

Abstract
Although many algorithms exist for estimating haplotypes from genotype data, none of them take full account of both the decay of linkage disequilibrium (LD) with distance and the order and spacing of genotyped markers. Here, we describe an algorithm that does take these factors into account, using a flexible model for the decay of LD with distance that can handle both “blocklike” and “nonblocklike” patterns of LD. We compare the accuracy of this approach with a range of other available algorithms in three ways: for reconstruction of randomly paired, molecularly determined male X chromosome haplotypes; for reconstruction of haplotypes obtained from trios in an autosomal region; and for estimation of missing genotypes in 50 autosomal genes that have been completely resequenced in 24 African Americans and 23 individuals of European descent. For the autosomal data sets, our new approach clearly outperforms the best available methods, whereas its accuracy in inferring the X chromosome haplotypes is only slightly superior. For estimation of missing genotypes, our method performed slightly better when the two subsamples were combined than when they were analyzed separately, which illustrates its robustness to population stratification. Our method is implemented in the software package PHASE (v2.1.1), available from the Stephens Lab Web site.
Introduction
At autosomal loci, the genetic material carried by a diploid individual can be thought of as being composed of two haplotypes, each containing the genetic information from one of the two homologous chromosomes. Knowledge of the haplotypes carried by sampled individuals would be helpful in many settings, such as linkage-disequilibrium (LD) mapping or attempting to make inferences regarding evolutionary mechanisms, such as selection or recombination, that may be acting on a region. However, although technologies continue to develop, current high-throughput approaches to genotyping do not provide haplotype information. Although, in some studies, collection of data from related individuals may allow haplotypes to be inferred, in general, such data may be costly or impossible to collect. These factors have generated considerable interest in computational and statistical approaches for inferring haplotypes from unphased genotype data in population samples.
All computational and statistical approaches to haplotype inference exploit LD, which is the nonrandom association of alleles among linked loci. LD tends to decay with distance; that is, there tends to be less LD between loci that are far apart than between loci that are close together. Earlier approaches to haplotype estimation—which include the algorithm of Clark (1990), maximum likelihood via the EM algorithm (Excoffier and Slatkin 1995; Hawley and Kidd 1995; Long et al. 1995), and the Bayesian approach of Stephens et al. 2001—ignore this fact, in that haplotype estimates from those approaches are independent of locus spacing and even of locus order.
As far as we are aware, the first haplotype-inference method to produce results that can depend on locus order was introduced by Niu et al. (2002). Those authors introduced the idea of “partition ligation” (PL), which approaches data sets containing several loci by first dividing them into segments containing a small number (∼8) of contiguous loci and then inferring haplotypes within each segment before iteratively combining results from adjacent segments. This framework was subsequently adopted by others, including Qin et al. (2002), Stephens and Donnelly (2003), and Lin et al. (2004), for different methods for inferring haplotypes within each segment and for combining segments. The most obvious benefits of PL are the reduction of computing times and the increase in the size of data set that can be tackled effectively. However, a side effect is that estimates of phase at any locus will depend more on data at nearby loci than on data at more-distant loci. Although this effect is presumably beneficial, it is unfortunately diluted by the fact that all existing approaches ignore the order and spacing of loci within segments formed.
Other recent methods for haplotype inference (e.g., Greenspan and Geiger [2004], Halperin and Eskin [2004], Kimmel and Shamir [2004]) also involve dividing data into segments of consecutive loci. However, unlike the methods described above, the division into segments is made with reference to observed patterns of LD. Motivated by the observation that some regions of the human genome appear to contain “blocks” of low haplotype diversity, these methods attempt to ensure that the segments formed correspond, in some sense, to “blocks” of high LD and of low haplotype diversity. Excoffier et al. (2003) take a different approach, using a window of neighboring loci when estimating phase at any given position, with window widths allowed to vary according to local levels of LD. None of these methods take into account locus spacing or the decay of LD within the blocks or windows formed.
Here, we describe a new algorithm for haplotype inference that modifies the approaches of Stephens et al. (2001) and Stephens and Donnelly (2003), to take explicit account of locus spacing and the decay of LD with distance. These previous approaches are Bayesian and make use of an “approximate coalescent” prior to reflect the fact that haplotypes in a population tend to group together in clusters of similar haplotypes. Our new algorithm can be thought of as modifying this prior to the “coalescent with recombination” (Hudson 1991), by use of recently developed models for how patterns of LD among multiple loci depend on the underlying recombination rate (Fearnhead and Donnelly 2001; Li and Stephens 2003). The assumptions underlying this coalescent process include the fact that the population has been evolving with a constant size for a long time and is randomly mating (so is in Hardy-Weinberg equilibrium) and that the locus under study is evolving neutrally. Since none of these assumptions will hold in real studies, it may be helpful to think of our prior not as an attempt to perform inference under a formal model but rather as an attempt to capture salient qualitative features of real data—notably, the tendency for haplotypes to cluster together (because of shared ancestry) and the fact that which haplotypes cluster together may change as one moves along the sequence (because of recombination). Since the underlying recombination rate is typically unknown and may vary on a fine scale (Crawford et al. 2004; McVean et al. 2004), we adopt a flexible model that estimates a different recombination rate in each SNP interval. In this way, we allow for both gradual decay of LD with distance and for more-abrupt decay of LD, as might be observed across a recombination hotspot, for example. As a result, the method is equally applicable, whether or not the region under study exhibits “blocklike” patterns of LD.
We compare the accuracy of this new algorithm with several of the numerous algorithms for haplotype inference now available (table 1). First, we assess accuracy of haplotype estimates, using X chromosome haplotypes determined from males (Lin et al. 2002). Second, we compare a subset of the methods on the haplotype data, determined from genotypes in trios in an autosomal region by Daly et al. (2001). Last, we assess the accuracy with which haplotype-inference methods are able to predict missing genotypes, using 50 genes resequenced by the University of Washington–Fred Hutchinson Cancer Research Center (UW-FHCRC) Variation Discovery Resource (Carlson et al. 2003). A considerable advantage of this last approach to comparing methods is that it requires only unphased genotype data, which are relatively abundant. Since real data almost invariably contain some missing genotypes, the accuracy with which such genotypes can be predicted from other (nonmissing) genotypes is of direct practical interest. Further, accuracy in predicting missing genotypes provides another test of how accurately the assumptions underlying each method capture patterns of real haplotype variation and, thus, an indirect way to assess which methods are likely to provide the most accurate haplotype estimates. Accurate methods for imputing missing data may also be helpful in developing methods for LD mapping and choosing haplotype-tagging SNPs (Johnson et al. 2001) (see the “Discussion” section). Despite these considerations, there appear to be few previous published comparisons of the accuracy of different methods for this task.
Table 1.
