

Abstract
Fatal congenital nonlysosomal cardiac glycogenosis has been attributed to a subtype of phosphorylase kinase deficiency, but the underlying genes and mutations have not been identified. Analyzing four sporadic, unrelated patients, we found no mutations either in the eight genes encoding phosphorylase kinase subunits or in the two genes encoding the muscle and brain isoforms of glycogen phosphorylase. However, in three of five patients, we identified identical heterozygous R531Q missense mutations of the PRKAG2 gene, which encodes the γ2-subunit of AMP-activated protein kinase, a key regulator of energy balance. Biochemical characterization of the recombinant R531Q mutant protein showed >100-fold reduction of binding affinities for the regulatory nucleotides AMP and ATP but an enhanced basal activity and increased phosphorylation of the α-subunit. Other PRKAG2 missense mutations were previously identified in patients with autosomal dominant hypertrophic cardiomyopathy with Wolff-Parkinson-White syndrome, characterized by juvenile-to-adult clinical onset, moderate cardiac glycogenosis, disturbed excitation conduction, risk of sudden cardiac death in midlife, and molecular perturbations that are similar to—but less severe than—those observed for the R531Q mutation. Thus, recurrent heterozygous R531Q missense mutations in PRKAG2 give rise to a massive nonlysosomal cardiac glycogenosis of fetal symptomatic onset and rapidly fatal course, constituting a genotypically and clinically distinct variant of hypertrophic cardiomyopathy with Wolff-Parkinson-White syndrome. R531Q and other PRKAG2 mutations enhance the basal activity and α-subunit phosphorylation of AMP-activated protein kinase, explaining the dominant nature of PRKAG2 disease mutations. Since not all cases displayed PRKAG2 mutations, fatal congenital nonlysosomal cardiac glycogenosis seems to be genetically heterogeneous. However, the existence of a heart-specific primary phosphorylase kinase deficiency is questionable, because no phosphorylase kinase mutations were found.
Introduction
Glycogen storage diseases (GSDs) with clinically prominent cardiac involvement can be caused by various gene defects. The lysosomal glycogenoses GSD II (Pompe disease [MIM #232300]) (with α-1,4-glucosidase deficiency) and GSD IIb (Danon disease [MIM #300257]) (with lysosome-associated membrane protein 2 [LAMP-2] deficiency) are systemic disorders that also affect either skeletal muscle, smooth muscle, and liver (GSD II) or skeletal muscle and the nervous system (GSD IIb). However, cardiac involvement usually dominates the clinical picture and is life limiting in GSD IIb and in the classic infantile form of GSD II, whereas the adolescent- and adult-onset forms of GSD II are typically governed by muscle involvement. The infantile form of GSD II has an autosomal recessive mode of inheritance, manifests perinatally, and leads to death within the 1st year of life (Hirschhorn and Reuser 2001), whereas GSD IIb can be X-linked dominant or recessive, has a juvenile or early-adult clinical onset, and leads to death, typically during the 2nd to 4th decades of life (Sugie et al. 2002; Arad et al. 2005). There appears to be a distinct third type of lysosomal cardiac and skeletal muscle GSD, with normal α-1,4-glucosidase and LAMP-2, an infantile-fatal course, and an unidentified gene defect (Yamamoto et al. 2001).
Among the nonlysosomal glycogenoses, GSD III (Cori or Forbes disease [MIM +232400]) (with debranching enzyme deficiency) and GSD IV (Andersen disease [MIM #232500]) (with branching enzyme deficiency) are also systemic disorders. Liver and/or (less frequently) muscle involvement usually dominates the clinical picture (Chen 2001). However, patients with prominent cardiac involvement (manifesting during the 1st or 2nd decade of life) have also been reported (Servidei et al. 1987; Lee et al. 1997; Ewert et al. 1999; Chen 2001).
Mutations in the PRKAG2 gene, encoding the γ2-subunit isoform of AMP-activated protein kinase (AMPK), give rise to a moderate, essentially heart-specific, nonlysosomal glycogenosis with clinical onset typically in late adolescence or in the 3rd decade of life, ventricular pre-excitation predisposing to supraventricular arrythmias, mild-to-severe cardiac hypertrophy, enhanced risk of sudden cardiac death in midlife, and autosomal dominant inheritance with full penetrance (familial hypertrophic cardiomyopathy with Wolff-Parkinson-White syndrome [FHC/WPWS {MIM #600858}]) (Blair et al. 2001; Gollob et al. 2001a, 2001b, 2002; Arad et al. 2002, 2005). The AMPK complex, which acts as a sensor of cellular energy status, is a heterotrimer comprising a catalytic α-subunit and regulatory β- and γ-subunits (Hardie 2003, 2004). The γ-subunits contain four tandem repeats of a sequence motif first recognized by Bateman (1997) and termed a “cystathionine β-synthase” (CBS) motif. These have recently been shown to act in pairs to form two binding sites (now termed “Bateman domains” [Kemp 2004]) for the regulatory nucleotides AMP and ATP (Scott et al. 2004). Binding of AMP (a signal of cellular energy deficiency [Hardie and Hawley 2001]) causes activation of the complex by triggering increased phosphorylation of a specific threonine residue (Thr-172) in the kinase domain on the α-subunit by a distinct upstream kinase, an effect antagonized by high concentrations of the inhibitory nucleotide ATP (Hawley et al. 1996). The major upstream kinase has recently been identified to be the tumor suppressor LKB1 (Hawley et al. 2003; Woods et al. 2003; Shaw et al. 2004). Once activated by cellular stresses that deplete ATP and raise the concentration of AMP, AMPK switches on various ATP-producing catabolic pathways (including glucose uptake and fatty-acid oxidation) and, conversely, down-regulates ATP-consuming processes (Hardie and Hawley 2001; Kemp 2004; Scott et al. 2004). All seven distinct FHC/WPWS mutations in the PRKAG2 gene identified to date were either heterozygous missense mutations or an insertion of a single amino acid within the Bateman domain region. At least four of the seven mutations have been shown to interfere with binding of the regulatory nucleotides AMP and ATP (Scott et al. 2004) and with activation of the complex by AMP (Daniel and Carling 2002; Scott et al. 2004). However, since these are loss-of-function effects, it has been difficult to explain why these mutations are dominant in vivo.
Rare, sporadic cases of severe prenatal- or neonatal-onset nonlysosomal cardiac glycogenosis—which leads to death within a few weeks to a few months after birth, through heart failure and respiratory compromise—have been attributed to a heart-specific variant of phosphorylase kinase (Phk) deficiency (MIM 261740) (Mizuta et al. 1984; Eishi et al. 1985; Servidei et al. 1988; Elleder et al. 1993; Regalado et al. 1999; Bührer et al. 2003). Phk is a regulatory protein kinase that stimulates glycogen breakdown in response to neural and endocrine signals by phosphorylating and activating the main glycogen-catabolizing enzyme, glycogen phosphorylase. It is composed of four subunits in a hexadecameric complex, (αβγδ)4, in which the δ-subunit is calmodulin, and it exists in numerous isoforms and splice variants. Two distinct genes encoding muscle and liver isoforms occur for both α and γ, a single gene occurs for β, and three genes encode identical calmodulin polypeptides. Genetic deficiency of Phk underlies a group of GSDs that differ in mode of inheritance and tissue involvement, depending on the mutant subunit gene. αM (PHKA1) mutations cause X-linked recessive muscle glycogenosis, αL (PHKA2) mutations cause X-linked recessive liver glycogenosis, β (PHKB) mutations give rise to autosomal recessive glycogenosis of both liver and muscle but with an essentially liver-specific clinical phenotype, and γTL (PHKG2) mutations underlie autosomal recessive liver glycogenosis with a particularly severe phenotype and a higher risk of cirrhosis (Kilimann 1997; Chen 2001). No mutations have been identified yet in the γM (PHKG1) gene, but they would be anticipated to cause an autosomal recessive muscle glycogenosis. A molecular explanation for an infantile-fatal, heart-specific, nonlysosomal glycogenosis with low Phk activity, however, has been lacking. Unlike the other forms of Phk deficiency, in which the tissue specificity of expression of the subunit isoforms provided guidance to the identification of the affected genes, no Phk gene sequences expressed specifically in the heart are known. However, particular missense mutations or genetic backgrounds could give rise to phenotypes of unanticipated tissue specificity, even though more widely expressed gene sequences are affected, as observed in other GSDs (Servidei et al. 1987; Ewert et al. 1999; Ziemssen et al. 2000; Chen 2001).
