

Abstract
Photodynamic therapy (PDT) is a promising treatment that uses light to excite photosensitizers in target tissue, producing reactive oxygen species and localized cell death. It is recognized as a minimally invasive, clinically approved cancer therapy with additional preclinical applications in arthritis, atherosclerosis, and infection control. A hallmark of ideal PDT is delivering disease‐specific cytotoxicity while sparing healthy tissue. However, conventional photosensitizers often suffer from non‐specific photoactivation, causing off‐target toxicity. Activatable photosensitizers (aPS) have emerged as more precise alternatives, offering controlled activation. Unlike traditional photosensitizers, they remain inert and photoinactive during circulation and off‐target accumulation, minimizing collateral damage. These photosensitizers are designed to “turn on” in response to disease‐specific biostimuli, enhancing therapeutic selectivity and reducing off‐target effects. This review explores the principles of aPS, including quenching mechanisms stemming from activatable fluorescent probes and applied to activatable photosensitizers (RET, PeT, ICT, ACQ, AIE), as well as pathological biostimuli (pH, enzymes, redox conditions, cellular internalization), and bioresponsive constructs enabling quenching and activation. We also provide a critical assessment of unresolved challenges in aPS development, including limitations in targeting precision, selectivity under real‐world conditions, and potential solutions to persistent issues (dual‐lock, targeting moieties, biorthogonal chemistry and artificial receptors). Additionally, it provides an in‐depth discussion of essential research design considerations needed to develop translationally relevant aPS with improved therapeutic outcomes and specificity.
Keywords: photodynamic therapy, photosensitizer, fluorescence, quenching, activation
Basic concepts of designing activatable photosensitizers are established in this Review by closely examining quenching mechanisms and bioresponsive constructs essential for their function. New strategies developed in the last five years that address the shortcomings of conventional activatable photosensitizers and which enhance selectivity are also summarized.

1. Introduction
1.1. Principles of Photodynamic Therapy
Photodynamic therapy (PDT) is a minimally invasive treatment modality that induces localized cell death through the interaction of light and photoactive agents called photosensitizers, yielding focal generation of reactive oxygen species (ROS). These ROS are products of electronic transitions incurred by photosensitizers when irradiated with light of an appropriate wavelength and intensity (Figure 1). Upon light absorption, photosensitizers are excited from the ground state (S0) to a short‐lived excited state (Sn) that decays rapidly through internal conversion to an excited singlet state (S1). A portion of these excited molecules undergo intersystem crossing (ISC), forming a more stable, long‐lived triplet state (T1). From T1, photosensitizers can follow two pathways to produce ROS: Type I or Type II reactions. In the Type I pathway, electron transfer occurs between the photosensitizers and surrounding biological substrates, generating free radicals. [1] These free radicals can further react with oxygen, producing superoxide anions (O2⋅−), which can be further converted to hydrogen peroxide (H2O2) or hydroxyl radicals (⋅OH). In Type II reactions, T1 photosensitizers interact directly with molecular oxygen to generate singlet oxygen (1O2). The produced ROS damages surrounding cellular substrates, instigating diverse cell death pathways depending on the photosensitizer cellular localization. For instance, plasma membrane accumulation primarily leads to necrosis, but can also induce ferroptosis and pyroptosis; [2] mitochondrial targeting can induce apoptosis and pyroptosis;[ 2 , 3 ] lysosomal localization may result in necrosis,apoptosis, ferroptosis, or autophagy;[ 2 , 4 , 5 ] Golgi apparatustargeting can trigger pyroptosis; [6] and endoplasmic reticulumlocalization typically triggers apoptotic cell death. [7]
Figure 1.
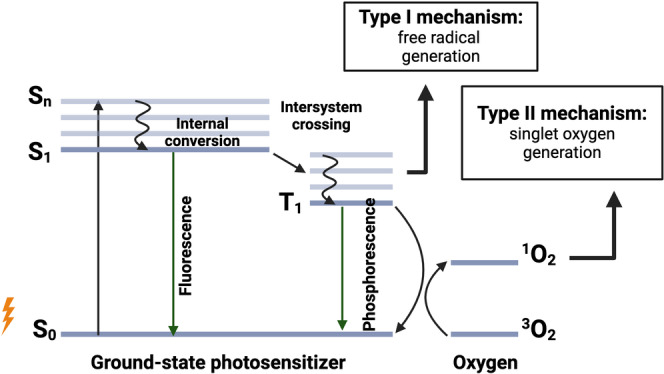
Jablonski diagram depicting photosensitizer electronic transitions that yield ROS during PDT.
Due to the short lifetimes and diffusion distances of 1O2 (~3.5 μs,<400 nm), [8] O2⋅− (~4 μs, <500 nm)[ 8 , 9 ] and ⋅OH (~8.7 ns, ~93 Å), [10] this PDT cellular damage/death is confined to areas of photosensitizer accumulation and light irradiation. This controlled, focal cell death lends PDT the lucrative therapeutic advantage of targeting irradiated pathological tissue while sparing healthy tissue. This capability is furthered by the propensity of the most prominent class of photosensitizers, porphyrins, to accumulate in malignant tissue faster and to greater extents than in healthy tissue. [11] As such, PDT has gained attention primarily as a targeted cancer therapy. [12] Its therapeutic utility has also extended to vascular (atherosclerosis, [13] age‐related macular degeneration, [14] transient blood–brain barrier opening) [15] and anti‐microbial applications. [16]
PDT bears a plethora of additional advantages compared to conventional therapies. Advances in laser light technology allow PDT to be administered in a minimally invasive manner that can replace or serve as an adjunct to surgery.[ 17 , 18 ] It is non‐ionizing and restricted to tissue areas exposed to light, leading to fewer concerns of systemic and off‐target toxicity versus radiation and chemotherapy. [19] PDT also exhibits immunomodulatory properties and mechanisms of action independent of variably expressed mutations or pathologic pathways susceptible to treatment resistance.[ 20 , 21 ] It can thus be easily integrated with conventional treatment modalities to overcome antibiotic, chemotherapy and radiation treatment resistance.[ 22 , 23 , 24 , 25 , 26 ] These collective unique advantages of PDT have thus catapulted preclinical and clinical explorations to maximize its remarkable therapeutic potential.
1.2. Evolution of Photosensitizers and Clinical Challenges
As a key element of PDT, photosensitizers must meet several requirements to ensure clinical safety and efficacy:[ 27 , 28 , 29 ] 1) systemically non‐toxic with low photoactivity in the absence of intended light exposure, 2) high ROS quantum yields upon light exposure, 3) preferential accumulation in pathological vs normal tissue, and 4) high molar extinction/absorbance at near‐infrared (NIR) wavelengths (700–1000 nm), situated ideally in the optical window corresponding to reduced light interference with biological substrates and thus deeper light penetration through tissue. The most clinically utilized class of photosensitizers are porphyrins (Table 1). Porphyrins are macrocyclic organic molecules with NIR Q‐bands. They are inherently benign unless exposed to light; a key to their safety profile, mitigating concerns of off‐target photosensitizer accumulation and toxicity in non‐irradiated tissue. The earliest porphyrin sensitizers comprised mixtures of porphyrin oligomers (e.g., Photofrin®). Although these photosensitizers were effective, they faced challenges of low extinction red (630 nm) Q‐bands, and skin phototoxicity due to prolonged skin retention (up to two months) and activation by ambient light.[ 29 , 30 ] Synthetic sister‐variants of porphyrins like chlorins and bacteriochlorins photosensitizers thus emerged, which displayed longer excitation wavelengths, higher extinction Q‐bands (650–800 nm, extinction coefficient: 1.4–4×104 M−1 cm−1), reduced skin retention (1–2 days), higher target tissue specificity and faster clearance.[ 31 , 32 ] Despite this evolution in clinical photosensitizer design, it is apparent from Table 1 that off‐target phototoxicity, and particularly skin photosensitivity, continues to remain a common PDT complication.
Table 1.
Clinically approved photosensitizers. Approved treatment indications, clinical side effects and preclinical biodistribution are presented.
|
Photosensitizer |
Approved treatment(s) |
Clinical off‐target effects |
Preclinical Biodistribution |
||||
|---|---|---|---|---|---|---|---|
|
Clinical indication |
PDT/ Controls |
Adverse events |
Model |
Dose/sacrificed time |
Biodistribution |
||
|
Porfimer sodium (Photofrin®) |
Esophageal, lung, endobronchial cancers |
Phase III: advanced esophageal cancer [30] |
PDT (300 J/cm): 110 Laser therapy: 108 |
Skin photosensitivity: 100 % Sun burn: 19 % |
Small cell lung carcinoma POVD mice [41] |
20 mg/kg, IV [a] 24 hours |
liver≫kidney>spleen>tumour >lung≈heart≈skin>blood ≈brain≈muscle |
|
Hematoporphyrin derivative (HiPorfin®) |
Cervical cancer, vaginal high‐grade squamous intra‐ epithelial lesions |
Phase I/II: advanced obstructive esophageal cancer [42] |
PDT (200–300 J/cm): 32 |
Acid reflux: 38 % Pain: 28 % Bleeding: 6 % Photosensitivity: 3 % |
Retinoblastoma athymic nude mice [43] |
20 mg/kg, IP [b] 24 hours |
liver≫kidney≫spleen>skin >lung>tumour>blood>brain |
|
5‐ALA (Levulan®) |
Mild to moderate actinic keratosis |
Phase III: Actinic Keratoses [44] |
PDT (10 J/cm2): 181 Vehicle PDT: 62 |
Erythema: 76 % Edema: 38 % |
Healthy rats [45] |
200 mg/kg. IV [a] 24 hours |
liver≫kidney>small bowel >blood≈skin≈muscle≈fat> lung≈bladder≈spleen≈colon ≈stomach>heart>brain |
|
Padoporfin (Tookad®) |
Locally advanced prostate cancer |
Phase II: Localized prostate cancer [46] |
PDT (200–300 J/cm): 85 |
Dysuria, urinary tractinfection: 48 % Constipation: 13 % Perineal pain: 12 % Inflammation and bleeding: 11 % |
Melanoma M2R mice [47] |
6 mg/kg, IV [a] 15 minutes |
liver≫kidney≈blood>lung ≈spleen≈tumour≈skin≈intestine ≈heart |
|
Methyl Aminolevulinate (Metvix®/ Metvixia®) |
Non‐hyperkeratotic actinic keratosis, basal cell carcinoma |
Phase III: Actinic Keratoses [48] |
PDT (75 J/cm2): 52 Surgery: 49 |
Skin burning sensation: 31 % Erythema: 13 % Pain: 13 % |
NA. Studies on topical cream. |
||
|
Temoporfin (Foscan®) |
Head & neck cancer |
Phase II: head & neck squamous cell carcinoma [49] |
PDT (20 J/cm2): 128 |
Pain: 21 % Skin photosensitivity: 19 % Edema: 11 % |
Mesothelioma H‐MESO1 mice [50] |
0.3 mg/kg, IV [a] 24 hours |
liver>lung≈kidney>heart ≈blood>tumour>skin>muscle |
|
Talaporfin (Laserphyrin®) |
Early centrally located lung cancer |
Phase II: esophageal cancer [51] |
PDT (100 J/cm2): 52 |
Pain: 44 % Nausea: 39 % Esophageal stenosis: 24 % |
Breast cancer mammary carcinoma mice [52] |
5 mg/kg, IV [a] 24 hours |
liver>adrenal gland>kidney >spleen≈tumour>skin≈intestine ≈lung>stomach≈heart>blood ≈brain |
|
Verteporfin (Visudyne®) |
Age‐related macular degeneration |
Phase III: Age‐related macular degeneration [53] |
PDT (50 J/cm2): 402 Placebo: 207 |
Visual disturbance: 18 % Injection adverse events: 13 % Photosensitivity: 3 % |
Mastocytoma P815 mice [54] |
80 μg, IV [a] 24 hours |
liver≫spleen≫intestines>kidney >skin>lung>heart≈blood >tumour≈muscle |
|
Cetuximab‐IR700 (Akalux®) |
Head & neck cancer |
Phase I&II: Recurrent head & neck squamous carcinoma [55] |
PDT (100 J/cm or 50 J/cm2): 39 |
Pain: 41 % Edema: 38 % Skin irritation: 26 % Dysphagia: 23 % Constipation: 20 % |
EGFR‐expressing A431 carcinoma mice [56] |
5 μg, IV [a] 24 hours |
tumour≫blood>spleen>liver >lung>heart>kidney≫intestine >muscle |
[a] Intravenous injection (IV). [b] Intraperitoneal injection (IP)
Localized delivery of photosensitizers (e.g., intratumoural injection) can reduce skin phototoxicity compared to intravenous injection while preserving PDT response. [33] However, it is not always feasible, nor does it necessarily overcome off‐target phototoxicity within tumour‐adjacent normal tissue that remain inside light irradiation zones. As aforementioned, many photosensitizers naturally accumulate to higher degrees in pathological versus healthy tissue. As such, in combination with careful treatment planning (dosimetry, photosensitizer‐light interval modulation, image‐guided light delivery) this specificity should theoretically spare disease‐adjacent healthy tissue. However, in reality, this may not be the case, particularly when treating vital organs and sensitive tissue volumes or those with baseline high healthy tissue photosensitizer accumulation even with higher accumulation in adjacent diseased tissue, this healthy tissue would remain susceptible to PDT.[ 34 , 35 ] For example, a phase II/III clinical trial of Photofrin‐PDT for malignant pleural mesothelioma reported frequent and serious cardiac and respiratory adverse events, including mortality 3/73 (4 %) caused by stroke and myocardial infarction with cardiac tamponade, atrial fibrillation 21/73 (28 %), respiratory failure 14/73 (19 %), deep venous thrombosis 17/73 (23 %), and persistent air leak 17/73 (23 %), likely due to off‐target Photofrin®‐PDT sensitivity.[ 34 , 35 , 36 , 37 ]
This tumour‐adjacent phototoxicity was also evidenced in a phase I trial testing Visudyne®‐PDT in vertebral bone metastases, where 10 % of the 30 patients experienced vertebral collapse after image‐guided PDT treatment. [38] This was despite using a sensitizer‐light interval (15 minutes) designed to maximize tumour‐to‐neural uptake in a rat bony metastasis model, achieving 5.5‐fold higher Visudyne accumulation in vertebrate metastasis than spinal cord.[ 39 , 40 ] However, this interval was suboptimal for tumour‐to‐adjacent vertebrate tissue selectivity, yielding a 2‐fold difference at 15 minutes, compared to a more optimal 5‐fold difference at 3 hours. This created room for photoactivation of Visudyne in tumour‐adjacent vertebral tissue, and off‐target PDT effects. These ongoing challenges of skin phototoxicity, target‐adjacent healthy tissue photosensitizer accumulation and associated off‐target photosensitivity require innovations in photosensitizer design that improve PDT selectivity.
1.3. Increasing PDT Selectivity via Activatable Photosensitizers
Two broad approaches have been explored to increase photosensitizer, and in turn PDT, selectivity. The first included association of ligands for biomarkers upregulated in diseased versus healthy cells with photosensitizers, creating targeted photosensitizers designed to be preferentially uptaken by diseased tissue. Photosensitizers can be conjugated directly to these targeting ligands or can be encapsulated within nanocarriers surface‐decorated with targeting ligands. An example is Akalux®, a clinically approved targeted photosensitizer in which the photosensitizer IR700 is conjugated to monoclonal antibodies that target human epidermal growth factor receptor (HER), commonly overexpressed in cancer. In mouse models, the HER‐targeted IR700 demonstrated three times higher fluorescence signal in HER1‐positive A431 tumours than HER1‐negative 3T3‐HER2 tumours 24 hours post intravenous injection. [57] Beyond antibodies, proteins, sugars and peptides have also been explored to generate targeted photosensitizers as has been reviewed cogently by several authors.[ 12 , 28 , 58 ]
A second approach to improving PDT selectivity is the construction of aPS. Unlike conventional “always on” photosensitizers or targeted photosensitizers, the photoreactivity and ROS generation abilities of aPS are turned off, i.e. quenched under dark or light‐exposed environments alike. As such, these aPS remain “locked” or inactive when circulating and distributing to healthy tissue, even under light exposure, thereby minimizing off‐target PDT effects. They are only rendered photosensitive or “switched‐on” after interacting with disease‐specific biomarkers and stimuli, such as abnormal pH, redox environment, or enzymes and nucleic acids that are elevated in pathological tissues.[ 59 , 60 , 61 ] In this sense, aPS are akin to pro‐drugs. Once “switched‐on” or “unlocked” by pathological biostimuli, the aPS can be excited by light to trigger PDT reactions specifically in diseased environments where the aPS is activated (a feasibility due to the short diffusion distances of ROS generated).
The term ‘activatable photosensitizer’ has been broadly applied in the literature with varying contextual definitions, encompassing: 1) carriers that release photosensitizing agents upon external stimulation, 2) substances that mediate drug release from supramolecular constructs via photodynamic or photothermal pathways, and 3) conventional photosensitizers that are occasionally misclassified as activatable compounds, whereby the term “activation” is misused to describe their inherent light‐activatable ROS generation ability. In this review, we define ‘activable photosensitizer’ as a photosensitizer construct with quenched photosensitivity that requires disease‐specific biostimuli to “activate” or unquench the photosensitizer photosensitivity prior to light irradiation. To this end, aPS contain three features: 1) photosensitizer, 2) a moiety that quenches the photosensitizer photosensitivity, and 3) a photosensitizer structural modification, linker or supramolecular assembly that enables this quenching and is labile to pathological biostimuli.
This review will summarize fundamentals of aPS, including quenching mechanisms, molecular and supramolecular aPS designs, common pathological biostimuli for aPS activation and associated advantages and ongoing challenges. It will also provide an overview of the progress made over the last five years in aPS construction, advanced activation strategies, expanded applications, guidance for future directions and study design considerations. While previous reviews have provided comprehensive overviews of design strategies and their importance,[ 62 , 63 , 64 , 65 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 ] our focus is to complement these works by delving into the mechanistic details of how tumour microenvironment (TME) characteristics influence selectivity and by identifying opportunities for further innovations to enhance therapeutic precision and minimize off‐target effects.