Summary of the Haplotype Inference Algorithms for Unrelated Individuals
| Algorithm | Description | Reference(s) |
| PHASE v2 recom (-MR) | Bayesian method with approximate “coalescent with recombination” prior, capturing the fact that each sampled haplotype tends to be similar to another haplotype or to a mosaic of other haplotypes. | Present study |
| PHASE v2 hybrid (-MQ) | Hybrid algorithm reduces computing time over -MR by assumption of no recombination for the majority of the computation, before incorporating recombination for the final steps. | Present study |
| PHASE v2 no recom (-MS) | Bayesian method with approximate “coalescent without recombination” prior, on the basis of the idea that sampled haplotypes look similar to other haplotypes. Ignores decay of LD with distance. | Stephens et al. 2001; Stephens and Donnelly 2003 |
| Bayes-Dirichlet | Bayesian method with Dirichlet prior on population haplotype frequencies. Ignores similarity of haplotypes. (Our own implementation of the algorithm in Haplotyper, mentioned below.) | Stephens et al. 2001; Niu et al. 2002 |
| Arlequin 3.0a (ELB) | Bayesian method. Prior takes some account of similarity of haplotypes. Bases inference for each locus on data in a window of nearby loci, which is allowed to vary in size on the basis of local levels of LD. | Excoffier et al. 2003 |
| Haplotyper | Bayesian method with a Dirichlet prior on the haplotype frequencies. Ignores similarity of haplotypes. | Niu et al. 2002 |
| PL-EM | A maximum-likelihood approach. Uses EM algorithm plus computational trick (PL) to obtain estimates of haplotype frequencies and uses these estimates to find most probable haplotypes for each individual. | Qin et al. 2002 |
| snphap v1.2 | Similar to PL-EM above, but uses different tricks to reduce computational demands. | David Clayton Software Web page |
| hap | Based on an ad hoc modification of the Dirichlet prior for population haplotype frequencies. | Lin et al. 2002 |
| hap2 | An improved implementation of hap above. | Lin et al. 2004 |
| HAP | Algorithm partitions loci into blocks of low haplotype diversity; assumes that haplotypes within blocks will conform approximately to a “perfect phylogeny” (i.e., no recombination or repeat mutation). | Eskin et al. 2003a, 2003b; Halperin and Eskin 2004 |
All our assessments suggest that haplotype-inference methods based on an approximate coalescent prior are consistently more accurate than are the other methods we consider. Of coalescent-based approaches, our new approach clearly outperforms the previous approaches of Stephens et al. (2001) and Stephens and Donnelly (2003) (which ignore the decay of LD with distance) for the autosomal data sets, whereas its performances in inferring the X chromosome haplotypes is only slightly superior.
Methods
We now give an overview of the algorithm and discuss associated theoretical issues. A more detailed description is given in appendix A.
Our algorithm is similar to those used by Crawford et al. (2004) and Ptak et al. (2004), although the focus here is different: in those articles, the unknown underlying haplotypes were treated as “nuisance parameters,” and the underlying recombination process was the object of interest, whereas here we treat the recombination process as a “nuisance parameter” and assess the accuracy of estimated haplotypes. Another difference is that, here, we adopt a more flexible model for the underlying recombination process.
Let G=(G1,…,Gn) denote the observed genotypes of the n individuals at L loci; let H=(H1,…,Hn) denote the actual (unobserved) haplotypes, where Hi is the pair of haplotypes of individual i. Note that Gi may be “missing” alleles at some loci, in which case H includes estimates of the unobserved alleles. Let ρ=(ρ1,…,ρL-1) denote the (unknown) vector of recombination rates between each pair of consecutive loci, scaled by the effective population size. That is,
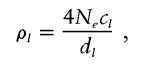 |
where Ne is the (unknown) diploid effective population size, cl is the (unknown) probability of recombination per generation between markers l and l+1, and dl is the physical distance between markers l and l+1, assumed known. Informally, ρldl measures the expected breakdown in LD across the interval spanned by markers l and l+1, with large values corresponding to a large expected breakdown in LD.
Our aim is to estimate H from the observed genotype data G, taking account of information in the data—particularly in the rate of decay of LD with distance—on the underlying recombination rates ρ. We do this by developing a Markov chain–Monte Carlo (MCMC) algorithm to sample from the conditional distribution of the haplotypes and recombination parameters, given the genotype data Pr(H,ρ|G). Using this algorithm, we can obtain a point estimate for H (e.g., the estimated posterior mode for the haplotype pair of each individual), together with measures of confidence in the accuracy of individual haplotype estimates.
In outline, our algorithm starts with initial guesses for H and ρ and a random ordering of the individuals ν and then iterates the following steps many times:
-
1.
for each individual i in turn (in the order given by the ordering ν), update the individual’s pair of haplotypes by sampling Hi from Pr(Hi|Gi,H-i,ρ), where H-i is the set of current guesses for the haplotypes of all individuals except i;
-
2.
propose a new value for ρ and accept it or reject it according to the Metropolis-Hastings (MH) acceptance probability (eq. [2]); and
-
3.
propose a new value for ν (by proposing to switch two randomly chosen individuals) and accept it or reject it according to the MH acceptance probability.
Step 1 can be thought of as estimating the haplotypes for individual i, taking into account the individual’s genotype data (Gi), the haplotypes of all other individuals (H-i), and the underlying patterns of LD (ρ). Several previous haplotype-inference algorithms (e.g., those of Stephens et al. [2001], Niu et al. [2002], and Stephens and Donnelly [2003]) adopt a similar structure, although without the parameter ρ, and accomplish this step in different ways, depending on the underlying assumptions. Here, we make use of the conditional distributions from Fearnhead and Donnelly (2001) and Li and Stephens (2003), which, in estimation of haplotypes in individual i, favors haplotypes that are similar to a mosaic of the haplotypes possessed by other individuals. Break points in such a mosaic can occur anywhere but are most likely to occur in marker intervals that are large or have a higher recombination rate (higher ρl); see figure 1 for illustration. Thus, in inferring the haplotypes, the method takes into account marker spacing and the decay of LD with distance.
Figure 1.

Illustration of how π(hk+1|h1,…,hk) builds hk+1 as an imperfect mosaic of h1,…,hk. The figure illustrates the case k=3 and shows three possible values (h4A, h4B, and h4C) for h4, given h1, h2, and h3. Each column of circles represents a SNP locus, with blackened and unblackened circles representing the two alleles. Each possible h4 can be thought of as having been created by “copying” parts of h1, h2 and h3. The shading in each case shows which haplotype was “copied” at each position along the chromosome and indicates whether the haplotype is most closely related to h1, h2, or h3. Changes in the shading along a haplotype represent ancestral recombination events; these are more likely to occur between SNPs that are farther apart (e.g., SNPs 2 and 3 or 4 and 5) or that have a higher rate of recombination between them. The imperfect nature of the copying process is exemplified at the third and fourth locus, where h4B has the blackened allele despite having “copied” h2, which has the unblackened allele. The occurrence of several such “imperfections” on a single chunk indicate that the haplotype is relatively highly diverged from the one that it copied in that region (see text).
More precisely, we use
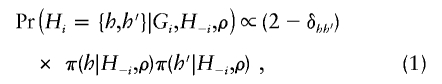 |
where δhh′=1 if h=h′ and =0 otherwise; the conditional distribution π is a modification of that of Fearnhead and Donnelly (2001), as described below. (The second term on the right side should be “π(h′|H-i,h,ρ),” but we omitted “h” from this conditioning for convenience of implementation and for the happy side effect of producing an expression that is symmetrical in h and h′, which, for our choice of π, is not otherwise guaranteed.)