Here, we report the molecular genetic analysis of a group of sporadic patients with fatal congenital nonlysosomal cardiac glycogenosis and a biochemical diagnosis of cardiac Phk deficiency. The complete coding sequences of all eight genes encoding Phk subunits (including calmodulin) and of the two genes for the muscle and brain isoforms of glycogen phosphorylase were analyzed. When we found no mutations in the Phk or phosphorylase genes, we also analyzed the PRKAG2 gene. In three of five cases, the disease was associated with a unique, recurrent heterozygous R531Q missense mutation in PRKAG2. Biochemical analysis of the recombinant mutant protein revealed stronger perturbations of molecular properties than those of any other PRKAG2 mutation characterized to date.
Subjects and Methods
Case Reports
Detailed accounts of patients A, B (Regalado et al. 1999), and E (Bührer et al. 2003) have been published elsewhere. In patient A (1200; case 1 in the study by Regalado et al. [1999]), a Hispanic American female, cardiomegaly was detected in utero by ultrasound and echocardiography, and worsening bradycardia prompted cesarean section at 31 wk of gestation. Shortly after birth, the infant developed cardiorespiratory problems requiring intubation. Electrocardiography (ECG) revealed a short PR interval, wide QRS, inverted T waves, and ST elevation. She experienced recurrent cardiac and respiratory complications throughout her hospital course and succumbed at age 2 3/4 mo to ventricular fibrillation. An autopsy 10 h postmortem showed massive cardiomegaly (136 g [normal 21 g]), with biventricular mural thickening and effacement of both chambers. Other notable features were macroglossia, faciocervical dysmorphism, enlarged and dysmorphic kidneys, and an increased number of pancreatic islets. Microscopically, periodic acid Schiff (PAS) stain–positive and amylase-sensitive vacuolar glycogen depositions were seen in heart tissue. Cardiac glycogen concentration was 3.3% (control 0.9%), and cardiac Phk activity was 0.001 U (control 0.34 U). Histological examination revealed no abnormal glycogen deposition in skeletal muscle, tongue, kidneys, spleen, or liver.
Patient B (1877; case 2 in the study by Regalado et al. [1999]), a white American male, was delivered by cesarean section because of breech presentation and nonreassuring heart rate at 37 wk of gestation. He remained ventilator dependent through most of his hospital course, which was characterized by progressive cardiac enlargement (first noted at day 4 by chest X-ray), secondary pulmonary compression, and oral feeding intolerance. Increasing respiratory distress led to death at age 2 mo. An autopsy at 11 h postmortem showed marked cardiomegaly (65 g [normal 24 g ± 5 g]), with biventricular mural thickening and effaced, slitlike chambers. Other notable features were macroglossia and facial dysmorphism, bilateral cataracts, enlarged kidneys, and hydrocephalus. Histological examination revealed abundant vacuolar, PAS-positive, amylase-sensitive glycogen deposition in the heart. Cardiac glycogen was 3.1% (control 0.7%), and cardiac Phk was 0.03 U (control 0.38 U). No histological or histochemical evidence of abnormal glycogen deposition was found in tissues other than the heart.
Patient C (JW), a white American male, was born at full term by cesarean section because of breech presentation. He developed cardiorespiratory failure within 30 min of life and required mechanical ventilation and, ultimately, extracorporeal membrane oxygenation. ECG showed a normal PR interval, no delta wave, low QRS voltages, and prominent ST wave elevations, consistent with ischemia and/or a lateral wall infarct. Echocardiogram revealed severe left ventricular dysfunction without hypertrophy or structural heart disease. His clinical course was complicated by renal failure secondary to hypotension. On day 10, he became less responsive and developed unusual movements. Electroencephalogram revealed a burst suppression pattern and seizure activity, indicating a dismal prognosis. Support was withdrawn, and he died on day 11. An autopsy at 2 h postmortem consisted of limited biopsies of heart, liver, lung, skeletal muscle, and kidney. Gross examination revealed no congenital malformations, but there was pulmonary congestion with bilateral pleural effusions, anasarca, ascites, a right occipital cephalohematoma, and incomplete, broad palmar creases. Light microscopy revealed diffuse vacuolization of the cardiac myofibers, but not of other tissues. Electron microscopy showed abundant non–membrane-bounded glycogen in cardiac myocytes and a slight increase in non–membrane-bounded glycogen in liver. Glycogen concentration was enhanced in the heart (3.2% [control 0.5%]) and was normal in skeletal muscle and liver. Phosphorylase kinase activity was 15% of the control value in heart and 50%–60% of control values in liver and skeletal muscle. Total phosphorylase and debranching enzyme activities were also ∼50% of control values in all three tissues. Family history was notable for a previous sister who had died at several hours of life from respiratory failure, despite mechanical ventilation. She had a loud systolic murmur, but no echocardiogram could be obtained. She also had dysmorphic features (flattened occiput, epicanthal folds, and nuchal folds, as well as possible cystic hygroma, single palmar crease, and clinodactyly), hypotonia, and large adrenals (determined by ultrasound). Karyotype was normal, and autopsy was declined. The parents (nonconsanguineous) and two other siblings are unaffected.
Patient D (RB), a white British female, was delivered at 37 wk of gestation by cesarean section because of fetal bradycardia. The parents are nonconsanguineous, and there are two previous healthy children. The baby had problems with feeding, bradycardia, and recurrent apnea in the 1st wk of life. She was not dysmorphic and had normal peripheral power and tone and normal primitive reflexes. There was no hepatomegaly. ECG showed evidence of preventricular hypertrophy with a short PR interval and very large QRS voltages in all leads. Chest X-ray revealed massive cardiomegaly, and echocardiogram confirmed a severe hypertrophic cardiomyopathy. Despite medical management, she developed progressive cardiac failure with pulmonary edema and hypotension and died at age 34 d. An autopsy at 6 h postmortem showed an extremely hypertrophic heart (124 g [normal 22 g]), which occupied the greater part of the thoracic cavity. The walls of both atria and ventricles appeared diffusely hypertrophic, smooth, and pale. The heart was otherwise anatomically normal. The lungs were edematous, and the liver was congested and focally hemorrhagic but not enlarged; the rest of the postmortem examination was unremarkable. Glycogen concentration was elevated both in heart (2.4%) and in skeletal muscle (2.9%). Phk was at 10% of the normal range mean in liver and at 1% of the normal range mean in muscle and was undetectable in heart, whereas total phosphorylase was in the normal range in all tissues. Branching enzyme was normal in liver and undetectable in muscle. α-Glucosidase in cultured fibroblasts was normal.
Patient E (Bührer et al. 2003), a white male, the first child of healthy nonconsanguineous German/Polish parents, was delivered at 28 wk of gestation by cesarean section after routine ultrasound examination detected persistent bradycardia. Heart rate remained between 90 and 120 beats per min, despite adequate oxygenation. An ECG revealed left-axis deviation, very short PQ intervals (0.06 s), enlarged QRS complexes with delta waves suggestive of WPWS, and depressed ST segments. Echocardiography showed atrial and biventricular hypertrophy without outflow obstruction and a small pericardial effusion. Respiratory distress necessitated mechanical ventilation nearly throughout the infant’s life, with the need for supplemental oxygen rising to 100% at day 14. Progressive cardial and pleural effusions, ascites, and pulmonary and peripheral edemas led to death at day 21. An autopsy was performed at 3 h postmortem. Light microscopy demonstrated massive subplasmalemmal, non–membrane-bounded, PAS-positive, and amylase-sensitive glycogen deposits in all tissues analyzed (heart, diaphragm, vastus lateralis muscle, and liver). Biochemically determined glycogen was also enhanced markedly in heart (10.8%) and muscle (6.7%/9.9% [normal 0.5%–2.0%]), and moderately in liver (7.0% [normal 2.0%–6.0%]) and erythrocytes (15 mg/dl [normal 0–10 mg/dl]). Phk activity was undetectable in heart, normal in muscle, slightly below the normal range in liver, and slightly above the normal range in erythrocytes. Phosphorylase-a was moderately below the normal range in heart, muscle, and liver and was normal in white blood cells; total phosphorylase was normal in all samples. α-Glucosidase in leukocytes was normal.
