

Abstract
Translocation of messenger RNAs through the nuclear pore complex (NPC) requires coordinated physical interactions between stable NPC components, shuttling transport factors, and mRNA-binding proteins. In budding yeast (y) and human (h) cells, Gle1 is an essential mRNA export factor. Nucleocytoplasmic shuttling of hGle1 is required for mRNA export; however, the mechanism by which hGle1 associates with the NPC is unknown. We have previously shown that the interaction of hGle1 with the nucleoporin hNup155 is necessary but not sufficient for targeting hGle1 to NPCs. Here, we report that the unique C-terminal 43 amino acid region of the hGle1B isoform mediates binding to the C-terminal non-FG region of the nucleoporin hCG1/NPL1. Moreover, hNup155, hGle1B, and hCG1 formed a heterotrimeric complex in vitro. This suggested that these two nucleoporins were required for the NPC localization of hGle1. Using an siRNA-based approach, decreased levels of hCG1 resulted in hGle1 accumulation in cytoplasmic foci. This was coincident with inhibition of heat shock-induced production of Hsp70 protein and export of the Hsp70 mRNA in HeLa cells. Because this closely parallels the role of the hCG1 orthologue yNup42/Rip1, we speculate that hGle1-hCG1 function in the mRNA export mechanism is highly conserved.
INTRODUCTION
A key question in the nuclear transport field involves delineating how an mRNA ribonucleoprotein (mRNP) crosses the aqueous channels formed by nuclear pore complexes (NPCs) in the nuclear envelope (NE). Architecturally conserved among eukaryotes, NPCs are large supramolecular complexes composed of ∼30 different proteins (termed nucleoporins or Nups; Rout et al., 2000; Cronshaw et al., 2002; Suntharalingam and Wente, 2003). In addition to nucleoporins, multiple other factors are required for the mRNA export mechanism. Current NPC translocation models for most mRNAs are based on soluble shuttling receptors that interact with both nucleoporins and RNA-binding proteins, enabling the threading and translocation of the mRNP particles through the central NPC channel (Lei and Silver, 2002; Reed and Hurt, 2002; Vinciguerra and Stutz, 2004). Because only fully processed mature mRNP complexes are targeted to the NPC, these transport factors may be recruited to mRNPs in a temporally defined, sequential manner (Dreyfuss et al., 2002; Lei and Silver, 2002; Jensen and Rosbash, 2003; Reed, 2003; Vinciguerra and Stutz, 2004). Further insights into the NPC translocation mechanism will require critical analysis of the interface between dynamic transport factors, nucleoporins, and RNA-binding proteins.
A subset of nucleoporins contains a domain(s) rich in phenylalanine-glycine (FG) amino acid repeats that bind directly to nucleocytoplasmic shuttling receptors (Suntharalingam and Wente, 2003). There are also non-FG binding sites for shuttling transport factors (Bailer et al., 1998; Shah and Forbes, 1998; Hodge et al., 1999; Pritchard et al., 1999; Schmitt et al., 1999; Pyhtila and Rexach, 2003). The karyopherin family of transport factors (importins, exportins, and transportin) require FG binding for the import and export of cargo such as proteins and tRNA (Macara, 2001; Bednenko et al., 2003; Suntharalingam and Wente, 2003). The NXF family of mRNA export factors, with Tap/Nxf1 in vertebrates and Mex67 in yeast, are novel FG-binding proteins that are structurally distinct from karyopherins (Izaurralde, 2002; Reed and Hurt, 2002). Although the Schizosaccharomyces pombe Mex67 is not strictly required (Yoon et al., 2000), Tap and Mex67 are essential for export of most cellular mRNAs in vertebrate and yeast Saccharomyces cerevisiae cells, respectively, and Tap is also required for export of retroviral RNAs bearing unique structural features (Segref et al., 1997; Gruter et al., 1998; Braun et al., 1999; Katahira et al., 1999; Bachi et al., 2000; Herold et al., 2003). Through two distinct regions, the ubiquitin association-like domain and the NTF2-like domain, Tap interacts with nucleoporin FG domains (Fribourg et al., 2001; Schmitt and Gerace, 2001; Grant et al., 2002, 2003). In addition, as a heterodimeric complex with the NTF2-like protein p15/Mtr2, Tap/Mex67's FG binding and mRNA export activity are enhanced (Black et al., 1999; Strasser et al., 2000; Guzik et al., 2001; Levesque et al., 2001; Katahira et al., 2002; Wiegand et al., 2002). Although capable of directly binding to RNA, Tap and Mex67 association with mRNA is mediated by the interaction with mRNA-binding proteins (Strasser and Hurt, 2000; Stutz et al., 2000; Huang et al., 2003; Gilbert and Guthrie, 2004; Vinciguerra and Stutz, 2004). Hence, through its dual function as an NPC- and mRNA-binding protein, it is thought Tap and Mex67 promote the translocation of mRNPs across NPCs in vertebrate and yeast cells.
The essential mRNA export factor Gle1 is also uniquely positioned to execute events required for the translocation of mRNPs through the NPC and their release in the cytoplasm. Gle1 is strictly required for the export of polyadenylated (poly(A)+) RNA in human, fission yeast, and budding yeast (Del Priore et al., 1996; Murphy and Wente, 1996; Watkins et al., 1998; Kendirgi et al., 2003). Moreover, nucleocytoplasmic shuttling of human (h) Gle1 is required for mRNA export in HeLa cells (Kendirgi et al., 2003). Gle1 has multiple potential functional domains (Rayala et al., 2004). Recent studies have uncovered a subset of interactions between Gle1, nucleoporins, mRNA-binding proteins, and other mRNA export factors. In budding yeast (y), yGle1 interacts with a novel protein Gfd1 that may serve as a bridging factor for association with mRNA-bound heterogeneous nuclear RNP (hnRNP) protein Nab2 (Hodge et al., 1999; Strahm et al., 1999; Suntharalingam et al., 2004). yGle1 also interacts with Dbp5, a highly conserved essential DEAD box protein with helicase activity (Snay-Hodge et al., 1998; Hodge et al., 1999; Strahm et al., 1999). The association of Dbp5 with yGle1 and nucleoporins is key for mRNA export (Hodge et al., 1999; Schmitt et al., 1999; Strahm et al., 1999; Zhao et al., 2002), and yGle1 and Dbp5 functions are regulated by the production of inositol hexakisphosphate (York et al., 1999; Miller et al., 2004). Finally, Gle1 and Tap/Mex67 are functionally linked to Rae1/Gle2, a factor with a specific NPC docking site and mRNA export roles (Brown et al., 1995; Murphy et al., 1996; Bailer et al., 1998; Pritchard et al., 1999; Yoon et al., 2000; Blevins et al., 2003). There is evidence that yGle1 and Mex67 act at distinct steps in the mRNA export pathway (Strawn et al., 2001; Miller et al., 2004). However, how the actions Gle1, Tap/Mex67, Dbp5, and Rae1/Gle2 are fully coordinated is unknown.
To further investigate the mRNA export mechanism, more detailed analysis of the Gle1 physical interaction partners will be required. To date, the only reported direct nucleoporin interactors include Nup42/Rip1 for yGle1 in S. cerevisiae cells and Nup155 for hGle1 in human cells (Murphy and Wente, 1996; Stutz et al., 1997; Strahm et al., 1999; Rayala et al., 2004). We recently characterized two hGle1 protein variants (hGle1A and hGle1B) that differ only at their extreme C-termini (Kendirgi et al., 2003; Figure 1). They both share an N-terminal 29 amino acid (aa) region that is necessary for interaction with Nup155 (Rayala et al., 2004); however, only hGle1B localizes at steady state to the NPC (Kendirgi et al., 2003). Thus, the interaction of hGle1A with Nup155 is not sufficient for NPC targeting. We speculated that the unique C-terminal 43 aa region of hGle1B plays a critical role in hGle1B-NPC interactions. Here, we report that the unique 43 aa C-terminal domain of hGle1B is necessary and sufficient for interaction with the nucleoporin hCG1 (also referred to as NPL1), the human orthologue of S. cerevisiae Nup42/Rip1 (Katahira et al., 1999; Strahm et al., 1999). Moreover, we have pinpointed roles for the hGle1B-hCG1 interaction in proper subcellular localization of hGle1 and in the export of mRNA required for heat shock protein 70 (Hsp70) production.
Figure 1.

Schematic representation of structural and functional features of hGle1B and hGle1A. Amino acid (aa) residues 1-29 delineate hNup155-binding domain (Nup155BD; Rayala et al., 2004), aa 105-360 the predicted coiled-coil domain, aa 444-483 the nucleocytoplasmic shuttling domain required for hGle1-mediated mRNA export (Kendirgi et al., 2003), aa 484-559 a novel nuclear localization region (NLR) with weak nuclear import activity (Kendirgi and Wente, unpublished results), and aa 655-698 the hCG1-binding domain (hCG1BD) which is absent from hGle1A (Kendirgi et al., 2003). The regions used in Figure 3B as His-tagged fusions (hGle1B548-698 and hGle1B655-698) are also shown.
