

Abstract
Once genetic linkage has been identified for a complex disease, the next step is often association analysis, in which single-nucleotide polymorphisms (SNPs) within the linkage region are genotyped and tested for association with the disease. If a SNP shows evidence of association, it is useful to know whether the linkage result can be explained, in part or in full, by the candidate SNP. We propose a novel approach that quantifies the degree of linkage disequilibrium (LD) between the candidate SNP and the putative disease locus through joint modeling of linkage and association. We describe a simple likelihood of the marker data conditional on the trait data for a sample of affected sib pairs, with disease penetrances and disease-SNP haplotype frequencies as parameters. We estimate model parameters by maximum likelihood and propose two likelihood-ratio tests to characterize the relationship of the candidate SNP and the disease locus. The first test assesses whether the candidate SNP and the disease locus are in linkage equilibrium so that the SNP plays no causal role in the linkage signal. The second test assesses whether the candidate SNP and the disease locus are in complete LD so that the SNP or a marker in complete LD with it may account fully for the linkage signal. Our method also yields a genetic model that includes parameter estimates for disease-SNP haplotype frequencies and the degree of disease-SNP LD. Our method provides a new tool for detecting linkage and association and can be extended to study designs that include unaffected family members.
Introduction
Positional cloning is widely used for identification of genes involved in human diseases. To date, hundreds of disease genes have been identified solely on the basis of their chromosomal position (Botstein and Risch 2003); examples include hemochromatosis (Feder et al. 1996), inflammatory bowel disease (Hugot et al. 2001; Ogura et al. 2001), and lactose intolerance (Enattah et al. 2002). The first step in a traditional positional-cloning approach involves a genomewide linkage analysis performed on a collection of families with multiple affected individuals. Often, linkage analysis results in a candidate region of 10–20 Mb. To localize the susceptibility allele more precisely, disease-marker association analyses with additional genetic markers specific to the linked region can be performed. With recent progress on high-throughput SNP genotyping (Sachidanandam et al. 2001; Syvanen 2001; Oliphant et al. 2002; Olivier et al. 2002) and the HapMap project (International HapMap Consortium 2003), these follow-up association studies are becoming less expensive and now routinely include hundreds or thousands of markers.
Association analysis often compares marker-allele frequencies between unrelated case and control subjects. In this design, only a subset of the samples originally collected for linkage analysis can be reused. As an alternative, family-based association methods have been developed. Family-based association tests offer a compromise between traditional linkage studies and case-control association studies. The classic family-based transmission/disequilibrium test was proposed to test for association in the presence of linkage in family trios containing two parents and one affected offspring (Spielman et al. 1993). This approach has been extended to discordant sib pairs (Curtis 1997; Boehnke and Langefeld 1998), sibships with multiple affected and unaffected sibs (Spielman and Ewens 1998), general pedigrees (Martin et al. 2000), and quantitative traits (Allison 1997; Rabinowitz 1997; Abecasis et al. 2000a, 2000b).
A shortcoming of these family-based association methods is that, although they test for association, they cannot distinguish between potentially causal SNPs and other variants showing weaker association, except in the case of quantitative traits (Cardon and Abecasis 2000). Göring and Terwilliger (2000) proposed a unified theoretical model for linkage and linkage disequilibrium (LD) analysis through the use of a “pseudomarker” locus, but their approach cannot accommodate information contributed by flanking markers. Horikawa et al. (2000) suggested a modified association approach by examining how the evidence of linkage was partitioned in accordance with the genotype at the associated SNP, but they did not explore the properties of this approach. Li et al. (2004) explored the relationship between family-specific weights based on the affected individuals’ genotypes and family-specific nonparametric linkage (NPL) scores, but their method does not quantify the relationship between SNP alleles and the linkage signal. Sun et al. (2002) developed an approach that identifies SNPs whose genotypes can fully explain the observed linkage signal, but their test does not identify SNPs that play a partial role in explaining the linkage signal.
In this article, we describe a statistical framework that identifies candidate SNPs that can fully or partly explain the observed linkage signal, through joint modeling of linkage and association with the use of affected sib pairs (ASPs). Our method uses genotype information contributed by both the candidate SNP and the flanking markers. When a candidate SNP is identified as being able to account for linkage, our approach estimates the degree of LD between the candidate SNP and disease alleles. The estimate of disease-SNP LD quantifies the degree to which the linkage signal can be explained by the candidate SNP. We summarize the available information using a simple likelihood of the marker data conditional on the trait data, with disease penetrances and disease-SNP haplotype frequencies as parameters. We estimate model parameters by maximum likelihood and propose two likelihood-ratio tests to characterize the relationship between the candidate SNP and the putative disease locus. We calculate LD between disease and SNP alleles on the basis of haplotype-frequency estimates. Our method can identify both associated and potentially causal SNPs. Here, we focus on the ASP study design, but our method can be readily extended to accommodate unaffected individuals and other family structures, as well as unrelated individuals.
Methods
Assumptions and Definitions
We assume that there is a set of ASPs typed for a candidate SNP and M⩾0 flanking markers that can help evaluate evidence for linkage. We wish to evaluate evidence for association at the candidate SNP and to estimate the degree of LD with the unobserved disease locus. If there are multiple SNPs, we consider them one at a time as the candidate SNP. We allow LD between the candidate SNP and the unobserved disease alleles, but we assume linkage equilibrium between the flanking markers and the candidate SNP. Our goal is to quantify the relationship between the candidate SNPs and the unobserved disease alleles. Our method assumes that a single diallelic polymorphism directly contributes to risk in each linked region. We address the implications of multiple disease variants in the “Discussion” section.
Consider a diallelic disease locus with disease-predisposing allele D (with frequency pD) and wild-type allele d (with frequency pd=1-pD) and a nearby diallelic SNP with alleles A (with frequency pA) and a (with frequency pa=1-pA). Denote the four disease-SNP haplotypes as DA, Da, dA, and da (with frequencies pDA, pDa, pdA, and pda, respectively). We assume Hardy-Weinberg equilibrium in the general population for all markers, including the superlocus formed by the combination of the disease and SNP loci. Let fg=P(affected|g) be the penetrance for a given genotype 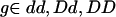 at the disease locus. By definition, the population prevalence of the disease, K, is equal to fddp2d+2fDdpdpD+fDDp2D, the attributable fraction equals K-fdd/K, and the genotype relative risk (GRR) equals fg/fdd.
at the disease locus. By definition, the population prevalence of the disease, K, is equal to fddp2d+2fDdpdpD+fDDp2D, the attributable fraction equals K-fdd/K, and the genotype relative risk (GRR) equals fg/fdd.
Let X=(X1,…,Xk,XSNP,Xk+1,…,XM) be the observed marker genotypes for the ASP, and let the probability of no change in identity-by-descent (IBD) status between consecutive markers be ψm=θ2m+(1-θm)2, where θm is the recombination fraction between markers m and m + 1 (1⩽m⩽M-1). Let Im, ISNP, and ID be the possibly unknown number of alleles shared IBD by an ASP at marker m, at the candidate SNP, and at the putative disease locus, respectively. For now, assume that there is no recombination between the candidate SNP and the disease locus, so that ISNP=ID. Denote disease locus IBD-sharing probabilities for an ASP by zi=P(ID=i|ASP), where i=0,1,2, and z=(z0,z1,z2). For ease of computation, we assume that there is no genetic interference, so that {Im} forms a hidden Markov chain.
