

Abstract
Integrin-linked kinase (ILK) is a multidomain protein involved in cell motility and cell-extracellular matrix interactions. ILK is found in integrin-containing focal adhesions in undifferentiated primary epidermal keratinocytes. Induction of keratinocyte differentiation by treatment with Ca2+ triggers formation of cell–cell junctions, loss of focal adhesions, and ILK distribution to cell borders. We now show that Ca2+ treatment of keratinocytes induces rapid (≤1 h) translocation to the cell membrane of the adherens junction (AJ) proteins E-cadherin and β-catenin. This is followed by slower (>6 h) localization of tight junction (TJ) proteins. The kinetics of ILK movement toward the cell periphery mimics that of AJ components, suggesting that ILK plays a role in the early formation of cell–cell contacts. Whereas the N terminus in ILK mediates localization to cell borders, expression of an ILK deletion mutant incapable of localizing to the cell membrane (ILK 191-452) interferes with translocation of E-cadherin/β-catenin to cell borders, precluding Ca2+-induced AJ formation. Cells expressing ILK 191-452 also fail to form TJ and sealed cell–cell borders and do not form epithelial sheets. Thus, we have uncovered a novel role for ILK in epithelial cell–cell adhesion, independent of its well-established role in integrin-mediated adhesion and migration.
INTRODUCTION
The formation of tissues during development and regeneration is crucially dependent on the balance of cell migration, cell–cell adhesion, and cell–matrix interactions. Cells generally migrate by creating and disassembling cell–cell and cell-matrix adhesions and by forming connections between these adhesions and their cytoskeleton, thus generating contractile forces required for motion. Central in this process are transmembrane proteins such as integrins and cadherins, which mediate cell–matrix and cell–cell interactions, respectively (Jamora and Fuchs, 2002; Brakebusch and Fassler, 2003; Watt, 2003).
The interactions between integrin-mediated migration/cell-matrix adhesion and cadherin-mediated cell–cell adhesion are complex. In some circumstances, such as during wound healing in tissues and in cultured cells, integrin-mediated cell motility can be opposed by cadherin-based cell–cell adhesion (Ojakian et al., 2001). In other circumstances, such as those seen during epithelial sheet migration during embryogenesis (Martin and Parkhurst, 2004), integrin-mediated cell-matrix adhesion and motility occur in the presence of cadherin-based intercellular adhesion. Recently, the existence of cross-talk pathways between integrin- and cadherin-mediated adhesion has become apparent. For example, focal adhesion kinase and paxillin regulate integrin-mediated adhesion and migration and positively modulate cell–cell adhesion mediated by N-cadherin in HeLa cells (Yano et al., 2004).
Complex epithelia, such as the epidermis, are composed of several cell layers containing distinct adhesion complexes. For example, the lowermost basal cells are linked to the extracellular matrix (ECM) via integrins. The upper suprabasal cells do not contact the matrix, but rather establish various cell–cell adhesion complexes, including desmosomes, adherens junctions (AJ), and tight junctions (TJ). An additional central element in the formation of cell–cell junctions is the actin cytoskeleton (Jamora and Fuchs, 2002; Morita and Miyachi, 2003).
Cell adhesion processes can be modeled in primary cultured epidermal keratinocytes by modulating the extracellular Ca2+ concentration. Cells cultured in <0.1 mM Ca2+ maintain characteristics of basal cells, such as proliferation and capacity to migrate and to establish cell-extracellular matrix contacts via αβ1 and other integrins. The β1 integrins are essential for proper directed and nondirected keratinocyte migration during wound healing, for proper remodeling of actin cables necessary for efficient migration and directed lamellipodial adhesion (Grose et al., 2002; Raghavan et al., 2003). Primary keratinocytes can be induced to differentiate by culture with extracellular Ca2+ concentrations above 0.1 mM (0.1–1.0 mM). Under these conditions, there is induction of terminal differentiation markers, integrin down-regulation, and establishment of strong cell–cell junctions (Dotto, 1999). Mechanistically, Ca2+-induced cell–cell adhesion begins when E-cadherin–β-catenin complexes translocate to the plasma membrane and recruit α-catenin (Bryant and Stow, 2004). These complexes simultaneously interact with actin filaments within the same cell and with E-cadherin complexes in adjacent cells, initiating the formation of AJ. Establishment of AJ is followed by formation of desmosomes, both of which are essential to establish epithelial sheets and cell polarity (Perez-Moreno et al., 2003).
Binding of integrins to extracellular matrix proteins results in recruitment of multiple intracellular proteins that activate signaling cascades and provide links to the actin cytoskeleton, including Integrin-linked kinase (ILK) (Hynes, 2002). ILK is essential for linking integrins to actin fibers, and it is an important modulator of cell migration (Zervas et al., 2001; Mackinnon et al., 2002; Sakai et al., 2003; Terpstra et al., 2003). In keratinocytes, Ca2+ treatment induces marked changes in ILK subcellular localization (Vespa et al., 2003). Specifically, ILK localizes to the cytoplasm and to areas associated with actin fibers and focal adhesions in undifferentiated cells, but it migrates to areas of cell–cell contact in keratinocytes cultured in the presence of ≥0.1 mM Ca2+. ILK localizes to areas of cell–cell adhesion in vicinity to AJ and associated actin fibers, and this pattern of distribution is dependent on an intact actin cytoskeleton. Given that ILK seems to be associated with both focal adhesions and cell–cell junctions, we explored the possibility that ILK plays a role in the formation of epithelial intercellular contacts. We now show that, in addition to its established role in cell–ECM interactions, ILK is an important modulator of Ca2+-dependent adherens and tight junction formation in epithelial cells.
MATERIALS AND METHODS
Cell Culture, Transfection, and Adenovirus Infections
Primary epidermal keratinocytes were isolated from 2-d-old CD-1 mice and cultured in Ca2+-free EMEM (Cambrex Bio Science Rockland, Rockland, ME) supplemented with 8% fetal bovine serum (FBS) previously treated with Chelex resin (Bio-Rad, Hercules, CA) and epidermal growth factor (10 ng/ml, Invitrogen, Carlsbad, CA), as described previously (D'Souza et al., 2001). Under these conditions, the extracellular Ca2+ concentration is ∼0.05 mM. All experiments were conducted 3–6 d after keratinocyte isolation. Formation of cell–cell junctions was induced by increasing the extracellular Ca2+ concentration to 1.0 mM for intervals indicated in individual experiments. For immunofluorescence experiments, keratinocytes were seeded onto HCl-washed glass coverslips incubated with poly-l-lysine (1 mg/ml; 1 h; 22°C) and coated with collagen-1 (5 μg/cm2; overnight; 22°C). IMDF fibroblasts (Apostolova et al., 2002) and HEK293 (American Type Culture Collection, Manassas, VA) cells were cultured in Advance DMEM (Invitrogen) supplemented with 3% FBS. Keratinocytes cultured in 0.05 mM extracellular Ca2+ were transfected with ExGen 500 (Fermentas, Hanover, MD). Junction formation was induced by increasing the extracellular Ca2+ concentration to 1.0 mM for 24 h. IMDF cells were transfected with Lipofectamine 2000 (Invitrogen), following the manufacturer's instructions.
Viral infections were conducted by incubating primary keratinocytes in serum- and Ca2+-free Eagle's minimum essential medium (EMEM) with adenovirus for 5 h, followed by culture in normal growth medium for times indicated in individual experiments. Under these conditions, we determined that ≥95% of primary mouse keratinocytes were infected, as confirmed by green fluorescent protein (GFP) fluorescence or immunofluorescence.
ILK Constructs and Recombinant Adenoviruses
Plasmids encoding human wild-type (wt) ILK and ILK E359K (Novack et al., 1998) were gifts of S. Dedhar (University of British Columbia, Vancouver, British Columbia, Canada). GFP-tagged rat ILK was a gift from C. Turner (SUNY Upstate Medical University, Syracuse, NY). Human ILK differs from the rat orthologue only in residues 197 (T in human, A in rat) and 259 (S in human, A in rat). Deletion and point mutant ILK cDNAs were generated by PCR. All ILK forms contain a V5 tag to allow detection. Expression vectors encoding AU1- or V5-tagged PINCH were generated by PCR, using as template a human expressed sequence tag (GenBank accession no. BE892419). All constructs were verified by dideoxy sequencing. Recombinant adenoviruses encoding GFP together with wt ILK or ILK E359K have been described previously (Vespa et al., 2003). The adenovirus encoding V5-tagged ILK 191-452 was generated using the AdEasy system (He et al., 1998). Viral stocks were purified through CsCl gradient centrifugation and titered by dilution assay in human embryonic kidney (HEK)293 cells and keratinocytes.
