incubation with the secondary antibody conjugated to horseradish peroxidase for 1 h at room temperature. Equal protein loading was confirmed by staining the filters with Ponceau S and/or reprobing with anti-β-actin mAb (Sigma). Immunoreactive bands were detected using enhanced chemiluminescence (Amersham) and visualized by autoradiography.
Cell fractionation and in vitro cell-free assay for caspase-induced cyt c release
Cells (5 × 106) were washed twice with ice-cold PBS and kept on ice in buffer containing 20 mM HEPES-KOH (pH 7.2), 100 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 250 mM sucrose, and protease inhibitors (1 mM PMSF and DTT, 10 mg/ml aprotinin, leupeptin, and pepstatin). Cells were irradiated at different time points and harvested at the same time for cell fractionation.27 The cell suspension was homogenized with a Dounce homogenizer (5–8 strokes), the homogenates were centrifuged at 750 × g for 5 min, and the supernatants were subjected to further centrifugation at 10 000 × g. The pellets containing mitochondria were designated as P10. The supernatants were then subjected to further ultracentrifugation at 100 000 × g for 45 min. The resulting pellets and supernatants, representing the endoplasmic reticulum and cytosolic fractions, were designated as P-100 and S-100, respectively. Cell fractions were examined by Western blotting for the presence of (MTCOI) or porin (mitochondria), calnexin (ER), and PCNA (nuclei), and no crosscontamination was detected.
The in vitro cell-free assay for caspase-induced cyt c release consisted of the P-10 fraction (30 μl, containing 2 μg/μl protein) mixed with recombinant caspase-3, as indicated, in the presence of 1 mM DTT and incubated at 30°C. In certain experiments, the samples containing caspase inhibitors (100 nM) were preincubated on ice for 30 min. The samples were then centrifuged and the pellets were assayed for cyt c, or as a control MTCO1, by immunoblotting.
Flow cytometric assays
To detect phosphatidylserine exposure on cell membranes, IM-9 cells were stained with FITC-Annexin V, simultaneously with dye exclusion of PI.27 This assay discriminates among intact cells (FITC−/PI−), early apoptotic cells (FITC+/PI−), and late apoptotic or necrotic cells (FITC+/PI+). For DNA analyses, control and irradiated IM-9 and IM-9 FLAG.Bcl-2 cells were fixed in 75% ethanol in PBS. Flow cytometric measurements were performed following addition of PI.29
2′,7′-Dichlorodihydrofluorescein diacetate (CM-H2DCFDA) and dihydroethidium (DHE, Molecular Probes), two oxidation-sensitive fluorescent dyes commonly used for detecting cellular H2O2 or superoxide anion, respectively, were employed for measuring intracellular ROS.10,11,24,46 DCFDA is the most widely used probe for measuring H2O2 levels. It is a cell-permeable, nonfluorescent molecule that provides an extremely sensitive method to follow redox changes over time.47 However, superoxide and NO generation are also capable of oxidizing DCFDA. Moreover, DCF, its oxidized fluorescent derivative, can leak out of cells over time, making difficult the detection of slow H2O2 production. Finally, cellular esterase activity is necessary for conversion of DCFDA to DCFH2, and in cells with low esterase activity DCFDA may underestimate the ROS levels. Nevertheless, use of both DCFDA and DHE provides a good estimate of ROS cellular changes. For determination of H2O2, cells were washed in serum-free medium twice, kept on ice, then stained in PBS (pH 7.2) with CM-H2 DCFDA (1 μM) at 37°C for 5 min, and examined by FACSp with excitation at 488 and emission at 535 nm. PI (to 10 μg/ml) was added 1 min before flow cytometry. For determination of superoxide anions, cells were stained with DHE (5 mM) in PBS (pH 7.2) at 37°C for 15 min and examined by FACS with excitation at 488 and emission at 620 nm. To validate the data, 0.1 mM H2O2 was added 1 h before the staining. Bivariate flow cytometry was performed with a FACS scan, and the data were analyzed with a Cell Quest software (Becton Dickinson, San Jose, CA, USA) from the cell population from which apoptotic cells were gated out against forward and side scatter or PI-positivity. Rhodamine 123 (R123) was used to determine Δψm27 in cells irradiated at different times, but all harvested at the same time, for staining. Cells were incubated with R123 (0.5 mM) for 10 min at 37°C before flow cytometry. To validate the data, m-chlorophenylhydrazone (mCCP; 1 μM) was used after 10 min staining of cells by the above dyes. In parallel experiments, 100 nM nonyl acridine orange (NAO) was used to determine mitochondrial mass.27
Measurement of PT in isolated mitochondria
Mitochondria were isolated from liver of Balb/c mice according to a standard protocol.35 Protein content of mitochondria was determined by the micro-biuret method, using BSA as standard. Mitochondrial swelling was monitored by the decrease of 90° light scatter at 520 nm in the PT-1 medium at 25°C, using a Jobin–Yvon FluoroMax-2 spectrofluorimeter.