Conditional Probability of Marker Data, Given an ASP
We wish to calculate P(X|ASP), the probability of the marker genotype data X for an ASP. By applying the forward and backward algorithms of Baum (1972), P(X|ASP) can be calculated as
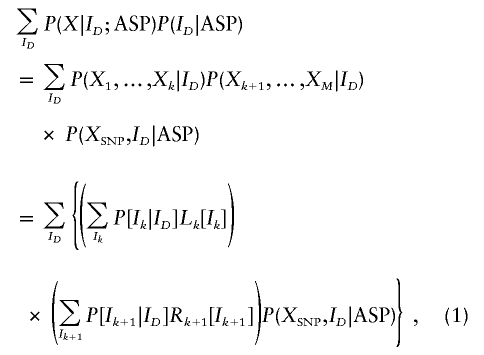 |
where k and k + 1 are the flanking markers on the left- and right-hand sides of the candidate SNP.
At an arbitrary marker m (1⩽m⩽M),
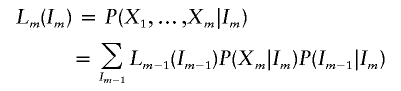 |
and
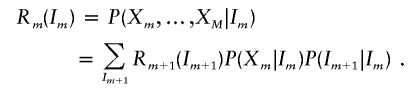 |
Special cases are L1(I1)=P(X1|I1) and RM(IM)=P(XM|IM). The conditional probabilities of the genotype data, given the number of alleles shared IBD by the sib pair at marker m, P(Xm|Im), are given in table 1 (Thompson 1975). IBD transition probabilities, P(Im+1|Im), are given in table 2 (Risch 1990). Recursive calculation of Lm(Im) and Rm(Im) allows the rapid evaluation of P(X|ASP) in a manner linear to the number of markers, M.
Table 1.
Conditional Probabilities P(Xm|Im) for Ordered Sib-Pair Genotypes Xm[Note]
|
P(Xm|Im) for |
|||
| Xm | Im=0 | Im=1 | Im=2 |
| (aa, aa) | p4a | p3a | p2a |
| (aa, ab) | 2p3apb | p2apb | 0 |
| (aa, bb) | p2ap2b | 0 | 0 |
| (aa, bc) | 2p2apbpc | 0 | 0 |
| (ab, ab) | 4p2ap2b | papb(pa+pb) | 2papb |
| (ab, ac) | 4p2apbpc | papbpc | 0 |
| (ab, cd) | 4papbpcpd | 0 | 0 |
Note.— a, b, c, and d are distinct alleles with frequencies pa, pb, pc, and pd, respectively.
Table 2.
IBD Transition Probabilities P(Im+1|Im) for a Sib Pair[Note]
|
P(Im+1|Im) for |
|||
| Im | Im+1=0 | Im+1=1 | Im+1=2 |
| 0 | ψ2m | 2ψm(1-ψm) | (1-ψm)2 |
| 1 | ψm(1-ψm) | ψ2m+(1-ψm)2 | ψm(1-ψm) |
| 2 | (1-ψm)2 | 2ψm(1-ψm) | ψ2m |
Note.— ψm=θ2m+(1-θm)2, where θm is the recombination fraction between loci m and m + 1.
To calculate P(XSNP,ID|ASP), let Gj denote the disease-SNP haplogenotype for sib j=1,2. Summing over all ordered haplogenotypes that are consistent with the observed SNP genotypes, we get
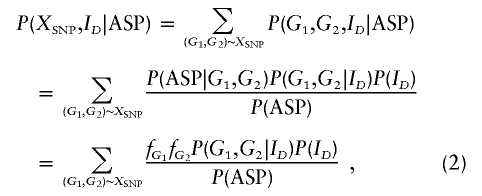 |
where P(G1,G2|ID) can be calculated from table 1 by regarding each haplogenotype as a genotype of the superlocus that has up to four alleles. For a sib pair, P(ID) takes the values (1/4, 1/2, 1/4). To illustrate how equation (2) is calculated, consider an ASP with SNP genotype A/A for the first sib and a/a for the second sib. The disease-SNP genotypes that are consistent with the observed SNP genotypes are G1 ∈ {DA/DA, DA/dA, dA/dA} and G2∈{Da/Da,Da/da,da/da}. If ID=0, then the numerator of equation (2) is
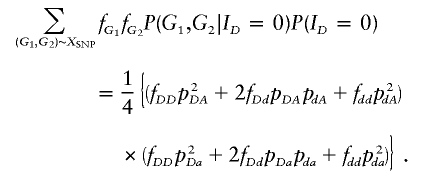 |
Similarly, we can obtain the probability of an ASP, where
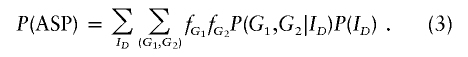 |
In the calculation of equations (2) and (3), we assume that the disease-affection statuses of the ASP are conditionally independent, given their genotypes at the disease locus. This is a common assumption for parametric likelihood calculation. It is exactly true when there are no other genetic or environmental risk factors shared among siblings, and it is a reasonable approximation when there are multiple disease-causing variants or shared environmental risk factors.
Our calculation allows analysis with missing genotypes. For example, to accommodate ASPs in which only one sib is genotyped at the candidate SNP, we sum over all possible SNP genotypes for the sib with missing genotype. Our calculation can also be readily extended to sib-pair samples that include unaffected individuals, by replacing fGj in equations (2) and (3) with 1-fGj for an unaffected individual.
The Relationship between Disease Locus and Candidate SNP
A useful measure of LD between two loci is the squared statistical correlation, defined as r2=(pDA-pDpA)2/[pD(1-pD)pA(1-pA)] in a sample of phased haplotypes. Multiplying r2 by the sample size yields the χ2 statistic for comparison of allele frequencies between cases and controls in a random population sample. r2 measures the degree of LD between the candidate SNP and the putative disease locus, as represented by the observed linkage signal, and can quantify the degree to which the linkage signal is explained by the candidate SNP. The candidate SNP and the putative disease locus can be in linkage equilibrium (r2=0), complete LD (r2=1), or partial LD (0<r2<1). Under linkage equilibrium, the candidate SNP is not associated with the putative disease locus and plays no causal role in the linkage signal. Under complete LD, the candidate SNP or a marker in complete LD with it can fully account for the linkage signal; we call this model “plausible causality.” Under partial LD, the candidate SNP partially accounts for the linkage signal.