Antibodies and Chemicals
The mouse monoclonal anti-V5 antibody was from Invitrogen. Rabbit polyclonal anti-occludin and mouse monoclonal anti-cyclin D1 antibodies were purchased, respectively, from Zymed Laboratories (South San Francisco, CA) and Cell Signaling Technology (Beverly, MA). Mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and rabbit polyclonal anti-GFP were from Biogenesis and Roche, respectively. Mouse monoclonal anti-AU1 and rabbit polyclonal anti-loricrin were from Covance (Berkeley, CA). The following reagents were from Sigma (St. Louis, MO): rhodamine-labeled phalloidin, rabbit polyclonal anti-β-catenin, mouse anti-γ-tubulin, and secondary Cy3-labeled antibodies. Secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 405 were from Molecular Probes (Eugene, OR). All other chemicals were purchased from Sigma.
Direct Fluorescence and Immunofluorescence Microscopy
Cells were rinsed with phosphate-buffered saline (PBS), followed by permeabilization and fixation with PBS containing 0.1% Triton X-100 and 4% paraformaldehyde for 20 min (22°C). The cells were rinsed twice with PBS and blocked overnight (4°C) with PBS containing 5% nonfat dry milk and 5% goat serum. After three PBS washes, the cells were incubated with primary antibody for 1 h (22°C) or with rhodamine-labeled phalloidin for 15 min (22°C). After three 20-min PBS washes, the cells were probed with the appropriate secondary antibody, at 22°C for 1 h. After removal of the secondary antibody, the cells were rinsed twice, incubated with Hoechst 33258 (10 μg/ml) in PBS for 5 min at 22°C, rinsed five times, and mounted in antifade medium (Fisher Scientific, Pittsburgh, PA). Cells expressing GFP-labeled proteins were processed for direct fluorescence in a similar manner, but omitting incubation with antibodies. Photomicrographs were obtained with a Leica DMIRBE or a Leica IRE2DM microscope, equipped with an Orca II digital camera (Hamamatsu Photonics, Hamamatsu City, Japan), using Openlab 3.5 software (Improvision, Conventry, United Kingdom). The results shown are representative of experiments conducted on duplicate samples at least three times.
Immunoprecipitations
IMDF cells (5.5 × 106 cells per 100-mm culture dish) were transfected and cultured for 48 h. The cells were rinsed with ice-cold PBS, harvested, and lysed in immunoprecipitation (IP) buffer (1% Triton X-100, 50 mM Tris-Cl, pH 7.6, 150 mM NaCl, 2 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride [PMSF], 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 2 μg/ml pepstatin). Cell debris in the lysates were removed by centrifugation, and samples containing 1 mg of protein were precleared with 10 μl of protein A/G-azlactone (Pierce Chemical, Rockford, IL) for 2 h at 4°C. After centrifugation, lysates were incubated with 1 μg of anti-V5 antibody (16 h; 4°C). Immune complexes were isolated by adding 10 μl of protein A/G-azlactone and incubating for 2 h at 4°C and washed 10 times with 1-ml aliquots of ice-cold IP buffer. Proteins bound to the beads were released by boiling in SDS sample buffer (50 mM Tris-Cl, pH 6.8, 1% SDS, 10% glycerol, 20 mM dithiothreitol [DTT], and 1% bromphenol blue) for 5 min. The proteins were resolved by SDS-PAGE, transferred to nitrocellulose membranes (Pall, East Hill, NY), and probed with antibodies indicated in individual experiments.
Immunoblotting
Keratinocytes or IMDF cells were harvested and lysed in buffer A (50 mM HEPES, pH 7.7, 250 mM KCl, 10% glycerol, 0.1% NP-40, 0.1 mM EDTA, 0.1 mM EGTA, 0.5 mM DTT, 0.4 mM NaF, 0.4 mM Na3VO4, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 1 μg/ml leupeptin, and 0.5 mM PMSF). Lysates were cleared by centrifugation (12,000 × g; 15 min) and used immediately or stored at –80°C. Gel electrophoresis and immunoblotting were conducted with 50 μg protein/sample, as described previously (Vespa et al., 2003). Immunoblots also were probed for GAPDH or γ-tubulin to normalize for protein loading. Unless otherwise indicated, the results shown in the immunoblots are representative of experiments conducted at least three times.
DNA Synthesis
DNA synthesis was measured estimating [3H]thymidine incorporation into acid-insoluble material as described previously (Chang et al., 2004).
RESULTS
Temporal Dissociation in the Kinetics of Adherens and Tight Junction Formation in Keratinocytes
Culture of primary murine epidermal keratinocytes with extracellular Ca2+ concentrations ≥0.1 mM triggers a complex series of responses, including formation of cell–cell junctions essential for acquisition of barrier function; up-regulation of differentiation markers, such as loricrin and involucrin; and irreversible exit from the cell cycle (Dotto, 1999). The formation of intercellular junctions in these cells is a complex process that involves formation of desmosomes, adherens, and tight junctions and reorganization of the actin cytoskeleton (reviewed in Perez-Moreno et al., 2003). We have demonstrated that Ca2+ also induces changes in the subcellular localization of ILK in these cells, promoting its migration to areas of cell–cell contact (Vespa et al., 2003).
To address the biological significance of ILK localization to cell borders, we first determined the temporal relationship of Ca2+-induced assembly of various intercellular junctions. We focused on the formation of AJ and TJ and associated changes in the actin cytoskeleton. We reasoned that this approach would allow us to correlate the formation of these junctions with the kinetics of ILK migration to the cell periphery, providing us clues as to the significance of the latter phenomenon.
Cells cultured in medium containing 0.05 mM Ca2+ exhibit a network of actin stress fibers and focal adhesions (Figure 1). Within 1 h of culture in medium with high (1.0) mM Ca2+, radial actin fibers begin to form along the cell borders, and progress to become actin bundles distributed in a punctate pattern by 3 h (Figure 1). These structures are maintained for at least 6 h, and after 12 h of culture in high Ca2+, a continuous ribbon of actin fibers is found along the cell periphery (Figure 1).
Figure 1.

Kinetics of adherens junction assembly in keratinocytes. Primary keratinocytes were cultured in medium containing 1.0 mM Ca2+. At the indicated times, cells were fixed and processed for indirect immunofluorescence microscopy using anti-β-catenin or anti-E-cadherin antibodies. As early as 1 h, a series of filopodia showing E-cadherin and β-catenin immunofluorescence form. Boxed areas are shown at higher magnification in d′, e′, and f′. Filopodia progress to a single ribbon containing adherens junctions proteins by 12 h. Bar, 25 μm.
Culture of keratinocytes in high Ca2+ medium also induces formation of filopodial extensions between adjacent cells, thus initiating formation of AJ (Vasioukhin et al., 2000). We analyzed the distribution of β-catenin and E-cadherin as a measure of AJ formation in our cultures. As early as 1 h after Ca2+ addition, we observed localization of both proteins in cellular processes extending across neighboring cells (Figure 1). Between 1 and 6 h, the number and size of these processes increased, allowing the establishment of more abundant punctate cell–cell contacts. Similar to actin fibers, by 12 h after Ca2+ addition, β-catenin and E-cadherin were uniformly distributed along cell borders.
Similar analysis of the time course of occludin distribution to the cell membrane revealed that TJs form with delayed kinetics relative to AJs. Specifically, occludin immunofluorescence is detectable in a punctate pattern throughout the cell, but not at borders, in cells cultured in low Ca2+, or during the first 6 h after Ca2+ addition (Figure 2). Occludin localization to the cell periphery only occurs by 12 h. Similarly, we observed claudin-1 immunoreactivity along cell junctions only in cells incubated with 1.0 mM Ca2+ for either 12 h or 24 h (our unpublished data). We also investigated Ca2+ induced changes in subcellular localization of ZO-1, a protein that can associate with E-cadherin and that is also found in TJ in polarized epithelial cells (Ando-Akatsuka et al., 1999). Within 1 h of culture in high Ca2+, ZO-1-positive areas around borders began to form in some cells (Figure 2). Areas of ZO-1 localization to the cell periphery were less abundant and were present in a lower proportion of cells than those exhibiting β-catenin or E-cadherin immunostaining (Figure 2). By 3 h after Ca2+ addition, ZO-1 was easily detected in the filopodial extensions formed between adjacent cells, in a pattern reminiscent to that described for AJ proteins (Figure 2). By 12 h after Ca2+ addition, ZO-1 was localized in a continuous pattern along sealed cell borders, similar to the AJ and TJ proteins described above. Thus, ZO-1 begins to localize to filopodial extensions slightly later than E-cadherin or β-catenin but before migration of occludin and, presumably, formation of TJ. Together, these data establish that migration of E-cadherin and β-catenin to the cell membrane occurs before that of ZO-1 and that both events precede formation of TJ.