MATERIALS AND METHODS
Yeast Two-hybrid Screen
The Matchmaker HeLa cell cDNA library (BD Biosciences Clontech, Mountain View, CA) was used to screen for protein interactors with GBD-hGle1B478-698 (pSW3018) in PJ69-4A yeast strain (James et al., 1996). Transformations were conducted using the lithium acetate method (Ito et al., 1983) and cells were plated on synthetic minimal (SM) media supplemented with 2% glucose (D) lacking leucine, uracil and histidine (LUH). Clones (n = 4.5 × 106) were screened, and colonies able to survive on LUH at 30°C were replica plated to SMD media lacking LUH and adenine (A). The 287 colonies that grew on SMD lacking LUHA were then tested for β-galactosidase expression by scoring for blue color in filter and liquid assays. All positive clones identified were subsequently tested for absence of self-activation and non-specific interaction with a GBD fusion to the coding sequence for Snf1 protein (Yang et al., 1992). In addition, only clones interacting with GBD-hGle1B (pSW3017) and not GBD-hGle1A (pSW938) were selected.
Recombinant Proteins
For expression of full-length hCG1 and hCG1 domains fused to glutathione-S-transferase (GST), Escherichia coli BL21 (DE3) cells were transformed with respective plasmids from Table 1. Recombinant protein expression was induced with 0.3 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG) for 12 h at 25°C. Bacterial pellets were frozen at -20°C before lysis in phosphate-buffered saline (PBS) containing 5 mM dithiothreitol (DTT), protease inhibitor cocktail (Roche, Indianapolis, IN), and 0.1 mg/ml lysozyme. Bacterial lysis was completed by French Press (11,000 psi, twice) and incubation for 30 min at 4°C in 1% Triton X-100. Lysates were subsequently cleared by centrifugation at 10,000 × g for 20 min. Supernatants were incubated with glutathione-Sepharose beads (GE Healthcare, Piscataway, NJ) for 1 h at 4°C, and beads were then washed in cold PBS buffer containing 0.2% Tween 20. GST-fusion proteins were eluted with 15 mM glutathione in 20 mM Tris, pH 8.5, containing 150 mM NaCl and 1 mM DTT. Proteins were concentrated using Centricon 30K (Millipore, Bedford, MA) and dialyzed against binding buffer (BB: 20 mM HEPES, pH 7.4, 110 mM KAc, 2 mM MgCl2, 0.1% Tween 20) overnight.
Table 1.
List of plasmids used in this study
| Plasmids | Fusion protein expressed | Description |
|---|---|---|
| Yeast two-hybrid plasmids | ||
| pSW3017 | GBD-hGle1B | Rayala et al. (2004) |
| pSW3018 | GBD-hGle1B478-698 | hGle1B aa 478-698 in fusion with GBD |
| pSW938 | GBD-hGle1A | Rayala et al. (2004) |
| CP3068 | GAD-hCG1 | hCG1 full-length in fusion with GAD |
| CP3069 | GAD-hCG1190-423 | hCG1 aa 190-423 in fusion with GAD |
| pSW3072 | GAD-hCG1366-423 | hCG1 aa 366-423 in fusion with GAD |
| pSW3073 | GAD-hCG11-165 | hCG1 aa 1-165 in fusion with GAD |
| pSW3074 | GAD-hCG1166-204 | hCG1 aa 166-204 in fusion with GAD |
| pSW3075 | GAD-hCG1205-365 | hCG1 aa 205-365 in fusion with GAD |
| CP77 | GBD-Snf1 | Gift from M. Carlson (Yang et al., 1992) |
| Prokaryotic expression plasmids | ||
| pSW3071 | GST-hCG1 | Full-length hCG1 in fusion with GST |
| pSW3077 | GST-hCG11-365 | hCG1 aa 1-365 in fusion with GST |
| pSW3084 | GST-hCG1366-423 | hCG1 aa 366-423 in fusion with GST |
| pSW3138 | His-hCG11-165 | hCG1 aa 1-165 in fusion with 6× His |
| pSW3101 | His-hGle1548-698 | hGle1B aa 548-698 in fusion with 6× His |
| pSW3102 | His-hGle1655-698 | hGle1B aa 655-698 in fusion with 6× His |
| pSW1413 | GST-hGle1483-559 | hGle1 aa 483-559 in fusion with GST |
| pSW3087 | GST-hCG1366-403 | hGle1 aa 366-403 in fusion with GST |
| pSW3086 | GST-hCG1366-383 | hGle1 aa 366-383 in fusion with GST |
| pSW3089 | GST-hCG1379-423 | hGle1 aa 379-423 in fusion with GST |
| pSW3088 | GST-hCG1403-423 | hGle1 aa 403-423 in fusion with GST |
| CP336 | GST-GFP | Gift from I. Macara (Richards et al., 1996) |
| Eukaryotic expression plasmids | ||
| pSW3099 | hNup155 | Full-length hNup155 |
| pSW3079 | hCG1-GFP | Full-length hCG1 in fusion with EGFP |
| pSW3110 | hGle1B | hGle1B full-length in pCDNA3 vector |
| pSW1406 | hGle1A | hGle1A full-length in pCDNA3 vector |
| pSW3080 | hCG11-365-GFP | hCG1 aa 1-365 in fusion with EGFP |
| pSW3081 | hCG1366-423-GFP | hCG1 aa 366-423 in fusion with EGFP |
His-tagged, 6×, recombinant protein expression was carried out in DH5α cells by induction with 0.3 mM IPTG for 3 h at 37°C. Cells were pelleted and lysed as described above in 50 mM Tris, pH 8.5, 5 mM BME, and 1 mM phenylmethylsulfonyl fluoride at 4°C. Cleared lysates were incubated with nickel-Sepharose beads (Ni-NTA, Qiagen, Chatsworth, CA) for 1 h at 4°C, extensively washed with 20 mM Tris, pH 8.5, 5 mM BME, 500 mM KCl, and 10 mM imidazole and eluted in wash buffer contain 100 mM imidazole. Eluted 6× His-tagged recombinant proteins were subsequently concentrated and dialyzed as described above.
In Vitro Binding Assays Using Recombinant GST or His-tagged Fusion Proteins
Radiolabeled proteins were translated from in vitro transcribed RNA using a T7 coupled transcription/translation reticulocyte lysate system (Promega, Madison, WI), in the presence of [35S]l-methionine (1000 Ci/mmol at 10 mCi/ml; ICN Biomedicals, Costa Mesa, CA), according to the manufacturer's instructions. For GST-based experiments, ∼5 μg of the respective GST-fusion protein was immobilized on glutathione-Sepharose beads (GE Healthcare) and incubated with the 35S-labeled protein(s) at 4°C for 2.5 h in binding buffer (110 mM KAc, 20 mM HEPES-KOH, pH 7.4, 2 mM NaAc, 5 mM Mg(Ac)2, 1 mM DTT, 10% glycerol, 0.2% Tween-20, and protease inhibitors). Beads were subsequently washed in Binding Buffer and resuspended in SDS-sample buffer. Proteins were separated by SDS-PAGE and analyzed by autoradiography, Coomassie staining or Western blotting as indicated in each figure. In experiments where [35S]hGle1B and [35S]hNup155 were used together, the radiolabeled proteins were first combined in transcription/translation buffer for 1 h at room temperature before addition of immobilized GST-hCG1 protein.
For His-tagged fusion protein experiments, 35S-labeled proteins were incubated with ∼5 μg of the respective purified His-tagged recombinant protein in 425 μl of Binding Buffer without DTT, and with complete EDTA-free protease inhibitors cocktail (Roche, Indianapolis, IN). Ni-NTA beads (10 μL; Qiagen) were subsequently added, and the incubation was carried out for an additional 1 h at 4°C. After extensive washing in Binding Buffer lacking DTT, bound complexes were eluted in SDS-sample buffer and analyzed as above.
Antibody Production and Western Blotting
Anti-peptide antibodies against hNup155 and hGle1B were raised in rabbits. The C-terminal residues of hNup155 (VQAITGNFKSLQAKLERLH) and the C-terminal residues of hGle1B (EKCLQHKDIPVPKGFL) synthetic peptides were synthesized and used as antigens and for affinity purification of the respective antibodies. Peptide synthesis, antibody production, and affinity purification were conducted by Bethyl Laboratories (Montgomery, TX). For Western blotting, after SDS-PAGE, proteins were transferred to nitrocellulose membranes using Mini Trans-Blot Cell (Bio-Rad, Hercules, CA), blocked in 5% nonfat milk in TBST, and incubated with rabbit anti-GFP (1:2000) (gift from M. Linder, Washington University School of Medicine, St. Louis, MO), mouse anti-β actin (1:10,000; Sigma), mouse mAb414 (1:20; Davis and Blobel, 1986), rabbit anti-Tap (1:1000; (Braun et al., 1999), mouse anti-Nup153 (1:1; Bodoor et al., 1999), rabbit anti-Nup155 (1:6000), mouse anti-Transportin 1 (1:1000; BD Transduction, Lexington, KY), rabbit anti-GLFG for Nup98 (1: 2500, WU956; Bucci and Wente, 1998), mouse anti-His (1:2000; Qiagen), or rabbit anti-hGle1B peptide antibodies (1:1000). Secondary alkaline phosphatase-conjugated anti-mouse or anti-rabbit antibodies (1:7000; Promega) were used followed by color development according to the manufacturer's recommendations.