2. Mechanisms of Activation
aPS are engineered to remain inactive in off‐target tissue and become activated only in the presence of specific stimuli in target tissue. This specificity is dependent on several important features. The first is high and stable quenching of aPS in its pro‐drug form, rendering an “off” state with low off‐target baseline photoreactivity. Quenching requires alteration of the photosensitizer′s energy dissipation pathways by inhibiting light absorption at its excitation wavelength or shifting its excitation wavelength. The second attribute is the use of aPS designs (e.g., structural modifications, linker‐based conjugations or supramolecular assemblies) that not only enables such quenching, but also robustly maintains it in off‐target milieu (e.g., amongst serum proteins and proteases) to minimize unintended adverse photosensitization. This construct must contain a bioresponsive element that enables precise and strong activation by a disease‐specific stimulus that restores the photosensitizer photoreactivity. This not only requires rational integration of bioresponsive groups with the photosensitizer, but also the selection of stimuli that are abundantly and specifically expressed in diseased cells or pathological tissue environments. This section overviews these quenching mechanisms, aPS constructs and activating biostimuli that facilitate precise control of aPS activation and associated PDT selectivity.
2.1. Photosensitizer Quenching Mechanisms
Mechanisms that govern photosensitizer quenching in aPS often stem from activatable fluorescent probe design principles. When a fluorescent probe is excited by light of an appropriate wavelength, it transitions from S0 to S1 and typically releases energy as fluorescence when returning to the ground state. [74] Mechanisms such as Förster resonance energy transfer (FRET), photoinduced electron transfer (PeT), intramolecular charge transfer (ICT), aggregation‐caused quenching (ACQ), and aggregation‐induced emission (AIE) are employed in activatable fluorescent probe to interfere with these light absorption or fluorescence decay pathways, keeping the fluorophore in an inactive state.[ 75 , 76 , 77 , 78 , 79 , 80 ] The quenching mechanisms underlying activatable fluorescent probe can and have been applied successfully to also interfere with photosensitizer ISC from S1 to T1, effectively silencing the photosensitizer fluorescence and ROS generation until it encounters a disease‐relevant stimulus.[ 81 , 82 ] Examples of aPS quenching mechanisms are provided in Figure 2 and will be overviewed in this section.
Figure 2.

Schematic overview of basic photophysical mechanisms underlying aPS quenching and activation.
2.1.1. Förster Resonance Energy Transfer (FRET)
FRET, named after Theodor Förster who developed its theoretical framework, [83] is the best‐known and most widely applied energy transfer mechanism in activatable fluorescence and PDT. FRET involves non‐radiative energy transfer from an excited donor to a ground‐state acceptor via long‐range dipole‐dipole interactions. It was first prominently utilized as a quenching mechanism to create activatable fluorescent probes by inhibiting energy release in the form of fluorescence from the excited donor.[ 84 , 85 ] FRET can also be employed in aPS design, where the excited photosensitizer transfers energy to a quencher instead of undergoing ISC to the T1, thereby preventing ROS generation.
The efficiency of FRET is critically dependent on three factors: the spectral overlap between the donor and acceptor molecules, their physical separation distance, and dipole orientation. [86] Specifically, efficient FRET within aPS requires high overlap between the emission spectrum of the donor and the absorption spectrum of the acceptor (Figure 2a). To this end, it is possible for photosensitizers with overlapping absorbance and emission spectra to self‐quench in a double‐photosensitizer construct (Homo‐FRET). However, most FRET‐based aPS designs make use of distinct quenchers and acceptors due to higher 1O2 quenching. [87] To maximize energy transfer efficiency, it is vital that the donor‐acceptor distance lie within a 10–100 Å. [88] This structural configuration can be commonly achieved by connecting the photosensitizer and quencher via a peptide or nucleic acid linker (Figure 2a).
The development of aPS began with the concept of ‘photosensitizing beacons’, which our group introduced to the PDT field by adapting the well‐established principle of molecular beacons that utilize FRET to control fluorescence emission in response to specific molecular markers. [89] In this work, we designed a system where a photosensitizer (FRET donor) was linked to a quencher (FRET acceptor) via a short peptide sequence specific to a protease overexpressed in cancer cells. The proximity of the photosensitizer and quencher, maintained by the peptide linker, enabled efficient FRET from the excited photosensitizer to the quencher. This transfer inhibited photosensitizer ISC into the T1 state, stopping ROS generating capability and thus suppressing the photosensitizer′s photodynamic activity. This activity was restored with enzymatic cleavage of the peptide sequence.
2.1.2. Photoinduced Electron Transfer (PeT)
PeT, like FRET, is an aPS quenching mechanism rooted in electron transfer between an acceptor and donor, and which also stems from activatable fluorescence probe design. Unlike FRET, where the excited fluorophore is always the donor, excited PeT fluorophores can act as either electron acceptors (a‐PeT) or electron donors (d‐PeT).[ 90 , 91 ] In a‐PeT, an electron from the highest occupied molecular orbital (HOMO) of a neighboring electron‐rich recognition moiety is transferred to the HOMO of the excited fluorophore, filling the vacancy created from fluorophore excitation. This prevents radiative decay (and therefore quenches the fluorescence) of the fluorophore′s excited electron back to ground state. In d‐PeT, the fluorophore′s excited electron in its lowest unoccupied molecular orbital (LUMO) transfers via non‐radiative decay to the slightly lower energy LUMO of a neighboring electron‐poor recognition moiety, leading to a charge transfer state, and quenched fluorescence (Figure 2b).
aPS utilizing the PeT mechanism require modification of the photosensitizer′s core structure to incorporate a PeT recognition moiety that acts as an electron donor or acceptor in healthy tissue environments, quenching photosensitizer fluorescence and photodynamic activity. This activity is restored when the recognition moiety encounters biostimuli that changes its electronic properties, preventing PeT. For instance, an amine group serves as a d‐PeT quencher in neutral pH environments. Under the acidic conditions of the TME, protonation of the amine group inhibits electron donation, thereby halting the PeT process and restoring the photosensitizer′s photodynamic activity.[ 92 , 93 , 94 ]
2.1.3. Intramolecular Charge Transfer (ICT)
Molecules capable of ICT consist of an electron donor and an electron acceptor connected by a π‐conjugated bridge that facilitates electron communication between the pair. This structure has been employed in fluorescent sensor designs because, upon excitation, charge redistribution between the electron donor and acceptor occurs via ICT, leading to a shift in the fluorescence spectrum toward longer wavelengths and providing a large Stokes shift. In these push‐pull systems, the charge transfer process is often promoted by structural rearrangements such as twisting, resulting in phenomena like twisted intramolecular charge transfer (TICT), which has applications in the field of aggregation‐induced emission (AIE) fluorescence probe. Although establishing a direct correlation between ICT and the enhancement of photodynamic activity presents a challenge, recent investigations have demonstrated that photoinduced charge separation‐charge recombination in orthogonal donor–acceptor organic systems can augment ISC efficiency in the absence of heavy‐atom effects. [95]
In cases where an ICT molecule functions as a photosensitizer, the design of ICT aPSs involves capping the electron donor or acceptor with a bioreactive protective group to suppress the ICT process (Figure 2c). Upon exposure to specific biostimuli, the protective group is removed, reactivating the ICT process and restoring the photosensitizer′s activity. For example, Zhai et al. developed an aPS comprising selenium‐substituted dicyanomethylene‐4H‐chromene as the electron acceptor and iodine‐substituted phenolate as the electron donor. [96] Caging the phenolate group with a bioresponsive moiety (e.g., alkaline phosphatase or peroxynitrite‐responsive groups) disrupts the ICT process and significantly inhibits red light absorption, leading to suppressed photoreactivity compared to unincarcerated aPS. Upon reaction with biostimuli, the protective group is removed, uncaging the phenolate, restoring ICT, and rapidly recovering photoreactivity at the wavelength of PDT light.
2.1.4. Aggregation‐Caused Quenching (ACQ)
ACQ is a photophysical property displayed by most photosensitizers and is one of the most straightforward mechanisms that can be leveraged for activatable PDT. Many photosensitizers comprise hydrophobic planar ring structures that induce photosensitizer aggregation in hydrophilic aqueous environments. [97] In their aggregated state, photosensitizer molecules interact with one another and form S0 complexes, resulting in altered fluorescence and reduced 1O2 quantum yields. This aggregation can be purposefully generated by assembling large numbers of photosensitizer in close proximity within nanostructures. [98] When the nanoparticles are intact, photoreactivity and 1O2 generation are quenched (Figure 2d). Upon interacting with a particular biomarker or process (e.g., cell‐surface receptors or endolysosomal uptake) the nanoparticles disassemble or degrade, releasing photosensitizers from aggregate form and restoring their fluorescence and PDT activity. For example, Li et al. conjugated phthalocyanine with 4‐sulfonatophenoxyl to form a self‐assembled nanoparticle that quenched phthalocyanine. [99] Specific binding of albumin and 4‐sulfonatophenoxyl would disassemble the nanoparticle to unquench phthalocyanine.
2.1.5. Aggregation‐Induced Emission (AIE)
AIE is an additional proximity‐based aPS mechanism. AIE‐based systems generally consist of large hydrophobic molecules with π‐conjugated structures. In their monomeric state, these molecules experience significant intramolecular motion, such as rotation and vibration, which leads to non‐radiative energy dissipation and inhibits efficient ROS generation under light exposure (Figure 2e). When aggregated, however, the close molecular stacking restricts these motions, enabling radiative relaxation to S0 or ISC to T1, yielding enhanced fluorescence and ROS production.[ 100 , 101 ] AIE was first displayed by Tang and colleagues in 2001. [102] The authors observed that that 1‐methyl‐1,2,3,4,5‐pentaphenylsilole showed a marked enhancement in photoluminescence upon aggregation, which they attributed to steric hindrance that prevents the molecule from assuming a planar conformation in its aggregated form. Since then, AIE have found applications in various biological fields, such as fluorescence imaging, DNA visualization, ion detection and activatable PDT.[ 103 , 104 ] AIE‐based aPS agents are designed to exhibit low fluorescence and photodynamic activity as monomers in solution. These aPS are then triggered to aggregate in response to biomarkers or environmental changes, leading to a “turn‐on” effect, boosting fluorescence and ROS generation. [105] The ability to control hydrophobicity, hydrophilicity, and local viscosity, ensuring photosensitizer aggregation at the desired site, is essential for designing activatable AIE‐based photosensitizers.[ 70 , 106 ] An example of an AIE photosensitizer is tetraphenylethene conjugated to a hyrophillic group via Cathepsin B‐cleavable peptide, reducing its hydrophobicity. Upon peptide cleavage, tetraphenylethene recovers its hydrophobicity, enabling AIE. [107]
2.2. Bioresponsive Constructs and Activation Stimuli
The fundamental principle of all aPS lies in the incorporation of bioreactive elements that enable quenching in healthy tissue, but drive photosensitizer activation specifically in response to pathological stimuli. These responsive elements can be incorporated through 1) use of a bioresponsive linker to conjugate the photosensitizer and quencher, 2) conjugating the photosensitizers directly to a bioresponsive quenching moiety, or 3) enabling assembly of photosensitizers within bioresponsive supramolecular structures. This section will overview the principles underlying bioresponsive moiety design, focusing on common pathological biostimuli exhibited in cancer, the most prominent application of PDT. Specifically, we will focus on common pathological features stemming from uncontrolled cancer growth (e.g., acidodic metabolic changes, hypoxia, oxidative stress, upregulated protein/gene expression, catalytic activity and cellular uptake) that distinguish tumours from healthy tissue, and which can be leveraged for cancer‐specific PDT activation. It should be noted that many of these features are shared with other inflammatory pathologies, including atherosclerosis and arthritis, the implications of which will be discussed in Section 4.
2.2.1. pH‐Responsive aPS Design
Unregulated tumour growth increases cell metabolic demands leading to tumour acidosis from increased H+ ion production; a result of glycolysis, lactate generation, and CO2 hydration, and exacerbated by inefficient clearance due to disorganized tumour neovasculature. [108] As a result, the extracellular pH (pHe) of solid tumours drops to about 6.7–7.1, while their intracellular pH (pHi) remains typically greater than 7.4. This makes TMEs more acidic than normal tissues (pHe normal cells ≈7.4, pHi normal cells ≈7.2) [109] Therefore, in theory, designing an aPS activated under mildly acidic conditions (pH 6.7–7.1) could enable tumour‐selective activation. However, achieving sensitivity to small pH changes of less than one unit remains a significant challenge. Consequently, most pH‐responsive photosensitizers are designed to be activated around pH 5.0 (Table 2) through three predominant means: 1) incorporation of pH‐responsive ionizable groups that facilitate quenching through PeT, 2) pH‐labile FRET linkers, and 3) pH‐dependent assembly/conformation of photosensitizer nanoassemblies, whereby strategy 3 is often combined with 1 or 2 via aPS nanoassembly.
Table 2.
Acid‐responsive aPS.
|
Responsive moiety |
Quenching mechanism |
Photosensitizer |
Fluorescence activation [c] |
ROS activation [d] |
pKa |
Subcellular localization |
Ref. |
|---|---|---|---|---|---|---|---|
|
Morpholine |
PeT, ACQ |
Phthalocyanine |
/ |
/ |
5.69 |
Lysosome; Pearson correlation coefficient 0.75 |
[147] |
|
Hydrazone linker |
FRET |
ZnPc |
ΦF: 0.12→ 0.28 (DMF) |
ΦΔ: 0.35→ 0.56 (DMF) |
Nanoparticle dispersion started at 6.5 |
– |
[111] |
|
Phenylboronate ester bond protonation of amino group |
ACQ |
Pheophorbide a |
×12 |
– |
Nanoparticle dispersion at 7.4–6.0 |
– |
[148] |
|
Protonation of tertiary amine group |
ACQ, PeT |
TPP |
×2.9 |
~×6.1 |
Nanoparticle dispersion started at 6.2. |
– |
[149] |
|
Intermolecular hydrogen bonding by protonated carboxyl groups |
ACQ |
Porphyrin |
/ |
×2–3 |
2.85 |
Lysosome |
[113] |
|
Amino group |
PeT |
BODIPY |
/ |
/ |
Nanoparticle dispersion 6.5–4.5 |
Lysosome |
[150] |
|
Schiff bases bond |
ACQ |
Pyrochlorophyll a |
– |
– |
Nanoparticle dispersion tested in 6.5 |
– |
[151] |
|
Amino linker |
ACQ |
BODIPY |
×12.8 |
×38.4 |
– |
Lysosome |
[152] |
“/” data reported but cannot be quantified; “–” data not reported.
With respect to the first approach, the incorporation of ionizable chemical groups, such as tertiary amines, directly into the photosensitizer molecule is a common approach for achieving pH sensitivity, often by suppressing photosensitizer activity through PeT mechanism. For example, Wang et al. demonstrated this approach by grafting morpholine, a protonatable group, onto silicon phthalocyanine (PcM) (Figure 3a). [110] The photoactivity of silicon phthalocyanine was inhibited by PeT via the morpholine. Under acidic conditions, the morphiline nitrogen atom becomes protonated, halting PeT and subsequently activating the phthalocyanine. By assembling the PcM with serum albumin, the authors also leveraged the third aforementioned pH‐responsive aggregation‐base strategy. The resulting pH‐responsive nanomaterial, NanoPcM (pKa 5.69) transitioned from non‐photoreactive aggregates to protonated photoreactive monomers in acidic environments, restoring fluorescence and 1O2 generation. Localization studies in 4T1 cells suggested that NanoPcMs are taken up via endocytosis and activated in lysosomes. Fluorescence imaging of 4T1 tumour‐bearing mice administered NanoPcM exhibited lower background fluorescence compared to control Methylene blue (MB), which displayed systemically diffuse and always‐on fluorescence. In PDT studies using the same animal model, treatment with NanoPcM PDT reduced tumour size by 25 % compared to MB PDT.
Figure 3.

Examples of pH‐responsive aPS. (a) pH responsive aPS design of PcM and NanoPcM. Reproduced with permission from Ref. [110]. Copyright 2022, American Chemical Society. (b) Chemical design of ZnPc‐Dox and Schematic diagram showing the molecular structures of the ZnPc‐Dox/TPZ@micelles. Reproduced with permission from Ref. [111]. Copyright 2021, The Royal Society of Chemistry. (c) Acid‐responsive property of PPTP and PPT NPs. Reproduced with permission from Ref. [112]. Copyright 2022, American Chemical Society. (d) A schematic illustration of self‐assembly and fibrillar transformation of the acid‐activated peptide‐porphyrin (PWG) nanoparticles. Reproduced with permission from Ref. [113]. Copyright 2020, Wiley‐VCH GmbH.
FRET‐based pH‐triggered designs employ a linker between the photosensitizer FRET donor and acceptor quencher that breaks in acidic environments. A common choice for such pH‐responsive linkers are acid‐sensitive Schiff bases, like hydrazone linkers. For instance, Guo et al. conjugated zinc phthalocyanine (ZnPc) to doxorubicin (Dox) via a hydrazone linker, forming a ZnPc‐Dox conjugate that partially quenched ZnPc′s photophysical activity through FRET with adjacent Dox (FRET efficiency 40 %) (Figure 3b). [111] Encapsulation of this conjugate into polymeric micelles, with or without tirapazamine (TPZ), further enhanced quenching (ZnPc‐Dox/TPZ@micelle and ZnPc‐Dox@micelle). The micelles remained stable in neutral conditions but degraded in acidic environments, releasing free ZnPc‐Dox. In HT29 cancer cells, acidic intracellular conditions facilitated micelle disintegration, restoring fluorescence from ZnPc and Dox, and enabling 37.5 % and 50 % higher cell death with (PDT+Dox cytotoxicity) and without (Dox cytotoxicity only) light treatment compared to hydrazone‐free controls. In vivo, ZnPc‐Dox/TPZ@micelles demonstrated potent antitumour effects in nude mice with HT29 tumours, highlighting the potential of multifunctional aPS systems to combine PDT and chemotherapy for enhanced efficacy. While these results suggest a synergistic interaction between Dox, TPZ, and PDT, the specific role of tumour‐site acidosis in improving PDT selectivity remained uncertain.