Figure 2.

Kinetics of tight junction assembly and ILK migration to cell borders in keratinocytes. Primary keratinocytes were cultured in medium containing 1.0 mM Ca2+. At the indicated times, cells were fixed and processed for indirect immunofluorescence microscopy using anti-occludin or anti-ZO-1 antibodies. Occludin, a component of tight junctions, exhibits a punctate cytoplasmic pattern during the first 6 h of incubation with Ca2+ and is found along cell borders by 12 h. In contrast, as early as 1 h, a series of filopodia showing ZO-1 immunofluorescence are visible, which progress to a single ribbon by 12 h. ILK immunostaining is found in the cytoplasm and in focal adhesions, indicated by the arrows, before the addition of Ca2+ (0 h). One hour after Ca2+ addition, focal adhesions are no longer visible, and ILK begins to localize to cell extensions, becoming more prominent by 3 h post-Ca2+. Boxed areas are shown at higher magnification in g′, h′, and i′. ILK localization to sealed cell borders is apparent by 12 h. Bar, 25 μm.
Temporal Relationship between ILK Migration to Cell Borders and AJ Formation
Keratinocytes cultured in low but not high Ca2+ medium express abundant integrins and form abundant focal contacts. In these cells, ILK is found in the cytoplasm as well as in focal adhesions (Figure 2). Within 1 h of culture in high Ca2+ medium, the pattern of ILK distribution to focal adhesions is lost and replaced by areas of ILK localization to nascent filopodial extensions between adjacent cells. With time, these areas increase in number and, by 3 h, ILK is readily detected in multiple regions involved in junction formation between neighboring cells, in a pattern similar to that observed for E-cadherin, β-catenin and ZO-1 (Figures 1 and 2), and concentrates along sealed cell borders by 12 h after Ca2+ addition to the culture medium. Notably, the timing of the initial ILK movement toward the developing junctions seems to be slightly delayed relative to that of E-cadherin and β-catenin and is more reminiscent of that observed for ZO-1. Together, these experiments show that ILK migrates to areas of AJ formation during Ca2+ induction of epithelial cell adhesion before formation of TJ and suggest a potential role for ILK in the early assembly of cell–cell junction complexes.
The N Terminus of ILK Is Required for Targeting to Cell Junctions
To begin to elucidate the mechanisms implicated in ILK localization to cell borders during epithelial sheet formation, we next focused on identifying the regions in ILK necessary for targeting to junctions. To this end, we generated a series of ILK deletion mutants and exogenously expressed them in keratinocytes cultured in medium containing 0.05 mM Ca2+. Twenty-four hours after transfection, the culture medium was adjusted to 1.0 mM Ca2+, and the subcellular localization of the transfected ILK forms was assessed after a further 24-h incubation period. ILK mutants lacking either 67 or 191 N-terminal residues (ILK 67-452 and 191-452) were incapable of localizing to cell borders. Rather, these N-terminal deletion mutants were distributed either throughout the cytoplasm or in a punctate pattern (Figure 3C). In these mutants, the pleckstrin homology and the kinase domains are intact, indicating that neither of these two regions is sufficient for ILK localization to cell junctions. In contrast, deletion of the C-terminal half (ILK 1-192) or introduction of a point mutation (ILK E359K) that abolishes the ability of ILK to interact with focal adhesion-associated proteins, such as paxillin, actopaxin, or affixin (Wu and Dedhar, 2001), yielded ILK forms that efficiently translocated to intercellular junctions (Figure 3C). Thus, the domains responsible for ILK distribution to cell borders during the establishment of intercellular adhesion encompass residues 1–192, in the N terminus of this protein, excluding involvement of the pleckstrin homology and the kinase domains.
Figure 3.

Subcellular localization of ILK deletion mutants during cell junction formation. (A) Schematic of ILK mutants exogenously expressed in primary murine keratinocytes. ILK domains are indicated at the top. A, putative ankyrin repeat; PH, pleckstrin homology domain. Positions at the right indicate the residues present in each mutant. X indicates the position of the E359K mutation. (B) Expression of the ILK mutants shown in A. Keratinocytes were transfected with expression vectors encoding the indicated V5-tagged ILK mutants. Lysates from transfected cells were resolved by SDS-PAGE and analyzed by immunoblotting with an anti-V5 antibody. The proteins visualized correspond to the expected size for each mutant. The blots also were probed with an antibody against GAPDH to provide a loading control. (C) Localization of ILK mutants in keratinocytes. Cells transfected with the indicated ILK mutants were cultured in 1.0 mM Ca2+ for 24 h to induce cell junction formation, fixed, and processed for immunofluorescence microscopy using anti-V5 antibodies. Bar, 25 μm.
To further delineate the N-terminal ILK domain involved in localization to cell borders, we tested a series of additional GFP-tagged ILK deletion mutants, schematically represented in Figure 4A. We verified that each mutant was expressed in the cells and confirmed presence of GFP-tagged proteins with the predicted molecular mass by immunoblot analysis of lysates prepared from cells transfected with plasmids encoding these ILK mutants (Figure 4B). We also confirmed that the presence of the GFP moiety did not alter the ability of ILK to migrate to cell borders (Figure 4C). N-terminal deletions lacking up to 54 amino acids also were able to localize to cell junctions, whereas mutants harboring further N-terminal deletions gradually lost this property (Figure 4 and Table 1). Specifically, whereas ILK 54-192 localized to borders as efficiently as wt ILK, ILK 57-192 and 60-192 exhibited weak localization to borders in only ∼20% of expressing cells, and strong cytoplasmic distribution in all expressing cells. Further N-terminal deletions, such as in ILK 62-192 completely abolished localization to the cell periphery. Thus, the first 54 residues in the N terminus of ILK are dispensable for membrane localization, but residues downstream of Ile54 are necessary. We next addressed the importance of amino acid residues around Glu192 for membrane localization of ILK. Thus, whereas ILK 1-192 efficiently localized to the cell periphery, we observed a decline in border localization of ILK 2-173 and ILK 2-160. Finally, ILK 2-144 did not migrate to cell borders at all (Table 1). Thus, the minimal domain that allows ILK to efficiently localize to cell borders comprises the region containing Ile54 to Glu192.
Figure 4.

Subcellular localization of GFP-tagged ILK mutants during cell junction formation. (A) Schematic of ILK mutants exogenously expressed in primary murine keratinocytes. The mutants have a GFP moiety in the N terminus. ILK domains are indicated at the top. A, putative ankyrin repeat; PH, pleckstrin homology domain. Positions at the right indicate the residues present in each mutant. X represents the position of the tetrapeptide motifs mutated in the ankyrin repeat regions. The following mutations were introduced: S, residues 36SPLH39→AAAA; T, residues 69TPLH72→AAAA, SV, residues 36SPLH39→AAAA and 102VPLH105→AAAA; STV, residues 36SPLH39→AAAA, 69TPLH72→AAAA and 102VPLH105→AAAA. (B) Expression of the ILK mutants shown in A. Keratinocytes were transfected with expression vectors encoding the indicated GFP-tagged ILK mutants. Lysates from transfected cells were resolved by SDS-PAGE and analyzed by immunoblotting with an anti-GFP antibody. The proteins visualized correspond to the expected size for each GFP-tagged ILK mutant. The blots also were probed with an antibody against GAPDH to provide a loading control. (C) Localization of ILK mutants in keratinocytes. Cells transfected with the indicated ILK mutants were cultured in 1.0 mM Ca2+ for 24 h to induce cell junction formation, fixed, and processed for direct fluorescence microscopy to visualize GFP. Bar, 25 μm.
Table 1.
Distribution of ILK mutant proteins to cell–cell junctions
| V5-tagged ILK proteins
|
||
| ILK mutant | Cell borders | |
| wt (1-452) | +++ | |
| 32-452 | +++ | |
| 67-452 | - | |
| 191-452 | - | |
| 1-192 | +++ | |
| E359K | +++ | |
| GFP-tagged ILK proteins
|
||
| ILK mutant | Cell borders | |
| wt (1-452) | +++ | |
| 42-192 | +++ | |
| 52-192 | +++ | |
| 54-192 | +++ | |
| 57-192 | + | |
| 60-192 | + | |
| 62-192 | - | |
| 2-173 | ++ | |
| 2-160 | + | |
| 2-144 | - | |
| S | +++ | |
| T | +++ | |
| ST | +++ | |
| SV | +++ | |
| STV | +++ | |
| GFP alone | - | |
Primary keratinocytes were transfected with plasmids encoding the indicated proteins. Twenty-four hours after transfection, the cells were cultured in medium containing 1.0 mM Ca2+ for 24 h to induce differentiation and formation of cell-cell junctions. The subcellular distribution of ILK proteins was visualized either by indirect immunofluorescence with an anti-V5 antibody (V5-tagged ILK proteins) or by direct fluorescence using the GFP moiety (GFP-tagged ILK proteins). +++ indicates strong staining at cell borders, + indicates weak staining at cell borders accompanied by strong cytoplasmic staining, and - indicates undetectable staining at cell borders and strong cytoplasmic staining. The data represent at least three experiments conducted in duplicate. Ankyrin repeat mutants are indicated as follows: S, 36SPLH39 → AAAA; T, 69TPLH72 → AAAA; ST, 36SPLH39 → AAAA and 69TPLH72 → AAAA; SV, 36SPLH39 → AAAA and 102VPLH105 → AAAA; STV, 36SPLH → AAAA, 69TPLH72 → AAAA, and 102VPLH105 → AAAA.