HeLa Cell Culture, Plasmid DNA Transfections, and Microscopy
CCL2 HeLa cells were maintained as previously described (Kendirgi et al., 2003). Transfection of expression plasmids was carried out using Polyfect (Qiagen) according to manufacturer's recommendations. Eighteen to 20 h posttransfection, cells were analyzed by direct fluorescence for green fluorescent protein (GFP) signal and by indirect immunofluorescence microscopy. Cells were fixed with 3% paraformaldehyde in PBS (pH 7.3) at room temperature for 20 min and permeabilized in PBS containing 0.5% Triton X-100 for 10 min. Cells were then blocked with 2% bovine serum albumin in PBS for 60 min, followed by incubation with primary antibodies diluted in blocking buffer for 2 h at room temperature (1:200, affinity-purified polyclonal rabbit anti-hGle1; Watkins et al., 1998) and mAb414 1:10 tissue culture supernatant (Davis and Blobel, 1986). Coverslips were then extensively washed with PBS and incubated with rhodamine-conjugated donkey anti-rabbit (1:200) and FITC-conjugated donkey anti-mouse (1:200) secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) for 60 min at room temperature, followed by washes in PBS. The coverslips were mounted in 90% glycerol containing paraphenylenediamine, and cells were observed using an Olympus BX50 microscope (Lake Success, NY). Digital images were recorded with the photometric CoolSNAP HQ camera (Roper Scientific, Tucson, AZ) using MetaVue 6.0 software (Molecular Devices, Downingtown, PA) and processed in Photoshop 7.0 (Adobe Systems, San Jose, CA).
Synthesis and Transfection of siRNA Pools
The sequence encoding the hCG1 C-terminal domain was cloned into Litmus 28i vector (NEB, pSW3104). hCG1C small interfering RNA (siRNA) pool was generated using ShortCut RNAi kit (New England Biolabs, Beverly, MA) according to the manufacturer's recommendation. Briefly, pSW3103 was linearized with EcoRI and XhoI separately before gel purification. Ten micrograms of each linearized construct was combined in a 1:1 ratio, and dsRNA was synthesized in vitro using T7 RNA polymerase. The reaction was incubated at 37°C overnight before addition of 2 U of RNAse-free DNAse I (RQ1 DNase, Promega). The dsRNA was purified by gel filtration and ethanol/salt precipitated. dsRNA, 10 μg, was subsequently used to synthesize siRNAs in vitro using RNase III nuclease provided in the ShortCut RNAi kit. The siRNA pool was ethanol/salt precipitated, resuspended in water and the concentration was estimated by OD260 before storing at -20°C. Control siRNA was synthesized using the Litmus 28i empty vector. The siRNA pools (30 nM) were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) as recommended by the manufacturer. Cells were grown in six-well plates at 30% confluence the day preceding the transfection. After transfection, cells were incubated in the presence of siRNAs/lipid carrier for 48 h before analysis (see below). When the siRNA pools were cotransfected with expression plasmids, 1.5 μg of plasmid DNA was used together with 30 nM of siRNA pools.
Heat-shock Assay
Heat-shock experiments were conducted 48 h after treatment with siRNA pools. HeLa cells in 35-mm dishes were rinsed with 1× PBS, and 1 ml of prewarmed (37 or 42°C) DMEM lacking methionine and containing 10% fetal bovine serum was added. After 30-min incubation in 5% CO2 at the appropriate temperature, [trans-35S] Label (100 μCi, 7 mCi/ml; ICN) was added, and cells were quickly returned to designated temperature for an additional 90 min. After metabolic labeling, cells were rinsed with 1× PBS, lysed in 150 μL of 1× SDS-sample buffer, and boiled. Samples were separated by SDS-PAGE, and gels were incubated with Amplify (GE Healthcare), dried, and placed on BioMAX MR (Eastman Kodak, Rochester, NY) film for autoradiography.
Cytoplasm/Nuclear RNA Isolation and RT-PCR
After siRNA treatment and heat shock as described above, HeLa cells were rinsed in PBS and covered with ice cold lysis buffer (1× PBS, 0.05% NP40, 1 mM DTT, 1U/μL RNasin; Promega). After a 10-min incubation on ice, the supernatant containing the cytoplasmic RNA fraction was collected and cleared by centrifugation at 13,000 rpm for 10 min at 4°C. The nuclear RNA fraction was isolated by addition of lysing buffer to the prelyzed cells together with Trizol LS (Invitrogen) reagent. Total RNA from each fraction was purified according to the manufacturer's recommendation. Genomic DNA contamination of the purified fractions was estimated by PCR before RT using the actin primers below. Each RNA fraction was subsequently reverse transcribed using ImProm-II reverse transcription system (Promega) as recommended by the manufacturer using actin-specific and Hsp70-specific reverse primers: 5′-CCATCTCTTGCTCGAAGTCC-3′ and 5′GTGCTTCGGAACAGGTCG-3′, respectively. Serial dilutions of the resulting cDNAs were made, and 10% of each was used in PCR studies. For actin and Hsp70 coamplification, primers sets 5′-GATTCCTATGTGGGCGACGAG-3′ (actin forward); 5′-CCATCTCTTGCTCGAAGTCC-3′ (actin reverse) and 5′-TTCACGGACACCGAGCG-3′ (Hsp70 forward); 5′GTGCTTCGGAACAGGTCG-3′ (Hsp70 reverse) were used in the same amplification reaction. Amplification was performed using RoboCycler Gradient 40 (Stratagene, La Jolla, CA) and the following conditions: 94°C for 30 s, 55°C for 1 min, and 72°C for 45 s (27 cycles). Forty percent of each reaction was separated by electrophoresis through a 1.2% agarose gel stained with ethidium bromide. Images of agarose gels were digitized, and band intensity was quantified corresponding to the Hsp70 and actin PCR products with ImageQuant (version 5.2) software (Molecular Dynamics, GE Healthcare). The dilution samples falling in the linear range were used to analyze the ratio of nuclear to cytoplasmic.
RESULTS
hGle1B Interacts with the Nucleoporin hCG1
To identify the protein(s) that interact specifically with hGle1B, a yeast two-hybrid screen was conducted. A bait plasmid was generated that harbored the sequence encoding the Gal4-binding domain (GBD) fused to the sequence encoding the C-terminal region of hGle1B from aa 478-698 (GBD-hGle1B478-698). This hGle1 region contained the unique 43 aa span as well as an additional 177 aa to account for structural versus primary binding site functions of the unique sequence. A yeast reporter strain harboring GBD-hGle1B478-698 was transformed with a HeLa cell cDNA library expressing Gal4-activation domain (GAD) fusions. Individual clones, 4.5 × 106, were screened, and positive interaction with GBD-hGle1B478-698 was scored based on the growth of yeast colonies on media lacking leucine, uracil, histidine, and adenine (-LUHA). A series of specificity tests were conducted (see Material and Methods). To discriminate between the positive isolates that could interact with full-length (FL) hGle1B versus FL hGle1A, direct two-hybrid assays were performed. We found 14 that interacted specifically with hGle1B. Restriction endonuclease digestion patterns and DNA sequence information revealed that the 14 clones encoded for the nucleoporin NLP1/hCG1 (Katahira et al., 1999; hereafter referred to as hCG1). Of these, 10 harbored the cDNA for the FL open reading frame (ORF) encoding hCG1 (aa 1-423), 2 encoded for hCG1 aa 7-423, and another 2 contained an N-terminal truncation of the hCG1 ORF encoding aa 190-423 (Figure 2).
Figure 2.

hGle1B and hCG1 interact in the yeast two-hybrid assay. A schematic representation of hCG1 domain structure is shown. Amino acid (aa) residues 1-24 form a Zinc finger domain (ZF), aa 165-203 a predicted coiled-coil domain (C-C), aa 204-365 the FG-repeat domain (harboring phenylalanine (F)-glycine (G) repeats), and aa 366-423 the carboxy (C)-terminal domain. Three classes of GAD-hCG1 clones were isolated in the yeast two-hybrid screen and are highlighted in the shaded area. Interaction in the two-hybrid assay was detected by growth on media lacking leucine, uracil, histidine, and adenine (-LUHA) (for GBD-hGle1 and GBD-Snf1) or lacking leucine, uracil, histidine, and tryptophan (for GBD-hGle1A) (unpublished data). Growth was scored with (-) representing no growth and (+) representing growth. Each GAD fusion was tested for specificity by cotransformation with GBD-Snf1 and for self-activation (unpublished data).
The Unique C-terminal 43 aa Region of hGle1B Is Necessary and Sufficient for the In Vitro Interaction with hCG1
To independently confirm the yeast two-hybrid results, we analyzed the interaction of hCG1 with hGle1A and hGle1B proteins by in vitro coprecipitation assays. Recombinant hCG1 fused with the glutathione-S-transferase protein (GST-hCG1) and GST alone were expressed and purified from bacteria. [35S]methionine-labeled hGle1B and hGle1A were generated in an in vitro rabbit reticulocyte lysate system and mixed with the recombinant GST fusion proteins and glutathione-Sepharose beads. Binding of [35S]hGle1A or [35S]hGle1B to GST-hCG1 was determined by eluting proteins from the glutathione beads in SDS sample buffer and separation by SDS-PAGE followed by autoradiography. As shown in Figure 3A, [35S]hGle1B coprecipitated specifically with GST-hCG1 and not with GST alone. Only background levels of [35S]-hGle1A bound to GST-hCG1 and GST. These results indicated that in contrast to hGle1B, hGle1A did not interact with hCG1 in vitro. This also correlated directly with the two-hybrid analysis and strongly suggested that the unique C-terminal region of hGle1B is required for the interaction with the nucleoporin hCG1.
