

Abstract
In addition to its role in nucleocytoplasmic transport, the nuclear pore complex (NPC) acts as a docking site for proteins whose apparent primary cellular functions are unrelated to nuclear transport, including Mad1p and Mad2p, two proteins of the spindle assembly checkpoint (SAC) machinery. To understand this relationship, we have mapped domains of yeast Saccharomyces cerevisiae Mad1p that interact with the nuclear transport machinery, including further defining its interactions with the NPC. We showed that a Kap95p/Kap60p-dependent nuclear localization signal, positioned in the C-terminal third of Mad1p, is required for its efficient targeting to the NPC. At the NPC, Mad1p interacts with Nup53p and a presumed Nup60p/Mlp1p/Mlp2p complex through two coiled coil regions within its N terminus. When the SAC is activated, a portion of Mad1p is recruited to kinetochores through an interaction that is mediated by the C-terminal region of Mad1p and requires energy. We showed using photobleaching analysis that in nocodazole-arrested cells Mad1p rapidly cycles between the Mlp proteins and kinetochores. Our further analysis also showed that only the C terminus of Mad1p is required for SAC function and that the NPC, through Nup53p, may act to regulate the duration of the SAC response.
INTRODUCTION
A defining feature of eukaryotic cells is the physical separation of the nuclear content from the remainder of the cell by the nuclear envelope (NE) membrane. All transport across this double membrane is regulated by massive protein structures termed nuclear pore complexes (NPCs). These highly conserved transport channels are composed of repetitive subunits that together form eightfold symmetrical structures that occupy transcisternal pores across the NE (Suntharalingam and Wente, 2003). In both yeast and mammalian cells, the NPCs are composed of ∼30 different proteins termed nucleoporins (nups) (Rout et al., 2000; Cronshaw et al., 2002), all of which are present in this structure in multiple copies. Macromolecular cargos destined to either enter or exit the nucleus contain specific sequences referred to as nuclear localization sequences (NLSs) or nuclear export sequences (NESs), respectively. These sequences are recognized by soluble cargo-binding proteins termed karyopherins (kaps) of which there are ∼14 in yeast. Kap/cargo complexes then bind to the NPC through interactions between the kaps and a subset of nups that contain phenylalanine-glycine repeats. These nups line the translocation channel, and, through an as yet ill-defined mechanism, facilitate transport through the NPC. A key energy-contributing player in the nuclear transport machinery is the small GTPase Ran. Both the nuclear localization of RanGTP and its conversion to RanGDP play key roles in transport termination steps, the recycling of kaps, and the formation of nuclear export complexes.
Recently, a growing body of evidence has accumulated implicating the NPC and NPC-associated components in additional functions outside of nuclear transport. For example, the Saccharomyces cerevisiae nucleoporin Nup2p and specific kaps have been suggested to influence the transcriptional state of genes by maintaining boundaries between transcriptionally active and inactive regions within the chromatin (Ishii et al., 2002). This has been proposed to occur by the physical tethering of reporter gene elements to the NPC by Nup2p. Consistent with the idea that the NPC functions in regulating chromatin structure, two proteins, Mlp1p and Mlp2p, have been shown to be associated with the nucleoplasmic face of the NPC and form fibers that extend into the nucleus (Strambio-de-Castillia et al., 1999). Nehrbass and colleagues (Galy et al., 2000; Feuerbach et al., 2002) have suggested these proteins physically link the NPCs to telomeres and facilitate the silencing of subtelomeric gene expression. Other studies also have implicated nups in transcriptional control on the basis of their apparent association with transcriptionally active genes (Casolari et al., 2004).
A separate body of evidence has implicated nucleoporins in pathways that control chromosome segregation and cell cycle progression. A specific subcomplex of the mammalian NPC, the Nup107/Nup160 complex, has been identified at mitotic kinetochores (Belgareh et al., 2001; Loïodice et al., 2004). In addition, two other NPC-associated proteins Ran-GAP1, a modulator of nuclear transport, and Nup358/RanBP2, another protein that binds the GTPase Ran, also concentrate at kinetochores during mitosis (Joseph et al., 2002; Salina et al., 2003). Nup358/RanBP2 seems to be required for proper kinetochore structure; however, the mechanistic basis for this is still unclear.
Other studies in budding yeast also have linked the functions of nups to mitotic events. Basrai and colleagues have shown that mutations in the gene encoding Nup170p cause defects in chromosome transmission and kinetochore integrity (Kerscher et al., 2001). It is possible that these phenotypes are the result of a specific nuclear transport defect or, alternatively, they reflect a separate function for Nup170p. Intriguingly, Nup170p is a member of a subcomplex of nups that interacts with two components of the spindle assembly checkpoint (SAC) machinery, Mad1p and Mad2p (Iouk et al., 2002). These proteins, along with other SAC components including Mad3p, Bub1p, Bub3p, and Mps1p, sense defects in the mitotic spindle and arrest cells in metaphase until the spindle is properly formed (for review, see Lew and Burke, 2003). Mad1p and Mad2p are concentrated at the NPCs throughout interphase in both yeast and metazoans (Campbell et al., 2001; Ikui et al., 2002; Iouk et al., 2002). The visualization of S. cerevisiae Mad1-green fluorescent protein (GFP) suggests its localization to NPCs is largely maintained during SAC activation, whereas most if not all of Mad2p is released from the NPCs and recruited to kinetochores (Iouk et al., 2002; Gillett et al., 2004). These localization dynamics are different from those of other yeast SAC proteins. Bub1p and Bub3p localize to kinetochores independent of SAC, whereas Mps1p localizes to spindle pole bodies and Mad3p displays a diffuse nuclear localization (Hardwick et al., 2000; Castillo et al., 2002; Kerscher et al., 2003; Gillett et al., 2004). The different localization patterns of these proteins coupled with observations that many of these proteins have been detected in complexes with one another, including, for example, complexes between Mad1p/Mad2p and Mad2p/Mad3p/Bub3p/Cdc20p (Chen et al., 1999; Fraschini et al., 2001), suggest these interactions are dynamic and contribute to the wait-anaphase signal that is produced when unattached kinetochores are detected. In vertebrates, the cycling of Mad2 on and off of a kinetochore associated Mad1/Mad2 complex has been suggested to sequester Cdc20 and inhibit progression into anaphase (De Antoni et al., 2005).
Of the SAC proteins only the Mad1p/Mad2p complex has been shown to be concentrated at the NPC. The association of this complex with the NPC is mediated by Mad1p, in part, through its interactions with the Nup53p-containing subcomplex, which contains Nup53p, Nup59p, Nup170p, and Nup157p (Iouk et al., 2002). However, the functional significance of the association of the Mad1p/Mad2p complex with the NPC is still unknown. We have investigated the physiological relevance of these interactions by analyzing the interactions of Mad1p with the NPC and karyopherins. Using truncation analysis, we have identified regions of Mad1p that are required for its targeting and binding to the NPC. We also show that Mad1p interacts with multiple nups, including Nup53p and a complex of Nup60p/Mlp1p/Mlp2p. Upon SAC activation, Mad1p cycles between the Mlp proteins and kinetochores. We have mapped the domains of Mad1p that control its binding to the NPC, to kinetochores, and its SAC activity. Moreover, we present evidence that the NPC may play a role in regulating the duration of the SAC.
MATERIALS AND METHODS
Yeast Strains and Media
All yeast strains (Table 1) were grown at 30°C in YPD (1% yeast extract, 2% bactopeptone, and 2% glucose) or synthetic media (SM) supplemented with the appropriate nutrients and 2% glucose unless otherwise noted (Adams et al., 1997). Benomyl plates were made as outlined in Straight and Murray (1997). Plasmid transformations were performed by electroporation as described in Delorme (1989). Genetic integrations of the GFP+ or RFP open reading frames (ORFs) were performed as described previously (Aitchison et al., 1995) using the plasmids pGFP+/HIS5, pRFP/HIS5, or pRFP/NatR (described below). Synthesis of the GFP fusions was confirmed by immunoblotting using anti-Mad1p or anti-GFP antibodies. Red fluorescent protein (RFP) fusions were confirmed with PCR followed by fluorescence.
Table 1.