Measurement of GSH and GSSG by HPLC
Cells (2 × 106) were washed twice with cold PBS, and 200 μl of 5% cold perchloric acid was added to precipitate proteins. Samples were then kept on ice for 15–20 min and centrifuged at 14 000 rpm for 15 min. The pellets were further used for protein assays. The supernatants were transferred and mixed with 20 μl of 240 mg/ml iodoacetic acid, followed by the addition of powder KHCO3 to neutralize pH. The samples were then kept at 20°C in dark for 30 min before 220 μl of 3% FDNB was added, and samples were analyzed at 365 nm by HPLC for reduced GSH or GSSG levels.
Acknowledgments
We thank Drs. SM Srinivasula, ES Alnemri (Thomas Jefferson University) for the caspase-9-DN, and G. Nunez (University of Michigan) for the pSFFV-FLAG.Bcl-2 constructs, and Ms. C Stanko (Cleveland Clinic Flow Cytometry Core) for help with flow cytometry. This work was supported by research grants from the National Institutes of Health to AA (CA81504 and CA82858) and GMC (HL29582).
References
- 1.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- 2.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 3.Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW, Zuniga-Pflucker JC, Kroemer G, Penninger JM. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 4.Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 5.Li W, Srinivasula SM, Chai J, Li P, Wu JW, Zhang Z, Alnemri ES, Shi Y. Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat Struct Biol. 2002;22:22. doi: 10.1038/nsb795. [DOI] [PubMed] [Google Scholar]
- 6.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 7.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 8.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 9.Voehringer DW, McConkey DJ, McDonnell TJ, Brisbay S, Meyn RE. Bcl-2 expression causes redistribution of glutathione to the nucleus. Proc Natl Acad Sci USA. 1998;95:2956–2960. doi: 10.1073/pnas.95.6.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 11.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 12.Voehringer DW. BCL-2 and glutathione: alterations in cellular redox state that regulate apoptosis sensitivity. Free Radic Biol Med. 1999;27:945–950. doi: 10.1016/s0891-5849(99)00174-4. [DOI] [PubMed] [Google Scholar]
- 13.Steinman HM. The Bcl-2 oncoprotein functions as a pro-oxidant. J Biol Chem. 1995;270:3487–3490. [PubMed] [Google Scholar]
- 14.Kluck R, Bossy-Wetzel E, Green D, Newmeyer D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Liu X, Bhalla K, Kim C, Ibrado A-M, Cai J, Peng T-I, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 16.Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- 17.Vander Heiden MG, Thompson CB. Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nat Cell Biol. 1999;1:E209–216. doi: 10.1038/70237. [DOI] [PubMed] [Google Scholar]
- 18.Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 19.Petit PX, Lecoeur H, Zorn E, Dauguet C, Mignotte B, Gougeon ML. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J Cell Biol. 1995;130:157–167. doi: 10.1083/jcb.130.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slater AF, Nobel CS, Orrenius S. The role of intracellular oxidants in apoptosis. Biochim Biophys Acta. 1995;1271:59–62. doi: 10.1016/0925-4439(95)00010-2. [DOI] [PubMed] [Google Scholar]
- 21.Jacobson MD, Raff MC. Programmed cell death and Bcl-2 protection in very low oxygen. Nature. 1995;374:814–816. doi: 10.1038/374814a0. [DOI] [PubMed] [Google Scholar]
- 22.Goossens V, Grooten J, De Vos K, Fiers W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci USA. 1995;92:8115–8119. doi: 10.1073/pnas.92.18.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li PF, Dietz R, von Harsdorf R. p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apoptosis blocked by Bcl-2. EMBO J. 1999;18:6027–6036. doi: 10.1093/emboj/18.21.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 25.Wong GH, Goeddel DV. Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science. 1988;242:941–944. doi: 10.1126/science.3263703. [DOI] [PubMed] [Google Scholar]