We parameterize our models by using three penetrances, fdd, fDd, and fDD, in addition to (1) allele frequencies pD and pA for the linkage equilibrium model, (2) single-allele frequency p=pD=pA for the complete LD model, and (3) haplotype frequencies pDA, pDa, and pdA for the general model. Given only ASPs, each of these models is identifiable, except the linkage equilibrium model, in which parameters (fdd, fDd, fDD, pD, and pA) are not all identifiable, because the data contain information for only pA and z=(z0,z1,z2), corresponding to a total of 3 df, since z0+z1+z2=1. To achieve an identifiable model, note that, under linkage equilibrium, P(XSNP,ID|ASP)=P(XSNP|ID)P(ID|ASP) and that P(XSNP|ID) depends on only pA. Thus, the linkage equilibrium model can be reparameterized in terms of (z0,z1,pA), resulting in a likelihood similar to the traditional maximum LOD score (MLS) linkage test (Risch 1990) but with an additional parameter, pA. Here, we assume that the candidate SNP is completely linked to the putative disease locus. In theory, one could allow recombination between the candidate SNP and the putative disease locus as well. However, there is confounding between recombination and IBD sharing at the SNP (Risch 1990). A commonly used approach to avoid confounding in multipoint MLS calculation is to assume no recombination. IBD-sharing probabilities z=(z0,z1,z2) should satisfy the triangle constraint 0⩽z1⩽0.5 and 0⩽z0⩽0.5z1 (Holmans 1993).
The previous models assume that the candidate SNP is completely linked to the putative disease locus. If the candidate SNP is unlinked, then IBD-sharing probabilities at the SNP should be z=(1/4,1/2,1/4), and the only estimable parameter is pA. As such, the relationship between the candidate SNP and the putative disease locus falls into one of four models (table 3).
Table 3.
Relationship of Disease Locus and Candidate SNP
| Likelihood | Linkage | Disease-SNPRelationship | r2 | Parametersa | Constraintsa |
| LUL | Unlinked | Linkage equilibrium | 0 | pA | 0<pA<1 |
| LLE | Completely linked | Linkage equilibrium | 0 | z0,z1,pA | 0⩽z1⩽0.5; 0⩽z0⩽0.5z1; 0<pA<1 |
| LLD | Completely linked | Complete LD | 1 | fdd,fDd,fDD,p=pD=pA | 0⩽fdd,fDd,fDD⩽1; 0<p<1 |
| LGM | Completely linked | Any level of LD | [0, 1] | fdd,fDd,fDD,pDA,pDa,pdA | 0⩽fdd,fDd,fDD⩽1; 0⩽pDA,pDa,pdA⩽1; 0⩽pDA+pDa+pdA⩽1 |
Disease penetrances fdd, fDd, and fDD are assumed to be not all equal.
For a sample of independent ASPs, the retrospective likelihood of the data is
where the product is taken over all independent ASPs. Here, we chose to use a retrospective likelihood because the data are ascertained through the disease-affection statuses of the ASPs. The use of a retrospective likelihood can avoid the problem of ascertainment bias so that the parameter estimates are valid for the general population. To maximize equation (4), we use a simplex algorithm (Nelder and Mead 1965), an optimization method that does not require derivatives. Below, we represent the maximum of a particular likelihood subject to its parameter constraints by 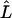 . In addition, we estimate r2 from frequency estimates of disease-SNP haplotype frequencies. The estimate of r2 is of particular interest in the case of partial disease-SNP LD; it reflects the degree to which a linkage result is explained by the candidate SNP.
. In addition, we estimate r2 from frequency estimates of disease-SNP haplotype frequencies. The estimate of r2 is of particular interest in the case of partial disease-SNP LD; it reflects the degree to which a linkage result is explained by the candidate SNP.
Likelihood-Ratio Statistic
Given different relationships between the candidate SNP and the disease locus, we can test for linkage, association, and plausible causality. We evaluate evidence for linkage with 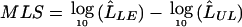 (see table 3 for explanations of LLE, LUL, LGM, and LLD). We evaluate evidence for association by testing whether the candidate SNP is in linkage equilibrium with the disease locus by use of the likelihood-ratio statistic
(see table 3 for explanations of LLE, LUL, LGM, and LLD). We evaluate evidence for association by testing whether the candidate SNP is in linkage equilibrium with the disease locus by use of the likelihood-ratio statistic 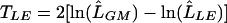 . Rejection of linkage equilibrium between the disease and SNP loci suggests the candidate SNP is associated with the disease locus and can account (in part) for the observed linkage signal. We examine plausible causality by testing whether the candidate SNP is in complete LD with the disease locus by use of the likelihood-ratio statistic
. Rejection of linkage equilibrium between the disease and SNP loci suggests the candidate SNP is associated with the disease locus and can account (in part) for the observed linkage signal. We examine plausible causality by testing whether the candidate SNP is in complete LD with the disease locus by use of the likelihood-ratio statistic 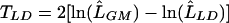 . Rejection of complete LD for an associated SNP suggests that the SNP cannot fully account for the observed linkage signal. If there is a single disease causal variant in the region, then it must be another SNP; otherwise, there might be other disease causal variants in the region.
. Rejection of complete LD for an associated SNP suggests that the SNP cannot fully account for the observed linkage signal. If there is a single disease causal variant in the region, then it must be another SNP; otherwise, there might be other disease causal variants in the region.
Empirical Null Distributions for Tests of Linkage Equilibrium and Complete LD
The asymptotic distributions of TLE and TLD under the null hypotheses might, in principle, be approximated by a mixture of χ2 distributions (Self and Liang 1987), but we have not derived the degrees of freedom and mixing parameters, because of the complexity of parameter constraints and boundaries. Alternatively, the significance of the tests can be assessed empirically by simulating marker genotypes under the null hypothesis and comparing the observed statistic with the simulated null distribution. One possibility would be to estimate disease-locus parameters and marker-allele frequencies under the null hypothesis and then to simulate genotypes for the candidate SNP and flanking markers conditional on the estimated parameters and observed phenotypes. In our preliminary investigations, this approach led to inflated type I error rates (data not shown), and we describe below, in detail, alternative strategies that may be theoretically less efficient but perform well in all the settings we examined.
For the TLE statistic, employed when the null hypothesis assumes linkage equilibrium between trait and marker loci, we sample SNP genotypes conditional on flanking-marker genotypes and estimated model parameters. In contrast, for the TLD statistic, employed when the null hypothesis assumes complete LD between the candidate SNP alleles and disease-susceptibility alleles, we sample flanking-marker genotypes conditional on the observed candidate SNP genotypes and estimated parameters.
For the linkage-equilibrium model, we use the observed data to obtain the SNP allele-frequency estimate 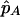 and the IBD-sharing probability estimates
and the IBD-sharing probability estimates 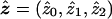 at the candidate SNP. To obtain a simulated sample under linkage equilibrium, for each ASP, we retain flanking-marker data and simulate the IBD configuration at the candidate SNP in accordance with
at the candidate SNP. To obtain a simulated sample under linkage equilibrium, for each ASP, we retain flanking-marker data and simulate the IBD configuration at the candidate SNP in accordance with
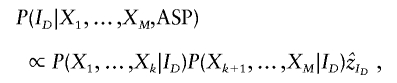 |
for ID = 0,1,2 and where P(X1,…,Xk|ID) and P(Xk+1,…,XM|ID) are the left- and right-chain probabilities calculated in equation (1). Given the IBD configuration at the candidate SNP, the ASP’s candidate-SNP genotypes can then be sampled on the basis of the estimated candidate-SNP allele frequency, 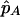 . Note that, when flanking-marker genotypes are not available, P(X1,…,Xk|ID) = P(Xk+1,…,XM|ID)=1, so that sampling is conditional on only the estimated parameter values and phenotypes. We obtain the null distribution of TLE by simulating a large number of replicates and calculating the statistic for each simulated data set.