Figure 3.

hCG1 interacts in vitro specifically with the unique hGle1B 43 aa C-terminal region. (A) Bacterially expressed/purified GST and GST-hCG1 (left) were incubated with in vitro transcribed-translated 35S-labeled FL hGle1A and hGle1B. Input and bound fractions were resolved by SDS-PAGE and detected by Coomassie staining or autoradiography. Each lane represents 20% of the input or bound fractions for Gle1. (B) In vitro transcribed-translated [35S]hCG1 was incubated with bacterially expressed and purified His-tagged hGle1548-698 and His-hGle1655-698. As a negative control, a His-fusion of hCG1 region spanning amino acids 1-165 was used. Samples were resolved by SDS-PAGE and analyzed by autoradiography or Western blotting. The Western blot with anti-His and anti-hGle1B peptide antibodies shows the recombinant protein used in the respective binding assay. Each lane represents 20% of the input or bound fractions for hCG1.
To test whether the unique 43 aa domain of hGle1B (aa 655-698; Figure 1) is sufficient for binding to hCG1, further in vitro binding assays were conducted (Figure 3B). Recombinant His-tagged hGle1B655-698 and His-tagged hGle1B478-698 were expressed and purified from bacteria. Each recombinant protein was incubated with [35S]hCG1 generated in a reticulocyte lysate system. Using nickel-Sepharose, the recombinant proteins were isolated and coisolation of [35S]hCG1 was determined (Figure 3B). In this assay, similar amounts of [35S]hCG1 were bound to His-hGle1B655-698 and His-hGle1B478-698 In contrast, minimal [35S]hCG1 was detected bound to the negative control His-hCG11-165. Thus, we concluded that the unique C-terminal 43 aa span of hGle1B is necessary and sufficient for the interaction with hCG1.
The non-FG C-terminal Region of hCG1 Mediates the Interaction with hGle1B
On the basis of the fusion proteins isolated in the two-hybrid screen, we speculated that the hGle1B-binding site in hCG1 resides in the C-terminal region of hCG1. To further map the region of hCG1 responsible, a panel of GAD-hCG1 deletion proteins was tested by the two-hybrid assay for interaction with GBD-hGle1B or GBD-Snf1. This included four GAD fusion constructs harboring the hCG1 coiled-coil domain (GAD-hCG1166-204), the hCG1 FG-repeat domain (GAD-hCG1205-365), the hCG1 C-terminal domain (GAD-hCG1366-423), or the hCG1 coiled-coil and FG-repeat domains(GAD-hCG1165-365). As shown in Figure 2, only yeast cells cotransformed with plasmids expressing GBD-hGle1B and GAD-hCG1366-423 or GAD-hCG1190-423 grew on -LUHA media. These results suggested that the hGle1B-binding domain in hCG1 was located within a 57 aa span (aa 366-423) at the C-terminal end of hCG1.
To independently confirm the yeast two-hybrid interaction between hGle1B and hCG1 and further delineate the binding domain in hCG1, in vitro binding assays were conducted using [35S]hGle1B from the reticulocyte lysate system and bacterially expressed, purified GST-hCG1 proteins. As shown in Figure 4A, [35S]hGle1B was isolated with GST-hCG1 and GST-hCG1366-423. However, [35S]hGle1B did not bind GST-hCG11-365 or GST alone. Furthermore, as shown in Figure 4B, the last 44 aa residues (379-423) of hCG1 showed significant levels of bound [35S]hGle1B. This binding was similar to the FL C-terminal domain (GST-hCG1366-423). In contrast, GST-GFP (as a negative control), GST-hCG11-365, and GST-hCG1366-403 showed minimal binding. Thus, we concluded that in vitro the last 44 aa residues of hCG1 are necessary and sufficient to mediate the interaction with hGle1B.
Figure 4.

hGle1B binds the non-FG C-terminal region of hCG1. Bacterially expressed, purified GST-hCG1 fusions or GST alone were incubated with in vitro-transcribed/translated [35S]hGle1B and glutathione-Sepharose beads. Bound proteins were eluted and analyzed by SDS-PAGE followed by Coomassie staining (A) or autoradiography (B). (C) Schematic representation of the hCG1 protein/domains and the various deletions of hCG1 C-terminal domain (hCG1366-423) generated as fusions with GST in bacteria. (D) Delineating the minimal region in hCG1 capable of binding hGle1B. [35S]hGle1B was expressed in a rabbit reticulocyte lysate system and incubated with the various GST-hCG1 fusions as well as with GST-hCG11-365 and GST-GFP (negative controls). Binding of [35S]hGle1B was only detected with GST-hCG1366-423 and GST-hCG1379-423. In A, B, and C, each lane represents 20% of the respective reaction. Asterisk (*) indicates degradation products.
hCG1 and hCG1366-423 Fused to GFP Colocalize at the Nuclear Envelope with hGle1 in HeLa Cells
If the hCG1366-423 region was sufficient for interacting with hGle1B, we speculated that GFP-tagged hCG1366-423 and FL-hCG1 should colocalize with endogenous hGle1 in HeLa cells. The subcellular distribution of ectopically expressed hCG1-GFP and GFP-hCG1366-423 was examined in Triton X-100 permeabilized/fixed HeLa cells using GFP direct fluorescence and indirect immunofluorescence with anti-hGle1 antibodies. Both hCG1-GFP and GFP-hCG1366-423 were detected at the NE coincident with hGle1 (Figure 5). In contrast, GFP alone did not colocalize with hGle1. Thus, the C-terminal domain of hCG1 mediates NE/NPC localization of hCG1.
Figure 5.

Localization of endogenous hGle1 and GFP-tagged hCG1 proteins in HeLa cells. HeLa cells were transiently transfected with plasmids expressing GFP, FL hCG1-GFP, hCG11-365-GFP, and GFP-hCG1366-423. Indirect immunofluorescence with anti-hGle1 polyclonal antibodies was used to detect endogenous hGle1. GFP and GFP-tagged proteins were detected by direct fluorescence. (arrows) highlight NE rim. Bar, 10 μm.
To determine whether the hCG1 C-terminal region is required for targeting hCG1 to the NE, we generated a GFP fusion construct expressing hCG1 lacking its C-terminal domain (hCG11-365, Figure 5, bottom panels). Interestingly, hCG11-365-GFP no longer localized with hGle1 at the NE. In contrast, the hCG11-365-GFP showed nuclear localization in a distinct punctate pattern. These results indicated that the hCG1 C-terminal domain, which also harbors the hGle1B-binding domain, is necessary and sufficient for targeting hCG1 to the NE.
hGle1 Can Interact Simultaneously with hNup155 and hCG1 In Vitro
We have previously shown that the binding of hGle1B to hNup155 is necessary but not sufficient for the NE localization of hGle1B. The hNup155-binding domain (Nup155BD) in hGle1 resides in the first 29 N-terminal residues of hGle1B (Rayala et al., 2004). This domain is distinct from the hCG1-binding domain (hCG1BD) found here in hGle1B (Figure 1). To test whether hGle1B could interact with hNup155 and hCG1 simultaneously, recombinant GST-hCG1 immobilized on glutathione-Sepharose beads was incubated with [35S]hGle1B and [35S]hNup155 generated in reticulocyte lysates. As shown in Figure 6A, [35S]hGle1B and [35S]hNup155 were coisolated with the GST-hCG1-coated beads. In the absence of hGle1B, minimal [35S]hNup155 was detected (Figure 6B). These results suggested that hGle1B can bind both nucleoporins at the same time. Therefore, we propose that in vitro hGle1B can form a trimeric complex with two nucleoporins, hCG1 and hNup155.
Figure 6.

hNup155, hCG1, and hGle1B form a heterotrimeric complex in vitro. In vitro transcribed-translated [35S]hNup155 was incubated with bacterially expressed/purified GST-hCG1 in the presence (A) or absence (B) of [35S]hGle1B. The bound proteins were isolated with glutathione-Sepharose, eluted, and separated by SDS-PAGE. Proteins were detected by Coomassie staining or autoradiography. Twenty percent of each reaction was analyzed.
hCG1 Knockdown in HeLa Cells Reduces Hsp70 Protein Levels and Inhibits Hsp70 mRNA Export
To analyze hCG1 function in cells, an RNA interference approach, siRNA was used to deplete hCG1 protein levels in HeLa cells. A pool of siRNA against the nucleotide sequence encoding the hCG1 C-terminal region from aa 366-423 (hCG1C) was generated using an in vitro synthesis strategy (Donze and Picard, 2002). A control siRNA pool that does not recognize human mRNA was also generated (see Materials and Methods). First, the activity of the hCG1C siRNA pool was confirmed by cotransfecting HeLa cells with the hCG1-GFP expression plasmid and the hCG1C siRNAs. The hCG1-GFP protein levels were analyzed by Western blotting with anti-GFP antibodies (Figure 7). Forty-eight hours after transfection with 30 nM of hCG1C siRNAs, the hCG1-GFP protein levels were significantly reduced compared with cells treated with control siRNAs (see Materials and Methods). Second, we tested the specificity of the hCG1C siRNA pool. The expression levels of various nucleoporins (Nup98, Nup153, p62, and Nup155) and transport factors (Tap, hGle1, and Trn1) in cells treated with either control or hCG1C siRNA pools were evaluated by Western blotting. As shown in Figure 7, the protein levels of all these nucleoporins and transport factors were not changed. This led us to conclude that the hCG1C siRNAs were specific for knocking down hCG1 protein levels.