Assembly‐based pH‐labile designs can be categorized into constructs that disassemble or change conformation under acidic environments, as evidenced by the previous examples. With respect to the first, many groups leverage the protonation of amine groups to trigger nanoparticle swelling and disassembly under acidic conditions. In this strategy, photosensitizers are attached to nanoparticle building blocks, quenching activity through ACQ. Protonation of amine groups in acidic conditions increases the polypeptide hydrophilicity, leading to nanoparticle swelling and disassembly, ultimately restoring photosensitizer activity. Zhang et al. exemplified this strategy by conjugating tetraphenylporphyrin (TPP) and an acid‐labile amine group, N,N‐diisopropylethylenediamine (DPA) to a polyglutamic acid backbone, forming micelles (PPTP‐NPs) loaded with paclitaxel dimers (PPTP@PTX2NPs) (Figure 3c). At acidic pH (<5.8), DPA protonation disrupted PeT‐mediated quenching and nanoparticle integrity, enhancing TPP fluorescence and 1O2 generation. [112] In solution, PPTP‐NPs dissociated under these conditions, increasing 1O2 yield by 6.1‐fold compared to neutral pH. In contrast, control micelles (PPT‐NPs) lacking DPA remained structurally intact and quiescent under acidic conditions, confirming the specificity of the DPA‐triggered response. In vitro, 4T1 cancer cells treated with PPTP‐NPs exhibited lysosomal uptake and activation. While control PPT NPs were not assessed for baseline activation, in vivo studies using 4T1 tumour‐bearing BALB/c mice revealed off‐target fluorescence from PPT@PTX2 NPs in the liver, lungs, and kidneys, indicating nonspecific particle disassembly. By comparison, PPTP@PTX2NPs achieved a 2.1‐fold higher fluorescence signal in tumours and exhibited greater penetration into tumour cores than control NPs, enhancing tumour‐specific activation.
Change in photosensitizer nano‐conformation via acidic environments can also be leveraged for photosensitizer activation. Sun et al. introduced an acid‐activated peptide‐based nano aPS system for the first time, demonstrating a unique design strategy that leverages pH‐responsive properties for tumour‐specific PDT. [113] The system utilized a peptide‐porphyrin conjugate (PWG), where a pH‐sensitive dipeptide (tryptophan‐glycine) was conjugated to a hydrophobic porphyrin core (Figure 3d). The tryptophan provided delocalized π‐electrons for fluorescence, while glycine contributed a carboxyl group that acted as the acid‐sensitive unit. Under physiological conditions, PWG self‐assembled into spherical nanoparticles. At lower pH (6.5 and 5.0), PWG nanoparticles underwent significant structural changes, transitioning from solid spheres to aggregated nanofibers due to protonation‐induced intermolecular hydrogen bonding. This transformation was confirmed by cryo‐TEM and molecular dynamics simulations. Notably, these fibrous structures enhanced 1O2 generation, with a 6.1‐fold increase at pH 5.0 compared to neutral pH, attributed to the preference for ISC in the fibrous state. In vitro studies in MCF‐7 breast cancer cells demonstrated lysosomal internalization of PWG nanoparticles and pH‐dependent phototoxicity, with an IC50 of 12.5 μg/mL at acidic pH versus 15 μg/mL at neutral pH.
2.2.2. Hypoxia‐Responsive aPS Designs
A hypoxic TME is a well‐established hallmark of locally advanced solid tumours, wherein 50–60 % of human cancers contain regions where the partial pressure of oxygen is less than 2.5 mmHg. [114] Hypoxia develops due to an imbalance between oxygen delivery and consumption, as rapid tumour growth outpaces neovascularization. [115] Since oxygen diffusion is limited to a critical distance of approximately 100 μm, [116] oxygen gradients emerge, worsening as tumour cells are positioned further from blood vessels and as tumour size increases. [117] In response to hypoxia, tumour cells activate the hypoxia‐inducible factor 1 family of transcription factors, which orchestrate the expression of genes that promote cellular adaptation and tumour progression. [118] A key mechanism of hypoxic adaptation involves a metabolic shift from oxidative phosphorylation to glycolysis, resulting in elevated NADH levels. This shift alters the redox potential of hypoxic cells, creating a reducing environment distinct from normal tissue. [119] Consequently, enzymes such as cytochrome P450 reductase, nitroreductase (NTR), azoreductase (AZR), and xanthine oxidase become overexpressed within the TME. Leveraging these hypoxia‐specific features, researchers are developing hypoxia‐activated prodrugs that selectively target hypoxic tumour cells. These prodrugs typically incorporate nitroaromatics, quinones, or benzotriazine dioxides, which are reduced in hypoxic conditions to produce cytotoxic effects. [120] Given the critical role of hypoxia in tumour progression, there is growing interest in imaging and targeting hypoxic cells. Fluorescent probes designed to assess hypoxia rely on reductases like NTR and AZR, which are overexpressed in hypoxic tumour cells. Similar principles guide the development of photosensitizers activated in hypoxic environments. Strategies often include incorporating nitro moieties reduced to amines or introducing azo bonds cleaved by AZR. These hypoxia‐responsive moieties are summarized in Table 3 and include utilization of azo benzene linkers that are reduced and broken by AZR and incorporation of NTR and AZR reactive groups into photosensitizer to facilitate quenching via ICT or PeT.
Table 3.
Hypoxia‐responsive aPS.
|
Stimuli |
Responsive Moiety |
Quenching Mechanism |
Photosensitizer |
Fluorescence activation [c] |
ROS activation [d] |
In vitro fold phototoxicity in hypoxia to normoxia |
Ref. |
|---|---|---|---|---|---|---|---|
|
NTR |
4‐Nitrobenzyl |
ICT |
Hemicyanine dye |
/ |
×2.5 |
Max ~4; 0–4 μM, 10 % vs 21 %, 4T1 |
[124] |
|
PeT |
Heptamethine aminocyanine |
/ |
/ |
Max ~2; 0–20 μM, 10 % vs 21 %, HeLa |
[153] |
||
|
PeT |
Fluorescein |
~×7 |
~×3.3 |
Max ~4.5; 0–20 μM, 10 % vs 21 %, HeLa, Detection limit 8 ng/ml |
[154] |
||
|
PeT, TICT |
Thio‐pentamethine cyanine |
Φfl 4 %→ 32 % (DCM) |
ΦΔ: 6 %→24.3 % (CH2Cl2) O2⋅−: C2‐NO2 ×2.8 →C2‐NH2 ×14.6 |
Max ~6; 0–4 μM, 2 % vs 21 %, HepG2 |
[155] |
||
|
2‐nitroimidazole |
ICT |
Synthesized molecule |
/ |
/ |
Max ~8; 0–2 μM, HeLa (hypoxic condition) |
[156] |
|
|
2‐methoxy‐ 4‐nitrophenyl |
ICT |
Benzophenothiazine |
/ |
~×4 |
Max ~5; 0–20 μM, EMT6 (CoCl2‐ simulating hypoxia) |
[157] |
|
|
AZR |
Azo benzene linker |
ultrafast IC |
Rhodamine |
Φfl 0.001→ 0.006 EtOH |
ΦΔ: 0.03→0.56 1 : 1 (v/v) buffer MeOH |
~2, 1 μM, 8 % vs 21 %, A549 |
[123] |
|
FRET |
PPa |
/ |
ΦΔ: Azo‐PDT 0.04→ ΦΔ: Pyro 0.63 (EtOH) |
~2, 2.5 μM, 1 % vs 21 %, BEL‐7402 |
[121] |
||
|
PeT, AIE |
AIEgen T‐2TM |
~×7 |
~×1.8 |
Only Fl reported (argon‐full cartridge‐simulating hypoxia) |
[122] |
||
|
Arylazo |
AIE |
TPFN |
~×54 |
– |
Max~5; 50 μM; MCF‐7 (hypoxic condition −) |
[158] |
“/” data reported but cannot be quantified; “–” data not reported. [c] Fluorescence intensity or quantum yield increasement compared to inactivated state. [d] ROS sensor signal intensity or ROS quantum yield increasement compared to inactivated state.
An example of a proof‐of‐concept design is the Azo‐PDT system developed by Wang et al. By linking pyrophosphate α (PPA, FRET donor) to the fluorescent NIR dye SiR‐665 (FRET acceptor) via an azo‐benzene linker, Azo‐PDT utilized the FRET mechanism to quench fluorescence and suppress 1O2 generation (Figure 4a). [121] In a hypoxia‐mimicking solution (NADPH and liver microsomes), cleavage of the azo bond restored fluorescence emission and 1O2 production. In BEL‐7402 cells (human liver cancer cells), Azo‐PDT showed enhanced fluorescence and photodynamic activity under hypoxic conditions compared to normoxic conditions. Specifically, Azo‐PDT demonstrated higher cytotoxicity in hypoxic environments (~60 % cell viability in normoxia, ~30 % cell viabilityin hypoxia), while PPA showed irradiation‐dependent cell viability regardless of oxygen levels. These results confirmed the hypoxia‐dependent photodynamic activity of Azo‐PDT. The cleavage of azo‐benzene linkers can be leveraged to trigger photosensitizer complex dissociation or nanoparticle degradation and can complement ACQ and AIE quenching strategies. For instance, a covalent organic framework created via azo‐benzene linkers quenched AIE luminogens (AIEgen)′s fluorescence and PDT activity via the PeT effect. It decomposed under hypoxic conditions, releasing AIEgens and enhancing their fluorescence and PDT properties (Figure 4b). [122] Additionally, the azo group can act as a direct quencher by inducing ultrafast intersystem crossing (IC) and reducing ISC efficiency (Figure 4c). [123]
Figure 4.

Examples of hypoxia‐responsive aPS. (a) Chemical structure of azo‐PDT and activation mechanism under tumour hypoxia situations. Reproduced with permission from Ref. [121]. Copyright 2020, Springer Nature. (b) Schematic illustration of the integration of AIEgen and DNA methyltransferase inhibitor into hypoxia‐responsive COFs for boosting image‐guided pyroptosis‐mediated cancer immunotherapy. Reproduced with permission from Ref. [122]. Copyright 2024, Elsevier Ltd. (c) Design strategy and chemical structures of azoSeR. Reproduced with permission from Ref. [123]. Copyright 2017, American Chemical Society. (d) Structures of CYNT‐1 and CYNT, including their mechanisms for sensing and PDT activation. Reproduced with permission from Ref. [124]. Copyright 2020, Wiley‐VCH GmbH.
Other hypoxia‐activatable photosensitizers utilize the reduction of various nitro groups, which are typically integrated directly into the photosensitizer molecule, quenching its activity through ICT or PeT mechanisms. For example, the aPS CYNT‐1′s PDT ability and fluorescence were fully quenched by PeT between two 4‐nitrobenzyl groups and the dye CYNT‐1(Figure 4d). [124] After confirming NTR‐selective activation of CYNT‐1 in solution, its activation by endogenous NTR in living HeLa cells was demonstrated. When HeLa cells were cultured under varying oxygen levels (21 %, 10 %, and 1 % O2) and treated with CYNT‐1, fluorescence intensity increased as oxygen levels decreased, suggesting CYNT‐1 can be activated by NTR, which is expressed more in low oxygen conditions. Notably, CYNT‐1′s PDT performance was effectively activated under mild hypoxia (10 % O2) but not under severe hypoxia (1 % O2) or normoxia, likely due to oxygen limitations in severe hypoxia.
2.2.3. Upregulated Enzyme‐Responsive aPS Designs
Mechanisms that govern photosensitizer quenching in aPS often stem from activatable fluorescent probe design principles. The malignant growth of cancer cells is accompanied by changes in enzymatic activity that contribute to tumour initiation, progression and metastasis (Table 4). As such, numerous enzymes are upregulated by cancer versus healthy cells, making them attractive targets for designing enzymatically degradable or activatable aPS systems. These enzyme‐specific systems also leverage the substrate specificity of enzymes to enhance the selectivity of aPS activation. The substrates used in aPS design can be divided into two broad categories: 1) enzyme cleavable peptidase, and 2) enzyme‐reactive chemical moieties.
Table 4.
Enzyme‐responsive aPS.
|
Enzyme |
Fold enzyme expression to healthy tissue |
Recognition Substrate |
Quenching mechanism |
Photosensitizer |
Fluorescence activation [c] |
ROS activation [d] |
Ref. |
|---|---|---|---|---|---|---|---|
|
MMP‐2 |
Breast cancer: ×2.5 [159] Lung cancer: ×1.5 [160] |
PLGVR peptide |
FRET |
DSBDP |
/ |
/ |
[126] |
|
MMP‐7 |
Breast cancer : ×1.5 [159] Colon cancer: ×1.5 [161] |
GPLGLARK peptide |
FRET |
PPa |
×12 |
×18 |
[125] |
|
MMP‐9 |
Breast cancer: ×2.5 [159] Colon cancer: ×1.5 [162] |
GRIGFLRTAKGG peptide |
FRET |
PPa |
/ |
/ |
[163] |
|
Cathepsin B |
Renal cancer: ×28 [164] (large SD) Lung cancer: ×5.6 [165] Late‐stage colorectal cancer: ×5 [166] |
GFLG peptide |
FRET |
DSBDP |
/ |
/ |
[126] |
|
AIE |
TPP |
– |
– |
[107] |
|||
|
ICT |
BODIPY |
/ |
/ |
[167] |
|||
|
GFLGK |
FRET, PeT |
ZnPc |
/ |
/ |
[168] |
||
|
GAGRRAAG |
FRET |
Pheophorbide a |
/ |
– |
[169] |
||
|
Val‐Cit |
FRET |
Hemicyanine |
– |
×6 |
[170] |
||
|
ALP |
Myeloma: ×7 [171] Seminoma: ×10 [172] |
Phosphate group |
AIE |
TPA |
×50 |
– |
[173] |
|
ICT |
Hemicyanine |
/ |
/ |
[174] |
|||
|
Hemicyanine |
×21.3 |
×3.8 |
[175] |
||||
|
Tetraphenylethane |
/ |
×140 |
[176] |
||||
|
Hemicyanine |
/ |
/ |
[177] |
||||
|
PeT |
Hetero‐rosamine |
×126 |
/ |
[178] |
|||
|
FpYGpYGpY |
AIE |
Hemicyanine |
/ |
/ |
[179] |
||
|
GGT |
Gastric cancer: ×1.5 [180] Prostate cancer: ×1.5 [181] Hepatocellular carcinoma: ×2 [182] |
acylamide bond (−CO−NH−) in glutamine |
PeT., SOCT‐ISC |
Aza‐BODIPY |
/ |
/ |
[183] |
|
γ‐glutamyl |
ICT |
Rhodamine |
/ |
/ |
[184] |
||
|
Synthetic photosensitizer |
/ |
/ |
[185] |
||||
|
Diketopyrrolopyrrole |
×80 |
/ |
[186] |
||||
|
NQO1 |
Breast cancer: ×3 [187] Lung cancer: ×14 [188] Breast cancer: ×9 [188] |
Quinone |
ICT |
Hemicyanine |
/ |
/ |
[189] |
|
Bromine cyanine |
/ |
/ |
[190] |
||||
|
DSBDP |
×45 |
/ |
[191] |
||||
|
β‐gal |
Ovarian cancer: ×4–13 [192] Prostate cancer: ×4 [193] |
Glycosidic linkage |
ICT |
Resorufin |
×22 |
/ |
[194] |
|
ACQ |
ZnPc |
×4 |
/ |
[195] |
“/” data reported but cannot be quantified; “–” data not reported. [c] Fluorescence intensity or quantum yield increasement compared to inactivated state. [d] ROS sensor signal intensity or ROS quantum yield increasement compared to inactivated state.
Proteases, such as cysteine cathepsins and matrix metalloproteinases (MMPs) in particular, leverage the first category. They have garnered considerable attention in cancer therapy and imaging due their upregulation in cancer cells and their ability to cleave target peptide sequences with high specificity. For example, the upregulation of matrix metalloproteinase‐7 (MMP‐7) has been leveraged by our group to create an MMP‐7‐activatable aPS. [125] This system (PP MMP7 B) is a conjugate of the photosensitizer Pyro and a Black Hole Quencher 3 (BHQ3) via an MMP‐7‐cleavable peptide sequence (GPLGLARK), enabling quenching through FRET. Upon MMP‐7‐mediated cleavage, Pyro was released, restoring fluorescence and 1O2 generation (19‐fold increase). In vitro studies demonstrated that PP MMP7 B was selectively activated in KB cells (MMP7+) but not in BT20 cells (MMP7−). Confocal microscopy revealed strong fluorescence in KB cells, while BT20 cells showed minimal signal. HPLC analysis confirmed MMP‐7‐dependent cleavage, producing activation fragments exclusively in KB cell medium. PDT studies demonstrated effective reduction of KB cell viability upon irradiation, with no effect on BT20 cells. In vivo studies using mice bearing KB tumours provided additional evidence of selectivity and efficacy. Fluorescence imaging showed no initial signal post‐injection of PP MMP7 B; however, fluorescence increased in the KB tumour after 20 minutes, reaching its peak at 3 hours. PDT treatment of the fluorescent tumour led to complete regression with no signs of regrowth over 30 days, while untreated tumours continued to grow. This demonstrates the high specificity and therapeutic potential of PP MMP7 B for MMP7+ tumours. Recently, Tam et al. have taken this concept further and reported a photodynamic molecular beacon (PMB) that links a distyrylboronic dipyrromethene (DSBDP)‐based photosensitizer with BHQ3 moiety via two peptide segments cleavable by MMP‐2 and cathepsin B (Figure 5a). [125] The ROS generation of this photosensitizer was quenched due to efficient FRET between the two components. This dual‐lock system required both MMP‐2 and cathepsin B enzyme upregulation for effective activation, minimizing off‐target effects and further enhancing tumour specificity. Dual‐lock systems, such as this, are discussed in further detail in Section 3.
Figure 5.

Examples of enzyme‐responsive aPS. (a) Schematic concept of PPMMP7B. Reproduced with permission from Ref. [125]. Copyright 2007, National Academy of Sciences. (b) Enzyme responsive aPS based on hetero‐rosamine scaffolds and pyridine caging/decaging strategy. Reproduced with permission from Ref. [127]. Copyright 2023, Wiley‐VCH GmbH. (c) Schematic illustration of TPE‐APP. Reproduced with permission from Ref. [128]. Copyright 2024 American Chemical Society. (d) Recognition mechanism of I‐HCy‐Q towards NQO1 and schematic illumination of I‐HCy‐Q for NQO1 imaging and mitochondria‐targeted PDT‐induced ICD. Reproduced with permission from Ref. [129]. Copyright Elsevier B.V. (e) Chemical structures of BDP1/BDP2 and Glu‐BDP1/Glu‐BDP2 as well as activation mechanism of Glu‐BDP1/Glu‐BDP2 by GGT. Reproduced with permission from Ref. [130]. Copyright 2022, Wiley‐VCH GmbH.