Yeast strains used in this study
| Name | Genotype | Source |
|---|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Brachmann et al., 1998 |
| Y3028 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 MAD1-GFP::HIS5 (BY4741) | This study |
| Y3029 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 MAD1-1-325-GFP::HIS5 (BY4741) | This study |
| Y3030 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 MAD1-1-250-GFP::HIS5(BY4741) | This study |
| Y3020 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 mad1::KanMX (BY4741) | This study |
| Y3021 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 mad1::HIS5 (BY4741) | This study |
| Mad1Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 mad1::KanMX (BY4742) | Giaever et al., 2002 |
| YMB1911 | MATa ura3-52 lys2-801 his3-200 leu2-3112 trp1-1 mad1::kanMX (DF5) | Iouk et al., 2002 |
| Y3031 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 mad1::KanMX MTW1RFP::HIS5 (BY4741) | This study |
| DF5a | MATa ura3-52 lys2-801 his3-200 leu2-3112 trp1-1 | |
| Kap121-34 | MATa ura3-52 lys2-801 his3-200 leu2-3112 trp1-1 kap121::HIS3::LEU2 pkap121-34::TRP1 (DF5) | Leslie et al., 2002 |
| Kap95-14 | MATa ura3-52 lys2-801 his3-200 leu2-3112 trp1-1 kap95::HIS3 pkap95-14::TRP1 (DF5) | Leslie et al., 2002 |
| Y3032 | MATa ura3-52 lys2-801 his3-200 leu2-3112 trp1-1 kap95::HIS3 pkap95-14::TRP1 mad1::KanMX (DF5) | This study |
| CPL32 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 nup53::kanMX (BY4741) | This study |
| nup59Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 nup59::KanMX (BY4742) | Giaever et al., 2002 |
| nup170Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 nup170::KanMX (BY4742) | Giaever et al., 2002 |
| nup157Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 nup157::KanMX (BY4742) | Giaever et al., 2002 |
| Y3034 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 nup60::KanMX (BY4742) | This study |
| nup2Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 nup2::KanMX (BY4742) | Giaever et al., 2002 |
| pom34Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 pom34::KanMX (BY4742) | Giaever et al., 2002 |
| gle1Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 gle1::KanMX (BY4742) | Giaever et al., 2002 |
| nup42Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 nup42::KanMX (BY4742) | Giaever et al., 2002 |
| gle2Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 gle2::KanMX (BY4742) | Giaever et al., 2002 |
| nup120Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 nup120::KanMX (BY4742) | Giaever et al., 2002 |
| nup100Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 nup100::KanMX (BY4742) | Giaever et al., 2002 |
| nup188Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 nup188::KanMX (BY4742) | Giaever et al., 2002 |
| nup116Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 nup116::KanMX (BY4742) | Giaever et al., 2002 |
| pom152Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 pom152::KanMX (BY4742) | Giaever et al., 2002 |
| mlp1Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 mlp1::KanMX (BY4742) | Giaever et al., 2002 |
| mlp2Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 mlp2::KanMX (BY4742) | Giaever et al., 2002 |
| mlp1Δ mlp2Δ | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 mlp1::KanMX mlp2::natR (BY4742) | This study |
| Y3036 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 nup2::KanMX MAD1-GFP::HIS5 (BY4742) | This study |
| Y3037 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 pom34::KanMX MAD1-GFP::HIS5 (BY4742) | This study |
| Y3040 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 nup60::KanMX MAD1-GFP::HIS5 (BY4741) | This study |
| Y3068 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 nup59::KanMX (BY4741) | This study |
| Y3058 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 nup60::KanMX (BY4741) | This study |
| Y3057 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 nup60::KanMX MAD1-GFP::HIS5 MTW1-RFP::NatR (BY4741) | This study |
| Y3061 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 mlp1::KanMX mlp2::natR MAD1-GFP::HIS5 (BY4742) | This study |
| Y3062 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 nup60::KanMX MAD1-GFP::HIS5 MLP2-RFP::NatR (BY4741) | This study |
| Y3064 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 mlp1::KanMX mlp2::natR MTW1-RFP::HIS5 (BY4742) | This study |
| Y3065 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 mlp1::KanMX MAD1-GFP::HIS5 (BY4742) | This study |
| Y3066 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 mlp2::KanMX MAD1-GFP::HIS5 (BY4742) | This study |
| Y3067 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 nup53::KanMX MAD1-GFP::HIS5 (BY4742) | This study |
| CPL61 | MATa ura3-52 lys2-801 ade2-101 his3-200 trp1-63 leu2-1 GAL1-NmycMPS1 (YPH499) | This study |
| CPL62 | MATa ura3-52 lys2-801 ade2-101 his3-200 trp1-63 leu2-1 GAL1-NmycMPS1 nup53Δ::kanMX (YPH499) | This study |
| CPL63 | MATa ura3-52 lys2-801 ade2-101 his3-200 trp1-63 leu2-1 GAL1-NmycMPS1 nup59Δ::kanMX (YPH499) | This study |
| CPL64 | MATa ura3-52 lys2-801 ade2-101 his3-200 trp1-63 leu2-1 GAL1-NmycMPS1 mad1Δ::kanMX (YPH499) | This study |
| YVG5 | MATa leu2-3112 his3-11,15 trp1Δ ade2-1 ura3-1 MLP1:: pA-HIS5 | Galy et al., 2004 |
| YVG7 | MATa leu2-3112 his3-11,15 trp1Δ ade2-1 ura3-1 MLP2:: pA-HIS5 | Galy et al., 2004 |
Plasmids
Plasmid inserts were constructed using PCR products produced with Expand High Fidelity PCR system (Roche Diagnostics, Laval, Quebec, Canada). pMad1-NLS-GFP3 was constructed by digesting pPho4140-166-GFP3 (Kaffman et al., 1998) with BglII and EcoRI liberating the nucleotides encoding Pho4140-166. A PCR product encoding Mad1p residues 499-533 (bipartite NLS plus 6 flanking residues N and C terminally) was introduced into this backbone with BglII and EcoRI linkers (note: ΔNLS constructs were deleted from the first residue of the predicted bipartite NLS (K506) to the last (K527). Plasmid-borne GFP fused to MAD1 truncations were made by digesting the CEN/URA3 plasmid pACPHO4-GFP (Kaffman et al., 1998) with BglII and EcoRI to remove PHO4 and inserting individual MAD1 cDNAs with BamHI and EcoRI linkers. These included the following plasmids (note: nucleotides of MAD1 are shown in parentheses where 1 is the A of the ATG start codon), pMad1-GFP, p250-325-GFP (750-975), p318-749-GFP (954-2247), and p475-749-GFP (1425-2247). Plasmids expressing the MAD1 ORF fragments lacking the NLS were similarly constructed from two separate DNA fragments and inserted into pRS316 or pACPHO4-GFP to produce the plasmids pmad1- ΔNLS-GFP, p318-749-ΔNLS-GFP, p475-749-ΔNLS-GFP. MAD1 gene fragments were inserted into the pYEX-BX plasmid (2μ/URA3) (BD Biosciences Clontech, Palo Alto, CA) to produce pMad1, pmad1-ΔNLS, p1-325, p318-749, and p475-749. The MAD1 cDNAs were inserted into pGEX-6P-1 (Amersham Biosciences, Baie D'Urfe, Quebec, Canada) to produce pGEX-Mad1 (1-2250), pGEX-1-325 (1-975), pGEX-318-749 (954-2250), and pGEX-475-749 (1425-2250). The plasmids pGFP+/HIS5 and pRFP/HIS5 were constructed by removing GFP from the pGFP/HIS5 (Dilworth et al., 2001) plasmid and replacing it with the GFP+ ORF (Scholz et al., 2000; a gift from Michael Niederweis, Erlangen, Germany) or the RFP ORF (a gift from Ray Truant, McMaster University, Hamilton, Ontario, Canada). The plasmid pRFP/NatR was a gift from Rick Rachubinski (University of Alberta, Edmonton, Alberta, Canada). The plasmids pNUP53 and pNUP53ΔKBD were constructed using pRS315 that includes the NUP53 promoter region with a BamHI site immediately 5′ and in frame with the ATG start codon and the NUF2 terminator with a BamHI site at its 5′ end. The wild-type NUP53 ORF was amplified by PCR and inserted using BamHI linkers. NUP53ΔKBD was PCR amplified with BamHI linkers from pΔ405-430-GFP (Lusk et al., 2002).
In Vitro Binding Assays
BL21 Escherichia coli cells expressing glutathione S-transferase (GST)-Mad1p, GST-1-325, GST-318-749, or GST-475-749 were grown to an OD600 of ∼1.0 and then treated with 1 mM isopropyl β-d-thiogalactoside for 6 h at 23°C. Cells were lysed with 0.67 mg/ml lysozyme in pH 7.5 lysis buffer (Lutzmann et al., 2002) and clarified lysates were incubated with glutathione Sepharose 4B beads (Amersham Biosciences). After washing with lysis buffer, beads were incubated with purified recombinant Nup53p or Nup170p isolated as described previously (Lusk et al., 2002; Makhnevych et al., 2003). After binding, the unbound fraction was collected, beads were washed extensively with lysis buffer, and bound proteins were eluted with SDS-PAGE sample buffer. Proteins in these samples were analyzed by SDS-PAGE and visualized with Bio-Safe Coomassie Blue (Bio-Rad, Montreal, Quebec, Canada).
Fluorescence Microscopy
All images of GFP and RFP fusion proteins were acquired in live cells using either a fluorescence microscope (Olympus BX-50) equipped with a SPOT digital camera or with a confocal microscope (LSM510; Carl Zeiss MicroImaging, Jena, Germany). Yeast strains harboring plasmid-borne fusions were grown to early log phase in the appropriate dropout media and then transferred to YPD for 2-3 h. Cells were then examined directly or after treatment with 12.5 μg/ml nocodazole (Sigma-Aldrich, Oakville, Ontario, Canada) for 1.5-2.5 h to arrest in G2/M phase. The subcellular distribution of Mad1-NLS-GFP3 was examined directly in logarithmically growing cells. Alternatively, cells expressing Mad1-NLS-GFP3 were treated with 10 mM NaN3 and 100 mM 2-deoxyglucose for 30 min at 30°C (Shulga et al., 1996; Iouk et al., 2002). Cells were then washed with CM-URA media (without glucose) and examined immediately. To examine the effects of metabolic poisons on the distribution of Mad1-GFP in G2/M-phase arrested cultures, logarithmically growing cells were first arrested in G1-phase with α-factor and released into 12.5 μg/ml nocodazole for 1 h in YPD, washed twice with YP (no glucose), and then incubated for 10 min at 30°C in YP containing 12.5 μg/ml nocodazole, 10 mM NaN3 and 100 mM 2-deoxyglucose before examination.
Laser Photobleaching of Mad1-GFP and Fluorescence Recovery after Photobleaching (FRAP) Analysis
The strain Y3057 was grown to an OD600 of ∼0.3 and arrested in G1 phase using α-factor for 2 h at 30°C. Cells were harvested and washed in YPD twice before release into YPD containing 12.5 μg/ml nocodazole at room temperature. After 1 h, cells were washed and resuspended in CM-URA containing 12.5 μg/ml nocodazole, then 1.2 μl of cell suspension was applied to a microscope slide with a thin agarose pad (Adames and Cooper, 2000; Fagarasanu et al., 2005) and sealed with valap (equal mixture of Vaseline, lanolin, and paraffin). The initial scan used both 488- and 543-nm light lines and was used to colocalize Mad1-GFP with Mtw1-RFP. Subsequent images did not use the 543-nm laser line (except for the final image). Kinetochore-associated Mad1-GFP was specifically bleached with 50 iterations of full intensity 488-nm light. Images were acquired every 12 s for the duration of each experiment. MetaMorph software was used to delineate concentric regions around both the kinetochore-Mad1-GFP focus and the Mlp-associated Mad1-GFP focus and integrated fluorescent values were determined and background subtracted. Data were initially logged to Microsoft Excel and then to Kaleidagraph. The first order rate constant, k, corresponds to the slope of a best-fit line through the points described by -LN[(Fmax - F(t))/(Fi - F0)], where Fmax is the bleached region at maximal recovery (Howell et al., 2004). The half-time of fluorescence recovery, t1/2, is calculated according to t1/2 = ln2/k.
4,6-Diamidino-2-phenylindole (DAPI) Staining
For experiments monitoring cell cycle progression of nup mutants in nocodazole, cells were arrested in G1 phase with 10 μg/ml α-factor and released into nocodazole at room temperature. Cells were sampled at 20-min intervals and fixed in 70% ethanol. Cells were washed out of ethanol and stained for ∼10 min with 1 μg/ml DAPI.
MPS1 Overexpression
The plasmid encoding GAL1-NmycMPS1 (pAFS120) was a gift from M. Winey (University of Colorado, Boulder, CO). The strain YPH499 was a gift from M. Basrai (NIH, Bethesda, MD). Mps1p overexpression was performed as outlined in Hardwick et al., 1996. Briefly, strains harboring inducible GAL1-MPS1 were grown overnight in YP containing 2% raffinose to an OD600 of ∼0.3. Cells were arrested in G1 phase with 10 μg/ml α-factor for ∼2 h and transferred to YP containing 2% raffinose and 3% galactose containing 10 μg/ml α-factor for another 2 h. Cells were released into the cell cycle by washing two times with YP containing 2% raffinose and 3% galactose. Samples were taken every 20 min for the duration of the experiment and fixed in 70% ethanol. Cell morphology was observed using light microscopy.