Figure 7.

Specific hCG1C siRNA depletion of hCG1 protein levels in HeLa cells. Forty-eight hours after cotransfection with hCG1-GFP expression plasmid and 30 nM of hCG1C siRNAs or control siRNAs, HeLa cells were lysed in SDS-sample buffer and fractions separated by SDS-PAGE for Western blotting. Membranes were blotted with anti-GFP (hCG1-GFP), anti-β actin (actin), mAb414 (p62), anti-Tap (Tap), anti-Nup153 (Nup153), anti-Nup155 (Nup155), anti-Transportin 1 (Trn1), anti-Nup116 GLFG (Nup98), and anti-hGle1B peptide anti-bodies (hGle1B).
Previous studies in budding yeast have demonstrated that the functional orthologue of hCG1, S. cerevisiae Nup42/Rip1, is required for export of heat shock-induced mRNA (Saavedra et al., 1997; Stutz et al., 1997; Strahm et al., 1999; Rollenhagen et al., 2004). We hypothesized that hCG1 may play a similar role in vertebrate cells and analyzed the ability of hCG1C siRNA-treated cells to produce Hsp70. A block in the export of transcripts encoding heat-shock proteins should result in decreased Hsp70 protein levels under heat shock conditions. HeLa cells transfected with 30 nM hCG1C siRNAs or control siRNAs were heat-shocked at 42°C for 2 h in the presence of [35S]methionine. For each set of siRNA transfections, protein expression patterns from 42°C heat shock-treated or 37°C control cells were analyzed by SDS-PAGE followed by autoradiography. As shown in Figure 8A, the Hsp70 protein levels were clearly elevated in heat-shocked cells transfected with control siRNAs (arrowhead). However, cells treated with hCG1C siRNAs showed a significant decrease in Hsp70 protein levels, whereas the overall protein expression levels remained unchanged.
Figure 8.
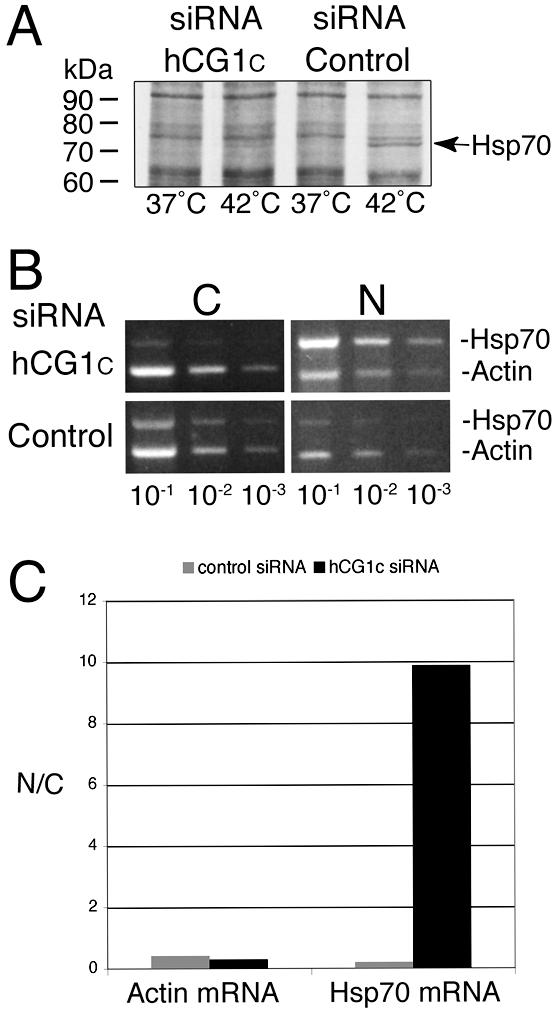
siRNA depletion of hCG1 inhibits Hsp70 protein production and nuclear export of Hsp70 mRNA. (A) Assessment of Hsp70 heat-shock protein expression levels in siRNA transfected cells. Forty-eight hours after transfection with 30 nM of hCG1C siRNAs or control siRNAs, HeLa cells were heat-shocked for 2 h in the presence of [35S]methionine. Total cell protein was subsequently extracted in SDS-sample buffer and resolved by SDS-PAGE followed by autoradiography. (B) Semiquantitative analysis of actin and Hsp70 mRNA distribution in subcellular fractions of siRNA treated HeLa cells after heat shock. After treatment with siRNA pools, cells were heat-shocked at 42°C for 2 h and total RNA from nuclear and cytoplasmic fractions was isolated and reverse-transcribed with actin- and Hsp70-specific primers. Serial dilutions were made and subsaturating PCR-based amplifications were performed. The products were separated on agarose gels and stained with ethidium bromide. (N, nuclear fraction; C, cytoplasmic fraction). (C) Quantitative analysis of the representative fractionation experiment in B. Bands corresponding to Hsp70 and actin PCR products in the linear dilution range (10-2 dilution) were quantified (see Materials and Methods), and the respective nuclear/cytoplasmic ratio was plotted as a bar graph.
To confirm that the decrease in Hsp70 protein levels was due to a defect in mRNA export, biochemical fractionation experiments were conducted with heat-shocked HeLa cells pretreated with siRNAs. The relative amounts of Hsp70 mRNA and actin mRNA were measured in cytoplasmic and nuclear fractions using a semiquantitative RT-PCR strategy. A representative trial is shown in Figure 8B. After treatment with the control siRNAs, the Hsp70 and actin mRNAs were found in both the cytoplasmic and nuclear fractions of the heat-shocked cells (Figure 8B, bottom row). Quantitative analysis was conducted (Figure 8C), with the control siRNA samples having nuclear/cytoplasmic ratios for the Hsp70 and actin mRNA levels at 0.21 and 0.42, respectively. The relative abundance of the actin mRNA was unchanged in the heat-shocked cells transfected with the hCG1C siRNAs, with the nuclear/cytoplasmic ratio at 0.30 for the representative experiment shown (Figure 8B, top row; Figure 8C). In sharp contrast, in the same hCG1C siRNA treated cells, the nuclear level of Hsp70 mRNA was significantly greater than the cytoplasmic level (Figure 8B, top row; Figure 8C). Quantitation of the experiment shown revealed a 60-fold increase in the nuclear to cytoplasmic Hsp70 mRNA level relative to the actin mRNA. Three independent trials confirmed the specific nuclear accumulation of the Hsp70 mRNA in the heat-shocked hCG1C siRNA transfected cells. Taken together, these results suggested that hCG1 is specifically required for the export of Hsp70 mRNA in HeLa cells.
Decreased hCG1 Levels Perturb hGle1 Localization in HeLa Cells
Given the protein-protein interaction between hGle1B and hCG1, we next studied the effect of hCG1C siRNAs on the intracellular distribution of endogenous hGle1 (Figure 9). Cells were transfected for 48 h with 30 nM of hCG1C siRNAs or control siRNAs, and indirect immunofluorescence with anti-hGle1 antibodies was performed. Interestingly, unlike cells treated with control siRNAs, the distribution of hGle1 was perturbed in cells transfected with hCG1C siRNAs. Strikingly, a significant fraction of hGle1 was found in distinct cytoplasmic foci. To test if NPCs were coincidently perturbed in hCG1C siRNAs-treated cells, the distribution of the nucleoporins recognized by the mAb414 antibody was examined. The mAb414 staining intensity and pattern in hCG1C siRNAs-treated cells was identical to control siRNAs-treated cells. In addition, the hGle1-containing foci did not appear to harbor Nups recognized by mAb414. Thus, NPCs were not globally affected by the hCG1C siRNAs. We concluded that depletion of hCG1 in HeLa cells results in specific mislocalization of endogenous hGle1.
Figure 9.

Analysis of endogenous hGle1 localization in siRNA-treated HeLa cells. Forty-eight hours after transfection with 30 nM hCG1C siRNAs or control siRNAs, cells were fixed and double indirect immunofluorescence microscopy was conducted. Endogenous hGle1 was localized using polyclonal rabbit anti-hGle1 antibodies, co-incident with NPC protein detection by monoclonal antibody 414. Ba, 10 μm.