Mutation Analysis
The study was approved by the ethics committee of the University of Bochum Medical School. The complete coding sequences of PHKA1, PHKA2, PHKB, PHKG1 (except patient C), CALM1-3, and PYGM were amplified by RT-PCR from total RNA purified from heart or skeletal muscle autopsy samples from patients A–D. The differentially spliced PHKA1 exons 27 and 28 (phosphorylation sites) and exon 19 (“α′ region”) were amplified, together with their flanking intron sequences, from genomic DNA. The PHKG1 promoter regions (∼1 kb) of patients A–D, the coding exons and flanking intron sequences of the PHKG1 gene of patient C, and the coding exons and flanking intron sequences of the PHKG2 genes of patients A–D were amplified from genomic DNA. All PCR products were analyzed by direct cycle sequencing with Applied Biosystems reagents and instruments. In addition, the intact overall organization of the PHKG1 genes of patients A–D was confirmed by bridging the four large introns by overlapping PCRs, and normal lengths of these PCR products were verified by agarose-gel electrophoresis. Mutation analysis of these genes has been described in more technical detail elsewhere: PHKA1, PHKB, PHKG1, CALM1-3, and PYGM (Burwinkel et al. 2003); PHKA2 (Burwinkel et al. 1996); and PHKG2 (Burwinkel et al. 1998). The PYGB coding sequences of patients A–D were amplified from total RNA (heart or skeletal muscle) in five overlapping intervals between −30 nt upstream of the start codon and +43 nt downstream of the stop codon. The PRKAG2 coding sequences of patients A–E were amplified by RT-PCR from RNA (2 + 1 overlapping intervals for the long splice variant and 1 + 1 intervals for the short splice variant) as well as by PCR from genomic DNA, and the PCR products were sequenced. Mutations were independently confirmed from RNA and genomic DNA.
Expression and Mutagenesis of AMPK Domains and Subunits and Their Analysis
Isolated Bateman domains from AMPK-γ2 were expressed as glutathione-S-transferase (GST) fusions in bacteria, and heterotrimeric AMPK complexes were expressed in mammalian cells as described elsewhere (Scott et al. 2004). All mutations were created using the QuikChange Site-Directed Mutagenesis system (Stratagene). Binding of nucleotides and assays of AMPK were performed as described elsewhere (Scott et al. 2004). Phosphorylation of Thr-172 on AMPK-α and of the AMPK phosphorylation site on acetyl-CoA carboxylase (ACC) was monitored with phosphospecific antibodies by use of an infrared scanner, as described elsewhere (Hawley et al. 2003).
Results
No Mutations in Phk and Glycogen Phosphorylase Genes
No Phk isoform genes or differentially spliced exons are known to be specifically expressed in the heart. Therefore, the mutation search could not be targeted directly to heart-specific Phk sequences. The muscle and liver isoform mRNAs of the α- and γ-subunits are coexpressed in comparable amounts in the heart, as are the three calmodulin genes (CALM1-3) and the muscle (PYGM) and brain (PYGB) isoforms of glycogen phosphorylase (Kilimann 1997). A search of the current genome and EST sequence databases, as well as our own experimental investigations (Burwinkel et al. 2003; the present study [data not shown]), did not indicate the existence of additional functional Phk subunit isoform genes or splice variants. To account for the possibility that, for example, certain missense mutations might affect the stability of—or functional interactions within—the cardiac isoform constellation of the Phk/phosphorylase protein complex in an unanticipated, heart-specific, and possibly dominant negative way, we analyzed the complete coding sequences of 10 candidate genes from patients A–D. However, we found no mutations or splicing abnormalities (table 1 [see the note to table 1 and the “Discussion” section for further comment]).
Table 1.
Mutation Analysis of Five Patients with Fatal Congenital Nonlysosomal Heart Glycogenosis and Low Phk Activity[Note]
|
Variant(s) Found in Patient |
|||||
| Gene | Aa | Bb | Cb | Da | Eb |
| PHKA1 | … | … | … | … | NA |
| PHKA2 | … | hemi. E38Qc | … | … | NA |
| PHKB | … | … | … | … | NA |
| PHKG1 + promoter | … | … | … | … | NA |
| PHKG2 | … | … | … | … | NA |
| CALM1 | … | … | … | … | NA |
| CALM2 | … | … | … | … | NA |
| CALM3 | … | … | … | … | NA |
| PYGM | het. G21Sd, het. R414Ge | … | … | … | NA |
| PYGB | … | … | het. A303Sf, het. D502Ng | het. A303S, het. D502N | NA |
| PRKAG2 | het. R531Qh | … | … | het. R531Qh | het. R531Qh |
Note.— Only nonsynonymous sequence variants are given. Numerous synonymous and intronic SNPs were also encountered; ellipses indicate that no nonsynonymous sequence variants were found; hemi. = hemizygous (PHKA2 is X chromosomal); het. = heterozygous; NA = not analyzed.
Female.
Male.
Polymorphism (GAG→CAG; 112G→C): frequency 0.025; three heterozygotes and one homozygote found in 200 mostly female control chromosomes (in the present study).
Rare or unique sequence variant (GGC→AGC; 61G→A), not found in 200 control chromosomes analyzed (in the present study).
Known polymorphism (CGG→GGG; 1240C→G) (dbSNP accession number rs11231866).
Known polymorphism (GCC→TCC; 907G→T) (dbSNP accession number rs3818199).
Known polymorphism (GAT→AAT; 1504G→A) (dbSNP accession number rs2227891); S303 and N502 seem to be on the same allele, since they cosegregate in family C with each other and with adjacent intronic polymorphisms and are also found in the same constellation in patient D.
Identical in patients A, D, and E (CGG→CAG; 1592G→A).
A Recurrent R531Q Mutation in the PRKAG2 Gene of Three Patients
FHC/WPWS is a cardiac glycogenosis with familial occurrence and a much less dramatic clinical manifestation than that of our patient group. However, to see whether our subjects might represent a particularly severe variant of FHC/WPWS that is possibly associated with a distinctive genotype, we analyzed the PRKAG2 gene. In patients A and D, and in the newly recruited patient E, we found identical, heterozygous R531Q missense mutations (fig. 1). Parental DNA was available only for patient E, but the R531Q mutation could be detected in neither the mother nor the father, indicating that it was a de novo mutation. This sequence variant was not found in 190 control chromosomes.
Figure 1.

Heterozygous R531Q missense mutation underlying fatal congenital nonlysosomal heart glycogenosis. A typical sequencer readout is shown (top). The high conservation of the mutant R531 residue is illustrated (middle) by alignments with the corresponding partial sequences from CBS4 of the human (“Hs”) γ1 and γ3 isoforms, pig γ3, and the Drosophila (“Dros”) and yeast AMPK paralogs. Alignments with partial sequences from other CBS domains of γ2 and γ3, in which mutations were identified in the corresponding arginine residues or the adjacent histidine residue, are shown (bottom). In the course of analysis of the PRKAG2 gene and cDNA, the following additional sequence features were noted: (1) RT-PCR product sequences displayed partial deletion of codon 251 (apparently leaky splicing) at the beginning of exon 6 in all five patients and a normal control individual and (2) SNPs include CDS-26C→T (allele frequencies: C, 0.89; T, 0.11 [n=190]), Ex6+36insA (allele frequency: A, 0.83 [n=12]), and Ex10-42C→T (allele frequencies: C, 0.83; T, 0.17 [n=12]).
Electron microscopy of cardiac tissue from the R531Q-mutation–positive patient E showed massive, subsarcolemmal, and intermyofibrillar deposition of nonlysosomal β-glycogen within enlarged myocytes (fig. 2). Glycogen rosettes (α particles) could not be detected. Sarcomeres were often multifocally destroyed, and some fibers were totally degenerated. However, large amylopectinlike polysaccharide aggregates, as reported in adult patients with FHC/WPWS and PRKAG2 mutations (Arad et al. 2002), were not observed.
Figure 2.

Image from electron microscopy of glycogen storage in myocardial fibers of R531Q-mutation–positive patient E. Large amounts of free monodispersed β-glycogen (“Gl”) accumulate between the myofibrils and beneath the sarcolemma. Many sarcomeres appear to be destroyed (arrows), and some fibers are totally degenerated (star). In most mitochondria (“M”), solitary vacuoles and electron-dense, thickened, and disoriented cristae can be observed. Numerous endomembranaceous vacuoles (“V”), some of them containing lipid, are seen in fibers with advanced degeneration (star). The scale bar is 1 μm.
Biochemical Characterization of the Mutant Protein
The R531Q mutation severely impairs AMP and ATP binding by the γ2-subunit
Among the previously characterized mutations, the most pronounced functional consequences—both molecular (Daniel and Carling 2002; Scott et al. 2004) and clinical—are associated with a different replacement in the same amino acid position (R531G), indicating that arginine 531 is of particular importance. The R531G-positive patient displayed severe arrhythmia since the age of 2 years but was still alive at age 43 years (Gollob et al. 2001b). We therefore employed the R531G mutant protein as an internal reference in the biochemical experiments of the present study.