Whole Cell Lysate Immunopurifications
Mlp1p and Mlp2p were C-terminally tagged with Staphylococcus aureus protein A (pA) and immunopurified from yeast whole cell lysates as described previously (Dilworth et al., 2001) with the following modifications: 1) cell disruption was achieved by grinding cell pellets under liquid nitrogen using the procedure described in Schultz et al. (1997); 2) lysate clarification consisted solely of low-speed centrifugation at 5000 × g for 15 min at 4°C; 3) after binding, the IgG-Sepharose beads were washed five times with 10 ml of wash buffer containing 50 mM MgCl2 to remove unbound proteins, and proteins that remained bound were eluted by the addition of SDS-PAGE sample buffer.
RESULTS
Regions of Mad1p That Mediate Its Interactions with the NPC and Kinetochores
Mad1p is a member of a group of proteins that function in the SAC (Lew and Burke, 2003). We have previously shown that the majority of Mad1p localizes to NPCs throughout the cell cycle (Iouk et al., 2002). In an effort to determine the functional significance of the association of Mad1p with the NPC and other components of the nuclear transport machinery, we constructed a series of Mad1p deletion mutants to define domains responsible for anchoring Mad1p to the NPC and targeting it to kinetochores. Our truncation mutants were, in part, designed to evaluate the function of three predicted coiled-coil domains positioned within amino acid residues 57-221, 264-320, and 390-585 (Figure 1).
Figure 1.

Truncation mutants of Mad1p. (A) schematic of N- and C-terminal truncations of Mad1p is shown. Amino acid residues are shown on the left. The SMART domain annotation resource (Schultz et al., 1998) predicts Mad1p contains three coiled-coil domains high-lighted in black with bordering residues indicated. The amino acid sequence of the Mad1p NLS and its position (hatched region) are shown.
The subcellular distributions of the Mad1p truncations were evaluated by C-terminally tagging various deletion mutants with GFP and monitoring their localization using confocal microscopy. The distribution of each of the truncations was evaluated in asynchronous cultures and cells arrested with nocodazole in G2/M phase (Figure 2A). Nocodazole destabilizes microtubules, causing an activation of the SAC machinery and subsequent cell cycle arrest. Under these conditions, most of the visible Mad1p remains associated with the NPC, whereas a pool of Mad1p seems to be recruited to kinetochores (Iouk et al., 2002; Gillett et al., 2004). To produce the GFP chimeras of full-length Mad1p and the C-terminal deletions containing amino acid residues 1-250 and 1-325, the GFP ORF was chromosomally integrated following the appropriate codon into the endogenous MAD1 gene to produce the Mad1-GFP, 1-250-GFP, or 1-325-GFP fusions. N-terminal deletions of MAD1 encoding amino acid residues 250-325, 318-749, and 475-749 were fused in frame with the GFP ORF, assembled in a yeast CEN plasmid, and introduced into a mad1Δ strain. Cellular levels of the plasmid encoded fusion proteins were similar or slightly higher than endogenous GFP tagged Mad1p, whereas the integrated 1-250-GFP and 1-325-GFP chimeras produced higher levels of protein (our unpublished data).
Figure 2.

In vivo localization of Mad1p truncation mutants. (A) The ORF encoding GFP was integrated after specific codons within the endogenous MAD1 gene in the strain BY4741 to produce chimeric ORFs encoding full-length Mad1-GFP (Y3028), 1-325-GFP (Y3029), and 1-250-GFP (Y3030). GFP-tagged N-terminal deletions of MAD1 encoding the truncation 250-325-GFP (pMad1-250-325-GFP), 318-749-GFP (pMad1318-749-GFP), or 475-749-GFP (pMad1-475-749-GFP) in a CEN/URA3 plasmid were introduced into mad1Δ cells (Y3021). Strains were grown to mid-log phase at 30°C and viewed directly (asynchronous [AS] cultures) or arrested in G2/M by treatment with 12.5 μg/ml nocodazole (NOC) for ∼2 h and then examined. Arrows indicate intranuclear foci and arrowheads indicate approximate nuclear position in representative cells. Fluorescent images were captured using a confocal microscope. Note, in addition to a diffuse nuclear signal, foci of 318-749-GFP and 475-749-GFP signal colocalized with the kinetochore marker Mtw1-RFP in 13 and 61% of asynchronous cells and 62 and 83% of nocodazole arrested cells, respectively. (B) Cells producing the tagged kinetochore protein Mtw1-RFP (Y3031) and either 318-749-GFP or 475-749-GFP were arrested in G1 phase using α-factor or in G2/M phase by treatment with 12.5 μg/ml NOC. Images of the two fluorescent proteins were obtained as in A. Merged images are shown on the right. Bars, 5 μm.
As shown previously, Mad1-GFP localized in a punctate pattern along the nuclear periphery, consistent with its NPC association, in both asynchronous and G2/M-arrested cells (Figure 2A; Iouk et al., 2002). Similarly, we observed that 1-325-GFP, containing the first and second predicted coiled coil regions of Mad1p, also concentrated at the nuclear periphery. The NPC association of this region was dependent on both predicted coiled-coil regions as the first (1-250-GFP) or the second (250-325-GFP) coiled-coil region alone exhibited severely reduced or undetectable levels of nuclear envelope signal, respectively. Instead these constructs were distributed throughout the cell (Figure 2A).
The NE signal observed with the 1-325-GFP construct also seemed reduced with higher levels of cytoplasmic signal compared with Mad1-GFP, suggesting that the C terminus of Mad1p may contribute to its association with the NPC. To evaluate this possibility, we examined the localization of N-terminal deletion mutants. Both 318-749-GFP and 475-749-GFP accumulated in the nucleus, but lacked any obvious NPC association (Figure 2A). Interestingly, 475-749-GFP also was visible at a single intranuclear focus in asynchronous cultures (Figure 2A). This pattern also was visible in cells arrested in G1 using α-factor or in G2/M after SAC activation induced by microtubule depolymerization using nocodazole (Figure 2, A and B). Coexpression with MTW1-RFP, encoding a kinetochore marker (Goshima and Yanagida, 2000), revealed that 475-749-GFP colocalized with kinetochores throughout the cell cycle and upon SAC activation. In contrast, 318-749-GFP was diffusely distributed throughout the nucleoplasm with no visible focal concentration in most cells grown asynchronously or arrested with α-factor (Figure 2, A and B). On SAC activation 318-749-GFP colocalized with kinetochores (Figure 2B). These results suggested that the region contained within residues 318-475 regulates the SAC induced association of Mad1p with the kinetochores.
Mad1p Contains a Classical NLS (cNLS) That Is Required for Its Efficient Targeting to the NPC
We hypothesized that an NLS could play a role in the efficiency of Mad1p targeting to the NPC and into the nucleus, in much the same way that an NLS-like region functions in the efficient targeting of Nup53p (Lusk et al., 2002) and the SUMO deconjugating enzyme Ulp1p (Panse et al., 2003) to NPCs. An NLS functioning in a similar manner and positioned in the third coiled-coil region of Mad1p might explain the decrease in NPC targeting of the 1-325 truncation and the nuclear accumulation of both the 318-749 and 475-749 truncations. A potential bipartite NLS (amino acid residues 506-527) was previously recognized by sequence analysis of Mad1p (Hardwick and Murray, 1995). To test whether this region functions as an NLS, a 499-533-GFP3 chimera containing the NLS and three tandemly repeated GFP ORFs was expressed in yeast, and the distribution of the fusion protein was evaluated by confocal microscopy. As shown in Figure 3A, this reporter concentrated in the nucleus. Moreover, its nuclear accumulation was dependent on energy, as treatment of cells with the metabolic poisons 2-deoxy-d-glucose and sodium azide (Shulga et al., 1996) inhibited its nuclear accumulation (Figure 3A). These data confirmed that this region of Mad1p is capable of functioning as an NLS.
Figure 3.
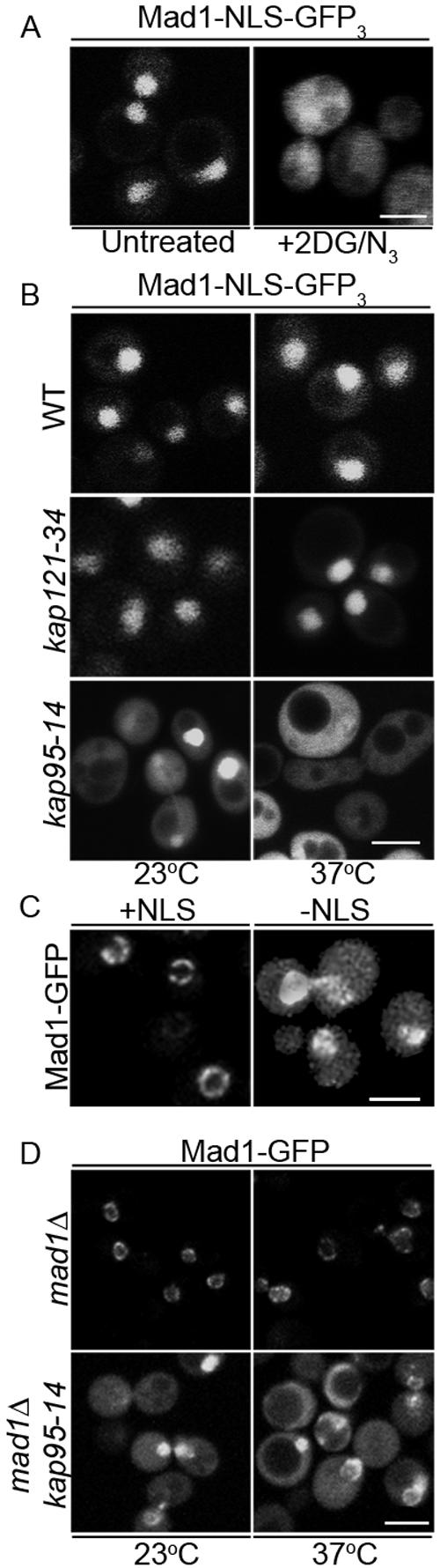
Mad1p contains a functional NLS. (A) The nucleotides encoding amino-acid residues 499-533 of Mad1p were inserted upstream of the ORFs encoding three tandemly repeated GFP in the plasmid pMad1-NLS-GFP3 (Mad1-NLS-GFP3) and introduced into wild-type yeast cells (BY4741). The localization of Mad1-NLS-GFP3 was then visualized using a confocal microscope before (untreated) and after treatment with the metabolic poisons 2-deoxy-d-glucose and sodium azide (+2DG/N3). (B) Import of the Mad1p NLS requires Kap95p. pMad1-NLS-GFP3 plasmid was introduced into a wild-type strain (DF5) and the temperature-sensitive karyopherin mutants kap121-34 and kap95-14. Localization of the NLS was examined at 23°C and 2.5 h after shifting cells to 37°C. (C) The NLS of Mad1p is required for the efficient localization of Mad1p to the NPC. Plasmids harboring MAD1-GFP (pMad1-GFP; +NLS) or mad1ΔNLS-GFP (pmad1ΔNLS-GFP; -NLS) were introduced into a mad1Δ strain (Y3021) and the localization of the fusion proteins was visualized with a confocal microscope. (D) Kap95p is required for the efficient localization of Mad1p to the NPC. A plasmid encoding Mad1-GFP was introduced into mad1Δ (YMB1911) and mad1Δkap94-14 (Y3032) strains. Localization of Mad1-GFP was compared in these two strains at 23°C and after cells were shifted to 37°C for 2.5 h. Bars, 5 μm.