Step 2 of the algorithm can be thought of as estimating recombination rates from patterns of LD in the current estimated haplotypes. The MH-acceptance probability is given by
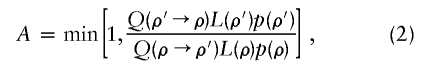 |
where Q(ρ→ρ′) is the probability of proposing a move to ρ′, given that we are currently at ρ, p(ρ) is a prior on ρ, and L(ρ)=Pr(H|ρ) is the likelihood for ρ. We use the PAC-B likelihood of Li and Stephens (2003) for L(ρ). (This likelihood depends on the order in which the haplotypes are considered; we use the order given by ν to order the individuals and order the two haplotypes within each individual alphabetically.) Our prior on ρ is similar to the “general recombination variation” model used by Li and Stephens (2003). Specifically, we assume that 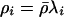 , where the prior on λi is that they are independent and identically distributed, with
, where the prior on λi is that they are independent and identically distributed, with 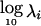 normally distributed, with mean 0 and SD 0.5, and the prior distribution for
normally distributed, with mean 0 and SD 0.5, and the prior distribution for 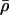 is uniform on a log scale, with the constraint
is uniform on a log scale, with the constraint 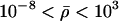 . Here,
. Here, 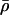 can be thought of as the background recombination rate across the region and λi the factor by which ρi deviates from this background. The prior on λi says that 95% of intervals between SNPs will have a recombination rate that is within a factor of 10 of the background rate. The range of allowed values for
can be thought of as the background recombination rate across the region and λi the factor by which ρi deviates from this background. The prior on λi says that 95% of intervals between SNPs will have a recombination rate that is within a factor of 10 of the background rate. The range of allowed values for 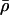 covers several orders of magnitude on either side of the average value for humans.
covers several orders of magnitude on either side of the average value for humans.
Step 3 of the algorithm is included partly to ensure that the algorithm (provided it is run long enough) will not depend on the individuals' entry order in the input file and partly to provide an order over which to compute the PAC-B likelihood in step 2. The approach is similar to that taken by Stephens and Donnelly (2003), except that they randomly ordered the individuals every iteration, whereas we use an MH proposal (with acceptance probability computed using a uniform prior on all possible orderings and the PAC-B likelihood).
The above outline omits many details. Perhaps the most important of these is that strict implementation of step 1 is not computationally tractable for data sets with a moderate number of loci, because it requires expression (1) to be evaluated for all haplotype pairs {h,h′} consistent with genotypes of individual i, and the number of such pairs may be huge. To avoid this problem, we make use of PL (Niu et al. 2002; Stephens and Donnelly 2003), wherein the data are first divided into small segments of consecutive loci, and the algorithm is then applied to each segment in turn before iteratively combining the results from adjacent segments. Each time the algorithm is applied to a segment, a list is made of the haplotypes that may “plausibly” occur (in at least one individual) within that segment. This list is typically short, compared with the total number of possible haplotypes. When two segments are combined, only haplotype pairs consistent with haplotypes on the corresponding lists are considered possible, and the fact that these lists are typically short keeps the computational burden manageable. Other details of the algorithm are given in appendix A.
Our algorithm, in common with algorithms of Stephens et al. (2001) and Stephens and Donnelly (2003), contains a “pseudo-Gibbs” step (Heckerman et al. 2000), in that the conditional distributions π used in our step 1 are defined directly, rather than being derived from an explicit prior distribution on H. Care must be taken with the use of algorithms containing such steps, since they are not, in general, guaranteed to converge to a stationary distribution. In previous work, we have defended this approach and, in particular, have shown that the similar algorithms of Stephens et al. (2001) and Stephens and Donnelly (2003) are guaranteed to converge to a stationary distribution because they are defined on a finite discrete space (see the appendix of Stephens and Donnelly [2003]). Unfortunately, this argument does not apply to this new algorithm, because step 2 includes a continuous parameter, ρ. Theoretical convergence of this algorithm, therefore, remains an open question; however, in our experience with a wide range of examples—both real data and simulations—we have seen no indication of convergence problems beyond those usually associated with these kinds of MCMC schemes, and, in our comparisons, the algorithm produces, on average, more-accurate haplotype estimates (by two different measures) than does the algorithm of Stephens and Donnelly (2003), which is guaranteed to converge.
Another theoretically dubious aspect of the algorithm is our use of different conditional distributions (those of Fearnhead and Donnelly [2001] and Li and Stephens [2003]) in steps 1 and 2 of the algorithm; the reason for this is explained below. We note that it would be possible to use the models of Li and Stephens (2003) to produce MCMC schemes that do not suffer from either of these theoretical weaknesses, but they would require substantially more computational time and seem to us unlikely to produce practically important gains in performance.
Formal Description of π
The conditional distribution π used to compute expression (1) is a slight modification of the one introduced by Fearnhead and Donnelly (2001) and is also similar to the conditional distribution πA of Li and Stephens (2003). For simplicity, we change notation slightly, and let h1,…,hk denote the (not necessarily distinct) haplotypes of k chromosomes sampled from a population, and let hk+1 denote the haplotype of another chromosome sampled from the same population. The conditional distribution π(hk+1|h1,…,hk,ρ) specifies the probability of observing a given value for hk+1, given the observed h1,…,hk and the values for the underlying recombination parameters ρ. It is based on the idea that hk+1 will look similar to a mosaic of h1,…,hk (fig. 1). Think of hk+1 as being, at each locus l=1,…,L, a (possibly imperfect) copy of one of h1,…,hk, and let Xl denote which haplotype hk+1 copies at locus l (so Xl∈{1,2,…,k}). In addition, we associate with the copying event at locus l a time Tl, which affects the probability that hk+1 will be identical to the allele it copied at locus l. Informally, one can think of Xl as indicative of which of the haplotypes h1,…,hk is most closely related to hk+1 at locus l and Tl (or, more precisely, Tl/k) as indicative of how closely related the two are. We allow Tl to take one of two possible values, which can be thought of as corresponding to “closely” or “distantly” related. Specifically, Tl can take values (t1,t2)=(0.586,3.414), with respective marginal probabilities (w1,w2)=(0.854,0.146). These numbers were chosen as a very rough approximation of a continuous exponential distribution with mean 1, on the basis of Gaussian quadrature with Laguerre polynomials (Evans 1993); see also the appendix of Stephens and Donnelly (2000). It is in the introduction of these Tj values that this conditional distribution differs from those of Li and Stephens (2003) and is similar to the one from Fearnhead and Donnelly (2001).
To mimic the effects of recombination, we model {(Xl,Tl):l=1,…,L} as a Markov chain on {1,…,k}, with Pr(X1=x,T1=tr)=(1/k)wr for (x,r)∈{1,…,k}×{1,2}, and
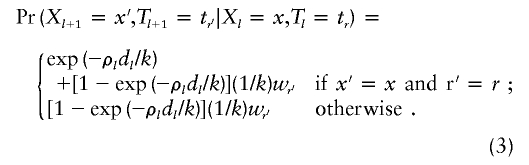 |
The idea here is that jumps in the mosaic process occur with probability 1-exp(-ρldl/k) (and so are most likely to occur in marker intervals in which ρldl is large) and, when a jump occurs, it is equally likely to jump to copying any of the chromosomes. Note that the jump probability we use differs slightly from that of Fearnhead and Donnelly (2001), who use ρldl/(k+ρldl). The numerical values of the two expressions are similar for small (typical) values of ρldl, but our expression has the desirable theoretical property that probabilities are unchanged by the addition of a locus, with a missing allele in hk+1, anywhere along the sequence.