In addition to proteases, various enzymes overexpressed in tumours, including ALP, quinone oxidoreductase‐1, NAD(P)H: Quinine oxidoreductase (NQO1), and γ‐glutamyl transpeptidase (GGT), catalyze reactions that can be exploited for aPS activation. Enzyme‐reactive probes designed to be substrates for these enzymes use catalytic substrates that alter the solubility or molecular structure of the photosensitizer, enhancing fluorescence emission and ROS generation. Examples of these enzymes and substrates used in aPS design are summarized in Table 5. For example, Lioret et al. reported a novel probe in which an ALP reactive group was introduced into a hetero‐rosamine dye with a 4‐pyridyl meso‐substituent. 4‐phosphoryloxybenzyl was used as the recognition moiety and PDT was inhibited via d‐PeT (Figure 5b). [127] The same ALP‐reactive functional group was introduced into the AIE probe TPE‐APP, which was designed to respond to the overexpression of ALP in cancer cells (Figure 5c). [128] The phosphate group of TPE‐APP is hydrolyzed in the presence of ALP, triggering the in situ formation of AIE aggregates. Dephosphorylation of TPE‐APP reduces intramolecular rotation and non‐radiative decay pathways, restoring fluorescence and ROS generation. Fluorescence activation occurred selectively in ALP‐overexpressing HeLa cells, with a high colocalization of TPE‐DMA fluorescence and ALP antibody (Alexa546‐Ab) signal, confirmed by a Pearson correlation coefficient of 94.4 %. In vitro PDT demonstrated over 9‐fold higher cytotoxicity in ALP+ HeLa cells compared to normal cells. In HeLa tumour‐bearing mice, TPE‐APP produced a strong fluorescence signal at the tumour site within 1 hour of intratumoural injection. In contrast, pretreatment with Van to inhibit ALP significantly reduced fluorescence, demonstrating that the fluorescence activation was due to ALP‐mediated unquenching. PDT experiments further demonstrated enhanced tumour inhibition in treated mice compared to Van pre‐treated controls.
Table 5.
Redox‐responsive aPS.
|
Stimuli |
Recognition group |
Quenching mechanism |
Photosensitizer |
Fluorescence Activation [c] |
ROS Activation [d] |
Ref. |
|---|---|---|---|---|---|---|
|
GSH |
Disulfide bond |
FRET, PeT |
ZnPc |
×8 |
×4–5 |
[196] |
|
FRET |
BODIPY |
ΦF: 0.02→ 0.09 (PBS) |
/ |
[133] |
||
|
FRET |
PPa |
×149 |
>×100 |
[134] |
||
|
ACQ |
MB |
×17.7 |
/ |
[197] |
||
|
ACQ |
Not indicated |
/ |
/ |
[198] |
||
|
2,4‐dinitrobenzene‐ sulfonyl moiety |
PeT |
ZnPc |
ΦF: 0.01→ 0.2 (DMF) |
/ |
[135] |
|
|
PeT |
BODIPY |
/ |
– |
[136] |
||
|
Nitro group |
ICT |
BODIPY |
×30 |
×8 |
[199] |
|
|
H2O2 |
Boronate ester |
AIE |
Tetraphenylpyrazine |
ΦF: 0.001→ 0.024 (DMSO) |
/ |
[200] |
|
PeT |
BODIPY |
/ |
/ |
[201] |
||
|
– |
Resorufin |
/ |
/ |
[202] |
||
|
Boronic acid |
ICT |
Ir(III) phenylpyridine |
– |
– |
[203] |
|
|
NO |
o‐phenylenediamine |
PeT |
Bis(phenylethynyl) benzene |
ΦF: 0.002→ 0.09 (PBS) |
ΦΔ: 0.001→ 0.82 (PBS) |
[204] |
[c] Fluorescence intensity or quantum yield increasement compared to inactivated state. [d] ROS sensor signal intensity or ROS quantum yield increasement compared to inactivated state.
NQO1, also known as DT‐diaphorase, is closely related to various carcinogenic processes and has been used as a biomarker for cancer diagnosis and targeted therapy. Cao et al. presented an NQO1‐activated NIR multifunctional therapeutic diagnostic probe, I‐HCy‐Q (Figure 5d). The NQO1 recognition group is directly attached to I‐HCy, which blocks ICT, resulting in decreased fluorescence and 1O2 quantum yield. [129] After catalytic hydrolysis by NQO1, I‐HCy is rapidly released and is particularly enriched in mitochondria, where NIR fluorescence emission and ROS production are restored. The linear detection range and detection limit of NQO1 of I‐HCy are 0.05–1.5 μg/mL and 5.66 ng/mL, respectively. It was found to be selective for NQO1 compared to other substrates such as glutathione (GSH) and bovine serum albumin (BSA) in solution, and it was reported that I‐HCy‐Q activated rapidly in 4T1 tumour‐bearing mice, while I‐HCy‐Q from 4T1 tumour‐bearing mice pretreated with intratumoural injection of dicoumarin (an NQO1 inhibitor) as a control emitted much weaker fluorescence.
Aside from cleavage, upregulated enzymes can also be used to activate aPS via conformational changes. One such example was presented by Miao et al. who developed heavy‐atom‐free boron dipyrromethene (BODIPY) derivative photosensitizers, incorporating bulky alkoxyaniline electron donors at the meso positions of BODIPY and distyryl BODIPY acceptors (Figure 5e). [130] Bulky alkoxyaniline electron donors were installed at the meso positions of the bodipy and distyryl bodipy acceptors, respectively, so that the electron donor and acceptor units are arranged in an orthogonal geometry (BDP1/BDP2). These orthogonally arranged donor‐acceptor units enhanced ISC efficiency via the SOCT‐ISC mechanism, prolonging the T1 and facilitating 1O2 generation. To enhance cancer specificity, γ‐glutamyl groups were attached to BDP1/BDP2 (Glu‐BDP1/Glu‐BDP2), activating the photosensitizers via GGT. Their cancer cell‐specific activation was evaluated in A549 and SHIN3 cancer cells or normal EA.hy926 and RAW264.7 cells demonstrating little cytotoxicity in both cancer and normal cells without light irradiation. After light irradiation, Glu‐BDP1/Glu‐BDP2 induced clear cell death in cancerous A549 and SHIN‐1 cells, with IC50 values up to 0. 40/0.41 μM and 0.39/0.37 μM, respectively, but were much less efficient in RAW264.7 and EA.hy926 normal cells (over 80 % cell viability in all condition). These cancer cell deaths were significantly inhibited by a GGT inhibitor (GGsTop, 100 μM), confirming that the activation ability was highly dependent on GGT activity. Although no tumour site‐specific activation relative to normal cells was identified in animal experiments, the effective PDT antitumour effect of Glu‐BDP2 was confirmed.
2.2.4. Redox‐Responsive aPS Design
Cancer cells generate elevated levels of ROS due to their high metabolic activity. Like healthy cells, they strive to maintain redox homeostasis by balancing ROS production with antioxidant defenses. Consequently, both ROS levels and the antioxidant GSH are typically higher in tumour cells compared to normal cells and can be leveraged for aPS activation.
Among the various ROS overexpressed in cancer cells, H2O2 is particularly stable under physiological conditions due to its non‐radical nature and high membrane permeability. These properties make H2O2 an attractive target for stimulus‐responsive systems. Boronate ester groups are widely used for H2O2 detection due to their ability to undergo oxidative hydrolysis triggered by H2O2. Li et al. developed a H2O2‐activated photosensitizer, DPP‐BPYS, based on a diketopyrrolopyrrole (DPP) core functionalized with triphenylamine (TPA) units for enhanced photostability and fluorescence (Figure 6a). [131] Featuring AIE properties and H2O2 responsiveness, DPP‐BPYS showed negligible fluorescence in aqueous solutions but displayed a rapid and specific fluorescence turn‐on upon H2O2 exposure, with a detection limit of 0.72 μM. DPP‐BPYS detected both exogenous and endogenous H2O2 in cancer cells and exhibited dose‐dependent fluorescence that correlated with H2O2 levels. In an inflammatory mouse model, DPP‐BPYS demonstrated an 8.2‐fold fluorescence enhancement, highlighting its potential for in vivo H2O2 imaging. However, its application in PDT remains unexplored.
Figure 6.

Representative work of H2O2‐ and GSH‐responsive aPS. (a) Schematic illustration of H2O2‐activated DPP‐BPYS. Reproduced with permission from Ref. [131]. Copyright 2022, Elsevier B.V. (b) Schematic illustration of proposed activation mechanism of 8a. Reproduced with permission from Ref. [133]. Copyright 2020, Elsevier Masson SAS. (c) Schematic Illustration of IND@RAL nanoparticle. Reproduced with permission from Ref. [134]. Copyright © 2019 American Chemical Society. (d) Chemical structure of of GSH‐responsive photosensitizer 2. Reproduced with permission from Ref. [135]. Copyright 2014, WILEY‐VCH Verlag GmbH & Co. KGaA. (e) Mode of operation for the GSH‐mediated activation of CyI‐S‐diCF3. Reproduced with permission from Ref. [137]. Copyright 2023 Royal Society of Chemistry.
Elevated intracellular GSH levels are characteristic of many tumour cells (e.g. Lung and breast cancer). GSH supports cellular adaptation to oxidative stress and confers resistance to cytotoxic drugs and radiation. [132] This protective mechanism allows cancer cells to survive and proliferate despite the hostile conditions of the TME, often leading to multidrug resistance, and presenting an opportunity to facilitate activatable PDT. GSH‐responsive photosensitizers have been extensively studied, as summarized in Table 6, for their potential to selectively activate within tumour environments. The most commonly employed GSH‐targeted aPS design involves incorporating disulfide bonds into photosensitizer, which can be cleaved by GSH, leading to activation. Cao et al. developed an aPS (8a) conjugating BODIPY derivatives via a disulfide bond, enabling quenching through intramolecular FRET (Figure 6b). [133] In its quenched state, 8a showed a fluorescence quantum yield (Φf) of 0.021 and a 1O2 quantum yield (ΦΔ) of 0.015, significantly lower than those of a single BODIPY unit (Φf 0.11 and ΦΔ 0.3). Under simulated intracellular conditions (10 mM GSH), 8a′s fluorescence steadily increased, recovering 77 % of its quantum yield after 720 minutes, while remaining stable in extracellular environments (2 μM GSH). Cellular studies revealed that 8a was selectively activated in GSH‐rich cancer cells (HeLa, A549, H22), exhibiting strong fluorescence and phototoxicity upon light irradiation, with IC50 values of 0.67 μM, 0.44 μM, and 0.48 μM, respectively. In contrast, normal HELF cells with lower GSH levels showed minimal fluorescence and no significant phototoxicity. Intravenous administration of 8a in mice with H22 hepatoma tumours demonstrated tumour‐specific uptake and activation. Fluorescence signal at tumour sites increased over time, while no significant signal was detected in major organs, confirming selective activation in the GSH‐rich TME. These results highlighted the potential of 8a for precise PDT in GSH‐overexpressing cancer cells.
Table 6.
Summary of biomarkers elevated in various diseases and normal cells/organelles, commonly used in activatable PDT.
|
Stimuli |
Pathology |
Normal cells/organelles |
|---|---|---|
|
Low pH |
cancer (extracellular pH ~6.7–7,1),[211] inflammation (extracellular pH ~6.5–7.0),[212] infections (open wounds extracellular pH ~6.5–8.5), [213] atherosclerosis (extracellular pH ~6.5–8.5), [214] neurodegenerative diseases (extracellular pH ~6.0–6.8) [215] |
skin cells (cellular pH: 4.0–5.8),[216] endosome/lysosome (pH 4.5–6.5) [217] |
|
Hypoxia |
cancer, [218] inflammation [219] ; atherosclerosis, [220] kidney disease, [221] liver disease, [222] diabetics,[223] osteoarthritis [224] |
bone marrow, [225] intestinal epithelium,[226] muscle cells [218] |
|
ROS |
cancer, [227] inflammation, [228] atherosclerosis,[229] neurodegenerative diseases, [230] diabetics,[231] liver disease [232] |
mitochondria, [233] peroxisome, [234] endoplasmic reticulum,[235] phagocytic immune cells,[236] |
|
Proteases (MMP, cathepsin B, etc) |
cancer, [239] inflammation, [240] atherosclerosis,[241] neurodegenerative diseases, [242] liver diseases, [243] osteoarthritis [244] |
digestive system cells, [245] immune cells,[246] |
The cleavage of disulfide bonds has emerged as a common stimulus to trigger the degradation of nanoparticles as well. Liu et al. demonstrated this approach by conjugating pyropheophorbide a (PPa) to lipids via disulfide bonds, forming conjugates that self‐assembled into liposomes (RALs) in aqueous solutions (Figure 6c). [134] In their intact state, RALs exhibited strong autofluorescence quenching of the PPa signal due to Homo‐FRET. Upon disassembly, the nanoparticles achieved a 149‐fold increase in fluorescence activation compared to the undisturbed state, with 1O2 generation closely correlating with fluorescence activation, showing an over 100‐fold enhancement. The GSH‐induced disruption of RALs was confirmed using TEM imaging and dynamic light scattering, which showed the structural disintegration of the liposomes.
Another approach to designing GSH‐reactive aPS involves inducing PeT by directly incorporating a quenching moiety into the photosensitizer that reacts with GSH. Hui et al. demonstrated this strategy by introducing a 2,4‐dinitrobenzenesulfonyl moiety, a strong electron‐withdrawing group, into ZnPc, effectively quenching its fluorescence and 1O2 generation via PeT (Figure 6d). [135] This modification resulted in a 20‐fold reduction in fluorescence quantum yield and a 7‐fold decrease in 1O2 generation efficiency compared to ZnPc. Upon pretreatment of MCF‐7 breast cancer cells with GSH monoester, fluorescence intensity increased 4‐fold, and ROS production was enhanced more than 3‐fold, confirming GSH‐triggered activation. Similarly, Turan et al. applied the same 2,4‐dinitrobenzenesulfonyl quenching moiety to a BODIPY‐based sensitizer, creating a GSH‐responsive aPS. [136] In normal MRC‐5 human fetal lung fibroblast cells, no significant phototoxicity or cytotoxicity was observed. However, in HCT116 colon cancer cells, the system exhibited an IC50 value of 20 nM, demonstrating its high selectivity and efficacy in targeting tumour cells. These results underscore the potential of GSH‐responsive aPS designs for mediating selective PDT.
Since PDT aims to trigger cell death by generating ROS in tumour cells, there is a concern that GSH′s ability to act as an antioxidant may hinder the efficacy of PDT. To this end, inducing apoptosis by disrupting cellular redox homeostasis is a promising antitumour strategy that is already being actively investigated in the field of oncology chemotherapeutics development. For this reason, several recent studies have introduced means of both activating PDT via GSH, and consuming GSH.
Fu and Wang et al. recently reported a strategy that not only activates PDT photoreactivity by GSH but also disrupts redox homeostasis by simultaneously reducing GSH levels (Figure 6e). [137] For the effective production of 1O2 species, an electron‐withdrawing phenyl sulfide unit of 3,5‐bis(trifluoromethyl)benzenethiol (BTBT) was incorporated into iodinated heptamethine cyanine. Fluorescence and 1O2 generation were quenched by more than 13‐fold by PeT from the BTBT moiety to heptamethine. Although the reaction did not result in complete quenching of ROS generation (ΦΔ of 0.45 % in the absence of GSH), it showed an increased quantum yield of 0.66 % after reaction with GSH. The corresponding aPS CyI‐S‐diCF3 reacted specifically with GSH via nucleophilic substitution, not only activating it but also consuming intracellular GSH. The intracellular GSH content was reduced by a ~22 % upon laser irradiation after CyI‐S‐diCF3 treatment compared to the untreated control, suggesting that CyI‐S‐diCF3 could promote GSH consumption as well as activatable PDT in tumour cells.
2.2.5. Cellular Internalization
Metabolic changes in cancer also lead to higher endolysosomal activity, passive and active cellular internalization/trafficking and nanoparticle extravasation relative to healthy tissue. Consequently, tumour tissue can display higher disassembly of uptaken supramolecular assemblies, like nanoparticles, compared to healthy tissue. This was exemplified by Rajora et al., who monitored the fluorescence unquenching of supramolecular porphyrin‐lipid assemblies delivered intravenously to 4T1 tumour‐bearing mice. [138] They observed that tumours displayed substantially higher fluorescence intensity per particle accumulation (in % ID/g) compared to healthy organs, tumour‐adjacent tissue and skin, suggesting that tumours exhibited preferentially higher fluorescence unquenching and thus particle disaggregation.