Due to the resemblance of the Mad1-NLS to a consensus bipartite, cNLS (Robbins et al., 1991; Makkerh et al., 1996), we predicted that its cognate karyopherin receptor was the heterodimer Kap60p/Kap95p. To address this possibility, we examined the localization of 499-533-GFP in cells harboring a temperature-sensitive allele of KAP95, kap95-14 (Leslie et al., 2002) as well as a separate karyopherin mutant (kap121-34) (Leslie et al., 2002). The 499-533-GFP accumulated in the nucleus in wild-type and kap121-34 cells at 23°C and nonpermissive temperature (37°C; Figure 3B). However, it was unable to accumulate in the nucleus of kap95-14 cells at 37°C, suggesting that the Kap95p/Kap60p complex plays an integral role in its import.
We also examined whether the cNLS of Mad1p and the Kap95p/Kap60p complex function in the efficient localization of Mad1p to the NPC. First, we compared the subcellular distribution of Mad1-GFP with a mutant mad1p protein lacking the cNLS. Plasmid-borne genes encoding both proteins were introduced into mad1Δ cells and Western blotting confirmed that both proteins were produced at similar levels (our unpublished data). As shown in Figure 3C, Mad1-GFP was detected exclusively at the nuclear periphery, whereas mad1ΔNLS-GFP showed increased levels of cytoplasmic fluorescence in addition to that visible at the NE, suggesting that mad1ΔNLS-GFP was inefficiently targeted to NPCs.
If the cNLS region of Mad1p functions in conjunction with karyopherins to facilitate the association of Mad1p with the NPC, a mutation in Kap95p or Kap60p that inhibits import would also alter Mad1p localization. This was what we observed. In a kap95-14 strain, which exhibits import defects at both permissive (23°C) and nonpermissive temperatures (37°C) (Leslie et al., 2002), Mad1-GFP showed a similar distribution as that observed with the mad1ΔNLS-GFP protein, including increased cytoplasmic fluorescence (Figure 3D). Moreover, we observed significant numbers of cells where Mad1-GFP had accumulated in the nucleus, suggesting that a loss of Kap95p function did not completely prevent its access to the nucleoplasm but clearly altered its ability to concentrate at the nuclear periphery.
Binding sites for Mad1p at the NPC
Once targeted to the NPC, the N-terminal regions of Mad1p (residues 1-325) mediated its interactions with the NPC. We have previously shown that the interactions of Mad1p with the NPC are, in part, mediated by its physical interactions with a subcomplex of nucleoporins containing Nup53p (Iouk et al., 2002). Using in vitro binding assays, we examined whether Mad1p and its truncations were capable of interacting directly with Nup53p. For these experiments, recombinant GST-Mad1p, GST-1-325, GST-318-479, GST-475-749, and GST alone were synthesized in E. coli, purified, and immobilized on glutathione-Sepharose beads. With the exception of the GST-Mad1p fusion, which was partially degraded, the bead-bound fraction consisted predominantly of single species. As shown in Figure 4A, recombinant Nup53p bound to GST-Mad1p and GST-1-325, but only weakly to GST-318-749. Nup53p failed to bind GST-475-749 and GST alone (Figure 4, A and B). Moreover, GST-Mad1p failed to bind recombinant Nup170p, another member of the Nup53p-containing subcomplex (our unpublished data). We conclude from these results that the 1-325 region of Mad1p that binds the NPC (Figure 2A) also is capable of binding Nup53p.
Figure 4.

Nup53p binds directly to Mad1p. (A) The recombinant proteins GST, GST-Mad1p, GST-1-325, or GST-318-749 were synthesized in E. coli (BL21) and purified using glutathione-Sepharose beads. The bead-bound proteins were incubated with (+) or without (-) purified, recombinant Nup53p (L). After this, the unbound fraction was collected, the beads were washed, and the bound fraction was eluted with SDS-PAGE sample buffer. Proteins present in equivalent proportions of the bound and unbound fractions were resolved using SDS-PAGE and visualized with Coomassie Blue staining. Molecular mass markers in kilodaltons are shown on the left. (B) Binding experiments were conducted using GST (our unpublished data), GST-1-325 or GST-475-749, and Nup53p as described in A). Note, GST-475-749 failed to bind Nup53p and was present in the unbound fraction. The lack of Nup53p in the bound fractions was only visible after extended periods of electrophoresis when the mobility of Nup53p is sufficiently different from that of GST-475-749 protein.
Neither Nup53p nor a structurally related nup, Nup59p, function as the sole binding site for Mad1p at the NPC. We have shown that null mutations of NUP53 or NUP59 contain NPC associated Mad1p at slightly reduced (Iouk et al., 2002) or near wild-type levels (Figure 5A). In an attempt to define other nups that bind to Mad1p, we examined the localization of Mad1p in various nup null mutants. Of the ∼30 NUP genes in yeast, the majority can be individually deleted without resulting in a lethal phenotype (Giaever et al., 2002). Of these, we detected a subset of nup and NPC associated protein mutant strains that exhibited altered growth characteristics on media containing the microtubule destabilizing drug benomyl. These data are summarized in Table 2. In each case, these null mutants showed an increased resistance to the growth inhibition caused by benomyl. One possible explanation for these results is that the SAC is altered in these mutants. In addition to mutations in genes that encode members of the Nup53p-containing complex (Iouk et al., 2002), three mutant strains, nup60Δ, nup2Δ, and pom34Δ, showed an increased resistance to growth inhibition caused by benomyl. Using these results as a guide, we focused on examining the effects of these null mutations on the localization of Mad1p. Deletion of the POM34 or NUP2 gene had no obvious effect on the association of Mad1p with the NPC (Figure 5A). In contrast, the NE concentration of the Mad1-GFP was eliminated in the nup60Δ mutant. The localization of Nup53p, however, was not effected in these cells (our unpublished data). Mad1-GFP was instead concentrated at a single intranuclear focus, which did not colocalize with the kinetochore marker Mtw1-RFP in asynchronous cultures (Figure 5B).
Figure 5.

The localization of Mad1-GFP is altered in mutants of nup60Δ, mlp1Δ, and mlp2Δ. (A) The GFP ORF was integrated after the last amino acid codon of the chromosomal copy of MAD1 in haploid strains containing null mutations in NUP53 (nup53Δ), POM34 (pom34Δ), NUP2 (nup2Δ), NUP60 (nup60Δ), MLP1 (mlp1Δ), MLP2 (mlp2Δ), and the MLP1 MLP2 double mutant (mlp1Δmlp2Δ) to produce the strains Y3067, Y3037, Y3036, Y3040, Y3065, Y3066, and Y3061, respectively. Logarithmically growing cultures of these strains and an isogenic wild-type counterpart (Y3028) were examined by fluorescence microscopy, and images were acquired using a confocal microscope. (B) The localization of Mad1-GFP was examined in the nup60Δ strain. Logarithmically growing asynchronous (AS) nup60Δ cells expressing genomically integrated MAD1-GFP and MTW1-RFP (Y3057) were directly visualized. Note, RFP and GFP foci did not colocalize. Y3057 cells also were synchronized in G1 with α-factor and then released into media containing 12.5 μg/ml nocodazole at 25°C. Cells were examined 60 min after addition of nocodazole. Arrows in the merged panel point to foci where Mtw1-RFP colocalizes with Mad1p-GFP. In cells containing two GFP and two RFP foci, colocalization of two foci is observed in 94% of cells. (C) In asynchronous cell cultures of the nup60Δ mutant, Mad1p colocalizes with Mlp2p. Logarithmically growing nup60Δ cells expressing genomically integrated MAD1-GFP and MLP2-RFP (Y3062) were visualized using fluorescence microscopy. Arrows indicate colocalization of Mad1-GFP with Mlp2-RFP in the merged panel. (D) Localization of Mad1-GFP in a mlp1Δmlp2Δ mutant. The plasmid pMad1-GFP was introduced into an mlp1Δmlp2Δ double mutant containing a genomically integrated MTW1-RFP (Y3064). Mad1-GFP and Mtw1-RFP were visualized in this background in both AS and in cells treated with 12.5 μg/ml nocodazole for 2 h. Bars, 5 μm.
Table 2.
Phenotypes of nupΔ and mlpΔ mutants on benomyl
| Query mutant sensitivity
|
||
|---|---|---|
| Mutant | Wild-type sensitivity | Reference |
| nup53Δ | Resistant | Iouk et al., 2002 |
| nup59Δ | Resistant | Iouk et al., 2002 |
| nup170Δ | Resistant | Iouk et al., 2002 |
| nup157Δ | Resistant | Iouk et al., 2002 |
| nup60Δ | Resistant | This study |
| nup2Δ | Resistant | This study |
| pom34Δ | Resistant | This study |
| gle1Δ | N/C | This study |
| nup42Δ | N/C | This study |
| gle2Δ | N/C | This study |
| nup120Δ | N/C (slow growth) | This study |
| nup100Δ | N/C | This study |
| nup188Δ | N/C | This study |
| nup116Δ | N/C | This study |
| pom152Δ | N/C | Iouk et al., 2002 |
| nup133Δ | N/C | This study |
| nup84Δ | N/C | This study |
| mlp1Δ | N/C | This study |
| mlp2Δ | N/C | This study |
| mlp1Δmlp2Δ | Resistant | This study |
The intranuclear Mad1-GFP foci observed in the nup60Δ mutant cells were reminiscent of the localization of Mlp1p and Mlp2p previously reported in this mutant (Feuerbach et al., 2002). These two structurally related proteins are attached to the nucleoplasmic face of the NPC and are thought to form fibers that extend into the interior of the nucleus (Strambio-de-Castillia et al., 1999). Removal of Nup60p causes Mlp1p and Mlp2p to be released from their NPC attachment and concentrate at a single focus in the nucleoplasm. An examination of Mad1-GFP and Mlp2-RFP in the nup60Δ mutant revealed that these proteins colocalize (Figure 5C, arrows). To assess whether Mlp1p and Mlp2p act as a binding site for Mad1p, we investigated the localization of Mad1-GFP in mutants lacking these nonessential proteins (mlp1Δ, mlp2Δ, and mlp1Δ mlp2Δ). As shown in Figure 5A, the NE concentration of Mad1-GFP was altered in each strain. Both the mlp1Δ and mlp2Δ mutants exhibited foci of concentrated Mad1-GFP as well as a diffuse nucleoplasm distribution, whereas only a diffuse intranuclear signal was present in the mlp1Δ mlp2Δ mutant. These results are consistent with the conclusion that Mlp proteins act as binding sites for Mad1p.