. Note that, when flanking-marker genotypes are not available, P(X1,…,Xk|ID) = P(Xk+1,…,XM|ID)=1, so that sampling is conditional on only the estimated parameter values and phenotypes. We obtain the null distribution of TLE by simulating a large number of replicates and calculating the statistic for each simulated data set.
Our procedure for simulating the null distribution of TLD is different. Note that, if the candidate SNP is in complete LD with the disease locus alleles (or it is the disease locus itself), then candidate SNP genotypes should be sufficient to explain IBD sharing in the region. This observation has previously been used by Sun et al. (2002), who calibrated the significance of their test by sampling flanking-marker genotypes conditional on the observed SNP genotypes for each ASP. For each ASP, we leave the candidate SNP genotypes for the ASP unchanged from their observed values. Then, we sample an IBD configuration at the candidate SNP conditional on the observed SNP genotypes for the ASP and the estimated parameters 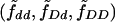 and
and 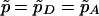 obtained from the complete LD model in accordance with
obtained from the complete LD model in accordance with
which can be obtained from equation (2). Finally, we sample genotypes for flanking markers, conditional on the IBD configuration at the candidate SNP. Specifically, we sample genotypes at marker k in accordance with transition probabilities P(Ik|ID) and the allele frequencies of marker k. The genotypes of marker k + 1 are sampled similarly but with transition probabilities P(Ik+1|ID). Moving left and right along the chromosome, we simulate flanking-marker genotypes on the basis of P(Im-1|Im) and P(Im+1|Im), respectively. We obtain the null distribution of TLD by simulating a large number of replicates and calculating the statistic for each simulated data set. This procedure for generating the empirical distribution of TLD has some limitations. In particular, when there are no flanking markers, our procedure leaves the original data unchanged, and so it is not possible to evaluate the significance of a particular value for TLD. Nevertheless, and as shown in the “Results” section, flanking markers provide most of the information required to distinguish between markers in complete LD and those in partial LD with the disease locus; thus, the distribution of TLD when there are no flanking markers is of little practical interest.
Simulations
We conducted a number of simulations to explore the properties of our proposed tests of no association and plausible causality and the resulting estimates of genetic model parameters. Table 4 describes the disease models we considered, which varied over a range of attributable fractions, disease-allele frequencies, GRRs, and sibling recurrence-risk ratio λs, defined as the recurrence risk for a sib of an affected individual divided by the population disease prevalence (Risch 1987). For all disease models, the population prevalence K of the disease was fixed at 2%.
Table 4.
Characteristics of Simulated Disease Models[Note]
|
λs=1.1 |
λs=1.3 |
|||||||||||
| Model | fdd | fDd | fDD | pD | AFa | GRR | fdd | fDd | fDD | pD | AFa | GRR |
| Dominant | .017 | .047 | .047 | .05 | .148 | 2.78 | .015 | .067 | .067 | .05 | .256 | 4.53 |
| .014 | .035 | .035 | .15 | .283 | 2.42 | .010 | .046 | .046 | .15 | .490 | 4.46 | |
| .010 | .029 | .029 | .30 | .478 | 2.79 | .003 | .036 | .036 | .30 | .828 | 10.41 | |
| Recessive | .019 | .019 | .261 | .05 | .030 | 13.48 | .019 | .019 | .438 | .05 | .052 | 23.12 |
| .018 | .018 | .094 | .15 | .085 | 5.15 | .017 | .017 | .149 | .15 | .148 | 8.72 | |
| .017 | .017 | .053 | .30 | .165 | 3.19 | .014 | .014 | .078 | .30 | .285 | 5.43 | |
| Additive | .017 | .046 | .075 | .05 | .145 | 2.70, 4.39 | .015 | .065 | .115 | .05 | .251 | 4.36, 7.71 |
| .015 | .032 | .050 | .15 | .266 | 2.21, 3.41 | .011 | .041 | .072 | .15 | .460 | 3.84, 6.68 | |
| .012 | .026 | .039 | .30 | .414 | 2.18, 3.36 | .006 | .030 | .053 | .30 | .717 | 5.23, 9.45 | |
Note.— Population disease prevalence K was fixed at 2%.
AF = attributable fraction.
In each model, we assumed the disease- and SNP-allele frequencies to be identical and, except where noted, used a map of 10 markers with eight equally frequent alleles (heterozygosity [H] of .875) evenly spaced at 11.16-cM intervals, corresponding to recombination fraction 0.10 under the no-interference map function of Haldane (1919). We centered the disease and SNP loci in the middle of the map and assumed zero recombination between them. We removed disease-locus genotypes prior to data analysis. For each of the disease models in table 4, we simulated 5,000 replicates of 500 ASPs under linkage equilibrium or complete LD to obtain null distributions and to determine critical values for each test. We simulated 2,000 replicates of 500 ASPs with various levels of disease-SNP LD to assess the empirical power of the corresponding tests. Here, we simulated the null distributions by using their generating values. In the “Discussion” section, we consider the impact of the use of null distributions estimated using our computationally intensive resampling procedures.
Results
Power to Reject Linkage Equilibrium (No Association)
Figure 1 displays the estimated power to reject the hypothesis of linkage equilibrium as a function of r2 for the disease models in table 4. As expected, the power of TLE increases as r2 increases, for all disease models, and it is at its maximum when r2=1. Figure 1 also shows that, for models with the same λs, it is relatively easier to detect association for a less-common disease allele than for a common one. More generally, we found that the power of the test is closely related to the GRR. We found that, for a fixed λs, lower disease-allele frequencies generally corresponded to a higher GRR for the disease models we considered.
Figure 1.

Power to reject linkage equilibrium (r2=0). Results are based on 2,000 replicates of 500 ASPs. All models have population disease prevalence K = 2% and sibling recurrence-risk ratio λs=1.1. Power was assessed at the 5% level.
These simulation results indicate that our method has good power to detect whether a candidate SNP is associated with the putative disease locus, even when the genetic effect is modest (λs=1.1). In each set of simulations, we found that the power of the test of linkage equilibrium to detect disease-SNP association does not depend on the magnitude of the observed linkage signal. Our results show that, given the same genetic effect as measured by λs, the power of TLE estimated using replicates with smaller MLS is nearly identical to the power of TLE estimated using replicates with larger MLS. For complex diseases with modest genetic effects, increased sharing near a disease locus can be overwhelmed by sampling variation in IBD estimates. Even when the evidence for linkage is absent, our method still yields a valid and useful test of association.