Binding of AMP and ATP was measured in vitro to GST fusion proteins containing Bateman domains A and B (GST-A/B [residues 274–569, CBS motifs 1–4]) or Bateman domain B only (GST-B [residues 430–556, CBS motifs 3–4]) of human γ2 (Scott et al. 2004), with either the normal arginine-531 residue, the R531Q mutation, or the R531G mutation (fig. 3 and table 2). The GST-B protein with the normal γ2 sequence bound one molecule of the activating nucleotide, AMP, and the affinity was reduced 16-fold by the R531G mutation, confirming previous findings (Scott et al. 2004). The R531Q mutation reduced the affinity for AMP even further (66-fold). The mutations also reduced the affinity for the inhibitory nucleotide ATP, and again the effect of R531Q (23-fold) was stronger than that of R531G (4.5-fold). The wild-type fusion protein containing both Bateman domains (GST-A/B) bound two molecules of AMP with high affinity and strong positive cooperativity, as observed previously (Scott et al. 2004). When the data were fitted to a two-site Hill plot model, the concentration giving half-maximal binding (B0.5) was 71 μM, and the Hill coefficient (h) was 1.8. The R531G mutant also bound two molecules of AMP with positive cooperativity but with a B0.5 of 2.3 mM, 33-fold higher than that for the wild type. The effect of the R531Q mutation was even more extreme, with a >100-fold increase in B0.5 and a loss of cooperativity (h=0.7). The loss of cooperative binding is evident from the much lower slope of the semilogarithmic plot in figure 3C. The results for ATP binding were analogous. Whereas the R531G mutation caused a 6-fold increase in the B0.5 for ATP but retained cooperativity, with the R531Q mutation, the effect was >200-fold, and cooperativity was lost.
Figure 3.

Binding of AMP (panels A and C) and ATP (panels B and D) by fusion proteins between GST and the Bateman domain B of γ2 (“GST-B” [panels A and B]) or between GST and both Bateman domains (“GST-A/B” [panels C and D]). In panels A and B, data were fitted to the binding model: Y = L/(Kd+L). In panels C and D, data were fitted to a two-site Hill plot model: Y = 2 × Lh/(B0.5h + Lh). “Y” represents fractional saturation, “L” represents ligand concentration, “B0.5” is the concentration of nucleotide that gives half-maximal binding, and “h” is the Hill coefficient. Best-fit parameters were estimated using GraphPad Prism (GraphPad software), and the curves were generated using the equation with those binding parameters.
Table 2.
Binding Parameters for AMP and ATP Measured with Wild-Type, R531G-Mutant, and R531Q-Mutant GST Fusion Proteins Containing Bateman Domain(s) B or A/B[Note]
| AMP |
ATP |
||||
| Fusion Typeand Protein | Na | Kd or B0.5b | h | Kd or B0.5b | h |
| GST-B: | |||||
| WT | 1 | 97 ± 7 | … | 396 ± 34 | … |
| R531G | 1 | 1,570 ± 120 | … | 1,790 ± 130 | … |
| R531Q | 1 | 6,400 ± 380 | … | 9,150 ± 140 | |
| GST-A/B: | |||||
| WT | 2 | 71 ± 5 | 1.8 ± .2 | 239 ± 12 | 1.8 ± .2 |
| R531G | 2 | 2,340 ± 220 | 1.4 ± .2 | 1,350 ± 66 | 2.9 ± .3 |
| R531Q | 2 | 7,320 ± 490 | .7 ± .1 | 53,000 ± 15,000 | .5 ± .1 |
Note.— WT = wild type.
N = number of binding sites assumed per protein molecule.
All values (in μM) are shown as mean ± SE of the mean. If a single binding site is assumed (N=1), the data were fitted to a simple binding equation Y = L/(Kd+L), where “Y” represents fractional saturation and “L” represents ligand concentration; the value given in this column is Kd. Where two binding sites are assumed (N=2), the data were fitted to the Hill equation, Y = 2 × Lh/(B0.5h+Lh), and the value given in this column is B0.5 (i.e., the concentration of ligand that gives half-maximal binding).
The R531Q and R531G mutations abolish AMP activation of the holoenzyme
To assess the effect of the R531Q mutation on AMP activation of the AMPK holoenzyme, we expressed the wild-type, R531G-mutant, and R531Q-mutant proteins of γ2 in HEK-293 cells, together with myc-tagged α1 and β1. The recombinant complexes were immunoprecipitated using anti-myc antibodies prior to assay. Extracts initially were made using a “slow lysis” procedure that involves scraping the cells off the culture plate, centrifuging them down, and resuspending them in lysis buffer. This procedure is known to cause maximal phosphorylation of Thr-172 on the α-subunit, because of undefined stresses occurring during the cell harvesting (Daniel and Carling 2002), and it allowed the effects of the mutations on allosteric activation by AMP to be observed independent of effects on phosphorylation. As observed elsewhere (Scott et al. 2004), the wild-type α1β1γ2 complex was activated 4-fold by AMP, with a half-maximal effect (A0.5) at 19 μM ± 11 μM (fig. 4). However, neither the R531G nor the R531Q mutant was activated significantly by AMP up to the maximal concentration of 100 μM (fig. 4), consistent with the strongly reduced binding affinity of these mutants for AMP (fig. 3). Instead, both mutants were slightly inhibited by AMP, which may be due to competition of AMP with ATP at the catalytic site on the kinase domain. The higher AMP affinity of holoenzyme activation, as compared with AMP binding to the isolated Bateman domain construct GST-A/B, confirms previous findings (Scott et al. 2004) and suggests that the integration of the Bateman domains into the holoenzyme context enhances nucleotide affinity.
Figure 4.

Activation of heterotrimeric α1β1γ2 complexes expressed in HEK-293 cells, with extracts made using the slow lysis procedure that causes maximal phosphorylation (Hardie et al. 2000). The complexes contained either wild-type (“WT”) γ2 or R531G and R531Q mutations of γ2 and were assayed in anti-myc immunoprecipitates at various concentrations of AMP. Data were fitted to the following equation: 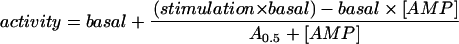 , where “basal” is the activity in the absence of AMP, “stimulation” is the maximal stimulation by AMP, and “A0.5” is the concentration of AMP that gives half-maximal activation. Best-fit parameters were estimated, and curves were generated as for figure 3.
, where “basal” is the activity in the absence of AMP, “stimulation” is the maximal stimulation by AMP, and “A0.5” is the concentration of AMP that gives half-maximal activation. Best-fit parameters were estimated, and curves were generated as for figure 3.
The R531Q and R531G mutations increase the basal phosphorylation and activity of AMPK
We next compared the activities (in the absence of AMP) of recombinant α1β1γ2 complexes that were prepared from HEK-293 cells by the “rapid” and “slow” lysis procedures (fig. 5A). The rapid lysis procedure involves lysing the cells in situ on the culture plate, and we believe that it preserves the phosphorylation status of AMPK that existed in the intact cells better than does the slow lysis procedure (Daniel and Carling 2002). Compared with the wild type, there was a significant increase (2.3-fold) in the basal activity (measured after rapid lysis) of the R531G mutant and an even greater increase (3.5-fold) in that of the R531Q mutant. Both mutants were further activated by the slow lysis procedure, but relatively less so than the wild-type enzyme, such that, after slow lysis, the activities of all three were similar. The increased basal activities of the R531G and R531Q mutants, as well as the further increase after slow lysis, correlated with an increased phosphorylation of Thr-172 on the α-subunit (fig. 5B) and an increased phosphorylation of the downstream target, ACC (fig. 5C).
Figure 5.

AMPK activity (A), phosphorylation of Thr-172 on the AMPK α-subunit (B), and phosphorylation of the downstream target ACC (C) in HEK-293 cells expressing heterotrimeric α1β1γ2 complexes, with or without γ2 mutations, as in figure 4. Cells were harvested by both the rapid and the slow lysis procedures, and AMPK activities were measured in anti-myc immunoprecipitates in the absence of AMP. Data are means ± SE of the mean, for results from duplicate culture plates.
Discussion
A Recurrent R531Q Missense Mutation in PRKAG2 Causes a Severe Congenital, Rapidly Fatal, Nonlysosomal Heart Glycogenosis
The present study identifies, for the first time, a molecular cause of congenital, rapidly fatal, nonlysosomal cardiac glycogenosis. Strikingly, we found three identical but independent heterozygous R531Q missense mutations in PRKAG2, in three of the five sporadic cases, all from diverse geographic origins. These findings extend the clinical spectrum of PRKAG2 mutations toward a very severe manifestation characterized by fetal onset (prenatal bradycardia was noted in all three patients, prompting preterm delivery by cesarean section, and prenatal cardiomegaly was noted in one patient), extreme cardiomegaly, and an infantile-fatal outcome. The R531Q-positive patients died of hemodynamic and respiratory failure secondary to hypertrophic nonobstructive cardiomyopathy but also had WPWS-like conduction anomalies. In patients with classic juvenile-/adult-onset FHC/WPWS, arrhythmia usually dominates the clinical picture (also in the few reported patients with symptomatic onset in early childhood [Blair et al. 2001; Gollob et al. 2001b]), and these patients virtually always survive into reproductive age. It will be of interest to investigate whether other PRKAG2 mutations associated with intermediate phenotypes can be found among children with hypertrophic cardiomyopathy and electrophysiological abnormalities (Strauss and Lock 2003; Arad et al. 2005). The phenotypes of the three R531Q-positive infants were very uniform, and there was no other PRKAG2 mutation in our group of five patients. Conversely, the R531Q mutation has never been reported in patients with classic FHC/WPWS, despite its tendency to arise recurrently, like the R302Q mutation in classic FHC/WPWS (see below). In accordance with this strict, mutually exclusive correlation between genotype and clinical phenotype, the molecular abnormalities of the R531Q mutant protein are more pronounced than those of any other PRKAG2 mutants characterized to date. Hence, the R531Q mutation by itself appears to be sufficient to cause the distinctive, very severe phenotype of its carriers, and neither the additional sequence variants (mostly polymorphisms) in the PYGM and PYGB genes of patients A and D (table 1) nor other, unidentified elements of genetic background need to be invoked.