DISCUSSION
In both yeast and human cells, the factors required for NPC localization of Gle1 have not yet been fully elucidated. Using a yeast two-hybrid strategy in combination with in vitro binding assays, we report here that the unique C-terminal region of hGle1B is both necessary and sufficient for interaction with the nucleoporin hCG1. Moreover, an siRNA knockdown strategy against hCG1 mRNA documented the physiological significance of the protein-protein interaction between hGle1 and hCG1 in human cells. The loss of hCG1 resulted in hGle1 accumulation in cytoplasmic foci. Coincidentally, the export of transcripts encoding Hsp70 was inhibited. This is the first evidence of a vertebrate protein with specific functions in the export of a heat-shock-induced mRNA (Hsp70) and closely parallels the mechanism in budding yeast. Finally, we further found that hGle1B can simultaneously bind hNup155 and hCG1 in vitro. We propose that hGle1 interactions with these nucleoporins represent steps in the mRNA export pathway.
hCG1 is the functional orthologue of the budding yeast Nup42/Rip1 nucleoporin (Strahm et al., 1999). The FG-repeat domains of hCG1 and Nup42/Rip1 constitute docking sites for Tap and Mex67, respectively (Katahira et al., 1999; Strahm et al., 1999). Moreover, the last 66 aa of Nup42/Rip1 physically interact with the C-terminal domain of yGle1 (Strahm et al., 1999). These observations are strikingly similar to the interaction reported here for hGle1B binding the C-terminal 44 aa region of hCG1. It is well documented that budding yeast mutants lacking Nup42/Rip1 (nup42/rip1Δ) are viable with no perturbations in bulk poly(A)+ RNA export from the nucleus. However, nup42/rip1Δ mutants have a marked defect in the export of heat-shock-induced mRNA as well as detectable loss of yGle1 at NPCs after heat shock (Saavedra et al., 1997; Stutz et al., 1997; Strahm et al., 1999; Rollenhagen et al., 2004). Our immunolocalization of endogenous hGle1 in hCG1 knockdown cells showed a redistribution of hGle1 to cytoplasmic foci, with no apparent bulk poly(A)+ export defects (unpublished data). However, we detected a significant decrease in Hsp70 levels and a nuclear accumulation of Hsp70 mRNA in cells treated with hCG1C siRNAs. This is highly similar to the role for Nup42/Rip1 in the export of heat-shock-induced transcripts, and supports the hypothesis that the Gle1-Nup42/hCG1 interaction and function is conserved between yeast and higher eukaryotes.
Despite binding both Tap/Nxf1/Mex67 and hGle1/yGle1, there may be differences between the respective Nup42/Rip1 and hCG1 functions. For example, Nup42/Rip1 is only required to retain yGle1 at NPCs after heat shock (Rollenhagen et al., 2004), whereas we observed perturbations in hGle1 localization in hCG1C siRNA cells at normal growth temperatures. hCG1 is distinct from Nup42/Rip1 in that it harbors predicted zinc finger (ZF) and coiled-coiled (CC) domains as well as a comparatively smaller FG-repeat domain (Katahira et al., 1999; Strahm et al., 1999; Le Rouzic et al., 2002). Here, we observed that hCG11-365-GFP harboring only the ZF, CC, and FG domains localizes in distinct intranuclear foci. Although the foci are smaller and more abundant, this phenotype is reminiscent of the reported intranuclear localization of Nup98 in GLFG bodies (Griffis et al., 2002), and future studies will be needed to characterize the hCG11-365-GFP foci.
The use of siRNAs to selectively deplete nucleoporins in vertebrate cells has become a useful tool in analyzing NPC function and assembly. It has been reported that the partial depletion of certain nucleoporins results in the coreduction of multiple other nucleoporins and decreased NPC density at the NE. This includes Nup107 (Boehmer et al., 2003; Walther et al., 2003), Nup133 (Walther et al., 2003), Nup98 (Ebina et al., 2004), and Nup85 (Harel et al., 2003). Interestingly, we found that the depletion of hCG1 does not produce a similar effect. Indeed, except for hGle1, none of the nucleoporins and transport factors tested were affected in hCG1C siRNA-treated HeLa cells. In particular, Nup153 and Nup98 whose protein levels are most often codepleted in other siRNA studies were not perturbed here. There are technical differences between our siRNA approach (in vitro synthesized pools) and those used in other reports (single chemically synthesized siRNA; Boehmer et al., 2003; Harel et al., 2003; Walther et al., 2003). However, the overall lack of general NPC perturbations in the hCG1-depleted cells argues that hCG1 is not required for NPC structural maintenance or assembly in the same way that Nup107 and Nup133 are required.
We previously reported that hGle1B localizes to NPCs at steady state in HeLa cells (Kendirgi et al., 2003). This localization is attributed in part to the 29-aa N-terminal domain that directly interacts with hNup155 (Rayala et al., 2004). However, although necessary, the Nup155BD alone is not sufficient for targeting hGle1B to NPCs. In fact hGle1A is not concentrated at the NE. We can now attribute the lack of hGle1A NPC localization directly to the lack of the unique 43 aa C-terminal region in hGle1A and to the lack of hGle1A interaction with hCG1. This study also further documents the role of non-FG-nucleoporins and non-FG regions in FG-nucleoporins in mediating interactions with transport factors. For example, yGle1 and yDbp5 bind to the non-FG domains of FG-nucleoporins, and a high-affinity binding site for the karyopherin Kap95 has been identified in the non-FG domain of Nup1 (Stutz et al., 1997; Hodge et al., 1999; Schmitt et al., 1999; Pyhtila and Rexach, 2003). For hGle1, the non-FG hNup155 (Rayala et al., 2004) and the non-FG region of hCG1 are both important for mediating hGle1B localization at NPCs.
We speculate that other domain(s) within hGle1 are required for NPC localization in addition to the N-terminal Nup155BD- and the C-terminal hGle1B-specific hCG1BD. An ectopically expressed chimeric protein Nup155BD-GFP-hCG1BD did not localize at the NE in HeLa cells (Kendirgi and Wente, unpublished results). The hGle1-shuttling domain is sufficient for mediating translocation through the NPC and may contribute to steady state NPC localization. This hypothesis warrants further future analysis of hGle1B functional domains to define additional NPC localization or nuclear transport regions that function in concert with the hNup155BD and hCG1BD. The presence of multiple domains within a shuttling transport factor that mediate interactions with nucleoporins is reminiscent of the mRNA export factor Tap/Mex67 (Izaurralde, 2002) and importin β/Kap95 (Bednenko et al., 2003).
The interaction of distinct hGle1B domains with hNup155 and hCG1 may constitute one or two independent steps in the translocation of hGle1B through NPCs. In vertebrates, the non-FG nucleoporin Nup155 is symmetrically distributed on both sides of NPCs (Radu et al., 1993; Cronshaw et al., 2002). If similar to the budding yeast Nup42/Rip1 (Rout et al., 2000), hCG1 may be located exclusively on the cytoplasmic NPC face. The ability of hGle1B to form an in vitro heterotrimeric complex with hCG1 and hNup155 may constitute one step in hGle1B translocation through NPCs with hGle1B bound to both Nups simultaneously. However, whether hGle1B forms a trimeric complex with hCG1 and hNup155 in HeLa cells is unknown. Thus, an alternative model is possible with hGle1B binding hCG1 and hNup155 in a mutually exclusive sequential manner as hGle1B moves through the NPC while shuttling in and out of the nucleus. If hCG1 is found on both faces of NPCs (Strahm et al., 1999), this is compatible with both models in that a hNup155-hGle1B-hCG1 heterotrimeric complex may form on both/either NPC side versus just the cytoplasmic. Interestingly, although a proteomic-based study shows both hNup155 and hCG1 are both components of an enriched vertebrate NPC fraction, hGle1 is absent and has not been biochemically identified as a vertebrate NPC component (Cronshaw et al., 2002). Thus, we predict the hGle1 association with hNup155 and hCG1 is transient during hGle1 nucleocytoplasmic shuttling dynamics and NPC translocation.
In conclusion, this study reveals that Gle1 protein-protein interactions in human and yeast cells may be highly conserved. Moreover, the function of Gle1 in yeast and human is likely highly similar. We speculate that although no evidence has been reported that yGle1 shuttles between the nucleus and cytoplasm, the shuttling activity of Gle1 in human cells (Kendirgi et al., 2003) may be conserved in yeast. Likewise, yGle1 has been connected to exporting mRNPs via RNA-binding protein Nab2 (Suntharalingam et al., 2004). Thus, although the link between hGle1 and mRNPs has not been revealed, we predict hGle1 will have a direct or bridged interaction with a RNA-binding protein. The further analysis of both human and yeast Gle1 will prove instrumental in delineating the function of Gle1 in the molecular cascade of events required for the mRNA export pathway.
Acknowledgments
We are grateful to Yingna Zhou for technical assistance. We thank M. Carlson (Columbia University) for providing us with the GBD-Snf1 construct, I. Macara (University of Virginia, Charlottesville, VA) for the GST-GFP construct, B. Burke (University of Florida, Gainesville, FL) for the anti-Nup153 antibodies, E. Izaurralde (EMBL, Heidelberg) for the anti-Tap antibodies, M. Linder (Washington University School of Medicine, St. Louis, MO) for the anti-GFP antibodies, and colleagues in the Wente lab for critical comments on the manuscript. This work was supported by GM072272 (to D.J.R.) and GM51219 (to S.R.W.) from the National Institutes of Health.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-11-0998) on July 6, 2005.