This upregulated particle uptake and disassembly by tumours can be leveraged to activate supramolecular ACQ‐enabled aPS. For example, our lab has generated porphysomes, liposomal nanoparticles resulting from the self‐assembly of pyropheophorbide‐a‐lipid (pyro‐lipid).[ 139 , 140 , 141 ] When intact, the pyro‐lipid moieties aggregate, leading to ACQ‐enabled quenching of the photosensitizer′s fluorescence and 1O2 generation capabilities. This photoreactivity and fluorescence is restored upon particle internalization by cancer cells in vitro and in vivo. This nanoconstruct and activatable nature has been applied for PDT in A549 tumour‐bearing mice, which displayed less post‐PDT skin irritation and more rapid skin healing (eschar resolved at day 15) compared to Photofrin‐PDT (eschar resolved at day 30), while demonstrating similar complete tumour response (25 % vs Photofrin‐PDT 15 %). [142] This activation can be modulated by altering the molar composition of pyro‐lipid, including EDTA moieties, inclusion of targeting moieties and altering lipid chain length. For example, Ho et al. reported that inclusion of EDTA‐lipids in porphysomes enhanced their intracellular uptake in KB cells by 25‐fold. [143] In KB subcutaneous tumour‐bearing mice and hamster cheek carcinoma models, EDTA‐lipid porphysomes demonstrated fast fluorescence‐enabled tumour delineation within minutes post‐injection and increased PDT potency (100 % survival rate) compared to porphysomes (60 %). Similarly, Jin and Cui et al. demonstrated enhanced (higher and faster) activation of porphysomes decorated with folate, whose receptor is overexpressed in many cancer cells. [144] Despite exhibiting lower tumour uptake, the folate porphysomes displayed substantially higher and more focal fluorescence activation in tumours and KB murine tumours and stronger PDT effects. We have found that pyro‐lipid lipoprotein mimicking nanoparticles decorated with apolipoprotein AI mimetic peptides (targeting SRBI) and apolipoprotein E3 (targeting LDLR) have also been shown to yield higher and faster tumour activation compared to porphysomes.[ 145 , 146 ] In addition to EDTA and targeting moieties, activation following tissue uptake can be hastened through the inclusion of C16 chain length pyro‐lipid and host phospholipid moieties vs longer C18 chain length lipids, while the longer chain length can confer more specific activation and higher tumour:adjacent muscle fluorescence ratios. [138] Overall, these works highlight the potential of using ACQ, pathological elevation of nanoassembly uptake and the advantages of simple particle composition modulation to achieve tumour activatable PDT and fluorescence imaging. As is evident across the aPS examples discussed thus far, ACQ and tumour cell internalization are ubiquitously used in combination with other aPS quenching mechanisms and biostimuli.
3. Limitations and Advances in aPS Designs
Despite their diverse design strategies, aPS continue to suffer from persistent limitations that need to be addressed to enhance their efficacy and specificity. In this chapter, we will build upon the design strategies discussed in Chapter 2 and critically analyze their effective application in the context of the TME to assess their potential for achieving increased PDT selectivity.
First, the selectivity of the biosensor construct needs to be improved. For example, GSH‐triggered aPS can also react with other molecules that contain this sulfhydryl group. Cysteine and homocysteine contain thio groups and are present in cells in micromolar amounts, and may contribute to GSH‐triggered aPS activation. [205] In the case of pH responsive aPS, the basis for the specificity of tumour acidity responsive aPS is that the extracellular pH of the TME is weakly acidic (~6.5) compared to healthy tissue (0.5 to 1 pH unit higher). However, as shown in Table 2, many pH‐responsive aPS do not report their pKa, and activation tests are often conducted at pH 5.0, which is more acidic than the TME. To effectively distinguish the subtle pH differences between the TME and normal tissues and achieve TME‐selective activation, the probe′s pKa should be close to 6.5, with a narrow response range that enables rapid switching between OFF and ON states. In fluorescent probe research, considerable effort has been directed toward developing probes that selectively react with specific biothiols[ 206 , 207 ] or exhibit sharp pH response ranges.[ 208 , 209 ] Insights from these studies can be leveraged to improve pH‐triggered aPS designs.
Second, the triggers used for aPS activation may not be as tumour‐specific as previously assumed. As described in Table 6, the stimuli used in aPS can be overexpressed in various tissues or in various pathologies. For example, some review papers suggest that difference in GSH concentrations between tumours and healthy tissue (thought to be up to 100 to 1000 times higher in tumours) is overestimated. In reality, the difference in GSH levels between tumour and normal cells is often modest, sometimes only by a factor of two. [210] Given that GSH is present in millimolar concentrations in most cells, using it as a trigger risks high background activation in healthy tissues, reducing the selectivity of the aPS. The reliance of GSH‐targeted aPS on disulfide bonds further limits selectivity as these bonds can be easily reduced by GSH in both tumour and normal cells. Furthermore, the TME is highly dynamic, with fluctuating GSH levels influenced by glutathione status and synthesis rates in cancer cells. This can lead to inconsistent aPS activation. Overall aPS designs centered around GSH face challenges in selectivity and efficiency that may be overcome in future studies by combining GSH‐responsive elements with tumour‐specific targeting groups or alternative bioresponsive activators.
Lastly, precise control over the location of aPS activation is crucial for maximizing therapeutic outcomes while minimizing off‐target effects, particularly for pH‐responsive aPS. If the bioresponsive moiety exhibits a pKa of pH of 5.5 or lower, it would be unlikely to activate within the TME and more likely to activate within intracellular lysosomes (pH 4.5–5.5). This can be problematic for several reasons. First, these studies were tailored to facilitate PDT in the TME, but often did not characterize localization of activation to support this intentional design and inform therapeutic mechanism of action, which in turn prevents strategic advancement of acid‐triggered aPS designs. Second, as lysosomal activity is present in both healthy and malignant cells, aPS that activate in lysosomes may not confer desired tumour‐specific PDT. It is true that many malignant cells exhibit larger, more active and more numerous endolysosomes/lysosomes, which could translate into more acid‐triggered aPS activation in tumours vs healthy tissue. However, in the absence of standard characterization of tumour vs healthy cell lysosomal activity, this mechanism of acid‐triggered aPS activation specificity remains unclear. Furthermore, the increased endolysosomal activity of tumour cells can also translate into higher aPS uptake in cancer vs healthy cells, making it unclear if enhancements observed in aPS‐mediated PDT were a result of higher tumour uptake, or from higher activation (or the combination thereof). Thus, validation and advancement of acid‐triggered aPS designs should include characterization of bioresponsive moiety pKa, activation localization, healthy vs tumour lysosomal activity and quantification of both aPS and associated control photosensitizer cancer cell absolute uptake and biodistribution.
For this reason, a comprehensive understanding of the distribution and concentration of activation triggers in both tumour and normal tissues, along with precise control over the activation site of aPS, is essential for the rational design of highly selective and effective photosensitizers. By addressing these limitations through targeted design and thorough characterization, we can enhance the specificity and efficacy of PDT, leading to improved clinical outcomes. This section discusses advanced designs aimed at improving the selectivity of aPS.
3.1. Dual‐Lock Activatable PDT
To minimize premature activation of aPS in complex biological systems and enhance selectivity, recent studies have increasingly supported the use of dual‐lock strategies that require the simultaneous recognition of multiple stimuli for activation. Dual‐lock aPS systems can be categorized into two types: 1) Parallel dual‐lock: These systems feature two independent bioresponsive moieties, and each moiety is exposed, both of which must react to activate the aPS. 2) Sequential dual‐lock: aPS contains two bioresponsive moieties linked in a sequential manner, requiring activation at the first site to enable subsequent activation at the second, thus unlocking the photosensitizer in a stepwise process.
With regards to parallel dual‐lock, Tam et al. designed a dual‐locked PMB based on the parallel logic gate, using a DSBDP‐based photosensitizer and a BHQ3, linked by two enzyme‐sensitive peptides (PLGVR for MMP‐2 and GFLG for cathepsin B) (Figure 7a). [126] Complete unquenching of the PMB was achieved only through dual enzyme cleavage, which disabled FRET. The MMP‐2 responsiveness was validated in buffer solution and in vitro, demonstrating that PMB exhibited greater phototoxicity in MMP‐2‐positive cancer cells than in MMP‐2‐negative cancer cells. The dual‐locking effect was assased using MMP‐2‐positive cells treated with an MMP‐2 inhibitor, a cathepsin B inhibitor, or both, showing that either inhibitor alone or in combination reduced photoactivity. In an A549 mouse model, only tumours treated with dual‐locked PMB PDT exhibited significant antitumour effects compared to co‐treatment with dual‐locked PMB and either enzyme inhibitor.
Figure 7.

Examples of dual‐lock aPS. (a) Molecular structure of the dual‐locked PMB 1 and its dual‐enzyme‐unlocked mechanism. Reproduced with permission from Ref. [126]. Copyright 2023, American Chemical Society. (b) Schematic illustration for the hierarchical disassembly of PEG=R‐BDP‐NEt NPs for enhanced tumour penetration and activatable PDT. Reproduced with permission from Ref. [150]. Copyright 2021, Elsevier Ltd.
Zhang et al. introduced a sequential dual‐lock strategy for TME and lysosome‐specific activation of aPS (Figure 7b). [150] They designed BODIPY‐loaded nanoparticles using a self‐assembled “one‐for‐all” building block (PEG=RGD‐BDP‐NEt), where the BODIPY photosensitizer was quenched via PeT from a conjugated diethylamino (Net) group. In the mildly acidic TME (pH ~6.5), hydrolysis of the Schiff base bond between polyethylene glycol (PEG) and cyclic Arg‐Gly‐Asp peptide (cRGD) caused a one‐step size reduction, exposing the cRGD group for integrin αvβ3 receptor‐mediated binding and internalization. Within lysosomes, a subsequent size reduction occurred as the diethylamino group was protonated, blocking PeT and restoring fluorescence and PDT activity. This hierarchical degradation‐delivery strategy enhanced internalization by exposing the targeting moiety and reducing nanoparticle size in the weakly acidic TME. The photosensitizer was subsequently activated within the lysosome, providing a more precisely controlled activation mechanism. Biodistribution studies in tumour‐bearing mice demonstrated that PEG=RGD‐BDP‐NEt nanoparticles achieved higher tumour accumulation and lower off‐target uptake compared to single‐lock cRGD‐BDP‐NEt (unprotected cRGD) and PEG‐BDP‐NEt (lacking cRGD). While the precise contribution of acidic activation to tumour‐specific delivery was not fully quantified, Zhang et al. demonstrated the efficacy of a stepwise acid‐activated design for enhanced photosensitizer delivery and activation in tumours.
3.2. Targeted aPS
An additional approach to improving aPS sensitivity is to decorate them with targeting ligands. This approach combines the strengths of aPS and targeted photosensitizers, faciliting both enhanced tumour‐specific accumulation and tumour‐specific activation to reduce off‐target effects. For example, nanoparticles can be engineered with an outer layer containing targeting ligands such as biotin or folic acid to enhance tumour‐selective uptake. The inner core houses an aPS that can be activated through many of the aforementioned quenching/biostiumi mechanisms. This approach may particularly be useful for addressing the ubiquitous presence of GSH in healthy and tumour tissue. Targeting moieties and supramolecular encapsulation of the aPS can facilitate intracellular aPS trafficking, preventing unintended activation by extracellular GSH and facilitating intracellular activation. As such combined use of aPS and targeting moieties may improve PDT selectivity at a macro and microscopic level. Several photosensitizers utilizing this combo strategy have been reported, often employing self‐assembled aPS nanoparticles with targeting moieties for ligands upregulated by tumours (Figure 8a, b).[ 251 , 252 , 253 ]
Figure 8.

Representative work of aPS with targeting moiety. (a) Molecular structure of MBP. Reproduced with permission from Ref. [251]. Copyright 2022, Elsevier Ltd. (b) Schematic diagram of DOX@FA‐PEG‐ss‐PLL (‐g‐Ce6) nanoparticles (DOX@FPLC NPs). Reproduced with permission from Ref. [252]. Copyright 2022, American Chemical Society. (c) Molecular structure of the biotinylated ZnPc–CA4 conjugate. Reproduced with permission from Ref. [254]. Copyright 2020, American Chemical Society.
Combining aPS targeting with a dual‐lock strategy can further improve specificity. Tam et al. developed an aPS system featuring a trimeric phthalocyanine scaffold with three ZnPc units, each substituted with a GSH‐responsive 2,4‐dinitrobenzenesulfonate (DNBS) quencher and linked via cathepsin B‐cleavable GFLG peptide chains (Figure 8c). [254] A cyclic peptide targeting the epidermal growth factor receptor (EGFR), overexpressed in many cancers, served as the targeting moiety. The photosensitizer, named PMB 16, was quenched due to PeT from the DNBS group and ACQ. Upon receptor‐mediated endocytosis, the presence of intracellular GSH and cathepsin B restored fluorescence emission and 1O2 production. The authors evaluated the contributions of EGFR‐mediated internalization and activation by cathepsin B and GSH. They found that the combination of all three factors resulted in a significant increase in intracellular activation, exceeding the sum of the individual effects. Fluorescence recovery was assessed under four conditions: (i) absence of both cathepsin B and GSH, (ii) exposure to cathepsin B only, (iii) exposure to GSH only, and (iv) exposure to both. Negligible fluorescence was observed in condition (i), while condition (iv) showed a 75 % fluorescence recovery, indicating the dominant role of the PeT quenching effect of the DNBS group. In vitro studies confirmed the binding affinity of the cyclic peptide to EGFR‐overexpressing cancer cells. EGFR‐positive cells exhibited intracellular fluorescence up to six times higher than EGFR‐negative cells. Further experiments using inhibitors and enhancers of cathepsin B and GSH demonstrated up to a 48‐fold increase in intracellular fluorescence when comparing all positive conditions to negative controls. This enhancement was significantly greater than previously reported systems activated by a single stimulus. Photocytotoxicity experiments showed a much lower survival rate in EGFR‐positive cells compared to EGFR‐negative cells, confirming the enhanced therapeutic effect due to activation by both GSH and cathepsin B. Although PMB 16 has not been tested in vivo, this study underscores the potential of combining targeting moieties with dual‐lock activation to improve activatable PDT specificity and efficacy.
3.3. Artificial Receptor‐Responsive aPS
An innovative approach to simultaneously achieve selectivity and targeted activation in PDT is the design of artificial receptor‐responsive aPS. Traditional methods often utilize intracellular proteases as activation stimuli; however, overexpression of these enzymes in tumour cells does not preclude their presence in normal cells. If the difference in expression levels between cancerous and normal cells is minimal, off‐target activation and associated cytotoxicity can still occur.
Bioorthogonal chemistry has opened new avenues for highly selective and efficient cancer therapy, particularly in PDT. [255] Way et al. developed a method to enhance the effectiveness of PDT by introducing bioorthogonal moieties into tumour acid‐activatable photosensitizers to anchor the photosensitizer to the cell membrane. [256] Their strategy involves anchoring an artificial receptor containing azide groups to the tumour cell membrane. pH‐sensitive nanoparticles modified with dibenzocyclooctyne (DBCO) and chlorin e6 (Ce6) accumulate in tumour tissue and degrade in the acidic environment, exposing DBCO and Ce6. A bioorthogonal click reaction between DBCO and the azide groups immobilizes Ce6 on the tumour cell surface. While this method improves cell membrane anchoring, it does not strictly activate the photosensitizer via bioorthogonal reactions, as fluorescence and ROS production are already initiated upon nanoparticle degradation.
In contrast, Linden et al. were the first to achieve ROS generation activated directly by bioorthogonal reactions (Figure 9a). [257] They designed tetrazine‐modified photosensitizers that are initially inactivated but can be reactivated through an inverse electron‐demand Diels–Alder reaction with a dienophile. The authors synthesized nine different tetrazine–BODIPY probes to explore the fluorescence quenching mechanism. Computational results suggested that the introduction of tetrazine decreased both the ΦF and ΦΔ due to an additional non‐radiative decay channel. Additionally, the tetrazine units enabled organelle‐specific localization of the photosensitizer, providing spatial control. This approach allows spatial control and organelle‐specific localization of the photosensitizer, enhancing selectivity in PDT.
Figure 9.

Representative work of artificial receptor‐responsive aPS. (a) Outline of the DNA‐targeted bioorthogonal strategy for activating the phototoxicity. Reproduced with permission from Ref. [256]. Copyright 2019, Wiley‐VCH Verlag GmbH & Co. KGaA. (b) Chemical structure and activation mechanism of dual‐locked enzyme‐activatable bioorthogonal targeting agent (DBTA) and bioorthogonally activatable probe (BAP). Reproduced with permission from Ref. [257]. Copyright 2024, American Chemical Society.
Building on this concept, Wang et al. utilized tetrazine moieties for selective imaging of senescent cancer cells by combining bioorthogonal targeting with dual‐lock strategies (Figure 9b). [258] They designed a tetrazine derivative trapped by two enzyme‐cleavable moieties associated with senescence and cancer, along with a bioorthogonally activatable fluorescent probe. This double‐locking enzyme‐activated bioorthogonal fluorescence turn‐on imaging probe was selectively activated in senescent cancer cells, exhibiting significantly higher fluorescence intensity compared to non‐senescent cancer cells (4.48‐fold) and non‐cancerous senescent cells (4.30‐fold). In mouse models, stronger NIR fluorescence was detected in tumours from mice treated with senescence‐inducing chemotherapy compared to untreated mice (1.65‐fold) and those receiving senescence‐abrogating therapy (2.29‐fold). While this work focused on imaging, it underscores the potential of developing dual‐locking enzyme‐activated bioorthogonal activatable PDT agents, inspired by the aforementioned work of Linden et al.
4. Summary and Outlook
aPS were first explored in the context of ‘photosensitizing beacons’ in 2004. [89] Since then, they have undergone much evolution to facilitate disease‐specific PDT. The overarching design principle of aPS is to quench a photosensitizer′s photoreactivity in off‐target tissue and then activate it through disease‐specific stimuli to turn this quenching mechanism off. As reviewed, quenching mechanisms underlying aPS include FRET, PeT, ICT, ACQ, and AIE. Originally derived from activatable fluorescent probe designs, these mechanisms focus on quenching fluorescence but also impact ISC efficiency and ROS production. Specifically, energy or electron transfer processes in FRET and PeT can influence the ISC pathway, while changes in absorption efficiency or wavelength due to ICT, ACQ, and AIE can interfere with excitation to the S1 state, thereby affecting ROS generation. Unquenching mechanisms reviewed include tumour‐specific biostimuli such as low pH, hypoxia in the TME, overexpressed enzymes in tumour cells, GSH, ROS, and cellular internalization. Bioresponsive constructs responding to these stimuli can be broadly classified into two main types: linker‐type constructs that cleave in response to a stimulus, and reactive moieties that are oxidized or reduced by the stimulus. Linker‐type constructs are mainly used in the form of connecting photosensitizers and quenchers via linkers that enable a FRET mechanism. On the other hand, oxidizing and reducing moieties are often directly connected to the photosensitizer, which itself acts as a quencher, and the photosensitizer is quenched by mechanisms such as PeT and ICT. A third construct strategy that is often used in conjunction with the above are supramolecular assemblies of aPS, predominantly in the form of nanoparticles, that are quenched via ACQ and activated through particle disassembly.