The concentration of Mad1p at the Mlp1p/Mlp2p foci in the nup60Δ strain allowed us to clearly visualize the recruitment of Mad1p from these structures to kinetochores upon SAC activation. As shown in Figure 5B, after arrest of nup60Δ cells with nocodazole, Mad1-GFP was visible at two distinct nuclear foci. One of these two foci colocalized with one of two kinetochore foci detected with Mtw1-RFP (arrows). The Mad1-GFP associated kinetochores likely represent those that have been dislodged from microtubules, whereas those lacking Mad1-GFP have not been released from the spindle (Gillett et al., 2004). Similar experiments were also conducted in the mlp1Δ mlp2Δ mutant. We observed that the diffuse nuclear Mad1-GFP concentrated at a single kinetochore cluster after treatment with nocodazole (Figure 5D). These observations are consistent with the results of the truncation analysis from which we concluded that the NPC binding domain of Mad1p is not required for kinetochore binding.
Mad1p Is Dynamically Associated with the Mlps
When the SAC is activated, two separate populations of Mad1p can be defined on the basis of their association with kinetochores and the NPC associated Mlp proteins. In the nup60Δ strain, these two pools are easily distinguishable. We have taken advantage of this phenotype to examine the dynamics of Mad1p movement between these two locations using FRAP analysis. For these experiments, nup60Δ cells producing Mad1-GFP and Mtw1-RFP were arrested in nocodazole-containing media, and the position of the Mad1p-containing unattached kinetochores was located and photo-bleached. Images were then acquired at 12-s intervals (Figure 6, A and B). Intensity measurements were collected from the bleached kinetochore and unbleached Mlp-associated Mad1-GFP. We observed that >60% of the kinetochore associated Mad1-GFP signal recovered with a t1/2 = 30 s. The <40% that failed to recover may have a slow turnover rate and/or there was an insufficient fluorescent pool for full recovery. The recovery of the kinetochore associated Mad1-GFP signal was accompanied by a corresponding decay of the Mlp-associated Mad1-GFP (Figure 6B), which was not detected in unbleached control cells (our unpublished data). In contrast, we detected no recovery of the Mtw1-RFP at the bleached kinetochore, consistent with previous observations that this protein is a component of the inner kinetochore (De Wulf et al., 2003; Pinsky et al., 2003). We interpret these results to reflect an exchange of bleached Mad1-GFP from kinetochores with unbleached Mad1-GFP associated with Mlp proteins.
Figure 6.

Mad1p dynamically associates with nocodazole treated kinetochores and requires energy for this association. (A) The strain Y3057 was arrested in G1 phase with α-factor and released into media containing 12.5 μg/ml nocodazole at 25°C. After 60 min, cells were harvested and placed on agarose slides. The kinetochore-associated Mad1-GFP was identified based on colocalization with Mtw1-RFP (prebleach). Kinetochore Mad1-GFP was bleached by applying 50 iterations of full intensity 488-nm light. GFP images only were acquired every 12 s. At the end of the experiment, Mtw1-RFP was once again visualized (8′). Arrows point to the bleached focus. Note, bleached Mtw1-RFP did not recover during the time course of the experiments. Bar, 2 μm. (B) Plots of the integrated fluorescent intensities at both kinetochore (circles) and Mlp protein foci (squares) are shown on the left, and the natural log plot of kinetochore recovery against time are shown on the right. The normalized fluorescent recovery of Mad1-GFP at kinetochores fit a single exponential function. The natural log plot was used to calculate the first order rate constant, k, and the t1/2 of Mad1-GFP recovery. (C) Y3057 cells were arrested in nocodazole containing medium (12.5 μg/ml) for 60 min. The culture was then split and harvested cells were suspended in media containing nocodazole and glucose (Noc + glucose) or with nocodazole, 2-deoxy-d-glucose and sodium azide (Noc + 2DG/N3) for 10 min. Images were collected using a confocal microscope. Bar, 5 μm.
The dynamic movement of Mad1p between the Mlp proteins and kinetochores prompted us to examine whether the association of Mad1p with these structures was dependent on energy. For these experiments, we again used the nup60Δ cells producing Mad1-GFP and Mtw1-RFP. After arrest with nocodazole, cells were treated for 10 min with the metabolic poisons 2-deoxyglucose and sodium azide to deplete cellular ATP and GTP (Shulga et al., 1996), and the distributions of Mad1-GFP and Mtw1-RFP were examined. Neither the distribution of Mtw1-RFP nor the Mlp protein associated Mad1-GFP was affected in treated cells (Figure 6C). However, the localization of Mad1-GFP at kinetochores was abolished. Similarly, the association of the C-terminal constructs of Mad1p (318-749 and 475-749) with kinetochores in arrested cells also was disrupted by metabolic poisons (our unpublished data). These results are consistent with the idea that the association of Mad1p with the kinetochores is dynamic and requires energy.
Role of the Nuclear Transport Machinery in Mad1p Function
We have used the various truncation mutants discussed above to evaluate the functional significance of the interactions of Mad1p with the nuclear transport machinery. To do this, we tested the ability of the various truncations to complement the benomyl sensitivity of a mad1Δ strain (Li and Murray, 1991). This strain, like other SAC mutants, loses viability in the presence of microtubule destabilizing drugs due to premature entry into anaphase in the absence of an intact spindle. Plasmids encoding various mad1p truncations, both untagged and GFP tagged, were introduced into a mad1Δ strain, and the resulting strains were serially diluted and plated on YPD plates containing 12.5 μg/ml benomyl and grown at 25°C to evaluate their ability to rescue wild-type growth rates. As shown previously, growth on benomyl-containing plates of the mad1Δ strain can be equally complemented by a plasmid-born copy of the MAD1 or the MAD1-GFP gene (Figure 7, A and B; Iouk et al., 2002). We observed that the NPC binding region of Mad1p alone (residues 1-325), or smaller truncations of this region, did not complement the benomyl sensitivity of the mad1Δ strain (Figure 7, A and B; our unpublished data). Truncations lacking the NPC-binding N-terminus, including the 318-749 and 475-749 constructs, rescued growth suggesting that the C terminus of Mad1p contained regions sufficient for SAC function. Of these truncations, we also detected a reproducible difference between the ability of the 318-749 and the 475-749 constructs to complement the mad1Δ benomyl sensitivity. Cells containing the 318-749 construct were more resistant to benomyl than those containing either the 475-749 construct or the full-length protein (Figure 7, A and B). This phenotype was similar to those observed in various nup null mutants (see above).
Figure 7.
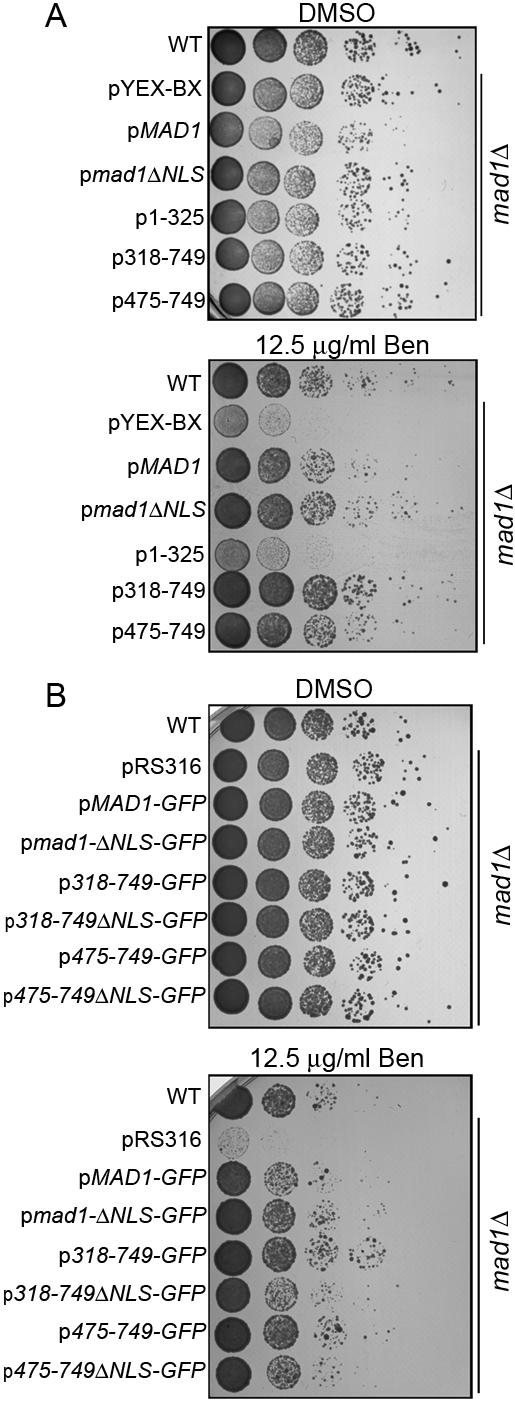
The C terminus of Mad1p is required for SAC function. Plasmids encoding Mad1p or the indicated truncations, either alone (A) or as fusions with GFP (B), were introduced into the mad1Δ strain Y3021. After overnight growth in liquid cultures, equal amounts of cells were serially diluted 10-fold onto YPD plates containing vehicle alone (dimethyl sulfoxide) or 12.5 μg/ml benomyl and grown at 25°C for 2-3 d. The growth characteristics of these strains were compared with wild-type (WT) cells (BY4741) and the mad1Δ strain containing the empty vector pYEX-Bx (A) or pRS316 (B). The ΔNLS constructs lack the coding region for amino acid residues 506-527 of Mad1p.
Each of the constructs that rescued the SAC function of the mad1Δ strain contained the cNLS peptide. Therefore, we tested whether the deletion of the NLS altered the function of the C-terminal constructs or full-length Mad1p. We observed that removal of the NLS from the full-length protein (mad1ΔNLS-GFP) did not seem to cause a reproducible change in its ability to complement the benomyl sensitivity of the mad1Δ strain (Figure 7, A and B). However, we did detect a difference in the 318-749ΔNLS and the 475-749ΔNLS constructs, which failed to fully restore benomyl resistance to the mad1Δ strain to that observed with their NLS-containing counterparts (Figure 7, A and B). These differences were not explained by differences in expression levels (our unpublished data). These results suggest that the cNLS contributes to the efficiency of the spindle checkpoint functions of these mutants.