To mimic the effects of mutation, we allow that the allele of hk+1 at locus l may not be an exact copy of the allele that it “copied.” Specifically, we assume that the allele at locus l is the result of applying a number (m, which may be 0) of mutations to the allele that it copied. We assume that m has a Poisson distribution with mean 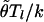 , where
, where 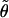 is a parameter the value of which controls the frequency of mutations. (Thus, consistent with the interpretation of Tl as a measure of divergence, mutations are more likely at loci where Tl is large.) The mutation mechanism at each locus l is specified by a matrix Pl, assumed to be known, whose (i,j)th element is the probability that an offspring is of type j, given that the progenitor is of type i and that a mutation occurs. Thus, if hi,l denotes the allele at locus l in haplotype i, then, given the copying process (X,T)=(X1,…,XL,T1,…,TL), the alleles hk+1,1,hk+1,2,…,hk+1,L are independent, with
is a parameter the value of which controls the frequency of mutations. (Thus, consistent with the interpretation of Tl as a measure of divergence, mutations are more likely at loci where Tl is large.) The mutation mechanism at each locus l is specified by a matrix Pl, assumed to be known, whose (i,j)th element is the probability that an offspring is of type j, given that the progenitor is of type i and that a mutation occurs. Thus, if hi,l denotes the allele at locus l in haplotype i, then, given the copying process (X,T)=(X1,…,XL,T1,…,TL), the alleles hk+1,1,hk+1,2,…,hk+1,L are independent, with
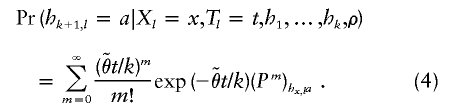 |
We approximate this infinite sum by the first 50 terms (which need be done only once for each locus, and the results tabulated).
In all the examples we consider here, the data consist of biallelic SNP loci. For these, we use, as did Stephens et al. (2001), 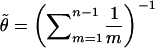 and a mutation process (P) in which a mutation at a locus always leads to a change from the current allele to the alternative allele. In software, we have also implemented a generalized stepwise mutation mechanism for microsatellite loci and a parent-independent mutation mechanism for other multiallelic loci, with
and a mutation process (P) in which a mutation at a locus always leads to a change from the current allele to the alternative allele. In software, we have also implemented a generalized stepwise mutation mechanism for microsatellite loci and a parent-independent mutation mechanism for other multiallelic loci, with 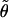 estimated as was done by Stephens et al. (2001 [see their appendix A]).
estimated as was done by Stephens et al. (2001 [see their appendix A]).
Note that when ρl=0 for all l (i.e., no recombination), the conditional distribution π described above simplifies to the conditional distribution used by Stephens et al. (2001).
Computation of π requires a sum over all possible values of (X,T):
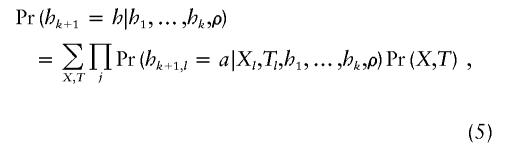 |
which can be accomplished efficiently by use of standard computational methods for hidden Markov models (as shown in Li and Stephens [2003]; e.g., their appendix A). The computational effort required to compute π by this approach increases linearly with the product of the number of loci and number of individuals.
Introducing T doubles the amount of computational effort required to compute each conditional distribution, compared with the distribution used by Li and Stephens (2003), but we believe it to improve phasing accuracy. For example, suppose that, at two closely linked SNP loci, we observe many homozygotes (e.g., genotypes AA and AA) and one double heterozygote (e.g., genotypes AC and AC). We would argue that the most likely explanation is that one of this individual’s two haplotypes is relatively highly diverged from the others in the sample and that the singleton mutations therefore lie together on this haplotype; that is, that this last individual most likely has one AA haplotype and one CC haplotype. Indeed, this is the solution favored by most haplotype-inference methods, including maximum likelihood via the EM algorithm, Clark’s algorithm, and previous versions of PHASE. However, without the use of the times T, our algorithm would randomize the phase of these singletons, independently at each site, and therefore consider the alternative solution (haplotypes AC and CA) equally plausible. With the Ts, our algorithm will identify one of the haplotypes as being highly diverged from the others and therefore tend to put the singletons all together on the same haplotype, as we argue it should. To what extent this improves the accuracy of phasing for nonsingleton SNPs is unclear.
Results
Our new algorithm is implemented with the software package PHASE v2 (v2.0 and subsequent versions include variants on the algorithm we describe here; results in this article are based on v2.1.1). The algorithm comes in two “flavors,” which we label “recom” and “hybrid,” or “-MR” and “-MQ,” after the switches used in the software to invoke them. The difference between them is that the first uses the “coalescent with recombination” at all times, whereas the second is a hybrid between this new recom algorithm and the algorithm of Stephens and Donnelly (2003) (in that it uses the coalescent without recombination until the final merge in the PL, when it switches to using the coalescent with recombination). The hybrid algorithm can be considerably faster and, as we demonstrate here, sacrifices relatively little in accuracy of haplotype estimates—although we caution that we have not assessed accuracy of recombination-rate parameter estimates for the hybrid algorithm. We compare the accuracy of these algorithms with the coalescent-based method of Stephens and Donnelly (2003), which does not take explicit account of the decay of LD and which we term “no recom” (invoked with the -MS switch in PHASE v2), and with several other available algorithms for haplotype inference (table 1).
Haplotype Inference: X Chromosome Data
Our first comparison uses SNP data analyzed and described by Lin et al. (2002). The data consist of X chromosome haplotypes derived from 40 unrelated males. The haplotypes comprise eight regions, which have a range of 87–327 kb and include 45–165 segregating sites. For each of the eight genes, we created 100 data sets, each consisting of 20 pseudoindividuals, created by randomly pairing the 40 chromosomes (as in previous comparisons using these data [Lin et al. 2002; Stephens and Donnelly 2003]).
We ran all computer programs except Arlequin 3.0a with their default or recommended settings, except as noted below. (Results for Arlequin 3.0a were kindly supplied by L. Excoffier.) For Haplotyper and PL-EM, “rounds” was set to 20. HAP was accessed via the HAP Webserver (accessed October 2003 and December 2003). For snphap, we reduced the lower posterior-trimming threshold to 0.001, in an attempt to obtain haplotype reconstructions for all individuals in the sample. The input format for hap2 (kindly provided by S. Lin) allows for the inclusion of family-level data; we input the individuals as unrelated parents with no offspring, and we used the following additional settings: 10,000 iterations, 5,000 burn-in, 20 thinning, (0, 0.5) minor-allele frequency limits, and |D′| blocks with 0.8 threshold.
To evaluate the accuracy of the haplotype reconstructions for the various methods, we calculated two error rates:
-
1.
the individual error rate, defined as the proportion of ambiguous individuals whose haplotypes are not completely correct, ignoring in each individual any positions with missing data, and
-
2.
the switch error, which measures the proportion of heterozygote positions whose phase is incorrectly inferred relative to the previous heterozygote position. This is 1 minus the switch accuracy defined by Lin et al. (2002).
For haplotypes with large numbers of SNPs or that cover larger genetic distances, it is difficult to correctly infer the entire haplotypes, so, in these cases, the switch error seems a more informative criterion for comparing methods.
Results for all methods are given in table 2. Results for the method of Stephens and Donnelly (2003) differ slightly from those given in that article, partly because we used a different 100 random pairings of the haplotypes and partly because we corrected errors in the locus order used by Stephens and Donnelly (2003) for two of the genes (GLA and TRP). For each gene, the ranking of the algorithms is generally similar for both the error measures we use. For six of eight genes, one of our new coalescent-based methods has the lowest individual-error rate; for seven of eight genes, one of them has the lowest switch-error rate. Next best is the coalescent-based method without recombination, followed by the Bayes-Dirichlet method (which is our implementation of an algorithm similar to the one underlying Haplotyper) and the method of Excoffier et al. (2003) implemented in Arlequin 3.0. One possible explanation for the superior performance of Bayes-Dirichlet compared with Haplotyper on these data is that the version of Haplotyper we used appeared to have problems accurately imputing missing data, which may have adversely affected its accuracy in some cases (although we excluded positions with missing data when scoring the algorithms). Another possible factor is that our implementation of PL used a different criterion for deciding how long the list of “plausible” haplotypes should be. Our criterion may tend to lead to longer lists, which should increase accuracy at the expense of extra computation. Overall, hap and hap2 exhibit performances slightly superior to those of PL-EM and Haplotyper, which are comparable with each other.