Abbreviations used: aa, amino acid; CC, coiled-coil; C, carboxy; FG, phenylalanine glycine; FL, full length; GAD, Gal4 activation domain; GBD, Gal4 DNA binding domain; GST, glutathione-S-transferase; GFP, green fluorescence protein; h, human; hnRNP, heterogeneous nuclear ribonucleoprotein; mRNP, mRNA ribonucleoprotein; NE, nuclear envelope; NPC, nuclear pore complex; ORF, open reading frame; poly(A)+, polyadenylated RNA; siRNA, small interfering RNA; y, budding yeast; ZF, zinc finger.
References
- Bachi, A. et al. (2000). The C-terminal domain of TAP interacts with the nuclear pore complex and promotes export of specific CTE-bearing RNA substrates. RNA 6, 136-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailer, S. M., Siniossoglou, S., Podtelejnikov, A., Hellwig, A., Mann, M., and Hurt, E. (1998). Nup116p and Nup100p are interchangeable through a conserved motif which constitutes a docking site for the mRNA transport factor Gle2p. EMBO J. 17, 1107-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednenko, J., Cingolani, G., and Gerace, L. (2003). Nucleocytoplasmic transport: navigating the channel. Traffic 4, 127-135. [DOI] [PubMed] [Google Scholar]
- Black, B. E., Levesque, L., Holaska, J. M., Wood, T. C., and Paschal, B. M. (1999). Identification of an NTF2-related factor that binds Ran-GTP and regulates nuclear protein export. Mol. Cell Biol. 19, 8616-8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins, M. B., Smith, A. M., Phillips, E. M., and Powers, M. A. (2003). Complex formation among the RNA export proteins Nup98, Rae1/Gle2 and TAP. J. Biol. Chem. 278, 20979-20988. [DOI] [PubMed] [Google Scholar]
- Bodoor, K., Shaikh, S., Salina, D., Raharjo, W. H., Bastos, R., Lohka, M., and Burke, B. (1999). Sequential recruitment of NPC proteins to the nuclear periphery at the end of mitosis. J. Cell Sci. 112, 2253-2264. [DOI] [PubMed] [Google Scholar]
- Boehmer, T., Enninga, J., Dales, S., Blobel, G., and Zhong, H. (2003). Depletion of a single nucleoporin, Nup107, prevents the assembly of a subset of nucleoporins into the nuclear pore complex. Proc. Natl. Acad. Sci. USA 100, 981-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, I. C., Rohrbach, E., Schmitt, C., and Izaurralde, E. (1999). TAP binds to the constitutive transport element (CTE) through a novel RNA-binding motif that is sufficient to promote CTE-dependent RNA export from the nucleus. EMBO J. 18, 1953-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J. A., Bharathi, A., Ghosh, A., Whalen, W., Fitzgerald, E., and Dhar, R. (1995). A mutation in the Schizosaccharomyces pombe rae1 gene causes defects in poly(A)+ RNA export and in the cytoskeleton. J. Biol. Chem. 270, 7411-7419. [DOI] [PubMed] [Google Scholar]
- Bucci, M., and Wente, S. R. (1998). A novel fluorescence-based genetic strategy identifies mutants of Saccharomyces cerevisiae defective for nuclear pore complex assembly. Mol. Biol. Cell 9, 2439-2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronshaw, J. M., Krutchinsky, A. N., Zhang, W., Chait, B. T., and Matunis, M. J. (2002). Proteomic analysis of the mammalian nuclear pore complex. J. Cell Biol. 158, 915-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, L. I., and Blobel, G. (1986). Identification and characterization of a nuclear pore complex protein. Cell 45, 699-709. [DOI] [PubMed] [Google Scholar]
- Del Priore, V., Snay, C. A., Bahr, A., and Cole, C. N. (1996). The product of the Saccharomyces cerevisiae RSS1 gene, identified as a high-copy suppressor of the rat7-1 temperature-sensitive allele of the RAT7/NUP159 nucleoporin, is required for efficient mRNA export. Mol. Biol. Cell 7, 1601-1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donze, O., and Picard, D. (2002). RNA interference in mammalian cells using siRNAs synthesized with T7 RNA polymerase. Nucleic Acids Res. 30, e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss, G., Kim, V. N., and Kataoka, N. (2002). Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 3, 195-205. [DOI] [PubMed] [Google Scholar]
- Ebina, H., Aoki, J., Hatta, S., Yoshida, T., and Koyanagi, Y. (2004). Role of Nup98 in nuclear entry of human immunodeficiency virus type 1 cDNA. Microbes Infect. 6, 715-724. [DOI] [PubMed] [Google Scholar]
- Fribourg, S., Braun, I. C., Izaurralde, E., and Conti, E. (2001). Structural basis for the recognition of a nucleoporin FG repeat by the NTF2-like domain of the TAP/p15 mRNA nuclear export factor. Mol. Cell 8, 645-656. [DOI] [PubMed] [Google Scholar]
- Gilbert, W., and Guthrie, C. (2004). The Glc7p nuclear phosphatase promotes mRNA export by facilitating association of Mex67p with mRNA. Mol. Cell 13, 201-212. [DOI] [PubMed] [Google Scholar]
- Grant, R. P., Hurt, E., Neuhaus, D., and Stewart, M. (2002). Structure of the C-terminal FG-nucleoporin binding domain of Tap/NXF1. Nat. Struct. Biol. 9, 247-251. [DOI] [PubMed] [Google Scholar]
- Grant, R. P., Neuhaus, D., and Stewart, M. (2003). Structural basis for the interaction between the Tap/NXF1 UBA domain and FG nucleoporins at 1A resolution. J. Mol. Biol. 326, 849-858. [DOI] [PubMed] [Google Scholar]
- Griffis, E. R., Altan, N., Lippincott-Schwartz, J., and Powers, M. A. (2002). Nup98 is a mobile nucleoporin with transcription-dependent dynamics. Mol. Biol. Cell 13, 1282-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruter, P., Tabernero, C., von Kobbe, C., Schmitt, C., Saavedra, C., Bachi, A., Wilm, M., Felber, B. K., and Izaurralde, E. (1998). TAP, the human homolog of Mex67p, mediates CTE-dependent RNA export from the nucleus. Mol. Cell 1, 649-659. [DOI] [PubMed] [Google Scholar]
- Guzik, B. W., Levesque, L., Prasad, S., Bor, Y. C., Black, B. E., Paschal, B. M., Rekosh, D., and Hammarskjold, M. L. (2001). NXT1 (p15) is a crucial cellular cofactor in TAP-dependent export of intron-containing RNA in mammalian cells. Mol. Cell Biol. 21, 2545-2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel, A., Orjalo, A. V., Vincent, T., Lachish-Zalait, A., Vasu, S., Shah, S., Zimmerman, E., Elbaum, M., and Forbes, D. J. (2003). Removal of a single pore subcomplex results in vertebrate nuclei devoid of nuclear pores. Mol. Cell 11, 853-864. [DOI] [PubMed] [Google Scholar]
- Herold, A., Teixeira, L., and Izaurralde, E. (2003). Genome-wide analysis of nuclear mRNA export pathways in Drosophila. EMBO J. 22, 2472-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge, C. A., Colot, H. V., Stafford, P., and Cole, C. N. (1999). Rat8p/Dbp5p is a shuttling transport factor that interacts with Rat7p/Nup159p and Gle1p and suppresses the mRNA export defect of xpo1-1 cells. EMBO J. 18, 5778-5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y., Gattoni, R., Stevenin, J., and Steitz, J. A. (2003). SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol. Cell 11, 837-843. [DOI] [PubMed] [Google Scholar]
- Ito, H., Fukuda, Y., Murata, K., and Kimura, A. (1983). Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153, 163-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaurralde, E. (2002). A novel family of nuclear transport receptors mediates the export of messenger RNA to the cytoplasm. Eur. J. Cell Biol. 81, 577-584. [DOI] [PubMed] [Google Scholar]
- James, P., Halladay, J., and Craig, E. A. (1996). Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144, 1425-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, T. H., and Rosbash, M. (2003). Co-transcriptional monitoring of mRNP formation. Nat. Struct. Biol. 10, 10-12. [DOI] [PubMed] [Google Scholar]
- Katahira, J., Straesser, K., Saiwaki, T., Yoneda, Y., and Hurt, E. (2002). Complex formation between Tap and p15 affects binding to FG-repeat nucleoporins and nucleocytoplasmic shuttling. J. Biol. Chem. 277, 9242-9246. [DOI] [PubMed] [Google Scholar]
- Katahira, J., Strasser, K., Podtelejnikov, A., Mann, M., Jung, J. U., and Hurt, E. (1999). The Mex67p-mediated nuclear mRNA export pathway is conserved from yeast to human. EMBO J. 18, 2593-2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendirgi, F., Barry, D. M., Griffis, E. R., Powers, M. A., and Wente, S. R. (2003). An essential role for hGle1 nucleocytoplasmic shuttling in mRNA export. J. Cell Biol. 160, 1029-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rouzic, E., Mousnier, A., Rustum, C., Stutz, F., Hallberg, E., Dargemont, C., and Benichou, S. (2002). Docking of HIV-1 Vpr to the nuclear envelope is mediated by the interaction with the nucleoporin hCG1. J. Biol. Chem. 277, 45091-45098. [DOI] [PubMed] [Google Scholar]