Despite this evolution, specificity and study design limitations challenge the further advancement of aPS. We discussed limitations of conventional aPS design strategies including non‐specific activation triggers, insufficient selectivity, inadequate control over activation localization, and means of overcoming such limitations. In addition to the advances described thus far, we hope to encourage readers to look towards the fields of medicinal chemistry and activatable fluorescence probes to apply lessons and advances to improve aPS designs. Additionally, though the focus of this review was placed on cancer, the most explored applications of aPS, it should be noted that the design constructs reviewed are relevant to other pathologies. We briefly summarize these potential future outlooks and study design considerations which may serve as next steps in advancing the field of activatable PDT.
4.1. Future Perspectives in aPS Development
While previous sections have discussed the design strategies of aPS and methods to overcome their limitations, broader developments in PDT offer opportunities to enhance overall therapeutic efficacy. In this section, we explore how these advancements can be integrated into aPS designs to improve therapeutic outcomes.
4.1.1. Embracing Type I PDT
PDT fundamentally involves three elements: light, photosensitizer, and oxygen. This inherent oxygen dependence limits the effectiveness of PDT in hypoxic TMEs. Although conventional hypoxia‐triggered aPS designs can improve photosensitizer specificity, they do not necessarily ensure effective PDT post‐activation in oxygen‐deficient environments. To overcome this limitation, several studies in the broader PDT field have turned towards Type I photosensitizers.[ 259 , 260 , 261 ] As a reminder, Type II PDT reactions produce 1O2, which react readily with unsaturated organic compounds, such as carbon‐carbon double bonds and aromatic hydrocarbons, through electrophilic addition and electron extraction. [262] On the other hand, type I PDT produces ⋅OH, which are stronger oxidants and are less restricted in their reaction profile than 1O2. For example, ⋅OH can react with a greater variety of biological molecules (including DNA, RNA, cholesterol, lipids, carbohydrates, proteins and antioxidants) at rate constants of 109 to 1010 M−1s−1. Thus, while both Type I and Type II PDT produce ROS in an oxygen‐dependent manner, the former can exert more potent cytotoxicity with the same amount of oxygen than the latter. Thus, application of photosensitizers that predominantly utilize Type I mechanisms is a logical strategy for developing more effective hypoxia‐specific aPS. As a result, work has been done to utilize Type I photosensitizers in aPS, and some of the work introduced in this review performs Type I PDT.[ 155 , 156 , 157 , 158 ]
4.1.2. Expanding Applications Beyond Cancer
PDT is not limited to tumour treatment but can also be applied to other diseases and antimicrobial applications that could benefit from aPS designs. In fact, as can be seen in Tables 4 and 6, many of the biostimuli used to trigger aPS for cancer therapy are shared by other pathologies. For example, MMP′s are upregulated not only in cancer but also in conditions like atherosclerosis and arthritis, where PDT could offer therapeutic benefits. Although limited studies have employed aPS for these conditions, activatable fluorophores have been investigated and could be adapted for activatable PDT. For instance, Walsh et al. designed a FRET‐based molecular beacon where the fluorescence and 1O2 generation of PPa were quenched by BHQ3 via a peptide linker substrate specific for MMP‐13, an enzyme upregulated in osteoarthritis. [263] Cleavage of the peptide by MMP‐13 led to fluorescence activation, with a 13‐fold increase in PPa fluorescence in solution and a 7‐fold higher fluorescence in the articular cartilage of mice in an osteoarthritis model. Although the differential activation between arthritic and healthy joints was not assessed, this example demonstrates the potential utility of applying aPS designs to non‐cancer pathologies.
In addition to the cancer‐specific biostimuli reviewed here, other targets may also be of interest when designing aPS.[ 264 , 265 ] For instance, hydrogen sulfide (H2S), alongside nitric oxide (NO), is a key gasotransmitter involved in regulating numerous physiological processes. Dysregulation of H2S levels has been implicated in various diseases, including cancer, Alzheimer′s disease, and diabetes, and is also associated with overexpression at sites of bacterial infection. A study from Wang et al. presented a multifunctional antimicrobial PDT platform activated by H2S. [266] Authors assembled copper(II) ions (Cu2+) with Ce6 to form a smart nanoassembly (Cu−Ce6 NA) exhibiting unique H2S‐activatable fluorescence and antimicrobial properties. The activation mechanism relied on the high reactivity between Cu2+ in the nanoassembly and excessive H2S produced by bacteria. This interaction induced degradation of the Cu−Ce6 NA within the bacterial infection microenvironment, leading to the release of Ce6 and restoration of its photodynamic capacity. Concurrently, the consumption of H2S amplified oxidative stress, enhancing the generation of 1O2 and boosting antimicrobial efficacy. Additionally, the generated copper polysulfides (CuxS) possessed intrinsic bactericidal effects, and the disturbance of bacterial sulfur metabolism provides further antimicrobial action. This report exemplified the utility of aPS for antimicrobial therapy and presents a compelling activation mechanism.
aPS, which enable spatiotemporal control of PDT, may also plays a crucial role in enhancing the efficacy of complementary non‐photoactive treatments. An example of this is in facilitating endosomal escape and improved drug delivery in target tissue. Li et al.′s work addresses key challenges in mRNA therapeutics by improving endosomal escape, a critical barrier to achieving high levels of protein expression. [267] The researchers developed a second near‐infrared (NIR‐II) lipid nanoparticle (LNP) system containing a pH‐activatable NIR‐II dye‐conjugated lipid (Cy‐lipid) to enhance mRNA delivery using a stimulus‐responsive photothermal‐promoted endosomal escape delivery strategy. In the acidic endosomal environment, Cy‐lipid becomes protonated, activating NIR‐II absorption for light‐to‐heat conversion upon 1064 nm laser irradiation. The heat generated induces morphological changes in the LNPs, enabling rapid endosomal escape, leading to a 3‐fold increase in enhanced green fluorescent protein mRNA translation compared to the non‐irradiated group. This broad applicability highlights the potential of activatable PDT beyond oncology.
4.1.3. Exploring Activation Beyond Light
A persistent limitation of PDT is the restricted tissue penetration of visible light, which hinders its application for treating deep‐seated tumours. To address this challenge, efforts have been directed toward developing PDT agents that can be activated by NIR II (1000–1700 nm) light[ 268 , 269 ] or leverage chemiluminescence,[ 269 , 270 ] Cherenkov light, or upconversion nanoparticle [271] to excite photosensitizers. Recent advancements in PDT field suggest the potential of using alternative external stimuli, such as ultrasound[ 272 , 273 ] and X‐ray, [274] to activate photosensitizers. While the precise mechanisms underlying these stimuli‐mediated activations remain unclear, their successful implementation could overcome the intrinsic limitations of PDT and expand its applicability to tumours located deep within tissues. Ultrasound in particular could also help overcome limitations in aPS target tissue delivery and specificity. When combined with microbubbles, therapeutic ultrasound has been shown to increase both the delivery and fluorescence activation of quenched photosensitizers in lipoprotein constructs within murine glioblastomas. [275] As such, therapeutic focused ultrasound may be used alone or in conjunction with photoexcitation as an advanced means of improving aPS activation. X‐rays can similarly be used alone or in concert synergistically with photoexcitation to facilitate more potent therapeutic effects.[ 276 , 277 ]
4.2. Study Design Consideration
The development and evaluation of aPS necessitate meticulous study design to accurately assess their efficacy, selectivity, and potential for clinical translation. Several critical factors must be considered to ensure that the observed outcomes truly reflect the performance of the aPS and are not confounded by experimental variables.
4.2.1. Distinction between Fluorescence and ROS Generation
Though fluorescence quenching is often conflated with ROS quenching, it is critical to note that fluorescence intensity does not directly indicate ROS production efficiency, as these represent competing decay pathways from the same initial excited state. The relative efficiency of ISC ultimately determines the balance between fluorescence emission and ROS generation. For example, in the Fu and Wang et al.′s paper, [137] fluorescence was over 13‐fold increased after activation but ROS was only 1.5‐fold increased. Since fluorescence emission and ISC are competitive processes sharing the S1 excited state, achieving simultaneously high fluorescence quantum yield and 1O2 quantum yield is inherently challenging. Therefore, researchers must clearly distinguish between the efficiency of fluorescence and ROS generation in aPS systems. Explicit reporting of both quantum yield or fold activation of ROS is essential for accurate characterization and comparison of aPS performance.
4.2.2. Use of Appropriate Non‐activatable Controls
The evaluation of aPS selectivity and activation efficiency requires the inclusion of appropriate non‐activatable controls in study designs. Often, the improvement in selectivity due to the activatable function is not clearly quantified because the control group is a conventional photosensitizer that is always active. While this provides a baseline comparison, it does not account for factors such as differences in cellular uptake, biodistribution, or inherent cytotoxicity unrelated to activation mechanisms. Including structurally similar, non‐activatable analogs of the aPS allows for a more accurate assessment of the specific advantages conferred by the activatable design. This approach helps to isolate the effects of activation from other variables and provides clearer insights into the selectivity and efficacy improvements attributable to the aPS. It also enables the identification of any unintended off‐target effects or background activity that may compromise therapeutic specificity.
4.2.3. Localization of Activation
Precise control over the location of aPS activation is crucial for maximizing therapeutic outcomes while minimizing off‐target effects. This is particularly important for pH‐triggered aPS, where the activation may occur extracellularly in the TME or intracellularly within acidic organelles like lysosomes. Studies should include detailed investigations into the localization of activation using appropriate imaging and analytical techniques. Determining the pKa of the bioresponsive moiety is essential to predict and control the activation site. For instance, a bioresponsive group with a pKa close to 6.5 is more likely to be activated in the mildly acidic TME, whereas a lower pKa may result in activation within lysosomes. Subcellular localization studies, possibly involving confocal microscopy or subcellular fractionation, can confirm where activation occurs. Understanding the activation localization informs the therapeutic mechanism and aids in optimizing aPS design for improved selectivity and efficacy.
4.2.4. Biodistribution and Pharmacokinetic Studies
Differentiating between enhanced therapeutic effects due to increased aPS activation versus increased uptake is critical for interpreting results accurately. Biodistribution and pharmacokinetic studies provide valuable information on the accumulation and retention of aPS in tumour and normal tissues over time. Quantitative analysis of aPS concentration in various organs helps delineate whether observed efficacy is a result of selective activation or merely due to higher uptake by tumour cells. Techniques such as tissue homogenate fluorescence spectroscopy, radiolabeling, or mass spectrometry can be employed to measure aPS distribution. Importantly, fluorescence imaging alone should not be used for biodistribution analysis as in activatable systems, it does not accurately quantify absolute delivery (it only demonstrates delivery of activated agent). Pharmacokinetic profiles reveal how quickly the aPS is cleared from the body and its potential to accumulate in off‐target tissues. These studies are essential for assessing the safety profile of the aPS and for designing dosing regimens that maximize tumour targeting while minimizing systemic exposure.
4.2.5. In Vivo PDT Studies
While in vitro experiments provide initial insights into aPS function, they cannot fully replicate the complexity of living organisms. In vivo PDT studies are essential to evaluate not only the therapeutic efficacy but also the safety and potential off‐target phototoxicity of aPS. It is important to assess both tumour regression and possible adverse effects on healthy tissues, particularly in areas adjacent to the tumour or in organs that are sensitive to light exposure. Off‐target phototoxicity is a key concern in PDT and a primary motivation for developing aPS; however, it is often underrepresented in studies. Evaluating skin photosensitivity and damage to vital organs near the tumour site provides a more comprehensive safety assessment. Additionally, the use of orthotopic tumour models is recommended over subcutaneous models. Orthotopic models more accurately mimic the TME and its interactions with surrounding vital structures such as the brain, heart, or lungs. This approach offers a realistic assessment of the aPS′s therapeutic window and its potential for clinical application.
4.3. Conclusion
Although PDT has been approved for cancer treatment for over 30 years, it has not yet become a standard treatment modality. A critical reason for this is the off‐target effects caused by photosensitizers that are constitutively active, leading to damage in healthy tissues. To address this issue, considerable efforts have been made to develop photosensitizers with targeting functions. Among these, aPS are particularly effective in reducing side effects by suppressing photosensitizer activity in non‐target areas.
In this review, we have established the basic concepts of designing aPS by closely examining quenching mechanisms and bioresponsive constructs essential for their function. We have also summarized new aPS strategies developed in the last five years that address the shortcomings of conventional aPS and which enhance selectivity. The activatable strategy may allow for wider clinical translation of photosensitizers, and combined with targeted delivery, offering the potential for highly selective cancer treatment while minimizing side effects.
Conflict of Interests
The authors declare no conflict of interest.
5.
Biographical Information
Dr. Nahyun Kwon is a postdoctoral researcher at the Princess Margaret Cancer Centre at the University Health Network. She earned her Ph.D in Chemistry and Nanoscience from Ewha Womans University in South Korea in 2020. Dr. Kwon′s research journey began with the development of fluorescent probes for disease diagnosis, and she now focuses on designing image‐guided drug delivery systems for photodynamic therapy. Her long‐term goal is to advance theranostic strategies that integrate disease diagnosis and treatment while minimizing side effects, contributing to innovative solutions in precision medicine.

Biographical Information
Dr. Maneesha Rajora is an M.D./Ph.D candidate within the University of Toronto Department of Medicine, former trainee and current research collaborator with Dr. Gang Zheng′s research group at the Princess Margaret Cancer Centre. She completed her B.Sc. (H) in Chemistry at Dalhousie University, M.A.Sc. and Ph.D. in Biomedical Engineering at the University of Toronto and Postdoctoral Fellowship in nanomedicine and microbubble‐enabled therapeutic focused ultrasound at the University Health Network.

Biographical Information
Hanyi Weng is a Ph.D candidate in Medical Biophysics at the University of Toronto. She completed her B.Sc. (H) in Neuroscience at the University of Toronto in 2022 and is completing her Ph.D. training under the supervision of Dr. Gang Zheng.

Biographical Information
Dr. Gang Zheng is a Professor and Tier 1 Canada Research Chair in Cancer Nanomedicine at the University of Toronto. He is also the Associate Research Director of the Princess Margaret Cancer Center. He obtained his BSc in 1988 from Hangzhou University and Ph.D. in 1999 from SUNY Buffalo. Following a postdoctoral training in P.D.T. at the Roswell Park Cancer Institute, he joined the University of Pennsylvania in 2001 as an Assistant Professor of Radiology and moved to Canada in 2006. His lab is best known for introducing activatable photosensitizers for P.D.T. and porphysome nanotechnology as cancer theranostics.

Acknowledgments
The author expresses gratitude to Juan Chen for her invaluable assistance in proofreading and providing insightful feedback. This work is supported by the Terry Fox New Frontiers Program Project Grant 1137, Canadian Institute of Health Research PJT190047 and 190093, Canada Research Chairs Program 950‐232468 and Princess Margaret Cancer Foundation.
Dedicated to Professor Brian C. Wilson on the occasion of his 80th birthday.
Kwon N., Weng H., Rajora M. A., Zheng G., Angew. Chem. Int. Ed. 2025, 64, e202423348. 10.1002/anie.202423348
Contributor Information
Dr. Maneesha A. Rajora, Email: maneesha.rajora@mail.utoronto.ca.
Dr. Gang Zheng, Email: gang.zheng@uhn.ca.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.W. M. Sharman, C. M. Allen, J. E. van Lier, in Methods in Enzymology, Academic Press, 2000, pp. 376–400. [DOI] [PubMed]
- 2. Deda D. K., Pavani C., Caritá E., Baptista M. S., Toma H. E., Araki K., J. Biomed. Nanotechnol. 2013, 9, 1307–1317. [DOI] [PubMed] [Google Scholar]
- 3. Guo W., Li Z., Huang H., Xu Z., Chen Z., Shen G., Li Z., Ren Y., Li G., Hu Y., ACS Appl. Mater. Interfaces 2022, 14, 17008–17021. [DOI] [PubMed] [Google Scholar]
- 4. Dos Santos Rodrigues B., Lakkadwala S., Kanekiyo T., Singh J., IJN 2019, 14, 6497–6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Domagala A., Stachura J., Gabrysiak M., Muchowicz A., Zagozdzon R., Golab J., Firczuk M., BMC Cancer 2018, 18, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hu Z.-C., Wang B., Zhou X.-G., Liang H.-F., Liang B., Lu H.-W., Ge Y.-X., Chen Q., Tian Q.-W., Xue F.-F., Jiang L.-B., Dong J., ACS Nano 2023, 17, 21153–21169. [DOI] [PubMed] [Google Scholar]
- 7. Yoon H.-E., Ahn M.-Y., Kim Y.-C., Yoon J.-H., J. Dent. Sci. 2022, 17, 1722–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmitt F.-J., Renger G., Friedrich T., Kreslavski V. D., Zharmukhamedov S. K., Los D. A., Kuznetsov V. V., Allakhverdiev S. I., Biochimica et Biophysica Acta (BBA) - Bioenergetics 2014, 1837, 835–848. [DOI] [PubMed] [Google Scholar]
- 9. Mikkelsen R. B., Wardman P., Oncogene 2003, 22, 5734–5754. [DOI] [PubMed] [Google Scholar]
- 10. Roots R., Okada S., Radiat. Res. 1975, 64, 306–320. [PubMed] [Google Scholar]
- 11. Orenstein A., Kostenich G., Roitman L., Shechtman Y., Kopolovic Y., Ehrenberg B., Malik Z., Br. J. Cancer 1996, 73, 937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Algorri J. F., Ochoa M., Roldán-Varona P., Rodríguez-Cobo L., López-Higuera J. M., Cancers 2021, 13, 4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin Y., Xie R., Yu T., Pharmaceutica 2024, 16, 729. [Google Scholar]
- 14. Ponnusamy C., Ayarivan P., Selvamuthu P., Natesan S., Curr. Drug Delivery 2024, 21, 683–696. [DOI] [PubMed] [Google Scholar]
- 15. Hirschberg H., Uzal F. A., Chighvinadze D., Zhang M. J., Peng Q., Madsen S. J., Lasers Surg. Med. 2008, 40, 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Youf R., Müller M., Balasini A., Thétiot F., Müller M., Hascoët A., Jonas U., Schönherr H., Lemercier G., Montier T., Gall T. L., Pharmaceutica 2021, 13, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Douplik A., Physics Procedia 2010, 5, 641–645. [Google Scholar]
- 18. Schipmann S., Müther M., Stögbauer L., Zimmer S., Brokinkel B., Holling M., Grauer O., Suero Molina E., Warneke N., Stummer W., J. Neurosurg. 2021, 134, 426–436. [DOI] [PubMed] [Google Scholar]
- 19. Dobson J., de Queiroz G. F., Golding J. P., The Veterinary Journal 2018, 233, 8–18. [DOI] [PubMed] [Google Scholar]
- 20. Lou J., Aragaki M., Bernards N., Kinoshita T., Mo J., Motooka Y., Ishiwata T., Gregor A., Chee T., Chen Z., Chen J., Kaga K., Wakasa S., Zheng G., Yasufuku K., Nat. Photonics 2021, 10, 3279–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lou J., Aragaki M., Bernards N., Chee T., Gregor A., Hiraishi Y., Ishiwata T., Leung C., Ding L., Kitazawa S., Koga T., Sata Y., Ogawa H., Chen J., Kato T., Yasufuku K., Zheng G., Biomaterials 2023, 292, 121918. [DOI] [PubMed] [Google Scholar]
- 22. Ito H., Shoji Y., Ueno M., Matsumoto K., Nakanishi I., Pharmaceutica 2023, 15, 2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aniogo E. C., George B. P., Abrahamse H., Int. J. Mol. Sci. 2021, 22, 13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spring B. Q., Rizvi I., Xu N., Hasan T., Photochem. Photobiol. Sci. 2015, 14, 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu X., Zhang H., Wang Y., Shiu B.-C., Lin J.-H., Zhang S., Lou C.-W., Li T.-T., Chem. Eng. J. 2022, 450, 138129. [Google Scholar]
- 26. Gilaberte Y., Rezusta A., Juarranz A., Hamblin M. R., Frontiers in Medicine 2021, 8, 788888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Allison R. R., Sibata C. H., Photodiagn. Photodyn. Ther. 2010, 7, 61–75. [DOI] [PubMed] [Google Scholar]
- 28. Zhou Z., Zhang L., Zhang Z., Liu Z., Asian J. Pharm. Sci. 2021, 16, 668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.C. Zane, G. D. Panfilis, P. Calzavara-Pinton, in Comprehensive Series in Photosciences (Eds.: P. Calzavara-Pinton, R.-M. Szeimies, B. Ortel), Elsevier, 2001, pp. 101–114.