Role of Putative Ankyrin Repeats in ILK Migration to Cell Junctions
In silico analysis of the human ILK sequence reveals the presence of three putative ankyrin repeats comprising residues 33–65, 66–98, and 99–131 (www.expasy.org), which have been proposed to be involved in ILK interactions with other proteins, such as PINCH1 and PINCH2 (Tu et al., 1999; Braun et al., 2003). Ankyrin repeats consist of a β-hairpin followed by two antiparallel α-helices and exhibit a highly conserved and absolutely essential TPLH tetrapeptide motif close to the initiation of the first α helix (Mosavi et al., 2002). Consistent with the in silico identified ankyrin repeats in this protein, three such motifs exist in ILK: 36SPLH39, 69TPLH72, and 102VPLH105. To determine the importance of these domains in ILK localization to cell borders, we ectopically expressed ILK 1-192 mutants in which one, two, or all three tetrapeptide motifs were mutated to alanines and found that they localized to cell junctions as efficiently as the wild-type protein (Figure 4 and Table 1). To verify that the mutations introduced had functional consequences on ILK, we also assessed the ability of various ectopically expressed ILK mutants to interact with PINCH1, which is known to require residues within the first putative ankyrin repeat. In agreement with previous observations (Tu et al., 1999; Braun et al., 2003), we were able to detect PINCH1 in immunoprecipitates from wild-type, E359K, and 1-192 ILK forms, but not from ILK 67-452 (Figure 5A). Importantly, although the ILK 1-192 mutant in which the SPLH tetrapeptide spanning residues 36–39 was converted to AAAA localized to cell junctions as efficiently as the wild-type protein, it failed to associate with PINCH1 in immunoprecipitation experiments (Figure 5B), indicating that this mutation indeed introduced functionally significant structural alterations in ILK. Thus, the ability of ILK to migrate to cell–cell junctions may involve novel regulatory mechanisms and/or protein–protein interactions and does not depend on its association with PINCH1 or on the presence of intact putative ankyrin repeats in ILK.
Figure 5.

Distinct domains in ILK mediate localization to cell–cell adhesions and interaction with PINCH1. (A) IMDF cells were transfected with the indicated V5-tagged ILK mutants and AU1-tagged PINCH1. Forty-eight hours later, lysates from transfected cells were prepared and subjected to immunoprecipitation with anti-V5 antibodies against the ILK forms expressed. The presence of ILK mutants or PINCH1 in immunoprecipitates was examined by immunoblotting with antibodies against V5 or AU1, respectively. (B) IMDF cells were transfected with AU1-tagged PINCH1 together with a V5-tagged ILK 1-192 mutant fused to GFP, in which residues 36SPLH39 were substituted by AAAA. Forty-eight hours later, lysates from transfected cells were prepared and subjected to immunoprecipitation with antibodies against PINCH1. The presence of the ILK mutant in PINCH1 immunoprecipitates was examined by immunoblotting with antibodies against the V5 tag in ILK.
Disruption of Cell–Cell Adhesion by ILK Mutants Lacking N-Terminal Junction-targeting Domains
To begin to address the significance of ILK localization to cell borders during cell junction formation, we analyzed the ability of the mutants examined previously (Table 1) to perturb intercellular adhesion. We reasoned that if ILK participates in junction formation, high levels of expression of ILK mutants unable to localize to cell borders might disrupt this process. Thus, when we expressed only the N-terminal region containing the junction-targeting domain (ILK 1-192), we found no detectable effect on cell–cell adhesion (Figure 3C; our unpublished data).
In stark contrast, those cells expressing ILK 67-452 or ILK 191-452, mutants incapable of localizing to borders, exhibited altered morphology and a marked impairment in formation of cell–cell contacts (shown in Figure 6 for ILK 191-452). Because ILK 191-452 induced a more pronounced phenotype, we focused on this mutant for subsequent experiments. We observed that, 24 h after Ca2+ addition to the extracellular medium, those cells expressing ILK 191-452 were incapable of forming sealed cell–cell borders, and large gaps between them remained (Figure 6A). Expression of ILK 191-452 also interfered with the localization of ZO-1 to cell borders. Specifically, 24 h after Ca2+ treatment, ZO-1 was detected in the periphery of cells adjacent to those in which ILK 191-452 was expressed, but it was undectable in those regions in direct contact with the ILK 191-452-expressing cells (Figure 6B). The disruption by ILK 191-452 of intercellular junction formation induced by Ca2+ is consistent with the concept that ILK positively contributes to the formation of epithelial cell–cell adhesion complexes and that ILK 191-452 seems to behave as a dominant-negative ILK form.
Figure 6.

Disruption of cell–cell adhesion by ILK 191-452. (A) Primary mouse keratinocytes were infected with a recombinant adenovirus encoding V5-tagged ILK 191-452, and were cultured for 24 h before addition of 1.0 mM Ca2+. The cells were cultured for an additional 24-h period to induce cell junction formation, and then they were fixed and processed for indirect immunofluorescence microscopy. Expression of ILK 191-452 (a–c) was visualized using an anti-V5 antibody. Boxed areas showing large intercellular gaps in the micrograph of a are shown at higher magnification in b and c. The localization of β-catenin also was assessed in these cells (d–f). Note the distribution of β-catenin in the cytoplasm of cells expressing ILK 191-452 (e and f). Treatment with Ca2+ induces these cultures to stratify into two to three cell layers, which results in a complex β-catenin pattern that also allows demarcation of the intercellular gaps indicated by the boxes in d and shown at higher magnification in e and f. (B) Primary keratinocytes were infected and processed as indicated in A, using anti-V5 or anti-ZO-1 antibodies as indicated. Hoechst 33258 was used to visualize the nuclei. The cell expressing ILK 191-452 is indicated by the arrows and exhibits severely reduced or absent ZO-1 immunostaining at the periphery. ZO-1 localization to cell junctions is not perturbed between two adjacent cells not expressing ILK 191-452. Bar, 50 μm (a and g) or 10 μm (b and c).
The morphological changes and alterations in ZO-1 observed in cells expressing ILK 191-452 prompted us to assess the patterns of localization of AJ and TJ proteins as well as the status of the actin cytoskeleton. We hypothesized that the reduced formation of cell–cell contacts in those cells would likely be associated with abnormal subcellular localization of junction proteins and/or alterations in F-actin. For these experiments, cells expressing wt ILK, ILK 191-452 or β-gal as a control were cultured for 24 h in 1.0 mM Ca2+, and the distribution of β-catenin and occludin as well as changes in F-actin organization were determined. As described above, primary keratinocyte monolayers cultured in 1.0 mM Ca2+ are polarized and exhibit cell junctions in a honeycomb-like pattern. The presence of 1.0 mM Ca2+ induced clear localization of β-catenin along cell borders in cells expressing β-gal or wt ILK indistinguishable from those observed in uninfected cells (Figure 7A). In contrast, expression of ILK 191-452 altered the organization of the cells in the monolayer and was accompanied by localization of β-catenin throughout the cell but not concentrated at cell borders. Similarly, a ribbon of occludin immunofluorescence was clearly visible in Ca2+-treated cells expressing β-gal or wt ILK, whereas a cytoplasmic distribution of occludin was apparent in ILK 191-452-expressing cells, which failed to form normal TJ (Figure 7B).
Figure 7.