The analysis of the Mad1p truncation mutants suggests that NPC binding is not required for the SAC function of Mad1p. These data are consistent with our observations that none of the NPC mutants we have implicated in binding Mad1p exhibit an increased sensitivity to benomyl. Instead, their more robust growth in the presence of this drug has led us to speculate that the association of Mad1p with the NPC could function to modulate its activity, potentially influencing exit from SAC arrest. To address this, wild-type BY4741 and various isogenic null mutant strains were arrested with α-factor and then released into media containing moderate levels of nocodazole (12.5 μg/ml). Under these conditions, BY4741 cells progress to M phase and are temporarily delayed in metaphase by the SAC machinery until they release into anaphase. Beginning at ∼100 min after nocodazole treatment, cells began to overcome arrest and an increasing number proceeded to telophase and were visible as large budded cells with two nuclei. However, we observed that progression from metaphase to telophase was strikingly delayed in the nup53Δ mutant. Whereas 50% of wild-type, nup59Δ, and nup60Δ large-budded cells contained two nuclei at 160 min, only 20% of the nup53Δ cells displayed this phenotype (Figure 8A), suggesting they are delayed in their progression through anaphase. This delay could be rescued by a plasmid-borne copy encoding wild-type Nup53p, but not a nup53p mutant lacking its Kap121p binding domain.
Figure 8.
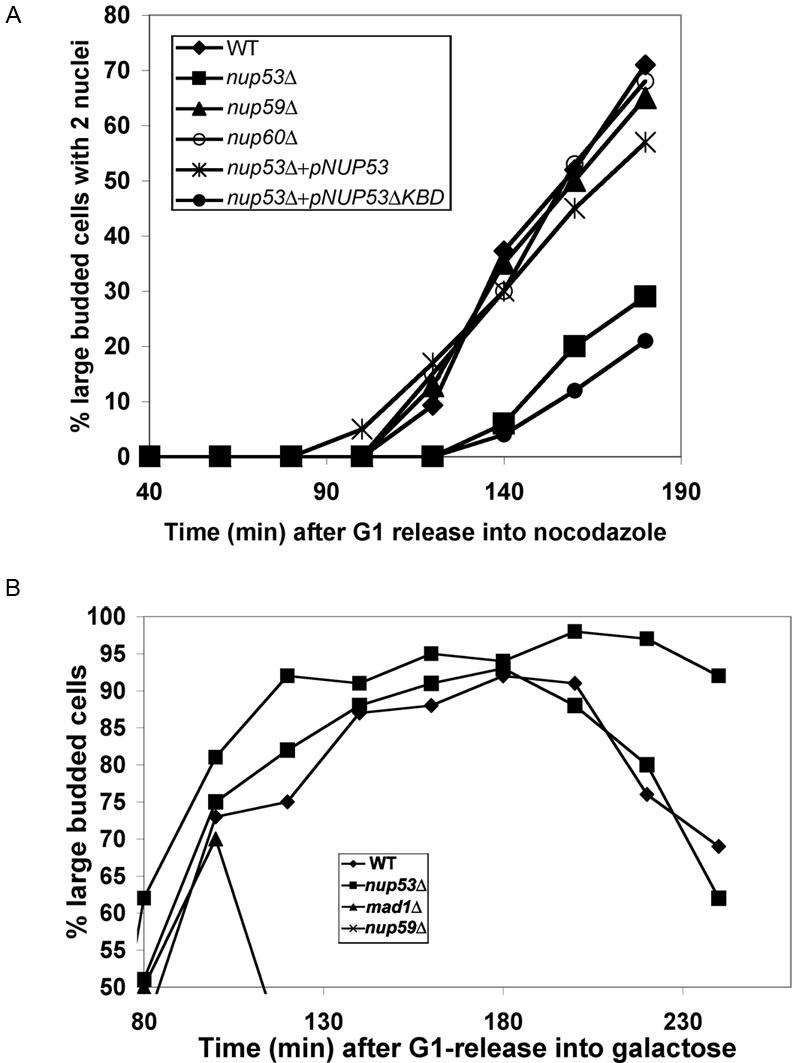
Termination of the SAC is delayed in nup53Δ cells. (A) WT (BY4741) cells and the mutant strains nup53Δ (CPL32), nup59Δ (Y3068), nup60Δ (3058), nup53Δ + pNUP53 (CPL32 containing pNUP53), and nup53Δ + pNUP53ΔKBD (CPL32 containing pNUP53ΔKBD) were synchronized in G1 phase using α-factor and released into media containing 12.5 μg/ml nocodazole. At the indicated time points, cells were fixed with 70% ethanol and stained with DAPI to visualize nuclei and monitor progression from metaphase to telophase. The number of large budded (G2/M) cells containing two DAPI-staining bodies was scored and is presented graphically as a percentage of total cells verses time (minutes) after release into nocodazole-containing media. (B) WT (CPL61), nup53Δ (CPL62), nup59Δ (CPL63), and mad1Δ (CPL64) strains containing an extra copy of the MPS1 ORF chromosomally integrated behind a galactose-inducible promoter were grown in media containing 2% raffinose. The cells were synchronized in G1 phase with α-factor at 30°C and released into media containing 2% raffinose and 3% galactose in the presence of α-factor to induce the synthesis of excess Mps1p. After 2 h, cells were released from G1 and G2/M-phase arrest was monitored by scoring the number of large budded cells at the indicated time points (minutes). These numbers are shown graphically as a percentage of total cells versus time.
The response of nup53Δ strains to SAC activation also was examined in cells overproducing the kinase Mps1p. Overexpression of Mps1p after release from α-factor arrest induces a SAC-dependent M-phase arrest (Hardwick et al., 1996). As shown in Figure 8B, under these conditions mad1Δ cells failed to arrest and exhibited only a transient increase in the number of large budded cells before exiting mitosis (Hardwick et al., 1996; Figure 8B). In contrast, wild-type, nup53Δ, and nup59Δ cultures accumulated as large budded cells until ∼200 min post-α-factor release, at which point the number of large budded wild-type and nup59Δ cells began to decrease as cells exited mitosis and progressed into G1 phase. However, as we observed with nocodazole-arrested cells, nup53Δ cultures remained arrested for an extended period, staying large budded through time points extending to 240 min (Figure 8B). These results are consistent with the conclusion that Nup53p plays a role in terminating the SAC checkpoint. Of note, we also attempted similar experiments using the nup60Δ strain. However, this strain failed to accumulate elevated levels of Msp1p and thus we were unable to use this assay.
DISCUSSION
An accumulating body of evidence has established that the NPC acts as a docking site for a variety of proteins whose apparent primary cellular functions are unrelated to nuclear transport. Two examples are the SAC proteins Mad1p and Map2p, which have been detected in association with NPCs in species from yeast to humans (Campbell et al., 2001; Iouk et al., 2002). These proteins bind tightly to one another (Chen et al., 1999), and, through Mad1p, this complex interacts with the NPC (Iouk et al., 2002). To understand the mechanisms that govern its association with the NPC and the kinetochores, we have mapped domains of Mad1p that interact with the nuclear transport machinery and further defined its interactions with NPC associated binding sites. In addition to Nup53p, these binding sites include a presumed Nup60/Mlp1/Mlp2 complex from which we propose Mad1p cycles to kinetochores when the SAC is activated. The interaction of Mad1p with the NPC occurs through an N-terminal region, which can be separated from a C-terminal region that controls both its SAC activity and kinetochore binding.
The efficient targeting of Mad1p to the NPC requires the karyopherin Kap95p and a cNLS positioned between residues 506 and 527 (Figure 3). Removing the cNLS from Mad1p or inhibiting Kap95p function reduces the efficiency with which Mad1p is targeted to the NPC (Figure 3, C and D). Karyopherin-assisted assembly of components of the NPC has previously been observed for both bona fide nups, such as Nup53p (Lusk et al., 2002), and other NPC associated proteins, including Ulp1p (Panse et al., 2003). In Ulp1p, its association with the NPC is proposed to be directly mediated by karyopherins. It is also possible that Kap95p/Kap60p performs a similar function for Mad1p or, perhaps more likely, the karyopherins facilitate the movement of Mad1p into the NPC and its nucleoplasmic face where it interacts with specific nups. Removal of the nup-binding domain, as in the 318-749 and 475-749 Mad1p constructs, seems to bypass this deposition step and leads to the release of the Mad1p truncations in the nucleoplasm (Figure 2A).
Once targeted to the NPC the association of Mad1p with the NPC is largely mediated by two predicted coiled-coil domains within its N-terminal 325 residues (Figure 2A). On the basis of detected physical associations and in vivo evidence, we predict that Mad1p is likely to interact directly with specific nups including Nup53p (Figures 4A and 5A; Iouk et al., 2002). Similarly, vertebrate Nup53 and Nup153, a potential vertebrate counterpart of Nup60p, play a role in the attachment of Mad1 to the NPC (Hawryluk-Gara et al., 2005; Hawryluk-Gara and Wozniak, unpublished data). In yeast these interactions may be transient in vivo and precede the eventual association of Mad1p with Mlp1p and Mlp2p. We have shown that strains lacking both Mlp1p and Mlp2p fail to concentrate Mad1p at the nuclear periphery (Figure 5, A and D). Moreover, in cells lacking Nup60p, Mad1p colocalizes with intranuclear clusters of Mlp proteins (Figure 5C).
A model where Mad1p interacts with Nup53p before its association with the Mlp proteins is consistent with accumulating data, suggesting that these proteins lie within proximity in the NPC. Nup53p is a member of a complex of nups, which includes its binding partners Nup157p and Nup170p, that is part of the core structures of the NPC (Marelli et al., 1998). This subcomplex interacts with Nup145Np (Lutzmann et al., 2005). The C terminus of this latter nup is required for the NPC association of Nup60p, which in turn is proposed to anchor the Mlp proteins to the NPC (Feuerbach et al., 2002). We also have obtained further data that support of this hierarchical model. Mass spectrometry was used to identify proteins associated with affinity-purified, protein A-tagged Mlp1 and Mlp2 (Supplemental Figure S1). The most abundant species identified included Nup170p, Nup157p, and Nup145p (both N- and C-terminal regions), supporting the idea that these nups are positioned near the Mlp proteins. Our analysis of the Mlp associated proteins, however, did not detect Mad1p suggesting this interaction may be labile under the conditions of these experiments. Nup60p and Nup53p also were not detected. However, this was not surprising as they are predicted to migrate in a region of the gel not analyzed due to contamination with the heavy chain of IgG.
The association of Mad1p with the NPCs throughout the cell cycle and upon SAC activation, as well as the proximity of kinetochores to the NE, precluded an unambiguous visual detection of Mad1p at kinetochores in our strain backgrounds. However, deletion of the NPC binding domain from Mad1p and the corresponding decrease in NE localization allowed us to clearly detect Mad1p truncations at kinetochores (Figure 2, A and B). Similarly, we could visualize the recruitment of full length Mad1p to unattached kinetochores after activation of the SAC in the nup60Δ strain (Figure 5B). These observations are consistent with chromatin immunoprecipitation experiments and data suggesting a partial colocalization of the Mad1-GFP with kinetochore markers (Gillett et al., 2004).