- Lei, E. P., and Silver, P. A. (2002). Protein and RNA export from the nucleus. Dev. Cell 2, 261-272. [DOI] [PubMed] [Google Scholar]
- Levesque, L., Guzik, B., Guan, T., Coyle, J., Black, B. E., Rekosh, D., Hammarskjold, M. L., and Paschal, B. M. (2001). RNA export mediated by tap involves NXT1-dependent interactions with the nuclear pore complex. J. Biol. Chem. 276, 44953-44962. [DOI] [PubMed] [Google Scholar]
- Macara, I. G. (2001). Transport into and out of the nucleus. Microbiol. Mol. Biol. Rev. 65, 570-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, A. L., Suntharalingam, M., Johnson, S. L., Audhya, A., Emr, S. D., and Wente, S. R. (2004). Cytoplasmic IP6 production is sufficient for mediating the Gle1-mRNA export pathway. J. Biol. Chem. 279, 51022-51032. [DOI] [PubMed] [Google Scholar]
- Murphy, R., Watkins, J. L., and Wente, S. R. (1996). GLE2, a Saccharomyces cerevisiae homologue of the Schizosaccharomyces pombe export factor RAE1, is required for nuclear pore complex structure and function. Mol. Biol. Cell 7, 1921-1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, R., and Wente, S. R. (1996). An RNA-export mediator with an essential nuclear export signal. Nature 383, 357-360. [DOI] [PubMed] [Google Scholar]
- Pritchard, C. E., Fornerod, M., Kasper, L. H., and van Deursen, J. M. (1999). RAE1 is a shuttling mRNA export factor that binds to a GLEBS-like NUP98 motif at the nuclear pore complex through multiple domains. J. Cell Biol. 145, 237-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyhtila, B., and Rexach, M. (2003). A gradient of affinity for the karyopherin Kap95p along the yeast nuclear pore complex. J. Biol. Chem. 278, 42699-42709. [DOI] [PubMed] [Google Scholar]
- Radu, A., Blobel, G., and Wozniak, R. W. (1993). Nup155 is a novel nuclear pore complex protein that contains neither repetitive sequence motifs nor reacts with WGA. J. Cell Biol. 121, 1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayala, H. J., Kendirgi, F., Barry, D. M., Majerus, P. W., and Wente, S. R. (2004). The mRNA export factor human Gle1 interacts with the nuclear pore complex protein Nup155. Mol. Cell Proteom. 3, 145-155. [DOI] [PubMed] [Google Scholar]
- Reed, R. (2003). Coupling transcription, splicing, and mRNA export. Curr. Opin. Cell Biol. 15, 326-331. [DOI] [PubMed] [Google Scholar]
- Reed, R., and Hurt, E. (2002). A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell 108, 523-531. [DOI] [PubMed] [Google Scholar]
- Richards, S. A., Lounsbury, K. M., Carey, K. L., and Macara, I. G. (1996). A nuclear export signal is essential for the cytosolic localization of the Ran binding protein, RanBP1. J. Cell Biol. 134, 1157-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollenhagen, C., Hodge, C. A., and Cole, C. N. (2004). The nuclear pore complex and the DEAD box protein Rat8p/Dbp5p have nonessential features which appear to facilitate mRNA export following heat shock. Mol. Cell Biol. 24, 4869-4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout, M. P., Aitchison, J. D., Suprapto, A., Hjertaas, K., Zhao, Y., and Chait, B. T. (2000). The yeast nuclear pore complex: composition, architecture, and transport mechanism. J. Cell Biol. 148, 635-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra, C. A., Hammell, C. M., Heath, C. V., and Cole, C. N. (1997). Yeast heat shock mRNAs are exported through a distinct pathway defined by Rip1p. Genes Dev. 11, 2845-2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, C. et al. (1999). Dbp5, a DEAD-box protein required for mRNA export, is recruited to the cytoplasmic fibrils of nuclear pore complex via a conserved interaction with CAN/Nup159p. EMBO J. 18, 4332-4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, I., and Gerace, L. (2001). In vitro analysis of nuclear transport mediated by the C-terminal shuttle domain of Tap. J. Biol. Chem. 276, 42355-42363. [DOI] [PubMed] [Google Scholar]
- Segref, A., Sharma, K., Doye, V., Hellwig, A., Huber, J., Luhrmann, R., and Hurt, E. (1997). Mex67p, a novel factor for nuclear mRNA export, binds to both poly(A)+ RNA and nuclear pores. EMBO J. 16, 3256-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, S., and Forbes, D. J. (1998). Separate nuclear import pathways converge on the nucleoporin Nup153 and can be dissected with dominant-negative inhibitors. Curr. Biol. 8, 1376-1386. [DOI] [PubMed] [Google Scholar]
- Snay-Hodge, C. A., Colot, H. V., Goldstein, A. L., and Cole, C. N. (1998). Dbp5p/Rat8p is a yeast nuclear pore-associated DEAD-box protein essential for RNA export. EMBO J. 17, 2663-2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahm, Y., Fahrenkrog, B., Zenklusen, D., Rychner, E., Kantor, J., Rosbach, M., and Stutz, F. (1999). The RNA export factor Gle1p is located on the cytoplasmic fibrils of the NPC and physically interacts with the FG-nucleoporin Rip1p, the DEAD-box protein Rat8p/Dbp5p and a new protein Ymr 255p. EMBO J. 18, 5761-5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser, K., Bassler, J., and Hurt, E. (2000). Binding of the Mex67p/Mtr2p heterodimer to FXFG, GLFG, and FG repeat nucleoporins is essential for nuclear mRNA export. J. Cell Biol. 150, 695-706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser, K., and Hurt, E. (2000). Yra1p, a conserved nuclear RNA-binding protein, interacts directly with Mex67p and is required for mRNA export. EMBO J. 19, 410-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strawn, L. A., Shen, T., and Wente, S. R. (2001). The GLFG regions of Nup116p and Nup100p serve as binding sites for both Kap95p and Mex67p at the nuclear pore complex. J. Biol. Chem. 276, 6445-6452. [DOI] [PubMed] [Google Scholar]
- Stutz, F., Bachi, A., Doerks, T., Braun, I. C., Seraphin, B., Wilm, M., Bork, P., and Izaurralde, E. (2000). REF, an evolutionary conserved family of hnRNP-like proteins, interacts with TAP/Mex67p and participates in mRNA nuclear export. RNA 6, 638-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutz, F., Kantor, J., Zhang, D., McCarthy, T., Neville, M., and Rosbash, M. (1997). The yeast nucleoporin Rip1p contributes to multiple export pathways with no essential role for its FG-repeat region. Genes Dev. 11, 2857-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntharalingam, M., Alcazar-Roman, A. R., and Wente, S. R. (2004). Nuclear export of the yeast mRNA-binding protein Nab2 is linked to a direct interaction with Gfd1 and to Gle1 function. J. Biol. Chem. 279, 35384-35391. [DOI] [PubMed] [Google Scholar]
- Suntharalingam, M., and Wente, S. R. (2003). Peering through the pore. Nuclear pore complex structure, assembly, and function. Dev. Cell 4, 775-789. [DOI] [PubMed] [Google Scholar]
- Vinciguerra, P., and Stutz, F. (2004). mRNA export: an assembly line from genes to nuclear pores. Curr. Opin. Cell Biol. 16, 285-292. [DOI] [PubMed] [Google Scholar]
- Walther, T. C. et al. (2003). The conserved Nup107-160 complex is critical for nuclear pore complex assembly. Cell 113, 195-206. [DOI] [PubMed] [Google Scholar]
- Watkins, J. L., Murphy, R., Emtage, J. L., and Wente, S. R. (1998). The human homologue of Saccharomyces cerevisiae Gle1p is required for poly(A)+ RNA export. Proc. Natl. Acad. Sci. USA 95, 6779-6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand, H. L., Coburn, G. A., Zeng, Y., Kang, Y., Bogerd, H. P., and Cullen, B. R. (2002). Formation of Tap/NXT1 heterodimers activates Tap-dependent nuclear mRNA export by enhancing recruitment to nuclear pore complexes. Mol. Cell Biol. 22, 245-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X., Hubbard, E. J., and Carlson, M. (1992). A protein kinase substrate identified by the two-hybrid system. Science 257, 680-682. [DOI] [PubMed] [Google Scholar]
- Yoon, J. H., Love, D. C., Guhathakurta, A., Hanover, J. A., and Dhar, R. (2000). Mex67p of Schizosaccharomyces pombe interacts with Rae1p in mediating mRNA export. Mol. Cell Biol. 20, 8767-8782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York, J. D., Odom, A. R., Murphy, R., Ives, E. B., and Wente, S. R. (1999). A phospholipase C-dependent inositol polyphosphate kinase pathway required for efficient messenger RNA export. Science 285, 96-100. [DOI] [PubMed] [Google Scholar]
- Zhao, J., Jin, S. B., Bjorkroth, B., Wieslander, L., and Daneholt, B. (2002). The mRNA export factor Dbp5 is associated with Balbiani ring mRNP from gene to cytoplasm. EMBO J. 21, 1177-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]