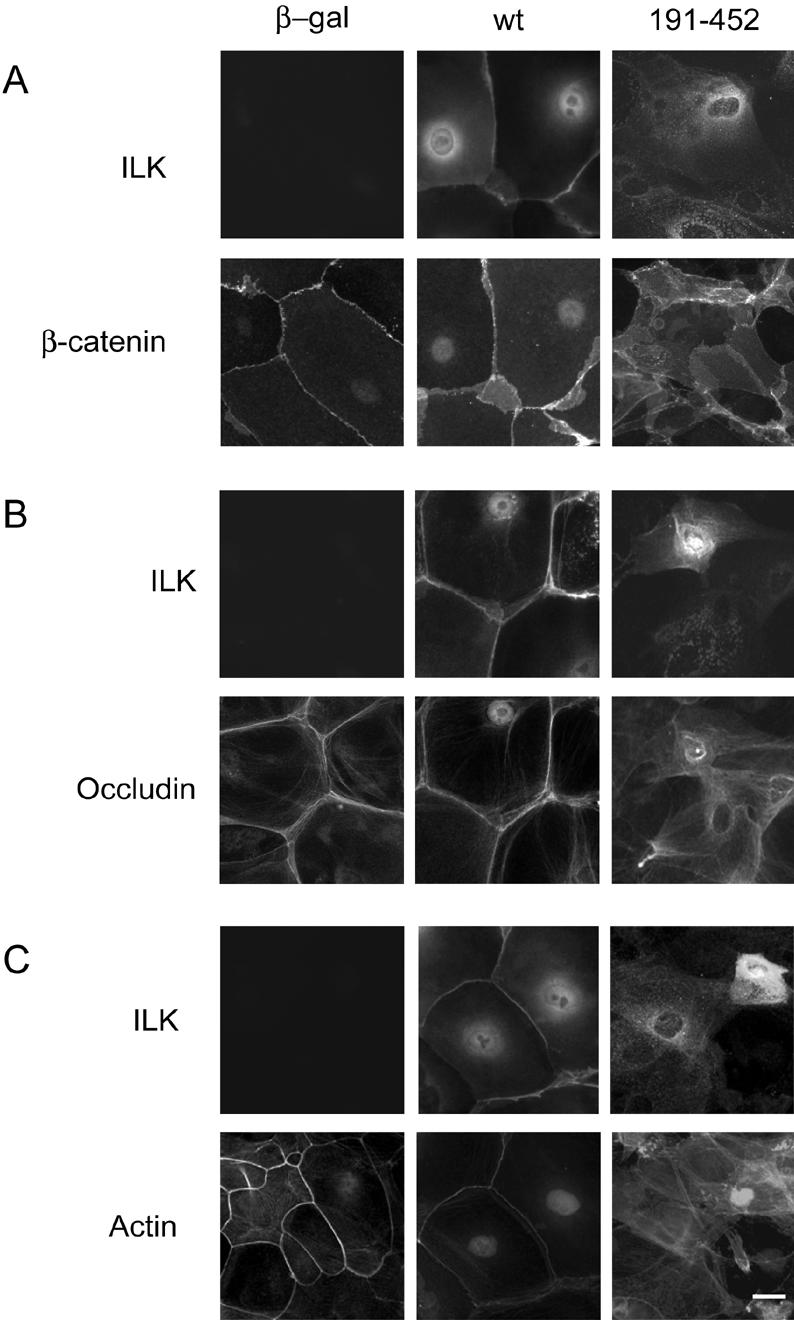
Expression of ILK 191-452 impairs cytoskeletal remodeling and localization of junction proteins to cell borders. Primary mouse keratinocytes were infected with a recombinant adenovirus encoding either β-galactosidase (β-gal), V5-tagged ILK (wt), or V5-tagged ILK 191-452 and cultured for 24 h before addition of 1.0 mM Ca2+. The cells were cultured for an additional 24-h period to induce cell junction formation and were fixed and processed for indirect immunofluorescence microscopy using an anti-V5 antibody to visualize wt or mutant ILK. The cells also were probed with antibodies against β-catenin or occludin, as indicated, or with rhodamine-labeled phalloidin to detect F-actin. In contrast to cells expressing β-gal or wt ILK, those cells expressing ILK 191-452 do not exhibit normal β-catenin or occludin staining in the cell periphery. Cortical actin fibers decorating cell borders are also absent in cells infected with virus encoding ILK 191-452 after 24 h of culture in 1.0 mM Ca2+. Bar, 25 μm.
The establishment of intercellular junctions in keratinocytes requires substantial remodeling of the actin cytoskeleton. This process involves assembly of radial actin fibers associated with nascent AJ, eventually resulting in the formation a cortical ribbon of actin fibers along the cell periphery. This type of cytoskeletal reorganization was readily visible in cells expressing β-gal or wt ILK (Figure 7C). In contrast, cortical actin fibers were not visible in cells expressing ILK 191-452. In these cells, actin filaments consisted of disorganized fibers which differed even from those visible in keratinocytes cultured in 0.05 mM Ca2+. Thus, expression of ILK 191-452 seems to disrupt cell–cell adhesion by mechanisms that include interference with the radial actin cable formation, without abolishing actin polymerization per se
Effect of ILK 191-452 on Expression of Junction Proteins and on Differentiation Responses
To investigate whether the observed alterations in junction formation consequent to ILK 191-452 expression are associated with reduced expression of AJ or TJ components, we determined the levels of E-cadherin, β-catenin, and occludin in cells infected with recombinant adenoviruses encoding either wt ILK or ILK 191-452. We observed no statistically significant changes in the levels of those proteins relative to uninfected controls, irrespective of the ILK form expressed in the cells (Figure 8, A–C). Although additional proteins participate in the formation of AJ and TJ, these results indicate that the mechanisms of impaired formation of intercellular contacts in cells expressing ILK 191-452 do not involve down-regulation of E-cadherin, β-catenin, or occludin. We also ascertained that impairment of junction formation in cells expressing ILK 191-452 was not consequent to apoptosis. Indeed, these cultures did not show evidence of fragmented nuclei or increased levels of activated poly(ADP ribose) polymerase (PARP) (a substrate for caspase-3 and a marker of apoptosis; Figure 8, A and C; our unpublished data).
Figure 8.

Effect of wt ILK and ILK 191-452 on cell junction proteins, differentiation markers, and DNA synthesis. (A and B) Primary mouse keratinocytes were infected with a recombinant adenovirus encoding V5-tagged wt ILK or V5-tagged ILK 191-452. Twenty-four hours after infection, the cells were cultured for an additional 24 h either in 0.05 or 1.0 mM Ca2+ to induce differentiation and cell junction formation. Lysates from infected cells were resolved by SDS-PAGE and analyzed by immunoblotting with antibodies against the indicated proteins. Recombinant ILK forms were visualized by probing with anti-V5 antibodies. The blots were also probed with an antibody against GAPDH to provide a loading control. In the blot probed for PARP, the band with lower mobility (indicated by *) represents the unprocessed precursor, and the arrow indicates the cleaved, apoptosis-associated form. (C) Levels of the indicated proteins in keratinocytes infected with the indicated ILK-encoding adenovirus were calculated from densitometric analysis of three experiments similar to those shown in A and B, except for cyclin D1, in which the average of two experiments is shown. The levels of each protein were normalized to GAPDH or γ-tubulin. The results are expressed as mean + SD, relative to protein levels in uninfected cells cultured in low Ca2+ medium (0.05 mM; set at 100%), except for loricrin, in which they are presented as a percentage of loricrin in uninfected cells cultured in 1.0 mM Ca2+ (set at 100%). (D) Keratinocytes were infected with recombinant adenoviruses encoding the indicated proteins. Twenty-four hours after infection, the cells were cultured in medium containing 1.0 mM Ca2+ for 0, 24, or 48 h, as indicated. The cells were incubated with [3H]deoxythymidine for 2 h before being harvested to determine incorporation of tritium in DNA. The results, expressed as a percentage of DNA synthesis in uninfected cells at t = 0 (set at 100%), are presented as the mean of three experiments conducted in triplicate. SEM values did not exceed 15% and are omitted from the graph for clarity. *p < 0.05 (Student's t test).
Treatment of epidermal keratinocytes with 0.1–1.0 mM Ca2+ triggers a differentiation program in which formation of intercellular junctions is but one aspect. In addition, Ca2+ induces up-regulation of loricrin expression and down-regulation of cyclin D1. Ca2+ also causes irreversible withdrawal from the cell cycle (Hohl et al., 1991; Ng et al., 1996; Martinez et al., 1999). We evaluated whether blockade of cell–cell junction formation in keratinocytes expressing ILK 191-452 was accompanied by alterations in other aspects of Ca2+-induced differentiation. We found that expression of ILK 191-452, but not of wt ILK, interfered with induction by Ca2+ of loricrin expression (Figure 8, A–C), similar to the reduced loricrin expression in the epidermis of mice with conditional inactivation of the E-cadherin gene, in which AJ do not form (Young et al., 2003). In contrast, the presence of ILK 191-452 had little, if any, effect on the reduction in cyclin D1 expression induced by Ca2+ (Figure 8, B and C), suggesting that, similar to what is observed for other epidermal differentiation markers, the mechanisms governing up-regulation of those two keratinocyte proteins are not identical.