Table 2.
Comparison of Accuracy of Methods for Reconstructing Haplotypes[Note]
|
Individual-Error Rate for Gene |
|||||||||
| Algorithm | GLRA2 | MAOA | KCND1 | ATR | GLA | TRPC5 | BRS3 | MECP2 | Average (± SE) |
| PHASE v2 recom (-MR) | .73 | .52 | .46 | .47 | .70 | .66 | .65 | .77 | .62 (±.015) |
| PHASE v2 hybrid (-MQ) | .76 | .53 | .47 | .47 | .69 | .63 | .65 | .78 | .62 (.015) |
| PHASE v2 no recom (-MS) | .77 | .53 | .48 | .48 | .70 | .63 | .66 | .79 | .63 (.015) |
| Bayes-Dirichlet | .82 | .58 | .56 | .57 | .76 | .61 | .74 | .84 | .68 (.015) |
| Arlequin (ELB) | .73 | .56 | .53 | .57 | .76 | .74 | .62 | .77 | .66 (.013) |
| hap2 | .78 | .62 | .72 | .58 | .88 | .79 | .70 | .84 | .74 (.013) |
| hap | .79 | .61 | .54 | .62 | .89 | .58 | .72 | .85 | .70 (.016) |
| PL-EM | .82 | .63 | .70 | NA | NA | NA | .76 | NA | .73a (.014) |
| snphap | .82 | .60 | .68 | .74 | .90 | NA | .74 | NA | .75a (.017) |
| Haplotyper | .89 | .76 | .72 | .72 | .79 | .72 | .79 | .64 | .75 (.009) |
| HAP | .93 | NA | .86 | .89 | .98 | NA | .82 | .92 | .90a (.009) |
|
Switch-Error Rate for Gene |
|||||||||
|
GLRA2 |
MAOA |
KCND1 |
ATR |
GLA |
TRPC5 |
BRS3 |
MECP2 |
Average (± SE) |
|
| PHASE v2 recom (-MR) | .08 | .07 | .14 | .17 | .11 | .17 | .12 | .16 | .12 (.005) |
| PHASE v2 hybrid (-MQ) | .09 | .06 | .14 | .17 | .11 | .15 | .10 | .16 | .12 (.005) |
| PHASE v2 no recom (-MS) | .10 | .06 | .14 | .18 | .12 | .15 | .10 | .17 | .13 (.005) |
| Bayes-Dirichlet | .11 | .08 | .17 | .25 | .14 | .14 | .14 | .20 | .15 (.007) |
| Arlequin (ELB) | .11 | .09 | .22 | .23 | .14 | .22 | .20 | .20 | .18 (.007) |
| hap2 | .09 | .07 | .20 | .23 | .14 | .19 | .11 | .17 | .15 (.007) |
| hap | .14 | .10 | .22 | .29 | .22 | .13 | .14 | .23 | .18 (.008) |
| PL-EM | .12 | .09 | .23 | NA | NA | NA | .14 | NA | .15a (.015) |
| snphap | .16 | .10 | .24 | .36 | .23 | NA | .14 | NA | .21a (.015) |
| Haplotyper | .16 | .12 | .27 | .32 | .16 | .20 | .15 | .19 | .20 (.008) |
| HAP | .18 | NA | .35 | .41 | .31 | NA | .18 | .31 | .29a (.015) |
Note.— The best performance in each column is highlighted in bold italics. The eight genes are the data sets on which methods were tested by Lin et al. (2002). The individual- and switch-error rates are defined in the text. Estimated SDs for the average error rate (final column) are given in parentheses (although, for assessment of the significance of an observed difference between two methods, it would be best to take account of the paired nature of the data by computing an SE for the difference in error rates). The data for Haplotyper and hap are taken from Lin et al. (2002). For two of the genes, indicated by “NA,” and for unknown reasons, HAP would not process the input files we submitted. Similarly, for four genes, also indicated by “NA,” PL-EM and snphap failed to complete runs for the majority of simulations.
Average computed with partial data.
One unfortunate aspect of data sets formed from randomly pairing X chromosomes is that they can include genotypes in which one allele—and only one allele—is known to be missing. This does not typically occur in real genotype data, and it may be thought to adversely affect relative performance of some of the methods, particularly HAP and snphap, since the versions of these programs we used were unable to use information on the single observed allele at such loci. To investigate this possibility, we reran PHASE-MR, ignoring the single observed allele at such loci. The results were almost identical to those obtained when those alleles were not ignored, suggesting that this unusual pattern of missing alleles is probably not the explanation for the relatively poor performance of HAP for these data. (It is worth noting that the comparisons of Halperin and Eskin [2004], in which HAP was competitive with PHASE, used an earlier version of PHASE, implementing the algorithm of Stephens et al. [2001]).
Haplotype Inference: Autosomal Data
A slightly surprising feature of our results is that our new algorithms appear to produce only small gains in performance over coalescent-based algorithms that do not explicitly model decay in LD. Since LD decays more quickly on the autosomes than on the X chromosome (Schaffner 2004), it is possible that relative performance of the methods might differ on autosomal data. We therefore applied our coalescent-based algorithms to the genotype data for the children in the 129 trios described by Daly et al. (2001) and compared estimated haplotypes with the actual haplotypes inferred from the parental genotypes (ignoring genotypes for which phase could not be so inferred) (kindly supplied to us in a convenient electronic form by G. Kimmel). Since Halperin and Eskin (2004) also used these data for comparisons, we included HAP in this comparison, using the results posted on the HAP Webserver.
For these data, our new algorithms (PHASE-MR and -MQ) produced more-accurate haplotype estimates than either the coalescent-based algorithm that ignored recombination (PHASE-MS) and HAP: respective individual error rates were 0.42, 0.39, 0.50, and 0.51; switch error rates were 0.030, 0.034, 0.043, and 0.057. Clearly, the relative performance here of HAP is better than for the X chromosome data: the switch-error rate is less than double that of our new approach, compared with more than double (on average) in the X chromosome comparisons. This might have been expected, since patterns observed in these data partly motivated some of the assumptions underlying HAP.
Missing Data
To assess the accuracy of the algorithms for imputing missing alleles, we used genotype data of 24 African Americans (AA) and 23 individuals of European descent (ED) at 50 genes that we randomly selected from the UW-FHCRC Variation Discovery Resource Web site (September 2003). These genes have been completely resequenced and contain 15–230 segregating sites, with an average of 85 per gene. For convenience, we ignored the few triallelic SNPs and multisite insertion-deletion polymorphisms in the data.
To assess how well the various haplotyping methods impute missing genotype data, we introduced into the data 5% artificial missingness (in addition to the 4.6% native missingness). We used two different patterns of missingness: missing alleles and missing genotypes. In the first, each observed allele was denoted as missing with probability 0.05, independent of all other genotypic data and missingness patterns. In the second, each observed genotype was denoted as missing with probability 0.05, again independently. Neither of these patterns is likely to capture all aspects of patterns of missingness in real data, in which variations in DNA quality or molecular effects can cause some individuals and some sites to have more than their fair share of missing data. For this reason, absolute accuracy of methods of this test may tend to be better than for real data. However, we would expect relative performances to be similar.
To provide a baseline against which to compare methods, we implemented a naive “straw man” approach to imputing the genotypes. For the alleles-missing pattern, this method imputes the most common allele in the sample at that site; for the genotypes-missing pattern, it imputes the most common genotype in the sample at that site. Thus, the straw man ignores LD when imputing missing data, and the size of the improvement of each method over the straw man indicates the effectiveness with which that method exploits patterns of LD in its imputation algorithm.