- 26.Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, Miziorko H, Epstein CJ, Wallace DC. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Q, Gong B, Almasan A. Distinct stages of cytochrome c release from mitochondria: evidence for a feedback amplification loop linking caspase activation to mitochondrial dysfunction in genotoxic stress induced apoptosis. Cell Death Differ. 2000;7:227–233. doi: 10.1038/sj.cdd.4400629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Q, Gong B, Mahmoud-Ahmed A, Zhou A, Hsi ED, Hussein M, Almasan A. Apo2L/TRAIL and Bcl-2-related proteins regulate type I interferon- induced apoptosis in multiple myeloma. Blood. 2001;98:2183–2192. doi: 10.1182/blood.v98.7.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Almasan A, Yin Y, Kelly RE, Lee EY, Bradley A, Li W-W, Bertino JR, Wahl GM. Deficiency of retinoblastoma protein leads to inappropriate S-phase entry, activation of E2F-responsive genes, and apoptosis. Proc Natl Acad Sci USA. 1995;92:5436–5440. doi: 10.1073/pnas.92.12.5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haliwell B and Gutteridge JMC (1989) In Free Radicals in Biology and Medicine, 2 ed., Oxford: Clarendon Press, pp 1–81
- 31.Gong B, Chen Q, Endlich B, Mazumder S, Almasan A. Ionizing radiation-induced, Bax-mediated cell death is dependent on activation of serine and cysteine proteases. Cell Growth Differ. 1999;10:491–502. [PubMed] [Google Scholar]
- 32.Armstrong JS, Steinauer KK, Hornung B, Irish JM, Lecane P, Birrell GW, Peehl DM, Knox SJ. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 2002;9:252–263. doi: 10.1038/sj.cdd.4400959. [DOI] [PubMed] [Google Scholar]
- 33.Higuchi M, Honda T, Proske RJ, Yeh ET. Regulation of reactive oxygen species-induced apoptosis and necrosis by caspase 3-like proteases. Oncogene. 1998;17:2753–2760. doi: 10.1038/sj.onc.1202211. [DOI] [PubMed] [Google Scholar]
- 34.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia T, Jiang C, Li L, Wu C, Chen Q, Liu SS. A study on permeability transition pore opening and cytochrome c release from mitochondria, induced by caspase-3 in vitro. FEBS Lett. 2002;510:62–66. doi: 10.1016/s0014-5793(01)03228-8. [DOI] [PubMed] [Google Scholar]
- 36.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai J, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 38.Tan S, Sagara Y, Liu Y, Maher P, Schubert D. The regulation of reactive oxygen species production during programmed cell death. J Cell Biol. 1998;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Green D, Kroemer G. The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol. 1998;8:267–271. doi: 10.1016/s0962-8924(98)01273-2. [DOI] [PubMed] [Google Scholar]
- 40.Susin SA, Zamzami N, Castedo M, Daugas E, Wang HG, Geley S, Fassy F, Reed JC, Kroemer G. The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J Exp Med. 1997;186:25–37. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 42.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 43.Robertson JD, Enoksson M, Suomela M, Zhivotovsky B, Orrenius S. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J Biol Chem. 2002;13:13. doi: 10.1074/jbc.M204185200. [DOI] [PubMed] [Google Scholar]
- 44.Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES. Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J Biol Chem. 2002;277:13430–13437. doi: 10.1074/jbc.M108029200. [DOI] [PubMed] [Google Scholar]
- 45.Lassus P, Opitz-Araya X, Lazebnik Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science. 2002;297:1352–1354. doi: 10.1126/science.1074721. [DOI] [PubMed] [Google Scholar]
- 46.Macho A, Hirsch T, Marzo I, Marchetti P, Dallaporta B, Susin SA, Zamzami N, Kroemer G. Glutathione depletion is an early and calcium elevation is a late event of thymocyte apoptosis. J Immunol. 1997;158:4612–4619. [PubMed] [Google Scholar]
- 47.Curtin JF, Donovan M, Cotter TG. Regulation and measurement of oxidative stress in apoptosis. J Immunol Methods. 2002;265:49–72. doi: 10.1016/s0022-1759(02)00070-4. [DOI] [PubMed] [Google Scholar]
- 48.Simonian PL, Grillot DA, Nunez G. Bcl-2 and Bcl-XL can differentially block chemotherapy-induced cell death. Blood. 1997;90:1208–1216. [PubMed] [Google Scholar]