We also measured the effect of ILK expression on DNA synthesis in differentiating keratinocytes. Keratinocytes respond to 1.0 mM Ca2+ treatments by failing to exhibit time-dependent increases in DNA synthesis, which are observed in exponentially proliferating, undifferentiated cells (D'Souza et al., 2001). Differentiating keratinocytes expressing ILK 191-452 or β-gal via adenovirus-mediated gene transfer did not show significant differences in DNA synthetic capacity relative to uninfected control cultures (Figure 8D). In contrast, DNA synthesis in wt ILK-expressing cells was almost fivefold greater than that measured in uninfected keratinocytes (Figure 8D). Several conclusions can be drawn from these results. First, they indicate that the alterations in normal ILK function likely resulting from ILK 191-452 overexpression disrupt expression of some differentiation markers and cell–cell adhesion induced by Ca2+. Second, they are consistent with the concept that it is possible to uncouple these aspects of differentiation from the proliferative response in cultured keratinocytes induced to terminally differentiate.
ILK 191-452 Impairs Cell–Cell Adhesion by Interfering with Early Stages of AJ Formation
The experiments outlined above established that expression of ILK 191-452 interferes with Ca2+-induced formation and/or maintenance of sealed cell junctions. However, they did not explore whether the inability of cells expressing ILK 191-452 to form sealed cell borders occurred consequent to a perturbation of AJ and/or TJ. To identify the process altered by ILK 191-452 during formation of epithelial sheets, we assessed the kinetics of AJ and TJ formation in keratinocytes infected with adenovirus encoding ILK 191-452 and compared them with those in neighboring cells not expressing this mutant. As discussed above, addition of Ca2+ to the extracellular medium triggers movement of β-catenin and E-cadherin to the cell membrane within 1 h in noninfected cells (Figure 9; our unpublished data). In contrast, β-catenin immunostaining was not detected at the periphery of ILK 191-452-expressing cells (Figure 9). Three hours after Ca2+ addition, β-catenin immunostaining had expanded to a larger proportion of the cell membrane in uninfected cells but not in those expressing mutant ILK. Notably, at this time, nonexpressing cells already exhibited substantial morphological changes induced by Ca2+, including presence of numerous filopodia, and acquisition of a polyhedral shape, whereas infected cells frequently showed a few long processes similar to those observed in migrating keratinocytes cultured in low Ca2+ concentrations (e.g., Figure 9, 3 h). We also observed a low degree of β-catenin immunostaining at the periphery of uninfected cells adjacent to mutant ILK-expressing cells, but we did not detect β-catenin in cell borders of two adjacent cells expressing ILK 191-452 as late as 24 h (Figures 7 and 9). Furthermore, occludin also failed to migrate to the plasma membrane in cells expressing ILK191-452 (Figure 7). Because the formation of TJ requires previous assembly of AJ, it is not known whether ILK 191-452 also impairs TJ establishment directly, or indirectly, as a consequence of its perturbation of AJ. Together, our data are consistent with the notion that ILK participates in early events of AJ assembly and cytoskeletal remodeling during epithelial sheet formation, a hitherto unidentified role for this important scaffold protein.
Figure 9.

Expression of ILK 191-452 disrupts Ca2+-induced migration of β-catenin to cell borders. Primary mouse keratinocytes were infected with a recombinant adenovirus encoding V5-tagged ILK 191-452 and were cultured for 24 h before addition of 1.0 mM Ca2+. The cells were cultured in medium containing 1.0 mM Ca2+ to induce cell junction formation. At the indicated times after Ca2+ addition, cells were fixed and processed for indirect immunofluorescence microscopy using an anti-V5 antibody to visualize ILK 191-452 or an anti-β-catenin antibody to assess AJ formation. Those cells expressing ILK 191-452 lack detectable β-catenin immunostaining at the periphery. The β-catenin localization to cell junctions is not perturbed between adjacent cells not expressing ILK 191-452. Bar, 25 μm.
DISCUSSION
ILK has multiple functional domains that mediate interactions with a variety of proteins. They include an N-terminal domain with putative ankyrin repeats, a pleckstrin-homology domain, and a C-terminal half with homology to kinases (Hannigan et al., 1996). ILK has not been crystallized; consequently, some of these domains (e.g., the ankyrin repeats) have not been experimentally demonstrated. ILK serves as a scaffold that brings together other proteins associated with the actin cytoskeleton. Specifically, the N terminus of ILK mediates interactions with PINCH and ILKAP, whereas the C-terminal half binds to integrins β1 and β3, paxillin, actopaxin, and affixin (reviewed in Wu and Dedhar, 2001). Our analysis of ILK mutants has defined a novel functional domain in ILK, which includes amino acid residues 54–192, excludes the putative first ankyrin repeat and is essential to mediate ILK localization to cell borders during junction formation in epidermal keratinocytes. This domain also functions in a manner independent of interactions with known ILK-binding proteins, such as PINCH, suggesting the possibility that ILK may interact with as yet unidentified proteins through its N-terminal half, thus creating cell adhesion multiprotein networks. Our results are consistent with the concept that the N-terminal half of ILK likely fulfills multiple roles and is capable of numerous interactions that require distinct domains. Finally, although the kinase properties of ILK remain the subject of investigation, the ability of ILK 1-192, which lacks the kinase domain, to localize to cell junctions excludes the possibility that ILK kinase activity, if any, is necessary for this process.
ILK changes its subcellular distribution during keratinocyte differentiation from focal adhesions to cell borders, although a cytoplasmic pool of this protein is maintained, irrespective of the differentiation status of the cells (Vespa et al., 2003). The mutants ILK E359K and ILK 1-192 are found in the cytoplasm and at cell–cell junctions, but they do not localize to focal adhesions (Hannigan et al., 1996; Nikolopoulos and Turner, 2001). This indicates that the domains required for ILK localization to cell–ECM contacts are separable from those involved in localization to cell borders. Our experiments do not exclude the possibility that endogenous ILK is transferred from focal adhesion complexes to cell–cell contact sites; however, our results are consistent with the concept that a movement from cytoplasmic pools to cell borders also may occur.
Our kinetic analyses of Ca2+-induced junction formation revealed that formation of AJ in keratinocytes can be temporally distinguished from TJ assembly, in contrast to the apparently coordinated formation of these structures in some simple epithelial cells (Fukuhara et al., 2003). In agreement with previous studies (Pummi et al., 2001), we observed a movement of ZO-1 toward cell borders before that of occludin. Remarkably, ZO-1 localized to the early filopodial extensions generated between adjacent cells (Figure 2), in a pattern very similar to that observed for E-cadherin and β-catenin, suggesting that ZO-1 participates in the establishment of early cell–cell contacts and in the formation of both AJ and TJ in keratinocytes as well as other epithelial cells. Our observations also indicate that important events mediating AJ formation occur within 1 h of Ca2+ addition to the extracellular medium, well before the migration of TJ proteins, such as claudin and occludin, to nascent junctions, which only occurs 6–12 h after Ca2+ addition.
ILK Localization to Filopodia at Early Stages of Junction Formation
ILK has been reported to distribute to focal adhesions in a variety of cell types (Wu and Dedhar, 2001). It has also been implicated in epidermal-mesenchymal transition and epithelial cell acquisition of a migratory phenotype (Oloumi et al., 2004). In epidermal keratinocytes, ILK seems to play fundamentally distinct roles, depending on the extracellular Ca2+ concentration, which also determines the differentiation status of the cells. Thus, in cells cultured in low Ca2+, which prevents formation of cell–cell junctions and induction of differentiation programs, we have detected ILK in focal adhesions. Under these conditions, ILK also promotes migration and localizes to lamelopodial extensions in motile cells (Vespa and Dagnino, unpublished observations). ILK in focal adhesions has an established role in linking integrins to the actin cytoskeleton and remodeling of the latter during formation or disassembly of cell contacts with the extracellular matrix (Hannigan et al., 1996; Nikolopoulos and Turner, 2001, 2002). Remarkably, Ca2+ induction of cell–cell contacts involves changes in the actin cytoskeleton and exhibits some similarity to the events involved in the initial formation of cell extensions during migration. Although the exact mechanisms are not fully understood, several steps in this process have been defined (reviewed in Perez-Moreno et al., 2003). The presence of adequate Ca2+ concentrations triggers movement of E-cadherin–β-catenin complexes to the cell membrane. Associated with this movement is the formation of filopodia, and the initial cell–cell contacts occur through interaction between E-cadherin complexes found in the filopodia of adjacent cells. A crucial step in this process is the anchoring of E-cadherin/catenin species to cortical actin filaments, which stabilizes the nascent adherens junctions and causes them to cluster in complexes that can be visualized as a series of puncta. During this process, actin fibers organize radially on each side of the punctum and anchor cortical actin rings. Multiple filopodia form this way, which interdigitate with those of adjacent cells. Ultimately, a network of actin fibers interconnected by adherens junctions assemble, giving rise to the sealed cell borders of epithelial sheets. Intriguingly, ILK also localizes to Ca2+-induced filopodial extensions, in proximity to E-cadherin–β-catenin complexes, and with similar kinetics to those of adherens junction formation, preceding formation of tight junctions. ILK has an important role in modulating the actin cytoskeleton in D. melanogaster, C. elegans and in mammals, through mechanisms dependent or independent of integrins (Brakebusch and Fassler, 2003). Specifically, ILK-deficient epiblasts exhibit abnormal actin accumulation and organization at sites of integrin adhesion, and fail to polarize (Sakai et al., 2003). ILK is a scaffold that recruits adaptor proteins such as α- and β-parvin, PINCH and paxillin, which also modulate the actin cytoskeleton (Wu and Dedhar, 2001; Brakebusch and Fassler, 2003).