- 30. Lightdale C. J., Heier S. K., Marcon N. E., McCaughan J. S., Gerdes H., Overholt B. F., Sivak M. V., Stiegmann G. V., Nava H. R., Gastrointestinal Endoscopy 1995, 42, 507–512. [DOI] [PubMed] [Google Scholar]
- 31. Mĺkvy P., Messmann H., Regula J., Conio M., Pauer M., Millson C. E., MacRobert A. J., Bown S. G., Neoplasma 1995, 42, 109–113. [PubMed] [Google Scholar]
- 32. Correia J. H., Rodrigues J. A., Pimenta S., Dong T., Yang Z., Pharmaceutica 2021, 13, 1332. [Google Scholar]
- 33. Baran T. M., Lasers Surg. Med. 2018, 50, 476–482. [DOI] [PubMed] [Google Scholar]
- 34. Hahn S. M., Putt M. E., Metz J., Shin D. B., Rickter E., Menon C., Smith D., Glatstein E., Fraker D. L., Busch T. M., Clin. Cancer Res. 2006, 12, 5464–5470. [DOI] [PubMed] [Google Scholar]
- 35. Sun H., Ong Y., Yang W., Sourvanos D., Dimofte A., Busch T. M., Singhal S., Cengel K. A., Zhu T. C., J. Biomed. Opt. 2024, 29, 018001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Friedberg J. S., Simone C. B., Culligan M. J., Barsky A. R., Doucette A., McNulty S., Hahn S. M., Alley E., Sterman D. H., Glatstein E., Cengel K. A., The Annals of Thoracic Surgery 2017, 103, 912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhu T. C., Liang X., Kim M. M., Finlay J. C., Dimofte A., Rodriguez C., Simone C. B., Friedberg J. S., Cengel K. A., Front. Phys. 2015, 3, 10.3389/fphy.2015.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fisher C., Ali Z., Detsky J., Sahgal A., David E., Kunz M., Akens M., Chow E., Whyne C., Burch S., Wilson B. C., Yee A., Clin. Cancer Res. 2019, 25, 5766–5776. [DOI] [PubMed] [Google Scholar]
- 39. Akens M. K., Yee A. J. M., Wilson B. C., Burch S., Johnson C. L., Lilge L., Bisland S. K., Photochem. Photobiol. 2007, 83, 1034–1039. [DOI] [PubMed] [Google Scholar]
- 40. Akens M. K., Hardisty M. R., Wilson B. C., Schwock J., Whyne C. M., Burch S., Yee A. J. M., Breast Cancer Res. Treat. 2010, 119, 325–333. [DOI] [PubMed] [Google Scholar]
- 41. Tronconi M., Colombo A., De Cesare M., Marchesini R., Woodburn K. W., Reiss J. A., Phillips D. R., Zunino F., Cancer Lett. 1995, 88, 41–48. [DOI] [PubMed] [Google Scholar]
- 42. Zeng R., Liu C., Li L., Cai X., Chen R., Li Z., Technol. Cancer Res. Treat. 2020, 19, 1533033820930335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gomer C. J., Rucker N., Mark C., Benedict W. F., Murphree A. L., Invest. Ophthalmol. Visual Sci. 1982, 22, 118–120. [PubMed] [Google Scholar]
- 44. Piacquadio D. J., Chen D. M., Farber H. F., J. F. Fowler Jr. , Glazer S. D., Goodman J. J., Hruza L. L., Jeffes E. W. B., Ling M. R., Phillips T. J., Rallis T. M., Scher R. K., Taylor C. R., Weinstein G. D., Arch. Dermatol. 2004, 140, 10.1001/archderm.140.1.41. [DOI] [PubMed] [Google Scholar]
- 45. van den Boogert J., van Hillegersberg R., de Rooij F. W. M., de Bruin R. W. F., Edixhoven-Bosdijk A., Houtsmuller A. B., Siersema P. D., Paul Wilson J. H., Tilanus H. W., J. Photochem. Photobiol. B 1998, 44, 29–38. [DOI] [PubMed] [Google Scholar]
- 46. Azzouzi A.-R., Barret E., Moore C. M., Villers A., Allen C., Scherz A., Muir G., De Wildt M., Barber N. J., Lebdai S., Emberton M., BJU Int. 2013, 112, 766–774. [DOI] [PubMed] [Google Scholar]
- 47. Mazor O., Brandis A., Plaks V., Neumark E., Rosenbach-Belkin V., Salomon Y., Scherz A., Photochem. Photobiol. 2005, 81, 342–351. [DOI] [PubMed] [Google Scholar]
- 48. Rhodes L. E., De Rie M., Enström Y., Groves R., Morken T., Goulden V., Wong G. A. E., Grob J.-J., Varma S., Wolf P., Arch. Dermatol. 2004, 140, 17–23. [DOI] [PubMed] [Google Scholar]
- 49. D'Cruz A. K., Robinson M. H., Biel M. A., Head Neck 2004, 26, 232–240. [DOI] [PubMed] [Google Scholar]
- 50. Cramers P., Ruevekamp M., Oppelaar H., Dalesio O., Baas P., Stewart F. A., Br. J. Cancer 2003, 88, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tamaoki M., Yokoyama A., Horimatsu T., Hirohashi K., Amanuma Y., Higuchi H., Mitani Y., Yoshioka M., Ohashi S., Muto M., Esophagus 2021, 18, 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ferrario A., Kessel D., Gomer C. J., Cancer Res. 1992, 52, 2890–2893. [PubMed] [Google Scholar]
- 53.Treatment of Age-related Macular Degeneration With Photodynamic Therapy (TAP) Study Group Arch. Ophthalmol. 1999, 117, 1329–1345. [PubMed]
- 54. Richter A. M., Cerruti-Sola S., Sternberg E. D., Dolphin D., Levy J. G., J. Photochem. Photobiol. B 1990, 5, 231–244. [DOI] [PubMed] [Google Scholar]
- 55. Cognetti D. M., Johnson J. M., Curry J. M., Kochuparambil S. T., McDonald D., Mott F., Fidler M. J., Stenson K., Vasan N. R., Razaq M. A., Campana J., Ha P., Mann G., Ishida K., Garcia-Guzman M., Biel M., Gillenwater A. M., Head Neck 2021, 43, 3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sato K., Watanabe R., Hanaoka H., Harada T., Nakajima T., Kim I., Paik C. H., Choyke P. L., Kobayashi H., Molecular Oncology 2014, 8, 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mitsunaga M., Ogawa M., Kosaka N., Rosenblum L. T., Choyke P. L., Kobayashi H., Nat. Med. 2011, 17, 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rajora M. A., Lou J. W. H., Zheng G., Chem. Soc. Rev. 2017, 46, 6433–6469. [DOI] [PubMed] [Google Scholar]
- 59. Lovell J. F., Liu T. W. B., Chen J., Zheng G., Chem. Rev. 2010, 110, 2839–2857. [DOI] [PubMed] [Google Scholar]
- 60. Luby B. M., Walsh C. D., Zheng G., Angew. Chem. Int. Ed. 2019, 58, 2558–2569. [DOI] [PubMed] [Google Scholar]
- 61. Li Z., Zhou Z., Wang Y., Wang J., Zhou L., Cheng H.-B., Yoon J., Coord. Chem. Rev. 2023, 493, 215324. [Google Scholar]
- 62. Wang X., Peng J., Meng C., Feng F., Chem. Sci. 2024, 15, 12234–12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nestoros E., Sharma A., Kim E., Kim J. S., Vendrell M., Nat. Chem. Rev. 2025, 9, 46–60. [DOI] [PubMed] [Google Scholar]
- 64. Zhao Y.-Y., Kim H., Nguyen V.-N., Jang S., Jun Jang W., Yoon J., Coord. Chem. Rev. 2024, 501, 215560. [Google Scholar]
- 65. Cheng P., Pu K., ACS Appl. Mater. Interfaces 2020, 12, 5286–5299. [DOI] [PubMed] [Google Scholar]
- 66. Zhou M., Liu X., Chen F., Yang L., Yuan M., Fu D.-Y., Wang W., Yu H., Biomed. Mater. 2021, 16, 042008. [DOI] [PubMed] [Google Scholar]
- 67. Han H.-H., Wang H.-M., Jangili P., Li M., Wu L., Zang Y., Sedgwick A. C., Li J., He X.-P., James T. D., Kim J. S., Chem. Soc. Rev. 2023, 52, 879–920. [DOI] [PubMed] [Google Scholar]
- 68. Zhang J., Ning L., Huang J., Zhang C., Pu K., Chem. Sci. 2020, 11, 618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lu S.-B., Wei L., He W., Bi Z.-Y., Qian Y., Wang J., Lei H., Li K., ChemistryOpen 2022, 11, e202200137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yu H., Chen B., Huang H., He Z., Sun J., Wang G., Gu X., Tang B. Z., Biosensors 2022, 12, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liu J., Shi J., Nie W., Wang S., Liu G., Cai K., Adv. Healthcare Mater. 2021, 10, 2001207. [DOI] [PubMed] [Google Scholar]
- 72. Zhang C., Pu K., Chem. Soc. Rev. 2020, 49, 4234–4253. [DOI] [PubMed] [Google Scholar]
- 73. Prajapati S., Hinchliffe T., Roy V., Shah N., Jones C. N., Obaid G., Pharmaceutica 2021, 13, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Luby B. M., Charron D. M., MacLaughlin C. M., Zheng G., Adv. Drug Delivery Rev. 2017, 113, 97–121. [DOI] [PubMed] [Google Scholar]
- 75. Sharma A., Verwilst P., Li M., Ma D., Singh N., Yoo J., Kim Y., Yang Y., Zhu J.-H., Huang H., Hu X.-L., He X.-P., Zeng L., James T. D., Peng X., Sessler J. L., Kim J. S., Chem. Rev. 2024, 124, 2699–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fujita K., Urano Y., Chem. Rev. 2024, 124, 4021–4078. [DOI] [PubMed] [Google Scholar]
- 77. Kobayashi H., Ogawa M., Alford R., Choyke P. L., Urano Y., Chem. Rev. 2010, 110, 2620–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li X., Kolemen S., Yoon J., Akkaya E. U., Adv. Funct. Mater. 2017, 27, 1604053. [Google Scholar]
- 79. Li X., Kim J., Yoon J., Chen X., Adv. Mater. 2017, 29, 1606857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ng K. K., Zheng G., Chem. Rev. 2015, 115, 11012–11042. [DOI] [PubMed] [Google Scholar]
- 81. Wu W., Shao X., Zhao J., Wu M., Adv. Sci. 2017, 4, 1700113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Li W., Zhang J., Gao Z., Qi J., Ding D., Coord. Chem. Rev. 2022, 471, 214754. [Google Scholar]
- 83. Förster T., Ann. Phys. 1948, 437, 55–75. [Google Scholar]
- 84. Wu L., Huang C., Emery B. P., Sedgwick A. C., Bull S. D., He X.-P., Tian H., Yoon J., Sessler J. L., James T. D., Chem. Soc. Rev. 2020, 49, 5110–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fang B., Shen Y., Peng B., Bai H., Wang L., Zhang J., Hu W., Fu L., Zhang W., Li L., Huang W., Angew. Chem. 2022, 134, e202207188. [DOI] [PubMed] [Google Scholar]
- 86. Loura L. M. S., Int. J. Mol. Sci. 2012, 13, 15252–15270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lovell J. F., Chen J., Jarvi M. T., Cao W.-G., Allen A. D., Liu Y., Tidwell T. T., Wilson B. C., Zheng G., J. Phys. Chem. B 2009, 113, 3203–3211. [DOI] [PubMed] [Google Scholar]
- 88. Corry B., Jayatilaka D., Rigby P., Biophys. J. 2005, 89, 3822–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chen J., Stefflova K., Niedre M. J., Wilson B. C., Chance B., Glickson J. D., Zheng G., J. Am. Chem. Soc. 2004, 126, 11450–11451. [DOI] [PubMed] [Google Scholar]
- 90. Niu H., Liu J., O'Connor H. M., Gunnlaugsson T., James T. D., Zhang H., Chem. Soc. Rev. 2023, 52, 2322–2357. [DOI] [PubMed] [Google Scholar]
- 91. de Silva A. P., Moody T. S., Wright G. D., Analyst 2009, 134, 2385. [DOI] [PubMed] [Google Scholar]
- 92. Escudero D., Acc. Chem. Res. 2016, 49, 1816–1824. [DOI] [PubMed] [Google Scholar]
- 93. Horiuchi H., Kuribara R., Hirabara A., Okutsu T., J. Phys. Chem. A 2016, 120, 5554–5561. [DOI] [PubMed] [Google Scholar]
- 94. Tian J., Ding L., Xu H.-J., Shen Z., Ju H., Jia L., Bao L., Yu J.-S., J. Am. Chem. Soc. 2013, 135, 18850–18858. [DOI] [PubMed] [Google Scholar]
- 95. Filatov M. A., Org. Biomol. Chem. 2020, 18, 10–27. [DOI] [PubMed] [Google Scholar]
- 96. Zhai W., Zhang Y., Liu M., Zhang H., Zhang J., Li C., Angew. Chem. Int. Ed. 2019, 58, 16601–16609. [DOI] [PubMed] [Google Scholar]
- 97. Baskaran R., Lee J., Yang S.-G., Biomaterials Research 2018, 22, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Li X., Lee S., Yoon J., Chem. Soc. Rev. 2018, 47, 1174–1188. [DOI] [PubMed] [Google Scholar]
- 99. Li X., Yu S., Lee Y., Guo T., Kwon N., Lee D., Yeom S. C., Cho Y., Kim G., Huang J.-D., Choi S., Nam K. T., Yoon J., J. Am. Chem. Soc. 2019, 141, 1366–1372. [DOI] [PubMed] [Google Scholar]
- 100. Hong Y., Lam J. W. Y., Zhong Tang B., Chem. Commun. 2009, 4332–4353. [DOI] [PubMed] [Google Scholar]
- 101. Zhao Z., Zhang H., Lam J. W. Y., Tang B. Z., Angew. Chem. Int. Ed. 2020, 59, 9888–9907. [DOI] [PubMed] [Google Scholar]
- 102. Luo J., Xie Z., Lam J. W. Y., Cheng L., Chen H., Qiu C., Kwok H. S., Zhan X., Liu Y., Zhu D., Tang B. Z., Chem. Commun. 2001, 1740–1741. [DOI] [PubMed] [Google Scholar]
- 103. Hong Y., Lam J. W. Y., Zhong Tang B., Chem. Soc. Rev. 2011, 40, 5361–5388. [DOI] [PubMed] [Google Scholar]
- 104. Mei J., Leung N. L. C., Kwok R. T. K., Lam J. W. Y., Tang B. Z., Chem. Rev. 2015, 115, 11718–11940. [DOI] [PubMed] [Google Scholar]
- 105. Hu F., Xu S., Liu B., Adv. Mater. 2018, 30, 1801350. [DOI] [PubMed] [Google Scholar]
- 106. Meng Z., Xue H., Wang T., Chen B., Dong X., Yang L., Dai J., Lou X., Xia F., J. Nanobiotechnol. 2022, 20, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yuan Y., Zhang C.-J., Gao M., Zhang R., Tang B. Z., Liu B., Angew. Chem. Int. Ed. 2015, 54, 1780–1786. [DOI] [PubMed] [Google Scholar]
- 108. DeBerardinis R. J., Chandel N. S., Nat. Metab. 2020, 2, 127–129. [DOI] [PubMed] [Google Scholar]
- 109. Corbet C., Feron O., Nat. Rev. Cancer 2017, 17, 577–593. [DOI] [PubMed] [Google Scholar]
- 110. Wang R., Kim K.-H., Yoo J., Li X., Kwon N., Jeon Y.-H., Shin S., Han S. S., Lee D.-S., Yoon J., ACS Nano 2022, 16, 3045–3058. [DOI] [PubMed] [Google Scholar]
- 111. Guo X., Jin H., Lo P.-C., Biomater. Sci. 2021, 9, 4936–4951. [DOI] [PubMed] [Google Scholar]
- 112. Zhang H., Pan J., Wang T., Lai Y., Liu X., Chen F., Xu L., Qu X., Hu X., Yu H., ACS Appl. Mater. Interfaces 2022, 14, 39787–39798. [DOI] [PubMed] [Google Scholar]
- 113. Sun B., Chang R., Cao S., Yuan C., Zhao L., Yang H., Li J., Yan X., van Hest J. C. M., Angew. Chem. Int. Ed. 2020, 59, 20582–20588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.P. Vaupel, A. Mayer, M. Höckel, in Methods in Enzymology, Academic Press, 2004, pp. 335–354. [DOI] [PubMed]
- 115. Challapalli A., Carroll L., Aboagye E. O., Clin. Transl. Imaging 2017, 5, 225–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Macdougall J. D. B., Mccabe M., Nature 1967, 215, 1173–1174. [DOI] [PubMed] [Google Scholar]
- 117. Wilson W. R., Hay M. P., Nat. Rev. Cancer 2011, 11, 393–410. [DOI] [PubMed] [Google Scholar]