Power to Reject Complete LD (Plausible Causality)
Intuitively, one would expect the power to reject the hypothesis of complete LD to increase as r2 decreases from 1, with maximum power when r2=0. Figure 2 shows that the simulation results agree well with this expectation for our recessive and additive models but not for our dominant models. In our simulations, we found that the magnitude of the TLD statistic is highly correlated with the MLS when r2 is low, and the dependence becomes less strong as r2 increases. To illustrate this effect, we estimated power using those replicate data sets in which MLS is >1 or >2.5 for a dominant model with pD=pA=0.30 and λs=1.3. Figure 3 suggests that our ability to detect complete LD is dramatically enhanced as the MLS increases. For example, when r2=0, the power is 46% given no minimum MLS requirement but increases to 88% when MLS >1 and increases to nearly 100% when MLS >2.5.
Figure 2.

Power to reject complete LD (r2=1). Results are based on 2,000 replicates of 500 ASPs. All models have population disease prevalence K = 2% and sibling recurrence-risk ratio λs=1.3. Power was assessed at the 5% level.
Figure 3.

Impact of linkage evidence on test of complete LD. Results are based on 2,000 replicates of 500 ASPs under a dominant model with population disease prevalence K = 2%, allele frequency pD=pA=0.30, and sibling recurrence-risk ratio λs=1.3. Power was assessed at the 5% level.
In general, determining that a SNP is not in complete LD with the disease allele is more difficult than detecting whether it is associated with the disease allele, and it generally requires a larger sample. Our simulation results suggest that, at the same level of genetic effect as measured by λs, it is easier to evaluate whether a SNP might be in complete LD with the disease allele if the disease is recessive. This might be because the ASPs are more likely to share two alleles IBD under a recessive model than under a dominant model, and such excess IBD sharing provides more information on linkage. Since the power of TLD depends strongly on the magnitude of the observed linkage signal, this situation provides greater power to evaluate whether a candidate SNP is plausibly causal.
Parameter Estimates
Our method yields maximum-likelihood estimates of the disease-SNP haplotype frequencies directly, and, from those, we can calculate estimates of LD measures, such as r2. Mean parameter estimates and empirical SDs for the additive model, given 500 and 2,000 ASPs, are listed in table 5. The bias of parameter estimates is similar for dominant and recessive models (data not shown).
Table 5.
Results for Additive Model[Note]
|
Mean (Empirical SD) Value |
||||||||
|
λs=1.1 |
λs=1.3 |
|||||||
| pD=pA,r2, and ASPs | 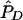 |
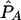 |
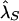 |
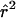 |
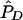 |
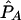 |
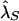 |
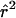 |
| pD=pA=.05: | ||||||||
| r2=.00: | ||||||||
| 500 | .28 (.17) | .09 (.04) | 1.08 (.07) | .28 (.36) | .23 (.13) | .08 (.06) | 1.22 (.10) | .17 (.29) |
| 2,000 | .25 (.12) | .07 (.02) | 1.08 (.04) | .16 (.23) | .20 (.08) | .07 (.03) | 1.24 (.06) | .13 (.25) |
| r2=.33: | ||||||||
| 500 | .22 (.22) | .07 (.02) | 1.11 (.06) | .31 (.33) | .11 (.15) | .07 (.02) | 1.28 (.12) | .40 (.28) |
| 2,000 | .16 (.09) | .06 (.02) | 1.10 (.04) | .35 (.29) | .05 (.06) | .06 (.02) | 1.27 (.06) | .38 (.22) |
| r2=.67: | ||||||||
| 500 | .09 (.13) | .06 (.02) | 1.13 (.07) | .55 (.37) | .05 (.03) | .06 (.02) | 1.30 (.09) | .56 (.32) |
| 2,000 | .06 (.03) | .06 (.01) | 1.11 (.03) | .65 (.30) | .04 (.02) | .06 (.01) | 1.29 (.05) | .56 (.28) |
| r2=1.00: | ||||||||
| 500 | .06 (.04) | .06 (.02) | 1.15 (.07) | .64 (.33) | .05 (.02) | .06 (.02) | 1.34 (.09) | .67 (.34) |
| 2,000 | .05 (.02) | .06 (.01) | 1.12 (.03) | .69 (.31) | .04 (.01) | .06 (.01) | 1.31 (.04) | .74 (.32) |
| pD=pA=.30: | ||||||||
| r2=.00: | ||||||||
| 500 | .48 (.22) | .34 (.08) | 1.06 (.05) | .40 (.33) | .47 (.18) | .34 (.07) | 1.17 (.11) | .20 (.24) |
| 2,000 | .45 (.12) | .34 (.03) | 1.03 (.03) | .33 (.28) | .44 (.07) | .33 (.03) | 1.16 (.05) | .08 (.11) |
| r2=.33: | ||||||||
| 500 | .33 (.15) | .33 (.06) | 1.08 (.06) | .48 (.31) | .31 (.15) | .32 (.06) | 1.21 (.10) | .44 (.27) |
| 2,000 | .31 (.10) | .32 (.03) | 1.05 (.03) | .46 (.27) | .30 (.11) | .32 (.03) | 1.20 (.05) | .38 (.18) |
| r2=.67: | ||||||||
| 500 | .28 (.12) | .33 (.06) | 1.12 (.06) | .56 (.30) | .28 (.10) | .32 (.06) | 1.27 (.10) | .65 (.27) |
| 2,000 | .27 (.08) | .32 (.03) | 1.09 (.03) | .64 (.24) | .29 (.06) | .32 (.03) | 1.26 (.05) | .68 (.18) |
| r2=1.00: | ||||||||
| 500 | .27 (.10) | .32 (.06) | 1.15 (.06) | .64 (.30) | .28 (.06) | .32 (.06) | 1.34 (.11) | .82 (.23) |
| 2,000 | .27 (.06) | .31 (.03) | 1.13 (.03) | .73 (.22) | .29 (.03) | .31 (.03) | 1.32 (.05) | .90 (.13) |
Note.— Results are based on 2,000 replicates of data sets simulated under an additive model with population disease prevalence K = 2%.
The bias of the maximum-likelihood allele-frequency estimates generally decreases as r2 increases to values close to 1, corresponding to greater information about the disease locus. Maximum-likelihood estimates are asymptotically unbiased under appropriate regularity conditions, notably when no null hypothesis parameter value is on the boundary of the parameter space. In our case, r2=1 results in two disease-SNP haplotypes with a frequency of 0, so our parameter estimates may be biased even in large samples. From table 5, we see that, when r2=1, pD can be underestimated and λs is slightly overestimated, with the bias decreasing as the sample size and magnitude of genetic effect increase.
Impact of Flanking Markers
To investigate the impact of flanking-marker data on our tests of disease-SNP LD, we simulated additional data sets using a map of 10 flanking markers, each with two equally frequent alleles (H=.50). We analyzed each of our data sets using 0, 2, 4, or all 10 flanking markers. Figure 4 suggests that having at least two flanking markers improves performance for both tests, but especially for the test of complete LD. Results based on 2, 4, or 10 flanking markers show only slight differences. Note that results are presented in figure 4B for the evaluation of the significance of TLD when there are no flanking markers. Although this is possible for a simulation study such as this (in which the true population parameter values are known and were used to simulate the null distribution of the statistic), it is not practical for analysis of real data, since our procedure for evaluating the empirical distribution of TLD requires information on flanking markers. In practice, this is not a serious limitation, because TLD has very low power when there are no flanking markers, and we recommend that it should be used only when at least two flanking markers are available.