The observed abnormalities in intercellular cell junctions in cells expressing ILK 191-452 suggests a potentially essential role for ILK in cell–cell adhesion, in addition to cell-extracellular matrix adhesion. Cells expressing this ILK mutant exhibit some filopodial extensions upon treatment with Ca2+, but they are disorganized and do not progress to the ordered, intercalating puncta containing E-cadherin complexes observed during normal junction formation. It is noteworthy that expression of ILK 191-452 also disrupts cortical F-actin filaments, in a manner independent of E-cadherin, β-catenin, and actin degradation. Although the expression of other junction components remains to be established, the above-mentioned observations are consistent with the concept that ILK may modulate actin reorganization and AJ formation induced by Ca2+. Given that ILK is a known modulator of the actin cytoskeleton, it is possible that in Ca2+-treated keratinocytes ILK serves as a molecular scaffold, participating in the formation of complexes containing junction proteins linked to the actin cytoskeleton. In this model, the N-terminus of ILK may anchor this protein to areas close to the plasma membrane, whereas the C terminus provides links to the actin fibers. Accordingly, ILK 191-452 may behave as a dominant negative mutant because it lacks the membrane-targeting domain. Alternatively, and consistent with the inability of AJ components to efficiently translocate from intracellular storage sites to the membrane, ILK also may play a role in this translocation event. Thus, ILK may fulfill dual functions in keratinocytes, participating in the cross-talk between integrin-mediated migration and adhesion to extracellular matrix, and cell–cell adhesion in epithelial sheets. Similar dual roles for the integrin-associated proteins focal adhesion kinase and paxillin have been recently described in epithelial cervical carcinoma cells (Yano et al., 2004).
Multiple changes in gene expression and DNA synthetic ability occur during terminal differentiation in keratinocytes. In particular, postmitotic keratinocytes exhibit various stages of differentiation, depending on whether they are traversing the spinous or the granular suprabasal layers. The transition through these different layers requires that expression of genes involved in early epidermal differentiation states be silenced, replacing them with expression of genes required during late stages of differentiation, while maintaining the cells in a quiescent state (Rutberg et al., 1997). As a result, complex and distinct mechanisms regulate expression of individual differentiation markers. Thus, the inhibitory effect of ILK 191-452 on expression of loricrin, without alterations in silencing of cyclin D1, likely reflect differences in the mechanisms that regulate these two genes.
Our studies also indicate that it is possible to dissociate Ca2+-induced cell junction formation and expression of loricrin from proliferative responses. The unexpected ability of wt ILK to induce DNA synthesis remains to be characterized, although we have determined that it does not involve changes in cyclin D1 expression. These observations contrast with the up-regulation of cyclin D1 reported in some mammary epithelial cell lines in which ILK was exogenously expressed (D'Amico et al., 2000). However, it also has been shown that cyclin D1 expression is dispensable for epidermal morphogenesis and keratinocyte proliferation. Specifically, cyclins D2 and D3 can mediate proliferative responses in these cells (Robles et al., 1998; Rodriguez-Puebla et al., 1999, 2000, 2002), and exogenous expression of E2F1 or E2F2 increases DNA synthesis without alterations in cyclin D1 levels in primary mouse keratinocytes (D'Souza et al., 2001). Thus, the enhanced DNA synthetic capacity of keratinocytes exogenously expressing ILK may occur through changes in cyclin D2 or D3, or alternatively, ILK may activate DNA synthetic pathways downstream from the D-type cyclins which remain to be identified. Irrespective of this aspect of ILK function, the results from our experiments position ILK as a central player in epithelial sheet formation and epidermal morphogenesis and integrity.
Acknowledgments
We thank Drs. D. Laird and E. H. Ball for helpful comments. L. D. is a Scientist of the Child Health Research Institute and the Lawson Health Research Institute, London, Canada. This work was supported with funding to L. D. and S.J.A.D. from the Canadian Institutes of Health Research (CIHR), and to L. D. from the National Science and Engineering Research Council of Canada (NSERC). A. V. is the recipient of a NSERC graduate studentship. During the course of this work, L. D. was a CIHR/Cancer Society New Investigator. S.J.A.D. is a CIHR New Investigator.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–02–0087) on June 22, 2005.
Abbreviations used: AJ, adherens junction(s); DTT, dithiothreitol; ECM, extracellular matrix; EMEM, Eagle's minimum essential medium; FBS, fetal bovine serum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GFP, green fluorescent protein; IP, immunoprecipitation; ILK, integrin-linked kinase; PBS, phosphate-buffered saline; PMSF, phenylmethylsulfonyl fluoride; TJ, tight junction(s).
References
- Ando-Akatsuka, Y., Yonemura, S., Itoh, M., Furuse, M., and Tsukita, S. (1999). Differential behaviour of E-cadherin and occludin in their colocalization with ZO-1 during the establishment of epithelial cell polarity. J. Cell. Physiol. 179, 115–125. [DOI] [PubMed] [Google Scholar]
- Apostolova, M. D., Ivanova, I. A., Dagnino, C., D'Souza, S.J.A., and Dagnino, L. (2002). Active import and export mechanisms regulate E2F-5 subcellular localization. J. Biol. Chem. 277, 34471–34479. [DOI] [PubMed] [Google Scholar]
- Brakebusch, C., and Fassler, R. (2003). The integrin-actin connection, an eternal love affair. EMBO J. 22, 2324–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, A., Bordoy, R., Stanchi, F., Moser, M., Kostka, G., Ehler, E., and Fassler, R. (2003). PINCH2 is a new five LIM domain protein, homologous to PINCH and localized to focal adhesions. Exp. Cell Res. 284, 239–250. [DOI] [PubMed] [Google Scholar]
- Bryant, D. M., and Stow, J. L. (2004). The ins and outs of E-cadherin trafficking. Trends Cell Biol. 14, 427–434. [DOI] [PubMed] [Google Scholar]
- Chang, W. Y., D'Souza, S.J.A., and Dagnino, L. (2004). The DP-1 transcription factor is required for keratinocyte growth and epidermal stratification. J. Biol. Chem. 279, 51343–51353. [DOI] [PubMed] [Google Scholar]
- D'Amico, et al. (2000). The integrin-linked kinase regulates cyclin D1 gene through glycogen synthase kinase 3β and cAMP-responsive element-binding protein-dependent pathways. J. Biol. Chem. 42, 32649–32657. [DOI] [PubMed] [Google Scholar]
- D'Souza, S.J.A., Pajak, A., Balazsi, K., and Dagnino, L. (2001). Ca2+ and BMP-6 signalling regulate E2F during epidermal keratinocyte differentiation. J. Biol. Chem. 276, 23531–23538. [DOI] [PubMed] [Google Scholar]
- Dotto, G. P. (1999). Signal transduction pathways controlling the switch between keratinocyte growth and differentiation. Crit. Rev. Oral. Biol. Med. 10, 442–457. [DOI] [PubMed] [Google Scholar]
- Fukuhara, A., Shimizu, K., Kawakatsu, T., Fukuhara, T., and Takai, Y. (2003). Involvement of nectin-activated Cdc42 small G protein in organization of adherens and tight junctions in Madin-Darby canine kidney cells. J. Biol. Chem. 278, 51885–51893. [DOI] [PubMed] [Google Scholar]
- Grose, R., Hutter, C., Bloch, W., Thorey, I., Watt, F. M., Fassler, R., Brackebusch, C., and Werner, S. (2002). A crucial role of β1 integrins for keratinocyte migration in vitro and during cutaneous wound repair. Development 129, 2303–2315. [DOI] [PubMed] [Google Scholar]