The phenotype of the nup60Δ mutants also allowed us to follow the dynamics of Mad1p association with kinetochores using photobleaching techniques. Our data show a rapid (t1/2 ∼30 s at 23°C) exchange of Mad1p at kinetochores, which is accompanied by similar movements of Mad1-GFP at the Mlp protein foci (Figure 6, A and B). The results of these experiments are consistent with the idea that Mad1p actively cycles between binding sites at the Mlp proteins and kinetochores. Because the nup60Δ strain does not exhibit a detectable SAC defect, it is likely that this cycling event occurs in wild-type cells where the Mlp proteins are associated with the NPC. Moreover, similar measurements performed in vertebrates also have detected that 20-30% of Mad1 exchanges on unattached kinetochores at a similar rate (t1/2 = ∼16 s) (Shah et al., 2004; Howell et al., 2004). However, the majority of Mad1 in this system seems to be largely immobile and believed to be stably associated with unattached kinetochores where it is thought to function as a catalytic platform for the wait-anaphase signal. Our results suggest that a larger percentage of Mad1p (>60%) is rapidly mobile in yeast and capable of cycling between kinetochores and NPC-associated structures. The functional relevance of this is unclear, but the prevalence of the mobile pool may suggest that it also plays a role in inhibiting progression into anaphase.
Strikingly, we observed that the association of Mad1p with the kinetochores, but not with the Mlp proteins (Figure 6C) was energy dependent. This does not seem to be the result of an inhibition of release of Mad1p from the Mlp proteins because we have observed a similar release from unattached kinetochores of Mad1p truncations that fail to associate with the NPC (our unpublished data). Moreover, it seems unlikely that the loss of kinetochore association is due to depletion of nuclear Mad1p caused by nuclear import inhibition because the Mlp protein-bound pool of Mad1p is not affected (Figure 6C). Thus, the most likely explanation is that the kinetochore binding site of the cycling pool of Mad1p is dependent on energy through an, as yet, undefined mechanism. It is important to note that the release of Mad1p from kinetochores was observed in experiments performed on nocodazole treated cell cultures (Figure 6C), suggesting microtubules do not play a role in this process. This is in contrast to observations examining the energy-dependent association of Mad2 with kinetochores in rat Ptk cells where release induced by metabolic poisons requires spindle microtubules (Howell et al., 2000).
We also noted that both full-length and the 318-749 region of Mad1p were recruited to kinetochores only upon activation of the SAC, whereas a further truncated fragment lacking residues 318-474 (truncation 475-749) was associated with kinetochores in asynchronous cultures, independent of activation of the SAC (Figure 2, A and B). A likely explanation is that residues 318-474 can inhibit kinetochore association by masking binding sites for kinetochore constituents that are present nearer the C terminus between residues 475-749. SAC activation is predicted to induce conformational changes in Mad1p or release a binding partner and expose the kinetochore binding site within residues 475-749. Which kinetochore-associated proteins act to anchor the Mad1p truncations to these structures is unclear. Our results would suggest that the Mad1p binding site is a constitutive kinetochore component. One candidate is the Ndc80p complex, which has been suggested to link the Mad1p interacting protein Mad2p to kinetochores when the SAC is activated (Gillett et al., 2004).
Deletion mutations of Mad1p discussed above also were used to evaluate the contribution of the nuclear transport apparatus to the function of Mad1p in the SAC. The benomyl sensitivity of mad1Δ strains could be suppressed by truncations containing the C-terminal, cNLS-containing region of Mad1p (318-749 or 475-749), but not by the N-terminal NPC binding domain (residues 1-325). The apparent requirement of the C terminus of Mad1p for SAC function also was supported by FACS analysis showing that in the presence of nocodazole these mutants arrest in G2/M similarly to wild-type cells (our unpublished data). The importance of this C-terminal domain in the SAC is likely linked to its ability to target to kinetochores (Figure 2) and bind Mad2p (Chen et al., 1999). Consistent with this idea, we have noted that introduction of the 318-749 and 475-749 truncations into mad1Δ cells, which normally contain a diffuse cellular pool of Mad2p (Iouk et al., 2002), induces the nuclear accumulation of Mad2p (our unpublished data).
The degree to which the C-terminal constructs were capable of rescuing the benomyl sensitivity of the mad1Δ strain was reduced, but not eliminated, when the cNLS was deleted (Figure 7B). These ΔNLS constructs are diffusely distributed throughout the cell (Supplemental Figure S2), presumably entering the nucleoplasm by diffusion in sufficient amounts for them to function in the SAC. However, they fail to accumulate at kinetochores suggesting threshold nuclear levels of the constructs must be reached for this event. Interestingly, inclusion of the NPC binding domain (e.g., the Mad1-ΔNLS construct) restored binding to unattached kinetochores (our unpublished data). Cumulatively, these results have led us to conclude that sequence elements that mediate the NPC association and karyopherin binding of Mad1p can function independently to establish a nuclear pool necessary for kinetochore recruitment.
The NPC also may play other roles in modulating SAC activity. Several pieces of data suggest that the NPC may negatively regulate the SAC, perhaps limiting the duration of the SAC. This function would explain the observation that a nup53Δ strain remains arrested in M phase for significantly longer periods of time than wild-type cells in the presence of moderate concentration levels of nocodazole (Figure 8A). Moreover, strains expressing the Mad1p 318-749 truncation (whose binding to the NPC and Nup53p is compromised) reproducibly exhibit an increased resistance to benomyl similar to that observed in nup53Δ strains (Figure 7). These phenotypes could be a consequence of extending the length of the SAC.
The involvement of Nup53p in SAC duration can be envisioned as occurring by different mechanisms. In one case, Nup53p, and possibly other NPC associated proteins may modulate the function of Mad1p, and possibly that of its binding partner Mad2p, by either directly regulating their interactions with other SAC components or by physically segregating them away from their sites of action including kinetochores. Recruitment of Mad2p back to the NPC is in fact what is observed after exit from the SAC (Iouk et al., 2002). Alternatively, it is possible that a regulatory role for Nup53p is linked to its ability to control Kap121p-mediated import in SAC-arrested cells and thus potentially the nuclear localization of key SAC control proteins (Makhnevych et al., 2003). This idea is consistent with our observation that a nup53p mutant lacking the Kap121p binding site is unable to complement the SAC duration defect of the nup53Δ strain (Figure 8A). However, we are careful to interpret these data because this mutation also inhibits the incorporation of Nup53p into the NPC (Lusk et al., 2002).
In conclusion, we envisage that the NPC, beyond its established role in nuclear transport, acts as a macromolecular hub from where the functions of a diverse spectrum of proteins, including SAC components are modulated. This role for the NPC could be accomplished by it acting as a reaction platform or through it functioning as a distribution center that controls access of proteins to their locations of action (e.g., accessibility of Mad1p or Mad2p to other elements of the SAC machinery and kinetochores). In addition to the SAC proteins, this role for the NPC is likely to extend to other classes of proteins. In species ranging from vertebrates and yeast, NPCs interact with proteins involved in diverse functions including, for example, the desumoylating enzyme Ulp1p (Panse et al., 2003) and a poly(ADP-ribose) polymerase tankyrase (Scherthan et al., 2000). The degree to which the dynamic interactions of these proteins with the NPC and karyopherins regulate their function represents a major challenge in understanding the role of the NPC in cellular physiology.
Supplementary Material
Acknowledgments
We thank Munira Basrai and members of the Wozniak laboratory for thoughtful discussion and critical reading of the manuscript. Many thanks to Andrei and Monica Fagarasanu for assistance with FRAP experiments and confocal microscopy. We are grateful to those cited in the text for providing reagents and to Honey Chan and Xuejun Sun for assistance with confocal microscopy. Funding for this work was provided by the Canadian Institutes for Health Research and by the Alberta Heritage Foundation for Medical Research.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05-01-0011) on July 6, 2005.