All R531Q-positive infants were sporadic cases, and analysis of the parents of patient E showed that R531Q was a newly arisen mutation in this family (the only family available for analysis). Given the complete penetrance of PRKAG2 mutations even in the less-severe, juvenile-/adult-onset FHC/WPWS, this suggests that R531Q mutations invariably cause an infantile-fatal disease, are therefore not passed on by their carriers, and always occur de novo. At the nucleotide level, these mutations are G→A replacements in a CpG context (fig. 1), which are known to occur at least 13-fold more frequently than other single-nucleotide replacements in the CG dinucleotide (Antonarakis et al. 2001). An identical replacement in the analogous position of the first CBS motif (arginine 531 is in the fourth CBS motif), R302Q (fig. 1), is the most common PRKAG2 mutation and has also arisen recurrently but is associated with milder clinical and molecular phenotypes (Gollob et al. 2001a, 2002; Arad et al. 2002; Daniel and Carling 2002; Scott et al. 2004). The occurrence of the R531Q mutation in a large proportion of cases with prenatal- or neonatal-onset severe nonlysosomal cardiac GSD offers a rapid, noninvasive diagnostic test in this fatal condition, and its apparently obligate de novo occurrence has corresponding implications for genetic counseling.
The clinical manifestation of PRKAG2 mutations is restricted to the heart. However, the γ2-subunit of AMPK is expressed in many tissues, and, even in the heart, it does not appear to be the predominant isoform, at least in rodents (Cheung et al. 2000; Mahlapuu et al. 2003). In patients with the severe R531Q mutation, glycogen accumulation was indeed seen in tissues other than the heart (patients D and E [see the “Case Reports” section]). Detailed examination can also reveal glycogen accumulation in noncardiac tissues in adult patients with FHC/WPWS and with milder PRKAG2 mutations (Murphy et al. 2005), and the systemic hypertension observed in two kindreds with FHC/WPWS (Gollob et al. 2001a, 2001b) may likewise be an extracardial manifestation. Specific aspects of the role of AMPK in cardiac metabolic physiology, however, may cause a particularly pronounced accumulation of glycogen in the heart.
The pronounced heart glycogenosis observed in patients with the R531Q mutation strengthens the view that glycogen storage is the primary pathological consequence of PRKAG2 mutations and that the conduction defects are secondary to this. This was first proposed after histological investigation of heart tissues of patients with FHC/WPWS and was corroborated by studies of transgenic mice overexpressing the N488I mutant of γ2 (Arad et al. 2002, 2003). The mice displayed massive cardiac glycogen deposition, ventricular pre-excitation, and an infiltration of the annulus fibrosus, which normally insulates the atria from the ventricles, by glycogen-filled myocytes. It was proposed that this infiltration gives rise to microscopic atrioventricular connections and causes the conduction abnormalities in these mice, in human patients with PRKAG2 mutations, and similarly also in patients with other cardiac glycogenoses (GSD II, GSD IIb, and GSD III) (Arad et al. 2002; Gollob et al. 2002).
The glycogen that accumulates in the cardiomyocytes of individuals with PRKAG2 mutations may in part be of abnormal structure. In the hearts of five adult patients with three different PRKAG2 mutations, Arad et al. (2002) found large, amorphous, amylase-resistant amylopectinlike polysaccharide aggregates, similar to those observed in patients with GSD IV. It is conceivable that the perturbation of AMPK function by these mutations directly or indirectly upsets the balance of the different enzyme activities that participate in glycogen synthesis and degradation and thus affects the structure of the branched glycogen polymer. However, in R531Q-mutation–positive patients A and E, the glycogen deposits were amylase sensitive (amylase was not applied in case D), and electron microscopy of the cardiac tissue of patient E (fig. 2) revealed normal glycogen β particles, with no evidence of the aggregates seen by Arad et al. (2002). Neither were abnormal polysaccharide aggregates reported in the hearts of transgenic mice overexpressing the FHC/WPWS mutation N488I, although these mice developed massive cardiac glycogenosis (Arad et al. 2003). The amylase-resistant molecular variant may be a minor side product, inert to physiological turnover, of the patients’ perturbed glycogen metabolism. It may accumulate only over the course of years or decades, and therefore it may be apparent only in adult patients.
Genetic Heterogeneity of Fatal Congenital Nonlysosomal Heart Glycogenosis
No mutations were found in patients B and C, either in PRKAG2 or in the Phk and phosphorylase genes. The hemizygous E38Q replacement in the PHKA2 gene of patient B is a polymorphism also found in controls (in both hetero- and homozygous constellations), and the heterozygous A303S and D502N replacements in the PYGB gene of patient C are common polymorphisms also detectable in his healthy parents (see the note to table 1). It remains to be seen whether the two PRKAG2 mutation–negative subjects are mutant in novel disease genes or whether they represent unusually severe manifestations of mutations in one of the other genes known to give rise to GSDs involving the heart. Their PHKA2 and PYGB polymorphisms might have mild functional consequences that aggravate the manifestation of a principal defect in another gene or influence its tissue specificity (Hansen et al. 1995; Westphal et al. 2002).
When the phenotypes of the three R531Q-positive subjects are compared with each other, with the two mutation-negative subjects, and with the three first-published cases of infantile-fatal nonlysosomal heart glycogenosis with low Phk activity and undefined genotype (Mizuta et al. 1984; Eishi et al. 1985; Servidei et al. 1988; Elleder et al. 1993), they all appear very similar. All but patient C display extreme cardiomegaly (heart weights 2.7–6.5-fold greater than normal), onset of cardiac or respiratory distress from before birth to within the first weeks of life, and death between age 3 wk and age 5 mo. The R531Q-negative patient C is set apart by a particularly malignant course and short lifespan (11 d), neurological symptoms, no WPWS-like ECG features or cardiomegaly, and a family history suggestive of autosomal recessive inheritance, probably reflecting a different genetic etiology of his condition.
The Existence of a Heart-Specific Primary Phk Deficiency Is Doubtful
Low-to-zero Phk enzyme activities were determined in heart autopsy samples from all five patients studied here, leading to their tentative identification as heart-specific Phk deficiency, an entity described previously (Mizuta et al. 1984; Eishi et al. 1985; Servidei et al. 1988; Elleder et al. 1993). However, we found no mutations or splicing abnormalities in three of the four patients whose Phk genes were analyzed, and the hemizygous E38Q replacement in the PHKA2 gene of the fourth patient is an innocuous polymorphism. We cannot exclude unusual mutation types not covered by our approach (such as a mutation in a transcription factor binding site affecting expression specifically in the heart), but it appears most likely that the low Phk activities in our patient group—and possibly in autopsy samples from infantile-fatal heart glycogenosis cases, in general—either are of secondary nature or are artifacts. AMPK is not believed to regulate Phk directly, but it is plausible that, in a metabolic status causing massive glycogen deposition, the activity or expression of Phk is down-regulated, so that low Phk activities in these cases may be authentic but secondary. It is noteworthy that, also in most patients with skeletal muscle glycogenosis and low Phk activity, we could not detect mutations either in Phk subunits or in PRKAG3 (the skeletal muscle isoform of the AMPK γ-subunit that is mutated in a mild muscle glycogenosis in pigs [Milan et al. 2000]) (Burwinkel et al. 2003). Phk is a notoriously unstable enzyme. In striated muscle tissues, it may be particularly sensitive to inactivation (e.g., by dephosphorylation or proteolysis). This could occur in vivo (i.e., in myopathic tissue chronically damaged by another primary defect) or after sampling (for further discussion, see the study by Burwinkel et al. [2003]). In the cardiac cases, in particular, samples were always obtained through autopsy several hours postmortem and would be particularly vulnerable to dephosphorylation or degradation.