- Hannigan, G. E., Leung-Hagesteijn, C., Fitz-Gibbon, L., Coppolino, M. G., Radeva, G., Filmus, J., Bell, J. C., and Dedhar, S. (1996). Regulation of cell adhesion and anchorage-dependent cell growth by a new beta 1-integrin-linked protein kinase. Nature 379, 91–96. [DOI] [PubMed] [Google Scholar]
- He, T. C., Zhou, S., da Costa, L. T., Yu, J., Kinzler, K. W., and Vogelstein, B. (1998). A simplified method for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 95, 2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl, D., Lichti, U., Breikreutz, D., Steinert, P. M., and Roop, D. R. (1991). Transcription of the human loricrin gene in vitro is induced by calcium and cell density and suppressed by retinoic acid. J. Investig. Dermatol. 96, 414–418. [DOI] [PubMed] [Google Scholar]
- Hynes, R. O. (2002). Integrins: Bidirectional, allosteric signaling machines. Cell 110, 673–687. [DOI] [PubMed] [Google Scholar]
- Jamora, C., and Fuchs, E. (2002). Intercellular adhesion, signalling and the cytoskeleton. Nat. Cell Biol. 4, 101–108. [DOI] [PubMed] [Google Scholar]
- Mackinnon, A. C., Qadota, H., Norman, K. R., Moerman, D. G., and Williams, B. D. (2002). C. elegans Pat-4/ILK functions as an adaptor protein within integrin adhesion. Curr. Biol. 12, 787–797. [DOI] [PubMed] [Google Scholar]
- Martin, P., and Parkhurst, S. M. (2004). Parallels between tissue repair and embryo morphogenesis. Development 131, 3021–3034. [DOI] [PubMed] [Google Scholar]
- Martinez, L. A., Chen, Y., Fischer, S. M., and Conti, C. J. (1999). Coordinated changes in cell cycle machinery occur during keratinocyte terminal differentiation. Oncogene 18, 397–406. [DOI] [PubMed] [Google Scholar]
- Morita, K., and Miyachi, Y. (2003). Tight junctions in the skin. J. Dermatol. Sci. 31, 81–89. [DOI] [PubMed] [Google Scholar]
- Mosavi, L. K., Minor, D.L.J., and Peng, Z.-Y. (2002). Consensus-derived structural determinants of the ankyrin repeat motif. Proc. Natl. Acad. Sci. USA 99, 16029–16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, D. C., Su, M.-J., Kim, R., and Bikle, D. D. (1996). Regulation of involucrin gene expression by calcium in normal human keratinocytes. Front. Biosci. 1, a16–a24. [DOI] [PubMed] [Google Scholar]
- Nikolopoulos, S. N., and Turner, C. E. (2001). Integrin-linked kinase (ILK) binding to paxillin LD1 motif regulates ILK localization to focal adhesions. J. Biol. Chem. 276, 23499–23505. [DOI] [PubMed] [Google Scholar]
- Nikolopoulos,S.N.,andTurner,C.E.(2002).Moleculardissectionofactopaxin-integrin-linked kinase-paxillin interactions and their role in subcellular localization. J. Biol. Chem. 277, 1568–1575. [DOI] [PubMed] [Google Scholar]
- Novack, A., Hsu, S. C., Leung-Hagesteijn, C., Radeva, G., Papkoff, J., Montesano, R., Roskelley, C., Grosschedl, R., and Dedhar, S. (1998). Cell adhesion and the integrin-linked kinase regulate the LEF-1 and β-catenin signaling pathways. Proc. Natl. Acad. Sci. USA 95, 4374–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojakian, G. K., Ratcliffe, D. R., and Schwimmer, R. (2001). Integrin regulation of cell–cell adhesion during epithelial tubule formation. J. Cell Sci. 114, 941–952. [DOI] [PubMed] [Google Scholar]
- Oloumi, A., McPhee, T., and Dedhar, S. (2004). Regulation of E-cadherin expression and beta-catenin/Tcf transcriptional activity by the integrin-linked kinase. Biochim. Biophys. Acta 1691, 1–15. [DOI] [PubMed] [Google Scholar]
- Perez-Moreno, M., Jamora, C., and Fuchs, E. (2003). Sticky business: Orchestrating cellular signals at adherens junctions. Cell 112, 535–548. [DOI] [PubMed] [Google Scholar]
- Pummi, K., Malminen, M., Aho, H., Karvonen, S.-L., Peltonen, J., and Peltonen, S. (2001). Epidermal tight junctions: ZO-1 and occludin are expressed in mature, developing and affected skin and in vitro differentiating keratinocytes. J. Investig. Dermatol. 117, 1050–1058. [DOI] [PubMed] [Google Scholar]
- Raghavan, S., Vaezi, A., and Fuchs, E. (2003). A role for αβ1 integrins in focal adhesion function and polarized cytoskeletal dynamics. Dev. Cell 5, 415–427. [DOI] [PubMed] [Google Scholar]
- Robles, A. I., Rodriguez-Puebla, M. L., Glick, A. B., Trempus, C., Hansen, L., Sicinski, P., Tennant, R. W., Weinberg, R. A., Yuspa, S. A., and Conti, C. J. (1998). Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 12, 2469–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Puebla, M. L., LaCava, M., Miliani de Marval, P. L., Jorcano, J. L., Richie, E., and Conti, C. J. (2000). Cyclin D2 overexpression in transgenic mice induces thymic and epidermal hyperplasia whereas cyclin D3 expression results only in epidermal hyperplasia. Am. J. Pathol. 157, 1039–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Puebla, M. L., Miliani de Marval, P. L., LaCava, M., Moons, D. S., Kiyokawa, H., and Conti, C. J. (2002). Ckd4 deficiency inhibits skin tumor development but does not affect normal keratinocyte proliferation. Am. J. Pathol. 161, 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Puebla, M. L., Robles, A. I., and Conti, C. J. (1999). Ras activity and cyclin D1 expression: an essential mechanism of mouse skin tumor development. Mol. Carcinog. 24, 1–6. [PubMed] [Google Scholar]
- Rutberg, S. E., Saez, E., Lo, S., Jang, S. I., Markova, N., Spiegelman, B. M., and Yuspa, S. H. (1997). Opposing activities of c-Fos and Fra-2 on AP-1 regulated transcriptional activity in mouse keratinocytes induced to differentiate by calcium and phorbol esters. Oncogene 15, 1337–1346. [DOI] [PubMed] [Google Scholar]
- Sakai, T., Li, S., Dicheva, D., Grashoff, C., Sakai, K., Kostka, G., Braun, A., Pfeifer, A., Yurchenco, P. D., and Fassler, R. (2003). Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion and controlling actin accumulation. Genes Dev. 17, 926–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terpstra, L., Prud'homme, J., Arabian, A., Takeda, S., Karsenty, G., Dedhar, S., and St-Arnaud, R. (2003). Reduced chondrocyte proliferation and chondrodysplasia in mice lacking the integrin-linked kinase in chondrocytes. J. Cell Biol. 162, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, Y., Li, F., Goicoechea, S., and Wu, C. (1999). The LIM-only protein PINCH directly interacts with integrin-linked-kinase and is recruited to integrin-rich sites in spreading cells. Mol. Cell. Biol. 19, 2425–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasioukhin, V., Bauer, C., Yin, M., and Fuchs, E. (2000). Directed actin polymerization is the driving force for epithelial cell–cell adhesion. Cell 100, 209–219. [DOI] [PubMed] [Google Scholar]
- Vespa, A., Darmon, A. J., Turner, C. E., D'Souza, S.J.A., and Dagnino, L. (2003). Ca2+-dependent localization of integrin-linked kinase to cell–cell junctions in differentiating keratinocytes. J. Biol. Chem. 278, 11528–11535. [DOI] [PubMed] [Google Scholar]
- Watt, F. M. (2003). Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J. 21, 3919–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C., and Dedhar, S. (2001). Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J. Cell Biol. 155, 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano, H., Mazaki, Y., Kurokawa, K., Hanks, S., Matsuda, M., and Sabe, H. (2004). Roles played by a subset of integrin signaling molecules in cadherin-based cell–cell adhesion. J. Cell Biol. 166, 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, P., Boussadia, O., Halfter, H., Grose, R., Berger, P., Leone, D. P., Charnay, P., Kemler, R., and Suter, U. (2003). E-cadherin controls adherens junctions in the epidermis and the renewal of hair follicles. EMBO J. 22, 5723–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zervas, C. G., Gregory, S. L., and Brown, N. H. (2001). Drosophila integrin-linked kinase is required at sites of integrin adhesion to link the cytoskeleton to the plasma membrane. J. Cell Biol. 152, 1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]