Figure 4.

Impact of the number of flanking markers. Results are based on 2,000 replicates of 500 ASPs simulated under an additive model with population disease prevalence K = 2%, allele frequency pD=pA=0.15, and sibling recurrence-risk ratios λs=1.1 (A) and 1.3 (B). Data were simulated using 10 flanking markers, each with two equally frequent alleles. Intermarker recombination fraction is 0.1. Power was assessed at the 5% level.
We next assessed the impact of flanking-marker heterozygosity on power estimation by conducting additional simulations using two flanking markers, each with two (H=.50), four (H=.75), or eight (H=.875) equally frequent alleles. We found that, for the test of linkage equilibrium, the power for two and four equally frequent alleles is only slightly lower than for eight equally frequent alleles, but the difference in power is more pronounced for the test of complete LD (fig. 5). The differences between power for 2, 4, and 10 flanking markers when flanking-marker H was ⩾0.75 were modest (data not shown), suggesting that even just two highly polymorphic flanking markers may provide substantial power to detect disease-SNP LD. The utility of even two flanking markers is especially helpful when linkage data are not available and additional genotyping is required.
Figure 5.

Impact of heterozygosity of flanking markers. Results are based on 2,000 replicates of 500 ASPs simulated under an additive model with population disease prevalence K = 2%, allele frequency pD=pA=0.15, and sibling recurrence-risk ratios λs=1.1 (A) and 1.3 (B). Data were simulated using two flanking markers, each with two, four, or eight equally frequent alleles. Intermarker recombination fraction is 0.1. Power was assessed at the 5% level.
We also evaluated the impact of flanking-marker densities on power (fig. 6) by simulating data sets using a map of 10 flanking markers, each with four equally frequent alleles (H=.75). Clearly, denser markers give greater power, but the increment of power is not substantial for the range of densities we considered. In practice, having flanking markers within ∼10 cM should be enough for the initial evaluation of a candidate SNP.
Figure 6.

Impact of intermarker recombination of flanking markers. Results are based on 2,000 replicates of 500 ASPs simulated under an additive model with population disease prevalence K = 2%, allele frequency pD=pA=0.15, and sibling recurrence-risk ratios λs=1.1 (A) and 1.3 (B). Data were simulated using 10 flanking markers, each with four equally frequent alleles. Power was assessed at the 5% level.
Comparison of GIST and STEPC
Li et al. (2004) examined whether a SNP might account in part for an observed linkage signal by testing for a correlation between SNP genotypes and family-specific NPL scores. Their method is implemented in the software package GIST. Their simulations show that GIST is useful for identifying SNPs that are in LD with the disease locus and suggest that GIST could also identify associated SNPs in the absence of evidence for linkage. We compared our test of r2=0 with GIST (table 6). We assessed the significance of TLE at the 5% significance level by comparing the observed statistic with the empirical null distribution simulated in accordance with the resampling procedure described in the “Methods” section. Results are based on 500 replicates of 500 ASPs. For each replicate, the empirical null distribution of TLE was obtained by resampling 1,000 times. The results indicate that our test has greater power than GIST for the models we considered. For example, our test has 89% power to reject linkage equilibrium when r2=.67 for an additive model with pD=pA=0.15 and λs=1.1 at the 5% significance level, whereas GIST has 76% power.
Table 6.
Comparisons with GIST and STEPC[Note]
|
r2=0 |
r2=.33 |
r2=.67 |
r2=1 |
|||||||||||||
| λs, Test, and Model | TLE | TLD | GIST | STEPC | TLE | TLD | GIST | STEPC | TLE | TLD | GIST | STEPC | TLE | TLD | GIST | STEPC |
| 1.1: | ||||||||||||||||
| Linkage equilibrium: | ||||||||||||||||
| Dominant | 5 | … | 5 | … | 78 | … | 47 | … | 100 | … | 75 | … | 100 | … | 90 | … |
| Recessive | 6 | … | 5 | … | 100 | … | 95 | … | 100 | … | 100 | … | 100 | … | 100 | … |
| Additive | 5 | … | 5 | … | 54 | … | 46 | … | 89 | … | 76 | … | 98 | … | 87 | … |
| 1.3: | ||||||||||||||||
| Complete LD: | ||||||||||||||||
| Dominant | … | 64 | … | 66 | … | 55 | … | 22 | … | 32 | … | 15 | … | 5 | … | 6 |
| Recessive | … | 97 | … | 98 | … | 82 | … | 62 | … | 57 | … | 27 | … | 5 | … | 5 |
| Additive | … | 56 | … | 58 | … | 44 | … | 26 | … | 24 | … | 12 | … | 6 | … | 5 |
Note.— Results are based on 500 replicates of 500 ASPs. All models have population disease prevalence K = 2% and allele frequency pD=pA=0.15. Significance of TLE and TLD is assessed by comparing the observed statistics with the empirical null distributions generated in accordance with the resampling procedures described in the “Methods” section. Power is assessed at the 5% level.
Sun et al. (2002) examined whether a SNP could fully explain the observed linkage signal and implemented their approach in the software package STEPC. The method of Sun et al. (2002) is based on the observation that if a SNP is the only variant in the region that influences the trait, then conditional on the affected relatives’ SNP genotypes, there should be no increased IBD sharing in the region among affected individuals. We compared our test of r2=1 with STEPC (table 6) by using 500 replicates of 500 ASPs. Again, the significance of TLD was assessed empirically using the empirical null distribution simulation procedure for TLD described in the “Methods” section. The two tests have nearly identical performance when r2=0. However, the power of STEPC drops quickly as r2 increases, so that, when SNPs are in moderate LD with the putative disease locus, our method has better resolving power and thus should identify a smaller set of potential explanatory SNPs.
Discussion
We have developed a statistical framework that quantifies the relationship between SNP alleles and unobserved trait alleles through joint modeling of linkage and association by use of ASP data. We described a parametric likelihood of the marker genotypes conditional on the trait data under the assumption that there is a single disease-causing variant in the region. Our unified likelihood framework naturally leads to two tests: (1) a test of whether a candidate SNP is in linkage equilibrium with the putative disease locus and (2) a test of whether the candidate SNP is in complete LD with the putative disease locus. In the first case, the rejection of linkage equilibrium suggests that the candidate SNP is associated with the putative disease locus and that the candidate SNP or one in LD with it accounts, at least in part, for the observed linkage signal. In the second case, the rejection of complete LD indicates that the candidate SNP cannot fully account for the linkage signal. Our method also yields estimates of interesting genetic parameters, including the disease-locus and SNP-allele frequencies, the locus-specific risk ratio λs, and the degree of disease-SNP LD. Our method uses ASPs and does not require parental genotypes. This feature is important for late-onset diseases, for which parents may not be available to study.