The R531Q and R531G Mutations Reduce the AMP and ATP Affinities but Enhance the Basal Activity and Phosphorylation of AMPK
All seven previously identified mutations in the γ2 gene that cause FHC/WPWS occur in the Bateman domain region (Gollob et al. 2002; Oliveira et al. 2003). Three of them (R302Q, R531G, and H383R) affect residues in (R302Q and R531G) or adjacent to (H383R) equivalent positions in the first, second, and fourth CBS motifs (fig. 1). These, together with a fourth mutation (T400N), cause defects in the binding of AMP and ATP to isolated Bateman domains, as well as defects in activation of the α1β1γ2 complexes by AMP (Daniel and Carling 2002; Scott et al. 2004). A fifth mutation that inserts a leucine residue between the first and second CBS motif has only a weak effect, if any, on these parameters (Daniel and Carling 2002; Scott et al. 2004), and the remaining two (Y487H and N488I) have not yet been tested. On the basis of structural modeling of the Bateman domains from γ2, we have proposed that the basic residues affected by the mutations are involved with binding of the phosphate group of AMP (Scott et al. 2004). Of the five PRKAG2 mutants previously tested, the R531G mutation caused the most severe defect in the binding of AMP and ATP to the GST fusion containing both Bateman domains (Scott et al. 2004), as well as the most severe defect in AMP activation of the intact heterotrimeric complex (Daniel and Carling 2002; Scott et al. 2004). Here, we show that the R531Q mutation causes even more severe defects in the binding of AMP and, especially, ATP. Binding of AMP or ATP to the construct containing both Bateman domains is highly cooperative (h=2), indicating that one of the sites is inaccessible to AMP until the nucleotide is bound to the other site. One explanation of the severe nature of mutations in Bateman domain B (R531G and R531Q) may be that this is the domain that binds the nucleotides first, so that mutations in this domain impair the binding of both molecules of the nucleotide. The R531Q mutation additionally reduces or abolishes the cooperative interactions between the two binding sites. The extension of our knowledge of Bateman domain mutations also has implications for the understanding of other Bateman domain–containing proteins, including several proteins involved in hereditary disorders, as well as for the design of drugs that target AMPK, for example, in type 2 diabetes (Kemp 2004; Scott et al. 2004).
Most, if not all, PRKAG2 mutations impair or abolish activation of the AMPK complex by AMP (i.e., they cause a loss of function). However, a loss-of-function mechanism is hard to reconcile with the dominant nature of these mutations, especially given a background of one wild-type γ2 allele plus the coexpressed γ1 isoform (under the assumption that the AMPK holoenzyme is an αβγ heterotrimer with no potential for dominant negative interactions between mutant and wild-type γ-subunits within a single holoenzyme complex). Arad et al. (2002) suggested, on the basis of equivalent mutations in the budding yeast homologue of the AMPK γ-subunits (Snf4), that two of the mutations (T400N and N488I) caused constitutive activation of the AMPK complex. Direct analysis of γ mutant proteins in mammalian cells has yielded conflicting results. Coexpression of an R70Q mutant in γ1 (equivalent to the R302Q mutant in γ2) with α1 and β1 in COS7 cells was reported to cause an increase in basal phosphorylation and activity of AMPK (Hamilton et al. 2001). Also, the coexpression of the R225Q mutant of γ3 (equivalent to R70Q in γ1 and to R302Q in γ2 [see fig. 1]) with α2 and β2 in COS cells led to an increase in basal activity and α-subunit phosphorylation (Barnes et al. 2004). Results from studies of transgenic mice overexpressing either the R225Q mutant γ3-subunit in skeletal muscle (Barnes et al. 2004) or the N488I mutant γ-subunit in the heart (Arad et al. 2003) also indicated increases in the basal activities of the mutant, compared with the wild-type enzymes. In contrast to these results, analysis of four (Daniel and Carling 2002) or five (Scott et al. 2004) FHC/WPWS mutations in human γ2, which are coexpressed with α1 and γ1 in CCL13 cells, exhibited reduced or absent activation by AMP but no evidence of an increase in the basal activity.
In the present study, we have reinvestigated the effect of the R531G mutation (as well as the R531Q mutation) on AMPK activity by studying expression in HEK-293, instead of CCL13, cells. Although the R531G and R531Q mutations both completely abolished AMP activation of the AMPK complex (fig. 4), they also caused an increase in the basal phosphorylation and activity of the heterotrimeric AMPK complex, determined using the rapid lysis method that we believe preserves the phosphorylation state (fig. 5). The latter was not observed in two previous studies of the R531G mutant (Daniel and Carling 2002; Scott et al. 2004), but this can now be ascribed to the use of CCL13 cells. Although originally derived from human liver (Chang 1954), the CCL13 cells available from the American Type Culture Collection are now thought to have resulted from contamination with HeLa cells (see the American Type Culture Collection Web site). Unlike HEK-293 cells, HeLa cells do not express the major upstream kinase for AMPK (i.e., LKB1) (Hawley et al. 2003), and we have now verified that CCL13 cells do not express LKB1 either (data not shown). We also confirmed (data not shown) that the increase in basal phosphorylation and activity, caused by the R531G and R531Q mutations when coexpressed with α1 and β1 in HEK-293 cells, is not observed upon expression in HeLa cells. Thus, the increased basal phosphorylation is observed only in cells expressing LKB1.
How can an increase in basal AMPK activity lead to glycogen storage? Activation of AMPK increases glucose uptake by causing acute activation or translocation, as well as increased expression, of both GLUT1 and GLUT4 (Hayashi et al. 1998; Kurth-Kraczek et al. 1999; Barnes et al. 2002; Fryer et al. 2002) in several tissues, including the heart (Russell et al. 1999). Although AMPK also phosphorylates the muscle isoform of glycogen synthase both in cell-free assays (Carling and Hardie 1989) and in vivo (Jørgensen et al. 2004) at a site that causes inactivation of the enzyme (Flotow and Roach 1989), the inactivation caused by phosphorylation at this site is overridden by high concentrations of the allosteric activator, glucose-6-phosphate (Flotow and Roach 1989). Increased glucose uptake caused by an elevated basal activity of AMPK over an extended period, in the absence of a proportional increase in consumption of glucose, may cause increased intracellular glucose-6-phosphate, leading to persistent allosteric activation of glycogen synthase and inhibition of glycogen phosphorylase. Increased glycogen content in the hearts of transgenic mice overexpressing wild-type γ2 (Arad et al. 2003) and reduced glycogen resynthesis in skeletal muscles of γ3-knockout mice after exercise (Barnes et al. 2004) also indicate that AMPK activity enhances glycogen synthesis; these data support the view that PRKAG2 disease mutations cause glycogen storage through basal activation of AMPK.
Correlations between Genotype, Molecular Properties, and Clinical Manifestation
The molecular properties of the mutant AMPK proteins can, in general terms, be correlated with their biological effects: the increase in the basal phosphorylation and activity of AMPK caused by disease mutations in PRKAG2 explains the dominant, gain-of-function–like character of these mutations, and the R531Q mutation is associated with both the most severe clinical phenotype and the most severe molecular abnormalities of all known PRKAG2 mutations. Yet, on closer inspection, we are left with paradoxes that indicate that we are still far from a coherent understanding of the molecular mechanisms of AMPK and of its physiology and pathophysiology in the heart; the particular severity of the R531Q mutation highlights these gaps in understanding.
Concerning the gain- versus loss-of-function issue, it remains striking that most disease-causing mutations have a specific impact on AMP and ATP affinity and occur very selectively—and even recurrently (R302Q and R531Q)—in strategic positions within the CBS motifs or in linker sequences between the motifs. In contrast, no mutations have been found in other parts of the γ-subunit or in the α- or β-subunits (Oliveira et al. 2003). This indicates a specific connection between nucleotide binding and the pathological effects of these mutations. However, it is believed that the phosphorylation of the α-subunit at Thr-172 occurs subsequent to AMP binding. It is therefore unclear how the observed increase of phosphorylation and basal activity in the rapid lysis cell culture system is brought about if the mutant proteins cannot bind AMP at physiological concentrations. This contradiction might be resolved if the mutations not only reduced nucleotide affinity but also simultaneously were “AMP-mimetic” (i.e., if they induced a more active conformation of the Bateman domains, even when no AMP is bound).