To measure the accuracy of imputed genotypes, we used the genotype-imputation error rate, which we define as the total number of imputed genotypes not identical to the original genotype, divided by the total number of imputed genotypes (both totals computed across all 50 genes, ignoring native missing genotypes).
We analyzed the 50 genes with each method, using identical missingness patterns for each method, in two different ways. First, we analyzed all 47 individuals together. Second, we analyzed the AA and ED samples separately.
Table 3 shows the accuracy of the genotypes estimated by each method. Although Arlequin (ELB) can be used to analyze data sets with missing data, it does not estimate the missing alleles and so was omitted from this comparison. For the majority of genes, PL-EM and Haplotyper failed to present resolved haplotypes for at least one of the three samples (AA, ED, and combined sample), which made it difficult to summarize results for those methods. We omitted results for Haplotyper but included the results for PL-EM for the eight genes that it successfully processed in all three samples. Similarly, snphap successfully processed only 34 of the 50 genes. HAP, hap2, and snphap appeared not to deal fully with genotypes for which only one allele is missing, and so we omitted them from the “alleles missing independently” set of comparisons.
Table 3.
Comparison of Accuracy of Methods for Imputing Missing Data[Note]
|
Missingness pattern for |
||||
| Alleles Analyzed Independently |
Whole Genotypes |
|||
| Algorithm | Separate | Combined | Separate | Combined |
| PHASE v2 recom (-MR) | .033 | .027 | .044 | .038 |
| PHASE v2 hybrid (-MQ) | .034 | .031 | .049 | .041 |
| PHASE v2 no recom (-MS) | .049 | .044 | .066 | .062 |
| Bayes-Dirichlet | .053 | .049 | .077 | .063 |
| PL-EM | .086 | .072 | .088 | .062 |
| HAP | NA | NA | .082 | .085 |
| hap2 | NA | NA | .101 | .117 |
| Straw man | .100 | .102 | .155 | .158 |
| snphap | NA | NA | .188 | .167 |
Note.— Each number is the genotype-imputation error rate (described in text). The “Separate” columns give the results for analysis of the 23 ED and 24 AD individuals separately; the “Combined” columns give the results for analysis of all 47 individuals together. The differences between our new methods (PHASE v2-MR and -MQ) versus all other methods are statistically significant. For the “Alleles Analyzed Independently” pattern, the results are based on a total of 19,021 missing alleles; for the “Whole Genotypes” pattern, the results are based on 18,958 missing alleles.
Our new algorithms outperformed all other methods, including the PHASE no recom method. Further examination revealed that this improvement is achieved by a consistent improvement across many genes rather than a large improvement in a small number of genes. For example, when analyzing the “genotype missing” data, with both samples together, the Bayes-Dirichlet method produced more-accurate results than did our new method for only 4/50 genes, and HAP produced more-accurate results for 1/50 genes.
For all of the PHASE methods (and some of the others), the accuracy of imputed genotypes was better when analyzing the ED and AA samples together than when analyzing them separately, although differences were small in absolute terms. On the basis of this, we speculate that, for these data, haplotype estimates obtained by PHASE v2 from all samples together will also tend to be slightly more accurate, on average, than those obtained by analyzing the two population samples separately.
Discussion
We have introduced a new algorithm for inferring haplotypes from population samples and have compared it in three ways with existing approaches. All comparisons point to the benefits of using models based on the coalescent to capture underlying patterns of haplotype variation. Although one might expect the effects of taking the decay of LD into account to be greatest over larger regions, the results shown in table 3 suggest that there are gains to be made even at the level of data collected over tens of kilobases. Further, the consistent improvement in accuracy we observed across all comparisons argues that the use of these kinds of models will be beneficial for most data sets—from most regions of the genome—and not beneficial only “on average.” Nevertheless, some users may worry that specific factors for their data set (such as unusual patterns of polymorphism due to selection or complex marker-ascertainment schemes) might make our approximate-coalescent model inappropriate. We suspect this to be rare but note that our experiments with missing data suggest a simple general strategy for comparing the likely accuracy of different haplotype-inference methods in such cases: delete a small proportion (e.g., 5%–10%) of the genotype data and see which inference method produces more-accurate genotype estimates (on average, over several repetitions of the process). This strategy depends, of course, on the use of a haplotype-inference method to impute missing genotypes that is similar to the one they use to estimate haplotypes. This is the case for all the methods implemented in PHASE—since they use the same statistical model for both tasks—but may not be the case for all other methods. Thus, although, for PHASE methods, the results in table 3 should reflect their relative accuracy in both imputing missing genotypes and inferring haplotypes, other methods may reflect relative accuracy only in the specific task of imputing missing genotypes.
The high accuracy with which our method can impute missing genotypes suggests that it could play a useful role in selecting which SNPs to genotype in large-scale association studies. As pointed out by Johnson et al. (2001), genotyping costs in such studies can be substantially reduced—without much loss in power—by genotyping only an appropriately chosen subset of SNPs (usually referred to as “tagSNPs”). One way of approaching this problem is to choose the tagSNPs so that genotypes at all other SNPs can be predicted with a high degree of accuracy from the typed SNPs. Most published methods along these lines involve fairly simple approaches to measuring how accurately genotypes at a subset of SNPs predict the genotypes at others (e.g., Chapman et al. [2003] use linear regression). It would be interesting to compare the accuracy of these simple approaches for predicting missing genotypes with our approach. If our approach provides more-accurate predictions, then, when coupled with appropriate analyses for identifying associations between genotype and phenotype, this could lead to more-efficient studies that require fewer tagSNPs.
For the data considered here, genotype-imputation estimates were more accurate for analysis of AA and ED samples together than for separate analysis. This is consistent with previous observations that haplotype inference methods are not especially sensitive to deviations from the assumption of Hardy-Weinberg equilibrium that underlies most of them (Fallin and Schork 2000; Stephens et al. 2001). However, in some studies, haplotype estimates may tend to be more accurate if samples from individuals of different ethnic backgrounds are analyzed separately rather than together. Specifically, this might be expected to occur if the different ethnicities are highly diverged in the region of study and/or the sample sizes available for each ethnicity are large. As discussed above, randomly deleting a fraction of known genotypes and determining which analysis approach (together or separate) provides more-accurate genotype estimates provides a strategy for deciding which approach is likely to provide the most-accurate haplotype estimates in a particular case. Note, though, that this strategy addresses only the expected accuracy of haplotype estimates and not issues of bias: we would always expect that analyzing samples from different backgrounds together would tend to systematically underestimate differences in haplotype frequencies among the groups, whereas analyzing samples separately would tend to systematically overestimate these differences.
Although the algorithms implemented in PHASE v2 are among the most-accurate algorithms for haplotype estimation available, they are also among the most computer intensive. The new algorithm we present here is the most computer-intensive of all of these. The most obvious reason for this is that computation of the conditional distribution π in expression (1) is substantially more time consuming than analogous computations that ignore recombination. However, another important factor is that allowance for recombination tends to increase the number of “plausible” haplotypes for each individual, which also increases the computational cost. Given the similar accuracy achieved by the other PHASE methods, particularly on the simulated X chromosome data, one might wonder whether the additional computing expenditure is worthwhile. Our view is that one should use the method that provides the best results within the time frame available. A single run of the slower of our new algorithms (-MR) on the largest of the data sets that we considered here (the data from Daly et al. [2001], with 103 SNPs for 129 individuals) took roughly 3 h of computing time on a 3-GHz CPU desktop machine, which remains well within the bounds of what we consider reasonable.