Simulation studies show that our method has good power to detect disease-SNP association, even when the sibling recurrence-risk ratio is as low as 1.1. We compared our test of linkage equilibrium with GIST (Li et al. 2004) and found our test to be more powerful in the models that we considered. The increase of power may come from the fact that our method is model-based, whereas GIST is nonparametric and is based on model-free NPL scores. Like GIST, the power of our test of linkage equilibrium does not depend on the overall strength of the linkage signal. Evidence of disease-SNP LD from our test also reveals underlying linkage, which might be overwhelmed by sampling variation in IBD estimates. This feature makes our method a useful tool for detecting linkage as well as association. We also compared our test of complete LD with STEPC (Sun et al. 2002) and found that the two tests have similar performance under linkage equilibrium, but our test has greater power to distinguish those SNPs that are in strong but incomplete LD with the putative disease locus.
In contrast to previous approaches (Sun et al. 2002; Li et al. 2004), our method yields disease-SNP haplotype-frequency estimates in the general population without the requirement of a separate control sample. These quantities lead to the estimate of disease-SNP r2, a measure that can be used to quantify the degree to which a linkage signal is explained by a candidate SNP. Our estimate of r2 also provides information about the distance between the candidate SNP and the unobserved disease locus and helps refine the region in which further candidate SNPs should be examined. The disease-allele frequency estimate may be helpful to researchers in selecting additional nearby SNPs, by focusing on those with frequencies close to the predicted disease-allele frequency. This approach becomes increasingly useful as r2 increases.
Our tests benefit from genotype information on flanking markers, which are available in many gene mapping studies. Our results show that even two highly polymorphic flanking markers can provide nearly as much information as many more markers for this purpose. Compared with other family-based association tests and a previous joint model of linkage and association (Göring and Terwilliger 2000), our model has the attractive feature of incorporating flanking-marker information when it is available. An alternative joint model of linkage and association was developed independently by Cantor et al. (2005). In contrast to our approach, theirs includes recombination as an additional parameter and fixes disease model parameters, including penetrances and the disease-allele frequency. Like the approach of Göring and Terwilliger (2000), the model of Cantor et al. (2005) does not readily incorporate genotype information contributed by flanking markers.
Since the test of linkage equilibrium does not depend strongly on the overall evidence of linkage, flanking-marker heterozygosity has less impact on power for this test than for the test of complete LD, which is highly dependent on the strength of linkage evidence. We assume linkage equilibrium between the flanking markers and the candidate SNP in our likelihood calculation. For flanking markers that are close together, they may show strong evidence of LD. For such data, we recommend selecting a small number of flanking markers in linkage equilibrium so that the linkage equilibrium assumption is satisfied.
The likelihood framework described in this article is applicable to dichotomous traits only. However, many disease-related traits—for instance, blood pressure and cholesterol level—are continuous in nature. Dichotomization can result in a loss of power of the corresponding tests. Fulker et al. (1999) developed a method that tests for linkage while simultaneously modeling allelic association by use of the variance-components framework. Their method was further extended to general pedigrees (Abecasis et al. 2000a; Cardon and Abecasis 2000). Attenuation of evidence for linkage when association to SNP alleles is modeled suggests that the candidate SNP accounts for linkage and provides information about unobserved trait alleles. Although these methods provide bounds on disease-allele frequencies and disease-marker LD (Cardon and Abecasis 2000), they provide no direct estimate of these quantities. To address the same question for quantitative traits, we plan to develop a statistical framework that summarizes the available information by a retrospective likelihood, with the putative trait locus and the SNP haplotype frequencies as parameters.
In most gene mapping studies, the ASPs are likely to be selected from a sample that was originally collected for linkage analysis and for which flanking-marker genotypes are available. Our likelihood calculation naturally allows for missing genotypes at the candidate SNP. Given fixed genotyping resources, researchers may initially type many SNPs in only one sib per ASP. This approach halves the genotyping costs, and additional simulations suggested that even one sib per ASP can provide meaningful information on whether a candidate SNP is associated with the disease locus. This suggests that, for an initial screen of SNPs, it may be cost effective to genotype only one sib per ASP, with genotyping of the other sibs done when a candidate SNP shows at least suggestive evidence of association.
In our simulations, we assessed the empirical power of TLE and TLD by simulating the null distributions generated using the true parameter values. For real data sets, these values are unknown, and the null distributions must be generated using the estimated parameter values. We described simulation procedures to obtain the empirical null distributions. For the test of linkage equilibrium, we simulate the SNP genotypes for each ASP, conditional on their flanking-marker genotypes, and leave the flanking-marker genotypes unchanged from their observed values. For the test of complete LD, we leave the SNP genotypes unchanged and simulate the flanking-marker genotypes, conditional on the observed SNP genotypes, to remove excess sharing explained by the flanking markers. The difference in these two simulation procedures is due to the inherent difference of the two tests. Under linkage equilibrium, the candidate SNP provides no information on the unobserved disease locus, and the SNP genotypes can be sampled by gene-dropping simulations. In contrast, under complete LD, the candidate SNP is statistically identical to the unobserved disease locus, and the SNP genotypes need to be preserved to retain complete information on the unobserved disease locus.
We examined the performance of our null distribution simulation procedures and found that the simulated null distributions for both tests agree well with those generated in accordance with their true parameter values at all levels of disease-SNP LD and that both simulation procedures give correct type I errors (fig. 7). Evaluation of the significance of our tests by use of their simulated null distributions can be computationally intensive in practice, especially when the sample size is large. For an initial screen of SNPs, one might choose to evaluate the significance of the linkage equilibrium test empirically only if TLE⩾3.84 (at the 5% significance level), since χ21 approximates the lower bound for the asymptotic distribution of TLE, and one may test whether a candidate SNP is potentially causal only if it shows significant evidence of association.
Figure 7.

Comparison of empirical null distributions. Results are based on 2,000 replicates of 500 ASPs. All models have population disease prevalence K = 2% and allele frequency pD=pA=0.15. The solid line in each plot is the density of the empirical null distribution simulated using true parameter values of the disease model. Dashed lines are density plots of the empirical null distributions generated using the resampling procedures described in the “Methods” section. The empirical null distribution was generated for each level of disease-SNP LD.
We described our likelihood framework in the context of ASPs, but our method can be readily extended to other study designs. We are extending our method to sibships of arbitrary size and disease-phenotype configuration and to include unrelated affected or unaffected individuals. Unaffected individuals are more representative of the general population and may help to infer the underlying genetic model parameters. We expect the power of our test of linkage equilibrium to increase when unrelated unaffected individuals are added to the study.
Despite its flexibility, our method has limitations. Like all statistical methods, ours is unable to distinguish the true disease causal variant from an allele that is in complete LD with it. We assume that there is a single disease causal variant in the candidate region. However, many complex diseases are influenced by multiple genetic variants and are possibly the result of gene-gene and gene-environment interactions. Individual variants may be neither necessary nor sufficient to explain the effect of a single locus on disease susceptibility—for example, three independent SNPs, a frameshift variant, and two missense variants of NOD2 were identified as determining susceptibility to inflammatory bowel disease (Hugot et al. 2001). If only one causal variant is assumed, then we expect our model to indicate that each variant is associated with the underlying disease loci but none is causal (i.e., for all, 0<r2<1). For complex diseases that are influenced by multiple genetic variants, fitting the data under the assumption of a single-locus disease model is equivalent to testing the marginal effect of a specific locus. If the marginal effect of that locus is modest, then we may have limited power to detect association. For those cases, it might be desirable to develop a method that allows the analysis of two-locus or even multilocus disease models.