Abbreviations used: NE, nuclear envelope; NPC, nuclear pore complex; SAC, spindle assembly checkpoint.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Adames, N. R., and Cooper, J. A. (2000). Microtubule interactions with the cell cortex causing nuclear movements in Saccharomyces cerevisiae. J. Cell Biol. 149, 863-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, A., Gottschling, D. E., Kaiser, C. A., and Stearns, T. (1997). Methods in Yeast Genetics, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Aitchison, J. D., Rout, M. P., Marelli, M., Blobel, G., and Wozniak, R. W. (1995). Two novel related yeast nucleoporins Nup170p and Nup157p: complementation with the vertebrate homologue Nup155p and functional interactions with the yeast nuclear pore-membrane protein Pom152p. J. Cell Biol. 131, 1133-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgareh, N., et al. (2001). An evolutionarily conserved NPC subcomplex, which redistributes in part to kinetochores in mammalian cells. J. Cell Biol. 154, 1147-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann, C. B., Davies, A., Cost, G. J., Caputo, E., Li, J., Hieter, P., and Boeke, J. D. (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115-132. [DOI] [PubMed] [Google Scholar]
- Campbell, M. S., Chan, G. K., and Yen, T. J. (2001). Mitotic checkpoint proteins HsMAD1 and HsMAD2 are associated with nuclear pore complexes in interphase. J. Cell Sci. 114, 953-963. [DOI] [PubMed] [Google Scholar]
- Casolari, J. M., Brown, C. R., Komili, S., West, J., Hieronymus, H., and Silver, P. A. (2004). Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell 117, 427-439. [DOI] [PubMed] [Google Scholar]
- Castillo, A. R., Meehl, J. B., Morgan, G., Schutz-Geschwender, A., and Winey, M. (2002). The yeast protein kinase Mps1p is required for assembly of the integral spindle pole body component Spc42p. J. Cell Biol. 156, 453-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, R. H., Brady, D. M., Smith, D., Murray, A. W., and Hardwick, K. G. (1999). The spindle checkpoint of budding yeast depends on a tight complex between the Mad1 and Mad2 proteins. Mol. Biol. Cell 10, 2607-2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronshaw, J. M., Krutchinsky, A. N., Zhang, W., Chait, B. T., and Matunis, M. J. (2002). Proteomic analysis of the mammalian nuclear pore complex. J. Cell Biol. 158, 915-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Antoni, A., et al. (2005). The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr. Biol. 15, 214-225. [DOI] [PubMed] [Google Scholar]
- De Wulf, P., McAinsh, A. D., and Sorger, P. K. (2003). Hierarchical assembly of the budding yeast kinetochore from multiple subcomplexes. Genes Dev. 17, 2902-2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme, E. (1989). Transformation of Saccharomyces cerevisiae by electroporation. Appl. Environ. Microbiol. 55, 2242-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilworth, D. J., Suprapto, A., Padovan, J. C., Chait, B. T., Wozniak, R. W., Rout, M. P., and Aitchison, J. D. (2001). Nup2p dynamically associates with the distal regions of the yeast nuclear pore complex. J. Cell Biol. 153, 1465-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagarasanu, M., Fagarasanu, A., Tam, Y. Y., Aitchison, J. D., and Rachubinski, R. A. (2005). Inp1p is a peroxisomal membrane protein required for peroxisome inheritance in Saccharomyces cerevisiae. J. Cell Biol. 169, 765-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerbach, F., Galy, V., Trelles-Sticken, E., Fromont-Racine, M., Jacquier, A., Gilson, E., Olivo-Marin, J. C., Scherthan, H., and Nehrbass, U. (2002). Nuclear architecture and spatial positioning help establish transcriptional states of telomeres in yeast. Nat. Cell Biol. 4, 214-221. [DOI] [PubMed] [Google Scholar]
- Fraschini, R., Beretta, A., Sironi, L., Musacchio, A., Lucchini, G., and Piatti, S. (2001). Bub3 interaction with Mad2, Mad3 and Cdc20 is mediated by WD40 repeats and does not require intact kinetochores. EMBO J. 20, 6648-6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy, V., Gadal, O., Fremont-Racine, M., Romano, A., Jacquier, A., and Nehrbass, U. (2004). Nuclear retention of unspliced mRNAs in yeast is mediated by perinuclear Mlp1. Cell 116, 63-73. [DOI] [PubMed] [Google Scholar]
- Galy, V., Olivo-Marin, J. C., Scherthan, H., Doye, V., Rascalou, N., and Nehrbass, U. (2000). Nuclear pore complexes in the organization of silent telomeric chromatin. Nature 403, 108-112. [DOI] [PubMed] [Google Scholar]
- Giaever, et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387-391. [DOI] [PubMed] [Google Scholar]
- Gillett, E. S., Espelin, C. W., and Sorger, P. K. (2004). Spindle checkpoint proteins and chromosome-microtubule attachment in budding yeast. J. Cell Biol. 164, 535-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima, G., and Yanagida, M. (2000). Establishing biorientation occurs with precocious separation of the sister kinetochores, but not the arms, in the early spindle of budding yeast. Cell 100, 619-633. [DOI] [PubMed] [Google Scholar]
- Hardwick, K. G., and Murray, A. W. (1995). Mad1p, a phosphoprotein component of the spindle assembly checkpoint in budding yeast. J. Cell Biol. 131, 709-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick, K. G., Weiss, E., Luca, F. C., Winey, M., and Murray, A. W. (1996). Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science 273, 953-956. [DOI] [PubMed] [Google Scholar]
- Hardwick, K. G., Johnston, R. C., Smith, D. L., and Murray, A. W. (2000). MAD3 encodes a novel component of the spindle checkpoint which interacts with Bub3p, Cdc20p, and Mad2p. J. Cell Biol. 148, 871-882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawryluk-Gara, L. A., Shibuya, E. K., and Wozniak, R. W. (2005). Vertebrate Nup53 interacts with nuclear lamina and is required for the assembly of a Nup93-containing complex. Mol. Biol. Cell 16, 2382-2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell, B. J., Hoffman, D. B., Fang, G., Murray, A. W., and Salmon, E. D. (2000). Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J. Cell Biol. 150, 1233-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell, B. J., Moree, B., Farrar, E. M., Stewart, S., Fang, G., and Salmon, E. D. (2004). Spindle checkpoint protein dynamics at kinetochores in living cells. Curr. Biol. 14, 953-964. [DOI] [PubMed] [Google Scholar]
- Ikui, A. E., Furuya, K., Yanagida, M., and Matsumoto, T. (2002). Control of localization of a spindle checkpoint protein, Mad2, in fission yeast. J. Cell Sci. 115, 1603-1610. [DOI] [PubMed] [Google Scholar]
- Iouk, T., Kerscher, O., Scott, R. J., Basrai, M. A., and Wozniak, R. W. (2002). The yeast nuclear pore complex functionally interacts with components of the spindle assembly checkpoint. J. Cell Biol. 159, 807-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii, K., Arib, G., Lin, C., Van Houwe, G., and Laemmli, U. K. (2002). Chromatin boundaries in budding yeast: the nuclear pore connection. Cell 109, 551-562. [DOI] [PubMed] [Google Scholar]
- Joseph, J., Tan, S. H., Karpova, T. S., McNally, J. G., and Dasso, M. (2002). SUMO-1 targets RanGAP1 to kinetochores and mitotic spindles. J. Cell Biol. 156, 595-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaffman, A., Rank, N. M., and O'Shea, E. K. (1998). Phosphorylation regulates association of the transcription factor Pho4 with its import receptor Pse1/Kap121. Genes Dev. 12, 2673-2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerscher, O., Hieter, P., Winey, M., and Basrai, M. A. (2001). Novel role for a Saccharomyces cerevisiae nucleoporin, Nup170p, in chromosome segregation. Genetics 157, 1543-1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerscher, O., Crotti, L. B., and Basrai, M. A. (2003). Recognizing chromosomes in trouble: association of the spindle checkpoint protein Bub3p with altered kinetochores and a unique defective centromere. Mol. Cell. Biol. 23, 6406-6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie, D. M., Grill, B., Rout, M. P., Wozniak, R. W., and Aitchison, J. D. (2002). Kap121p-mediated nuclear import is required for mating and cellular differentiation in yeast. Mol. Cell. Biol. 22, 2544-2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew, D. J., and Burke, D. J. (2003). The spindle assembly and spindle position checkpoints. Annu. Rev. Genet. 37, 251-282. [DOI] [PubMed] [Google Scholar]
- Li, R., and Murray, A. W. (1991). Feedback control of mitosis in budding yeast. Cell 66, 519-531. [DOI] [PubMed] [Google Scholar]
- Loïodice, I., Alves, A., Rabut, G., Van Overbeek, M., Ellenberg, J., Sibarita, J. B., and Doye, V. (2004). The entire Nup107-160 complex, including three new members, is targeted as one entity to kinetochores in mitosis. Mol. Biol. Cell 15, 3333-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusk, C. P., Makhnevych, T., Marelli, M., Aitchison, J. D., and Wozniak, R. W. (2002). Karyopherins in nuclear pore biogenesis: a role for Kap121p in the assembly of Nup53p into nuclear pore complexes. J. Cell Biol. 159, 267-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutzmann, M., Kunze, R., Buerer, A., Aebi, U., and Hurt, E. (2002). Modular self-assembly of a Y-shaped multiprotein complex from seven nucleoporins. EMBO J. 21, 387-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutzmann, M., Kunze, R., Stangl, K., Stelter, P., Toth, K. F., Bottcher, B., and Hurt, E. (2005). Reconstitution of Nup157 and Nup145N into the Nup84 complex. J. Biol. Chem. 280, 18442-18451. [DOI] [PubMed] [Google Scholar]
- Makhnevych, T., Lusk, C. P., Anderson, A. M., Aitchison, J. D., and Wozniak, R. W. (2003). Cell cycle regulated transport controlled by alterations in the nuclear pore complex. Cell 115, 813-823. [DOI] [PubMed] [Google Scholar]
- Makkerh, J. P., Dingwall, C., and Laskey, R. A. (1996). Comparative mutagenesis of nuclear localization signals reveals the importance of neutral and acidic amino acids. Curr. Biol. 6, 1025-1027. [DOI] [PubMed] [Google Scholar]
- Marelli, M., Aitchison, J. D., and Wozniak, R. W. (1998). Specific binding of the karyopherin Kap121p to a subunit of the nuclear pore complex containing Nup53p, Nup59p, and Nup170p. J. Cell Biol. 143, 1813-1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panse, V. G., Kuster, B., Gerstberger, T., and Hurt, E. (2003). Unconventional tethering of Ulp1 to the transport channel of the nuclear pore complex by karyopherins. Nat. Cell Biol. 5, 21-27. [DOI] [PubMed] [Google Scholar]
- Pinsky, B. A., Tatsutani, S. Y., Collins, K. A., and Biggins, S. (2003). An Mtw1 complex promotes kinetochore biorientation that is monitored by the Ipl1/Aurora protein kinase. Dev. Cell 5, 735-745. [DOI] [PubMed] [Google Scholar]
- Robbins, J., Dilworth, S. M., Laskey, R. A., and Dingwall, C. (1991). Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite nuclear targeting sequence. Cell 64, 615-623. [DOI] [PubMed] [Google Scholar]
- Rout, M. P., Aitchison, J. D., Suprapto, A., Hjertaas, K., Zhao, Y., and Chait, B. T. (2000). The yeast nuclear pore complex: composition, architecture, and transport mechanism. J. Cell Biol. 148, 635-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salina, D., Enarson, P., Rattner, J. B., and Burke, B. (2003). Nup358 integrates nuclear envelope breakdown with kinetochore assembly. J. Cell Biol. 162, 991-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherthan, H., Jerratsch, M., Li, B., Smith, S., Hulten, M., Lock, T., and de Lange, T. (2000). Mammalian meiotic telomeres: protein composition and redistribution in relation to nuclear pores. Mol. Biol. Cell 11, 4189-4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz, O., Thiel, A., Hillen, W., and Niederweis, M. (2000). Quantitative analysis of gene expression with an improved fluorescent protein. p6. Eur. J. Biochem. 267, 1565-1570. [DOI] [PubMed] [Google Scholar]
- Schultz, M. C., Hockman, D. J., Harkness, T. A., Garinther, W. I., and Altheim, B. A. (1997). Chromatin assembly in a yeast whole-cell extract. Proc. Natl. Acad. Sci. USA 94, 9034-9039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, J., Milpetz, F., Bork, P., and Ponting, C. P. (1998). SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl. Acad. Sci. USA 95, 5857-5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, J. V., Botvinick, E., Bonday, Z., Furnari, F., Berns, M., and Cleveland, D. W. (2004). Dynamics of centromere and kinetochore proteins; implications for checkpoint signaling and silencing. Curr. Biol. 14, 942-952. [DOI] [PubMed] [Google Scholar]
- Shulga, N., Roberts, P., Gu, Z., Spitz, L., Tabb, M. M., Nomura, M., and Goldfarb, D. S. (1996). In vivo nuclear transport kinetics in Saccharomyces cerevisiae: a role for heat shock protein 70 during targeting and translocation. J. Cell Biol. 135, 329-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight, A. F., and Murray, A. W. (1997). The spindle assembly checkpoint in budding yeast. Methods Enzymol. 283, 425-440. [DOI] [PubMed] [Google Scholar]
- Strambio-de-Castillia, C., Blobel, G., and Rout, M. P. (1999). Proteins connecting the nuclear pore complex with the nuclear interior. J. Cell Biol. 144, 839-855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntharalingam, M., and Wente, S. R. (2003). Peering through the pore: nuclear pore complex structure, assembly, and function. Dev. Cell 4, 775-789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.