Results for the X chromosome data in table 2 highlight the fact that, in some settings, haplotypes estimated by even the best available methods may be wrong for the majority of sampled individuals. The low overall accuracy in this case is presumably due to several factors, including the small sample size, the large number of markers, and the fact that the markers are spread over reasonably large regions. These results might lead one to question the relevance of statistical methods for estimating haplotypes in such settings. Fortunately, however, for many analyses, conclusions will not be critically dependent on every detail of each individual’s haplotypes. (Indeed, it seems prudent to design analyses of statistically reconstructed haplotypes in such a way that this is the case.) For this reason, although measuring the accuracy—by various criteria—of estimated haplotypes provides a convenient way to compare the relative merits of different algorithms, it seems impossible to translate such results into an absolute measure of their usefulness. In practice, what is needed is a way to determine whether, for a particular data set, haplotypes can be estimated with sufficient accuracy to draw reliable conclusions. For this, it seems essential that haplotype-reconstruction algorithms provide not only point estimates for the haplotypes but also a reliable measure of the uncertainty in estimated haplotypes. As noted by Stephens and Donnelly (2003), Bayesian approaches seem to have an advantage here, since the Bayesian framework provides a natural way of assessing uncertainty, via the conditional distribution of the haplotypes, given the genotype data.
Among Bayesian approaches, the new algorithms we present here should better capture the uncertainty in estimated haplotypes than do previous methods, because the assumptions underlying our approach are more realistic (as evidenced by the reduced error rates shown in tables 2 and 3). In particular, explicit allowance for recombination tends to increase the number of “plausible” haplotypes for an individual and thus leads to greater uncertainty in the estimated haplotypes. It may seem counterintuitive to claim that a method with greater uncertainty in estimated haplotypes is superior to a method with less uncertainty. However, methods that provide for less uncertainty risk being overconfident in their assessments of estimated haplotypes, with the resulting danger of being overconfident in subsequent conclusions. There are, however, situations for which our new algorithm, as we have applied it here, may be underconfident in its estimated haplotypes. This can occur when analyzing small data sets (few markers and/or few individuals), because, for such data sets, the method may tend to overestimate the recombination rates: with few markers, inference of the recombination rate will depend heavily on the prior distribution for the recombination parameter, and the rather flat prior that we use here perhaps allows too much weight on unrealistically large values. Fortunately, this problem is easily solved by use of a more realistic prior for the recombination rate, although appropriate choice of this prior will typically depend on the organism and the region under study; we wanted to avoid this additional subjectivity here. The prior can, however, be altered as a parameter in the PHASE v2 software package, which is available from the Stephens Lab Web site.
Acknowledgments
We thank two anonymous reviewers for their helpful and constructive comments. This work was supported by National Institutes of Health grants 1RO1HG/LM02585-01 (to M.S.) and T32 HG00035 (to P.S.).
Appendix A : Further Algorithmic Details
We refer readers to the publication by Stephens and Donnelly (2003) for details of the PL and for more details of that aspect of the algorithm. We focus on providing further details of steps 1 and 2 of our algorithm, outlined in the text.
Step 1: Updating Individuals
Updating the haplotype pair of individual i (including alleles at loci with missing genotypes) proceeds as follows:
-
1.
Make a list ℋi of all haplotype pairs that are both compatible with the observed genotypes of individual i, and occur in the current list L of plausible haplotypes (this list is created by the PL procedure; see Stephens and Donnelly [2003] for more details).
-
2.
For each haplotype pair in ℋi, compute the probability of the pair using expression (1).
-
3.
Sample a random pair in ℋi according to these probabilities and set the haplotypes of i to this pair.
-
4.
Impute missing positions: for each of the two haplotypes of i (one at a time), sample alleles at the positions with missing genotypes from their conditional distribution, given the current imputed values of the alleles at positions with nonmissing genotypes. This can be done efficiently, by using the forward-backward algorithm (Rabiner 1989) to first simulate the values of the hidden Markov chain (X,T) for the haplotype and then imputing the missing alleles independently at each locus, conditional on the simulated value of (X,T). If the new haplotype pair of i is not in ℋi, add it.
Note that step 4 is not strictly necessary, in that the missing alleles are also imputed by the other steps. It was included to improve mixing (although we have not performed a detailed study of its effectiveness).
Step 2: Updating Recombination Parameters
The recombination parameters, 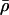 and λ=(λ1,…,λL-1), are initialized to be
and λ=(λ1,…,λL-1), are initialized to be 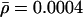 (on the basis of approximate genomewide average for humans) and λl=1. They are then updated, using Langevin MH proposals on a log scale (Besag 1994). Langevin MH updates are MH updates that use information from the derivative of the posterior density to propose moves in “sensible” directions that are likely to have a reasonable chance of being accepted. The standard MH acceptance probability is used. Specifically, the update proposals for
(on the basis of approximate genomewide average for humans) and λl=1. They are then updated, using Langevin MH proposals on a log scale (Besag 1994). Langevin MH updates are MH updates that use information from the derivative of the posterior density to propose moves in “sensible” directions that are likely to have a reasonable chance of being accepted. The standard MH acceptance probability is used. Specifically, the update proposals for 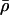 and λ are:
and λ are:
-
1.
Update
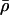 by proposing to add
by proposing to add 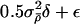 to
to 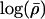 , where
, where 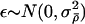 and
and 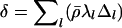 and Δl is the partial derivative of the PAC-B likelihood (Li and Stephens 2003), with respect to
and Δl is the partial derivative of the PAC-B likelihood (Li and Stephens 2003), with respect to 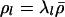 . (These derivatives can be efficiently computed analytically by use of the forward–backward algorithm for hidden Markov models (Rabiner 1989).
. (These derivatives can be efficiently computed analytically by use of the forward–backward algorithm for hidden Markov models (Rabiner 1989). -
2. Update λ, by proposing to add 0.5σ2λδl+ε to each log(λl), where
(the last term coming from the N(0,1.152) prior on log(λl)) and ε∼N(0,σ2λ).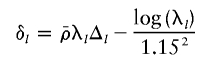
Here, 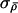 and σλ control the expected size of proposed jumps and need to be tuned to the particular data set being analyzed, to ensure reasonable mixing behavior. We adopt a rather simplistic procedure to perform this automatic tuning that is based on the idea that
and σλ control the expected size of proposed jumps and need to be tuned to the particular data set being analyzed, to ensure reasonable mixing behavior. We adopt a rather simplistic procedure to perform this automatic tuning that is based on the idea that 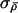 and σλ should be set so that acceptance probabilities are not too close to 0 or 1. Initially, we set
and σλ should be set so that acceptance probabilities are not too close to 0 or 1. Initially, we set 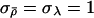 . Then, halfway through the burn-in iterations, better values can be found by repeating the following until acceptance rates for both ρ and λ are in the range 0.3–0.7:
. Then, halfway through the burn-in iterations, better values can be found by repeating the following until acceptance rates for both ρ and λ are in the range 0.3–0.7:
-
1.
update
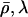 10 times each, using current values of
10 times each, using current values of 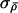 and σλ, and
and σλ, and -
2.
if the proportion of acceptances of
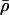 (respectively λ) was not in the range 0.3–0.7, then divide or multiply
(respectively λ) was not in the range 0.3–0.7, then divide or multiply 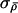 (respectively σλ) by 1+u, where u is random uniform on [0,1).
(respectively σλ) by 1+u, where u is random uniform on [0,1).