In summary, we have developed a unified likelihood framework to estimate useful genetic parameters and to test for both linkage equilibrium and complete LD between a candidate SNP and the putative disease locus. Results from these two tests complement each other in answering the question of whether the candidate SNP can account in part or in full for the observed linkage signal. An estimate of the disease-SNP LD provides a measure to quantify the degree of contribution of the candidate SNP to linkage evidence. Together with the disease locus and the SNP-allele frequency estimates, our method will be valuable in helping researchers to evaluate the role of a candidate SNP in disease susceptibility and to fine-map disease genes. We have implemented our method in a C++ program, which can be downloaded from the University of Michigan Center for Statistical Genetics Web site.
Acknowledgments
This research is supported by National Institutes of Health grants HG00376 (to M.B.) and HG02651 (to G.R.A.). M.L. is currently supported by a University of Michigan Rackham predoctoral fellowship. We gratefully thank two anonymous reviewers for their valuable comments.
Electronic-Database Information
The URL for data presented herein is as follows:
- University of Michigan Center for Statistical Genetics, http://csg.sph.umich.edu/
References
- Abecasis GR, Cardon LR, Cookson WO (2000a) A general test of association for quantitative traits in nuclear families. Am J Hum Genet 66:279–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abecasis GR, Cookson WO, Cardon LR (2000b) Pedigree tests of transmission disequilibrium. Eur J Hum Genet 8:545–551 10.1038/sj.ejhg.5200494 [DOI] [PubMed] [Google Scholar]
- Allison DB (1997) Transmission disequilibrium tests for quantitative traits. Am J Hum Genet 60:676–690 [PMC free article] [PubMed] [Google Scholar]
- Baum LE (1972) An inequality and associated maximization technique in statistical estimation for probabilistic functions of Markov processes. Inequalities 3:1–8 [Google Scholar]
- Boehnke M, Langefeld CD (1998) Genetic association mapping based on discordant sib pairs: the discordant-alleles test. Am J Hum Genet 62:950–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D, Risch N (2003) Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex diseases. Nat Genet Suppl 33:228–237 10.1038/ng1090 [DOI] [PubMed] [Google Scholar]
- Cantor RM, Chen GK, Pajukanta P, Lange K (2005) Association testing in a linked region using large pedigrees. Am J Hum Genet 76:538–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardon LR, Abecasis GR (2000) Some properties of a variance components model for fine-mapping quantitative trait loci. Behav Genet 30:235–243 10.1023/A:1001970425822 [DOI] [PubMed] [Google Scholar]
- Curtis D (1997) Use of siblings as controls in case-control association studies. Ann Hum Genet 61:319–333 10.1017/S000348009700626X [DOI] [PubMed] [Google Scholar]
- Enattah NS, Sahi T, Savilahti E, Terwilliger JD, Peltonen L, Jarvela I (2002) Identification of a variant associated with adult-type hypolactasia. Nat Genet 30:233–237 10.1038/ng826 [DOI] [PubMed] [Google Scholar]
- Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, et al (1996) A novel MHC class I–like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 13:399–408 10.1038/ng0896-399 [DOI] [PubMed] [Google Scholar]
- Fulker DW, Cherny SS, Sham PC, Hewitt JK (1999) Combined linkage and association sib-pair analysis for quantitative traits. Am J Hum Genet 64:259–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göring HHH, Terwilliger JD (2000) Linkage analysis in the presence of errors IV: joint pseudomarker analysis of linkage and/or linkage disequilibrium on a mixture of pedigrees and singletons when the mode of inheritance cannot be accurately specified. Am J Hum Genet 66:1310–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS (1919) The combination of linkage values and the calculation of distances between the loci of linked factors. J Genet 8:299–309 [Google Scholar]
- Holmans P (1993) Asymptotic properties of affected-sib-pair linkage analysis. Am J Hum Genet 52:362–374 [PMC free article] [PubMed] [Google Scholar]
- Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, et al (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26:163–175 10.1038/79876 [DOI] [PubMed] [Google Scholar]
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411:599–603 10.1038/35079107 [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium (2003) The International HapMap Project. Nature 426:789–796 10.1038/nature02168 [DOI] [PubMed] [Google Scholar]
- Li C, Scott LJ, Boehnke M (2004) Assessing whether an allele can account in part for a linkage signal: the genotype-IBD sharing test (GIST). Am J Hum Genet 74:418–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ER, Monks SA, Warren LL, Kaplan NL (2000) A test for linkage and association in general pedigrees: the pedigree disequilibrium test. Am J Hum Genet 67:146–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelder JA, Mead R (1965) A simplex method for function minimization. Comput J 7:308–313 [Google Scholar]
- Ogura Y, Bonen DK, Inohara N, Nicolae DN, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411:603–606 10.1038/35079114 [DOI] [PubMed] [Google Scholar]
- Oliphant A, Barker DL, Stuelpnagel JR, Chee MS (2002) BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotechniques Suppl:56–58, 60–61 [PubMed] [Google Scholar]
- Olivier M, Chuang LM, Chang MS, Chen YT, Pei D, Ranade K, de Witte A, Allen J, Tran N, Curb D, Pratt R, Neefs H, de Arruda Indig M, Law S, Neri B, Wang L, Cox DR (2002) High-throughput genotyping of single nucleotide polymorphisms using new biplex invader technology. Nucleic Acids Res 30:e53 10.1093/nar/gnf052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz D (1997) A transmission disequilibrium test for quantitative traits. Hum Hered 47:342–350 [DOI] [PubMed] [Google Scholar]
- Risch N (1987) Assessing the role of HLA-linked and unlinked determinants of disease. Am J Hum Genet 40:1–14 [PMC free article] [PubMed] [Google Scholar]
- ——— (1990) Linkage strategies for genetically complex traits. II. The power of affected relative pairs. Am J Hum Genet 46:229–241 [PMC free article] [PubMed] [Google Scholar]
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, et al (2001) A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409:928–933 10.1038/35057149 [DOI] [PubMed] [Google Scholar]
- Self SG, Liang KY (1987) Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under non-standard conditions. J Am Statist Assoc 82:605–610 [Google Scholar]
- Spielman RS, Ewens WJ (1998) A sibship test for linkage in the presence of association: the sib transmission/disequilibrium test. Am J Hum Genet 62:450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ (1993) Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52:506–516 [PMC free article] [PubMed] [Google Scholar]
- Sun L, Cox NJ, McPeak MS (2002) A statistical method for identification of polymorphisms that explain a linkage result. Am J Hum Genet 70:399–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvanen AC (2001) Assessing genetic variation: genotyping single nucleotide polymorphisms. Nat Rev Genet 2:930–942 10.1038/35103535 [DOI] [PubMed] [Google Scholar]
- Thompson EA (1975) The estimation of pairwise relationships. Ann Hum Genet 39:173–188 [DOI] [PubMed] [Google Scholar]