Of all the PRKAG2 mutations characterized to date, R531Q has the strongest effect on nucleotide binding and basal activation, but there is no abrupt transition in the nature of the molecular impacts between the R531G and R531Q mutations (figs. 3, 4, and 5). In contrast, the clinical phenotype of patients with R531Q is dramatically more severe than that of patients with the other mutations associated with classic FHC/WPWS, including R531G. The two patient groups differ strikingly in terms of (1) age at onset, course, and outcome; (2) degree of cardiomegaly; (3) the fact that hemodynamic and respiratory failure, as opposed to arrhythmia and conduction anomalies, dominate the clinical picture; and (4) in sporadic versus familial occurrence reflecting the profound difference in survival. It is possible that the molecular parameter in which the R531Q protein differs most markedly from the other mutants—and which most faithfully correlates with the biological impact of PRKAG2 mutations in general—has not yet been characterized. In this context, the R109insL mutation is also of interest, because it only slightly affects AMP binding (Scott et al. 2004) but still results in a severe adult phenotype (Blair et al. 2001); it may affect a mechanistic step between AMP binding and basal activation.
There are two known molecular effects in which R531Q is indeed unique among the PRKAG2 mutations characterized to date: it abolishes the cooperativity of nucleotide binding, and it has a particularly large impact on ATP binding. The latter effect makes the R531Q mutant the only γ2 mutant protein that is probably unoccupied by either AMP or ATP under in vivo conditions, whereas the wild type and the other mutant γ2-subunits are predicted to be occupied by ATP under resting conditions. The R531Q-mutant γ2-subunit, unoccupied by ATP, might be in a partially active conformation. If this is correct, AMPK might be regarded an “ATP-inhibited” as much as an “AMP-activated” kinase. Another aspect that deserves consideration is the highly cooperative, “ultrasensitive” response of AMPK, through which a small increase in the AMP:ATP ratio over a critical range of concentrations can translate into a strong activation of AMPK (Hardie et al. 1999). Shifting this narrow “window of sensitivity” to different degrees by different mutations could thus lead to profoundly nonlinear consequences. Also, the overexpression of the wild type and the N488I mutant γ2-subunits at similar levels in transgenic mice led to a much more severe phenotype in the N488I mice (Arad et al. 2003). All these observations suggest that the underlying pathomechanism is a form of AMPK dysregulation specifically connected with its interaction with AMP and ATP, which subsequently leads to an increase of basal phosphorylation and activity by the upstream kinase. However, the detailed molecular mechanisms linking the two phenomena remain to be fully elucidated.
Acknowledgments
We thank J. J. Regalado (Miami) and V. Worthington, A. W. Bates, and J. V. Leonard (London), for their contributions to the procurement and analysis of human specimens, and D. Carling, for the original plasmids encoding AMPK subunits. This work was supported by grants from the Deutsche Forschungsgemeinschaft (Ki 324/11), the University of Bochum Medical School intramural research grant program (FoRUM), and the Fonds der Chemischen Industrie (to M.W.K.), as well as by Research and Technological Development contract QLG1-CT-2001-01488 from the European Commission and a program grant from the Wellcome Trust (to D.G.H.). Tissue samples from patients A and B were obtained from the University of Miami Brain and Tissue Bank for Developmental Disorders, which is funded by National Institutes of Health contract NO1-HD-8-3284.
Electronic-Database Information
Accession numbers and URLs for data presented herein are as follows:
- American Type Culture Collection, http://www.atcc.org/
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/ (for 1240C→G [accession number rs11231866], 907G→T [accession number rs3818199], and 1504G→A [accession number rs2227891])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for Pompe disease, Danon disease, Cori or Forbes disease, Andersen disease, FHC/WPWS, and Phk deficiency)
References
- Antonarakis SE, Krawczak M, Cooper DN (2001) The nature and mechanisms of human gene mutation. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease, 8th ed. McGraw-Hill, New York, pp 343–377 [Google Scholar]
- Arad M, Benson DW, Perez-Atayde AR, McKenna WJ, Sparks EA, Kanter RJ, McGarry K, Seidman JG, Seidman CE (2002) Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest 109:357–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arad M, Maron BJ, Gorham JM, Johnson WH Jr, Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, Seidman JG (2005) Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med 352:362–372 [DOI] [PubMed] [Google Scholar]
- Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez-Atayde AR, Sawyer DB, Walter M, Li GH, Burgon PG, Maguire CT, Stapleton D, Schmitt JP, Guo XX, Pizard A, Kupershmidt S, Roden DM, Berul CI, Seidman CE, Seidman JG (2003) Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 107:2850–2856 [DOI] [PubMed] [Google Scholar]
- Barnes BR, Marklund S, Steiler TL, Walter M, Hjalm G, Amarger V, Mahlapuu M, Leng, Y, Johansson C, Galuska D, Lindgren K, Abrink M, Stapleton D, Zierath JR, Andersson L (2004) The 5′-AMP-activated protein kinase γ3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J Biol Chem 279:38441–38447 [DOI] [PubMed] [Google Scholar]
- Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, Fryer LG, Foufelle F, Carling D, Hardie DG, Baldwin SA (2002) Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK). J Cell Sci 115:2433–2442 [DOI] [PubMed] [Google Scholar]
- Bateman A (1997) The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci 22:12–13 [DOI] [PubMed] [Google Scholar]
- Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, Salmon A, Ostman-Smith I, Watkins H (2001) Mutations in the γ2 subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 10:1215–1220 [DOI] [PubMed] [Google Scholar]
- Bührer C, van Landeghem FKH, Felderhoff-Mueser U, Stadelmann C, Obladen M (2003) Fetal bradycardia at 28 weeks of gestation associated with cardiac glycogen phosphorylase b kinase deficiency. Acta Paediatr 92:1352–1353 [DOI] [PubMed] [Google Scholar]
- Burwinkel B, Hu B, Schroers A, Clemens PR, Moses SW, Shin YS, Pongratz D, Vorgerd M, Kilimann MW (2003) Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases. Eur J Hum Genet 11:516–526 [DOI] [PubMed] [Google Scholar]
- Burwinkel B, Shin YS, Bakker HD, Deutsch J, Lozano MJ, Maire I, Kilimann MW (1996) Mutation hotspots in the PHKA2 gene in X-linked liver glycogenosis due to phosphorylase kinase deficiency with atypical activity in blood cells (XLG2). Hum Mol Genet 5:653–658 [DOI] [PubMed] [Google Scholar]
- Burwinkel B, Shiomi S, Al-Zaben A, Kilimann MW (1998) Liver glycogenosis due to phosphorylase kinase deficiency: PHKG2 gene structure and mutations associated with cirrhosis. Hum Mol Genet 7:149–154 [DOI] [PubMed] [Google Scholar]
- Carling D, Hardie DG (1989) The substrate and sequence specificity of the AMP-activated protein kinase: phosphorylation of glycogen synthase and phosphorylase kinase. Biochim Biophys Acta 1012:81–86 [DOI] [PubMed] [Google Scholar]
- Chang RS (1954) Continuous subcultivation of epithelial-like cells from normal human tissues. Proc Soc Exp Biol Med 87:440–443 [DOI] [PubMed] [Google Scholar]
- Chen YT (2001) Glycogen storage diseases. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease, 8th ed. McGraw-Hill, New York, pp 1521–1551 [Google Scholar]
- Cheung PCF, Salt IP, Davies SP, Hardie DG, Carling D (2000) Characterization of AMP-activated protein kinase γ subunit isoforms and their role in AMP binding. Biochem J 346:659–669 [PMC free article] [PubMed] [Google Scholar]
- Daniel TD, Carling D (2002) Functional analysis of mutations in the γ2 subunit of AMP-activated protein kinase associated with cardiac hypertrophy and Wolff-Parkinson-White syndrome. J Biol Chem 277:51017–51024 [DOI] [PubMed] [Google Scholar]
- Eishi Y, Takemura T, Sone R, Yamamura H, Narisawa K, Ichinohasama R, Tanaka M, Hatakeyama S (1985) Glycogen storage disease confined to the heart with deficient activity of cardiac phosphorylase kinase: a new type of glycogen storage disease. Hum Pathol 16:193–197 [DOI] [PubMed] [Google Scholar]
- Elleder M, Shin YS, Zuntova A, Vojtovic P, Chalupecki V (1993) Fatal infantile hypertrophic cardiomyopathy secondary to deficiency of heart specific phosphorylase b kinase. Virchows Archiv A Pathol Anat 423:303–307 [DOI] [PubMed] [Google Scholar]