If necessary, this tuning can be repeated after the burn-in iterations have been completed, before the main iterations are performed.
Appendix B : List of Genes Used for Results Shown in Table 3
Genes are listed by HUGO Gene Nomenclature Committee abbreviation: CD36, CEBPB, CRF, CRP, CSF3, CYP4A11, CYP4F2, DCN, EPHB6, F11, F2RL3, F3, IFNG, IGF2, IL1B, IL2RB, IL4, IL5, IL10A, IL11, IL19, IL20, IL21R, IL22, IL24, KEL, LTA, LTB, MAP3K8, MMP3, PFC, PLAUR, PLG, PROC, PTGS2, SCYA2, SELE, SELL, SELPLG, SERPINC1, SFTPA1, SMP1, STAT6, TFPI, THBD, TNF, TNFAIP2, TNFAIP3, TRAF6, and VCAM1.
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- David Clayton Software page, http://www-gene.cimr.cam.ac.uk/clayton/software/
- HAP Webserver,http://www1.cs.columbia.edu/compbio/hap
- HUGO Gene Nomenclature Committee, http://www.gene.ucl.ac.uk/nomenclature/
- Stephens Lab Web site, http://www.stat.washington.edu/stephens/software.html (for PHASE)
- UW-FHCRC Variation Discovery Resource, http://pga.gs.washington.edu
References
- Besag JE (1994) Discussion on the paper by Grenander and Miller. J R Stat Soc B 56:591–592 [Google Scholar]
- Carlson C, Eberle M, Rieder M, Smith J, Kruglyak L, Nickerson D (2003) Additional SNPs and linkage-disequilibrium analyses are necessary for whole-genome association studies in humans. Nat Genet 33:518–521 10.1038/ng1128 [DOI] [PubMed] [Google Scholar]
- Chapman J, Cooper J, Todd J, Clayton D (2003) Detecting disease associations due to linkage disequilibrium using haplotype tags: a class of tests and the determinants of statistical power. Hum Hered 56:18–31 10.1159/000073729 [DOI] [PubMed] [Google Scholar]
- Clark AG (1990) Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol 7:111–122 [DOI] [PubMed] [Google Scholar]
- Crawford D, Bhangale T, Li N, Rieder M, Nickerson D, Stephens M (2004) Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat Genet 36:700–706 10.1038/ng1376 [DOI] [PubMed] [Google Scholar]
- Daly M, Rioux J, Schaffner S, Hudson T, Lander E (2001) High-resolution haplotype structure in the human genome. Nat Genet 29:229–232 10.1038/ng1001-229 [DOI] [PubMed] [Google Scholar]
- Eskin E, Halperin E, Karp R (2003a) Efficient reconstruction of haplotype structure via perfect phylogeny. J Bioinform Comput Biol 1:1–20 10.1142/S0219720003000174 [DOI] [PubMed] [Google Scholar]
- ——— (2003b). Large scale reconstruction of haplotypes from genotype data. In: Proceedings of the seventh annual international conference on research in computational molecular biology (RECOMB 2003), pp 104–113 [Google Scholar]
- Evans G (1993) Practical numerical integration. Wiley and Sons, Chichester, United Kingdom [Google Scholar]
- Excoffier L, Laval G, Balding DN (2003) Gametic phase estimation over large genomic regions using an adaptive window approach. Hum Genomics 1:7–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Slatkin M (1995) Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol 12:921–927 [DOI] [PubMed] [Google Scholar]
- Fallin D, Schork NJ (2000) Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization algorithm for unphased diploid genotype data. Am J Hum Genet 67:947–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnhead PN, Donnelly P (2001) Estimating recombination rates from population genetic data. Genetics 159:1299–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan G, Geiger D (2004) Model-based inference of haplotype block variation. J Comput Biol 11:493–504 10.1089/1066527041410300 [DOI] [PubMed] [Google Scholar]
- Halperin E, Eskin E (2004) Haplotype reconstruction from genotype data using imperfect phylogeny. Bioinformatics 20:1842–1849 10.1093/bioinformatics/bth149 [DOI] [PubMed] [Google Scholar]
- Hawley M, Kidd K (1995) HAPLO: a program using the EM algorithm to estimate the frequencies of multi-site haplotypes. J Hered 86:409–411 [DOI] [PubMed] [Google Scholar]
- Heckerman D, Chickering DM, Meek C, Rounthwaite R, Kadie C (2000) Dependency networks for inference, collaborative filtering, and data visualization. J Machine Learning Res 1:49–75 10.1162/153244301753344614 [DOI] [Google Scholar]
- Hudson RR (1991) Gene genealogies and the coalescent process. In Futuyma D, Antonovics J (eds) Oxford surveys in evolutionary biology. Vol 7, pp 1–44. Oxford University Press, Oxford, United Kingdom [Google Scholar]
- Johnson GC, Esposito L, Barratt BJ, Smith AN, Heward J, Genova GD, Ueda H, Cordell HJ, Eaves IA, Dudbridge F, Twells RC, Payne F, Hughes W, Nutland S, Stevens H, Carr P, Tuomilehto-Wolf E, Tuomilehto J, Gough SC, Clayton DG, Todd JA (2001) Haplotype tagging for the identification of common disease genes. Nat Genet 29:233–237 10.1038/ng1001-233 [DOI] [PubMed] [Google Scholar]
- Kimmel G, Shamir R (2004) Maximum likelihood resolution of multi-block genotypes. In: Proceedings of the Eighth Annual International Conference on Research in Computational Molecular Biology (RECOMB’04). The Association for Computing Machinery, San Diego, pp 2–9 [Google Scholar]
- Li N, Stephens M (2003) Modelling linkage disequilibrium, and identifying recombination hotspots using SNP data. Genetics 165:2213–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Chakravarti A, Cutler DJ (2004) Haplotype and missing data inference in nuclear families. Genome Res 14:1624–1632 10.1101/gr.2204604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Cutler DJ, Zwick ME, Chakravarti A (2002) Haplotype inference in random population samples. Am J Hum Genet 71:1129–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JC, Williams RC, Urbanek M (1995) An E-M algorithm and testing strategy for multiple locus haplotypes. Am J Hum Genet 56:799–810 [PMC free article] [PubMed] [Google Scholar]
- McVean GAT, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P (2004) The fine-scale structure of recombination rate variation in the human genome. Science 304:581–584 10.1126/science.1092500 [DOI] [PubMed] [Google Scholar]
- Niu T, Qin ZS, Xu X, Liu JS (2002). Bayesian haplotype inference for multiple linked single-nucleotide polymorphisms. Am J Hum Genet 70:157–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak SE, Roeder AD, Stephens M, Gilad Y, Pääbo S, Przeworski M (2004) Absence of the TAP2 human recombination hotspot in chimpanzees. PLoS Biol 2:849–855 10.1371/journal.pbio.0020155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin ZS, Niu T, Liu JS (2002) Partial-ligation–expectation-maximization algorithm for haplotype inference with single-nucleotide polymorphisms. Am J Hum Genet 71:1242–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabiner LR (1989) A tutorial on hidden Markov models and selected applications in speech recognition. Proc IEEE 77:257–286 10.1109/5.18626 [DOI] [Google Scholar]
- Schaffner SF (2004) The X chromosome in population genetics. Nat Rev Genet 5:43–51 10.1038/nrg1247 [DOI] [PubMed] [Google Scholar]
- Stephens M, Donnelly P (2000) Inference in molecular population genetics. J R Stat Soc Ser B 62:605–655 10.1111/1467-9868.00254 [DOI] [Google Scholar]
- Stephens M, Donnelly P (2003) A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73:1162–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]