- Ewert R, Gulijew A, Wensel R, Dandel M, Hummel M, Vogel M, Meyer R, Hetzer R (1999) Glycogenosis type IV as a seldom cause of cardiomyopathy. Z Kardiol 88:850–856 [DOI] [PubMed] [Google Scholar]
- Flotow H, Roach PJ (1989) Synergistic phosphorylation of rabbit muscle glycogen synthase by cyclic AMP-dependent protein kinase and casein kinase I: implications for hormonal regulation of glycogen synthase. J Biol Chem 264:9126–9128 [PubMed] [Google Scholar]
- Fryer LG, Foufelle F, Barnes K, Baldwin SA, Woods A, Carling D (2002) Characterization of the role of the AMP-activated protein kinase in the stimulation of glucose transport in skeletal muscle cells. Biochem J 363:167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollob MH, Green MS, Tang ASL, Gollob T, Karibe A, Hassan AS, Ahmad F, Lozado R, Shah G, Fananapazir L, Bachinski LL, Roberts R (2001a) Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med 344:1823–1831 [DOI] [PubMed] [Google Scholar]
- Gollob MH, Green MS, Tang ASL, Roberts R (2002) PRKAG2 cardiac syndrome: familial ventricular preexcitation, conduction system disease, and cardiac hypertrophy. Curr Opin Cardiol 17:229–234 [DOI] [PubMed] [Google Scholar]
- Gollob MH, Seger JJ, Gollob TN, Tapscott T, Gonzales O, Bachinski L, Roberts R (2001b) Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation 104:3030–3033 [DOI] [PubMed] [Google Scholar]
- Hamilton SR, Stapleton D, O’Donnell JB, Kung JT, Dalal SR, Kemp BE, Witters LA (2001) An activating mutation in the γ1 subunit of the AMP-activated protein kinase. FEBS Lett 500:163–168 [DOI] [PubMed] [Google Scholar]
- Hansen L, Hansen T, Vestergaard H, Björbaek C, Echwald SM, Clausen JO, Chen YH, Chen MX, Cohen PTW, Pedersen O (1995) A widespread amino acid polymorphism at codon 905 of the glycogen-associated regulatory subunit of protein phosphatase-1 is associated with insulin resistance and hypersecretion of insulin. Hum Mol Genet 4:1313–1320 [DOI] [PubMed] [Google Scholar]
- Hardie DG (2003) Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 144:5179–5183 [DOI] [PubMed] [Google Scholar]
- ——— (2004) The AMP-activated protein kinase pathway—new players upstream and downstream. J Cell Sci 117:5479–5487 [DOI] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA (2001) AMP-activated protein kinase: the energy charge hypothesis revisited. BioEssays 23:1112–1119 [DOI] [PubMed] [Google Scholar]
- Hardie DG, Salt IP, Davies SP (2000) Analysis of the role of the AMP-activated protein kinase in the response to cellular stress. Methods Mol Biol 99:63–75 [DOI] [PubMed] [Google Scholar]
- Hardie DG, Salt IP, Hawley SA, Davies SP (1999) AMP-activated protein kinase: an ultrasensitive system for monitoring cellular energy charge. Biochem J 338:717–722 [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG (2003) Complexes between the LKB1 tumor suppressor, STRADa/b and MO25a/b are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2:28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG (1996) Characterization of the AMP-activated protein kinase kinase from rat liver, and identification of threonine-172 as the major site at which it phosphorylates and activates AMP-activated protein kinase. J Biol Chem 271:27879–27887 [DOI] [PubMed] [Google Scholar]
- Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ (1998) Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes 47:1369–1373 [DOI] [PubMed] [Google Scholar]
- Hirschhorn R, Reuser AJJ (2001) Glycogen storage disease type II: acid α-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease, 8th ed. McGraw-Hill, New York, pp 3389–3420 [Google Scholar]
- Jørgensen SB, Nielsen JN, Birk JB, Olsen GS, Viollet B, Andreelli F, Schjerling P, Vaulont S, Hardie DG, Hansen BF, Richter EA, Wojtaszewski JF (2004) The α2-5′AMP-activated protein kinase is a site 2 skeletal muscle glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes 53:3074–3081 [DOI] [PubMed] [Google Scholar]
- Kemp BE (2004) Bateman domains and adenosine derivatives form a binding contract. J Clin Invest 113:182–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilimann MW (1997) Glycogen storage disease due to phosphorylase kinase deficiency. In: Swallow DM, Edwards YH (eds) Protein dysfunction in human genetic disease. Bios Scientific Publishers, Oxford, United Kingdom, pp 57–75 [Google Scholar]
- Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW (1999) 5′ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes 48:1667–1671 [DOI] [PubMed] [Google Scholar]
- Lee PJ, Deanfield JE, Burch M, Baig K, McKenna WJ, Leonard JV (1997) Comparison of the functional significance of left ventricular hypertrophy in hypertrophic cardiomyopathy and glycogenosis type III. Amer J Cardiol 79:834–838 [DOI] [PubMed] [Google Scholar]
- Mahlapuu M, Johansson C, Lindgren K, Hjälm G, Barnes BR, Krook A, Zierath JR, Andersson L, Marklund S (2003) Expression profiling of the γ-subunit isoforms of AMP-activated protein kinase suggests a major role for γ3 in white skeletal muscle. Am J Physiol Endocrinol Metab 286:E194-E200 [DOI] [PubMed] [Google Scholar]
- Milan D, Jeon JT, Looft C, Amarger V, Robic A, Thelander M, Rogel-Gaillard C, Paul S, Iannuccelli N, Rask L, Ronne H, Lundström K, Reinsch N, Gellin J, Kalm E, Roy PL, Chardon P, Andersson L (2000) A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288:1248–1251 [DOI] [PubMed] [Google Scholar]
- Mizuta K, Hashimoto E, Tsutou A, Eishi Y, Takemura T, Narisawa K, Yamamura H (1984) A new type of glycogen storage disease caused by deficiency of cardiac phosphorylase kinase. Biochem Biophys Res Commun 119:582–587 [DOI] [PubMed] [Google Scholar]
- Murphy RT, Mogensen J, McGarry K, Bahl A, Evans A, Osman E, Syrris P, Gorman G, Farrell M, Holton JL, Hanna MG, Hughes S, Elliot PM, Macrae CA, McKenna WJ (2005) Adenosine monophosphate-activated protein kinase disease mimicks hypertrophic cardiomyopathy and Wolff-Parkinson-White syndrome: natural history. J Am Coll Cardiol 45:922–930 [DOI] [PubMed] [Google Scholar]
- Oliveira SMJ, Ehtisham J, Redwood CS, Ostman-Smith I, Blair EM, Watkins H (2003) Mutation analysis of AMP-activated protein kinase subunits in inherited cardiomyopathies: implications for kinase function and disease pathogenesis. J Mol Cell Cardiol 35:1251–1255. [DOI] [PubMed] [Google Scholar]
- Regalado JJ, Rodriguez MM, Ferrer PL (1999) Infantile hypertrophic cardiomyopathy of glycogenosis type IX: isolated cardiac phosphorylase kinase deficiency. Pediatr Cardiol 20:304–307 [DOI] [PubMed] [Google Scholar]
- Russell RR, Bergeron R, Shulman GI, Young LH (1999) Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol 277:H643–H649 [DOI] [PubMed] [Google Scholar]
- Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG (2004) CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest 113:274–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servidei S, Metlay LA, Chodosh J, DiMauro S (1988) Fatal infantile cardiopathy caused by phosphorylase b kinase deficiency. J Pediatr 113:82–85 [DOI] [PubMed] [Google Scholar]
- Servidei S, Riepe RE, Langston C, Tani LY, Bricker JT, Crisp-Lindgren N, Travers H, Armstrong D, DiMauro S (1987) Severe cardiopathy in branching enzyme deficiency. J Pediatr 111:51–56 [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC (2004). The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA 101:3329–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss A, Lock JE (2003) Pediatric cardiomyopathy—a long way to go. N Engl J Med 348:1703–1705 [DOI] [PubMed] [Google Scholar]
- Sugie K, Yamamoto A, Murayama K, Oh SJ, Takahashi M, Mora M, Riggs JE, Colomer J, Iturriaga C, Meloni A, Lamperti C, Saitoh S, Byrne E, DiMauro S, Nonaka I, Hirano M, Nishino I (2002) Clinicopathological features of genetically confirmed Danon disease. Neurology 58:1773–1778 [DOI] [PubMed] [Google Scholar]
- Westphal V, Kjaergaard S, Schollen E, Martens K, Grunewald S, Schwartz M, Matthijs G, Freeze HH (2002) A frequent mild mutation in ALG6 may exacerbate the clinical severity of patients with congenital disorder of glycosylation Ia (CDG-Ia) caused by phosphomannomutase deficiency. Hum Mol Genet 11:599–604 [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13:2004–2008 [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Morisawa Y, Verloes A, Murakami N, Hirano M, Nonaka I, Nishino I (2001) Infantile autophagic vacuolar myopathy is distinct from Danon disease. Neurology 57:903–905 [DOI] [PubMed] [Google Scholar]
- Ziemssen F, Sindern E, Schröder JM, Shin YS, Zange J, Kilimann MW, Malin JP, Vorgerd M (2000) Novel missense mutations in the glycogen-branching enzyme gene in adult polyglucosan body disease. Ann Neurol 47:536–540 [PubMed] [Google Scholar]