- 118. Zoa A., Yang Y., Huang W., Yang J., Wang J., Wang H., Dong M., Tian Y., Translational Cancer Research 2022, 11, 3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sormendi S., Wielockx B., Front. Immunol. 2018, 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Yuan X., Xie Z., Zou T., Bioorg. Chem. 2024, 144, 107161. [DOI] [PubMed] [Google Scholar]
- 121. Wang C., Wang S., Wang Y., Wu H., Bao K., Sheng R., Li X., Sci. Rep. 2020, 10, 12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Liu D., Liang M., Tao Y., Liu H., Liu Q., Bing W., Li W., Qi J., Biomaterials 2024, 309, 122610. [DOI] [PubMed] [Google Scholar]
- 123. Piao W., Hanaoka K., Fujisawa T., Takeuchi S., Komatsu T., Ueno T., Terai T., Tahara T., Nagano T., Urano Y., J. Am. Chem. Soc. 2017, 139, 13713–13719. [DOI] [PubMed] [Google Scholar]
- 124. Xu F., Li H., Yao Q., Ge H., Fan J., Sun W., Wang J., Peng X., Chem. Sci. 2019, 10, 10586–10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zheng G., Chen J., Stefflova K., Jarvi M., Li H., Wilson B. C., Proc. Natl. Acad. Sci. USA 2007, 104, 8989–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Tam L. K. B., Chu J. C. H., He L., Yang C., Han K.-C., Cheung P. C. K., Ng D. K. P., Lo P.-C., J. Am. Chem. Soc. 2023, 145, 7361–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lioret V., Renault K., Maury O., Romieu A., Chem. Asian J. 2023, 18, e202300756. [DOI] [PubMed] [Google Scholar]
- 128. Xiong L.-H., Yang L., Geng J., Tang B. Z., He X., ACS Nano 2024, 18, 17837–17851. [DOI] [PubMed] [Google Scholar]
- 129. Cao C., Li J., Zhang X., Zhang X., Gong X., Wang S., Talanta 2024, 272, 125786. [DOI] [PubMed] [Google Scholar]
- 130. Miao J., Huo Y., Yao G., Feng Y., Weng J., Zhao W., Guo W., Angew. Chem. Int. Ed. 2022, 61, e202201815. [DOI] [PubMed] [Google Scholar]
- 131. Li X., Xu W., Yang Z., Li S., Gu X., Yuan T., Li C., Wang Y., Hua J., Sens. Actuators B 2023, 375, 132892. [Google Scholar]
- 132. Bansal A., Simon M. C., J. Cell Biol. 2018, 217, 2291–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Cao J.-J., Zhang M.-S., Li X.-Q., Yang D.-C., Xu G., Liu J.-Y., Eur. J. Med. Chem. 2020, 193, 112203. [DOI] [PubMed] [Google Scholar]
- 134. Liu D., Chen B., Mo Y., Wang Z., Qi T., Zhang Q., Wang Y., Nano Lett. 2019, 19, 6964–6976. [DOI] [PubMed] [Google Scholar]
- 135. He H., Lo P., Ng D. K. P., Chem. A Eur. J. 2014, 20, 6241–6245. [DOI] [PubMed] [Google Scholar]
- 136. Turan I. S., Cakmak F. P., Yildirim D. C., Cetin-Atalay R., Akkaya E. U., Chem. A Eur. J. 2014, 20, 16088–16092. [DOI] [PubMed] [Google Scholar]
- 137. Fu D., Wang Y., Lin K., Huang L., Xu J., Wu H., RSC Adv. 2023, 13, 22367–22374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Rajora M. A., Dhaliwal A., Zheng M., Choi V., Overchuk M., Lou J. W. H., Pellow C., Goertz D., Chen J., Zheng G., Adv. Sci. 2024, 11, 2304453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zhang Z., Cao W., Jin H., Lovell J., Yang M., Ding L., Chen J., Corbin I., Luo Q., Zheng G., Angew. Chem. Int. Ed. 2009, 48, 9171–9175. [DOI] [PubMed] [Google Scholar]
- 140. Lovell J. F., Jin C. S., Huynh E., MacDonald T. D., Cao W., Zheng G., Angew. Chem. Int. Ed. 2012, 51, 2429–2433. [DOI] [PubMed] [Google Scholar]
- 141. MacDonald T. D., Zheng G., Photonics Lasers Med. 2014, 3, 183–191. [Google Scholar]
- 142. Guidolin K., Ding L., Chen J., Wilson B. C., Zheng G., Nat. Photonics 2021, 10, 3161–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Ho T., Guidolin K., Makky A., Valic M., Ding L., Bu J., Zheng M., Cheng M. H. Y., Yau J., Chen J., Zheng G., Angew. Chem. Int. Ed. 2023, 62, e202218218. [DOI] [PubMed] [Google Scholar]
- 144. Jin C. S., Cui L., Wang F., Chen J., Zheng G., Adv. Healthcare Mater. 2014, 3, 1240–1249. [DOI] [PubMed] [Google Scholar]
- 145. Cui L., Lin Q., Jin C. S., Jiang W., Huang H., Ding L., Muhanna N., Irish J. C., Wang F., Chen J., Zheng G., ACS Nano 2015, 9, 4484–4495. [DOI] [PubMed] [Google Scholar]
- 146. Rajora M. A., Ding L., Valic M., Jiang W., Overchuk M., Chen J., Zheng G., Chem. Sci. 2017, 8, 5371–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Wang R., Kim K.-H., Yoo J., Li X., Kwon N., Jeon Y.-H., Shin S., Han S. S., Lee D.-S., Yoon J., ACS Nano 2022, 16, 3045–3058. [DOI] [PubMed] [Google Scholar]
- 148. Song R., Li T., Ye J., Sun F., Hou B., Saeed M., Gao J., Wang Y., Zhu Q., Xu Z., Yu H., Adv. Mater. 2021, 33, 2101155. [DOI] [PubMed] [Google Scholar]
- 149. Zhang H., Pan J., Wang T., Lai Y., Liu X., Chen F., Xu L., Qu X., Hu X., Yu H., ACS Appl. Mater. Interfaces 2022, 14, 39787–39798. [DOI] [PubMed] [Google Scholar]
- 150. Zhang Y., Zhao R., Liu J., Kong H., Zhang K., Zhang Y.-N., Kong X., Zhang Q., Zhao Y., Biomaterials 2021, 275, 120945. [DOI] [PubMed] [Google Scholar]
- 151. Yang Y., Zhu W., Cheng L., Cai R., Yi X., He J., Pan X., Yang L., Yang K., Liu Z., Tan W., Chen M., Biomaterials 2020, 246, 119971. [DOI] [PubMed] [Google Scholar]
- 152. Chen S., Li B., Yue Y., Li Z., Qiao L., Qi G., Ping Y., Liu B., Adv. Mater. 2024, 36, 2404296. [DOI] [PubMed] [Google Scholar]
- 153. Zheng J., Liu Y., Song F., Jiao L., Wu Y., Peng X., Chem. Commun. 2020, 56, 5819–5822. [DOI] [PubMed] [Google Scholar]
- 154. Liu Z., Song F., Shi W., Gurzadyan G., Yin H., Song B., Liang R., Peng X., ACS Appl. Mater. Interfaces 2019, 11, 15426–15435. [DOI] [PubMed] [Google Scholar]
- 155. Han F., Abbas Abedi S. A., He S., Zhang H., Long S., Zhou X., Chanmungkalakul S., Ma H., Sun W., Liu X., Du J., Fan J., Peng X., Adv. Sci. 2024, 11, 2305761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Zhang J., Zhang Y., Zhang H., Zhai W., Shi X., Li C., J. Mater. Chem. B 2023, 11, 4102–4110. [DOI] [PubMed] [Google Scholar]
- 157. Zeng Q., Li X., Li J., Shi M., Yao Y., Guo L., Zhi N., Zhang T., Adv. Sci. 2024, 11, 2400462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Tian J., Li B., Zhang F., Yao Z., Song W., Tang Y., Ping Y., Liu B., Angew. Chem. Int. Ed. 2023, 62, e202307288. [DOI] [PubMed] [Google Scholar]
- 159. Köhrmann A., Kammerer U., Kapp M., Dietl J., Anacker J., BMC Cancer 2009, 9, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Han L., Sheng B., Zeng Q., Yao W., Jiang Q., BMC Pulm. Med. 2020, 20, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Meng N., Li Y., Jiang P., Bu X., Ding J., Wang Y., Zhou X., Yu F., Zhang Y., Zhang J., Xia L., Front. Oncol. 2022, 12, 10.3389/fonc.2022.916907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Yu J., He Z., He X., Luo Z., Lian L., Wu B., Lan P., Chen H., Front. Oncol. 2021, 11, 10.3389/fonc.2021.771099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Stallivieri A., Colombeau L., Devy J., Etique N., Chaintreuil C., Myrzakhmetov B., Achard M., Baros F., Arnoux P., Vanderesse R., Frochot C., Bioorg. Med. Chem. 2018, 26, 688–702. [DOI] [PubMed] [Google Scholar]
- 164. Rudzinska-Radecka M., Frolova A. S., Balakireva A. V., Gorokhovets N. V., Pokrovsky V. S., Sokolova D. V., Korolev D. O., Potoldykova N. V., Vinarov A. Z., Parodi A., Zamyatnin A. A., Cells 2022, 11, 1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Werle B., Lötterle H., Schanzenbächer U., Lah T. T., Kalman E., Kayser K., Bülzebruck H., Schirren J., Krasovec M., Kos J., Spiess E., Br. J. Cancer 1999, 81, 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Abdulla M.-H., Valli-Mohammed M.-A., Al-Khayal K., Shkieh A. A., Zubaidi A., Ahmad R., Al-Saleh K., Al-Obeed O., McKerrow J., Oncol. Rep. 2017, 37, 3175–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Lu S., Lei X., Ren H., Zheng S., Qiang J., Zhang Z., Chen Y., Wei T., Wang F., Chen X., ACS Appl. Bio Mater. 2020, 3, 3835–3845. [DOI] [PubMed] [Google Scholar]
- 168. Wang Q., Yu L., Wong R. C. H., Lo P.-C., Eur. J. Med. Chem. 2019, 179, 828–836. [DOI] [PubMed] [Google Scholar]
- 169. Jain M., Bouilloux J., Borrego I., Cook S., van den Bergh H., Lange N., Wagnieres G., Giraud M.-N., Pharmaceuticals 2022, 15, 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Cheng Z., Benson S., Mendive-Tapia L., Nestoros E., Lochenie C., Seah D., Chang K. Y., Feng Y., Vendrell M., Angew. Chem. 2024, 136, e202404587. [DOI] [PubMed] [Google Scholar]
- 171. Nageshwari B., Merugu R., Indian J. Clin. Biochem. 2018, 34, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Hirano K., Domar U. M., Yamamoto H., Brehmer-Andersson E. E., Wahren B. E., Stigbrand T. I., Cancer Res. 1987, 47, 2543–2546. [PubMed] [Google Scholar]
- 173. Lam K. W. K., Chau J. H. C., Yu E. Y., Sun F., Lam J. W. Y., Ding D., Kwok R. T. K., Sun J., He X., Tang B. Z., ACS Nano 2023, 17, 7145–7156. [DOI] [PubMed] [Google Scholar]
- 174. Zhao C., Sun W., Huang X., Liu Y., Wang H.-Y., J. Med. Chem. 2024, 67, 13383–13391. [DOI] [PubMed] [Google Scholar]
- 175. Zhang Y., Zhao M., Miao J., Gu W., Zhu J., Cheng B., Li Q., Miao Q., ACS Materials Lett. 2023, 5, 3058–3067. [Google Scholar]
- 176. Xiong L.-H., Yang L., Geng J., Tang B. Z., He X., ACS Nano 2024, 18, 17837–17851. [DOI] [PubMed] [Google Scholar]
- 177. Li Y., Zhang C., Wu Q., Peng Y., Ding Y., Zhang Z., Xu X., Xie H., Angew. Chem. Int. Ed. 2024, 63, e202317773. [DOI] [PubMed] [Google Scholar]
- 178. Lioret V., Renault K., Maury O., Romieu A., Chem. Asian J. 2023, 18, e202300756. [DOI] [PubMed] [Google Scholar]
- 179. Ji S., Gao H., Mu W., Ni X., Yi X., Shen J., Liu Q., Bao P., Ding D., J. Mater. Chem. B 2018, 6, 2566–2573. [DOI] [PubMed] [Google Scholar]
- 180. Luo Z., Chen Y., Chen B., Zhao Z., Wu R., Ren J., BMC Medical Genomics 2024, 17, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Kawakami K., Fujita Y., Matsuda Y., Arai T., Horie K., Kameyama K., Kato T., Masunaga K., Kasuya Y., Tanaka M., Mizutani K., Deguchi T., Ito M., BMC Cancer 2017, 17, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Yao D., Jiang D., Huang Z., Lu J., Tao Q., Yu Z., Meng X., Cancer 2000, 88, 761–769. [PubMed] [Google Scholar]
- 183. Miao J., Huo Y., Yao G., Feng Y., Weng J., Zhao W., Guo W., Angew. Chem. Int. Ed. 2022, 61, e202201815. [DOI] [PubMed] [Google Scholar]
- 184. Li J., Wang T., Jiang F., Hong Z., Su X., Li S., Han S., J. Mater. Chem. B 2021, 9, 5829–5836. [DOI] [PubMed] [Google Scholar]
- 185. Liu F., Zhu D., Li Y., Kong M., Li Y., Luo J., Kong L., Sens. Actuators B 2022, 363, 131838. [Google Scholar]
- 186. Yang Z., Xu W., Wang J., Liu L., Chu Y., Wang Y., Hu Y., Yi T., Hua J., J. Mater. Chem. C 2020, 8, 8183–8190. [Google Scholar]
- 187. Yang Y., Zhang Y., Wu Q., Cui X., Lin Z., Liu S., Chen L., J. Exp. Clin. Cancer Res. 2014, 33, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Marín A., Lopez de Cerain A., Hamilton E., Lewis A. D., Martinez-Peñuela J. M., Idoate M. A., Bello J., Br. J. Cancer 1997, 76, 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Cao C., Li J., Zhang X., Zhang X., Gong X., Wang S., Talanta 2024, 272, 125786. [DOI] [PubMed] [Google Scholar]
- 190. Li Z., Feng Q., Hou J., Shen J., Bioorg. Chem. 2024, 143, 107021. [DOI] [PubMed] [Google Scholar]
- 191. Xue E. Y., Kang F., Zhou Y., Ng D. K. P., Chem. Commun. 2023, 59, 7056–7059. [DOI] [PubMed] [Google Scholar]
- 192. Asanuma D., Sakabe M., Kamiya M., Yamamoto K., Hiratake J., Ogawa M., Kosaka N., Choyke P. L., Nagano T., Kobayashi H., Urano Y., Nat. Commun. 2015, 6, 6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Wagner J., Damaschke N., Yang B., Truong M., Guenther C., McCormick J., Huang W., Jarrard D., PLoS One 2015, 10, e0124366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Almammadov T., Elmazoglu Z., Atakan G., Kepil D., Aykent G., Kolemen S., Gunbas G., ACS Appl. Bio Mater. 2022, 5, 4284–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Chu J. C. H., Xiong J., Wong C. T. T., Wang S., Tam D. Y., García-Fernández A., Martínez-Máñez R., Ng D. K. P., J. Med. Chem. 2024, 67, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Shi W., Ng D. K. P., Zhao S., Lo P., ChemPhotoChem 2019, 3, 1004–1013. [Google Scholar]
- 197. Zeng Q., Li X., Xie S., Xing D., Zhang T., Biomaterials 2022, 290, 121867. [DOI] [PubMed] [Google Scholar]
- 198. Li B., Tian J., Xie X., Zhang F., Wu C., Shan Y., Qi G., Song W., Ping Y., Liu B., Adv. Funct. Mater. 2024, 34, 2309524. [Google Scholar]
- 199. Li Z., Liu Y., Chen L., Hu X., Xie Z., J. Mater. Chem. B 2017, 5, 4239–4245. [DOI] [PubMed] [Google Scholar]
- 200. Wu Q., Li Y., Li Y., Wang D., Tang B. Z., Mater. Chem. Front. 2021, 5, 3489–3496. [Google Scholar]
- 201. Wang Z.-W., Su D., Li X.-Q., Cao J.-J., Yang D.-C., Liu J.-Y., Molecules 2018, 24, 32.30577688 [Google Scholar]
- 202. Almammadov T., Kolemen S., Dyes Pigm. 2021, 193, 109499. [Google Scholar]
- 203. Barretta P., Ponte F., Mazzone G., J. Mol. Liq. 2024, 402, 124761. [Google Scholar]
- 204. Hu W., Xie M., Zhao H., Tang Y., Yao S., He T., Ye C., Wang Q., Lu X., Huang W., Fan Q., Chem. Sci. 2018, 9, 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Yin C.-X., Xiong K.-M., Huo F.-J., Salamanca J. C., Strongin R. M., Angew. Chem. Int. Ed. 2017, 56, 13188–13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Niu L.-Y., Chen Y.-Z., Zheng H.-R., Wu L.-Z., Tung C.-H., Yang Q.-Z., Chem. Soc. Rev. 2015, 44, 6143–6160. [DOI] [PubMed] [Google Scholar]
- 207. Jung H. S., Chen X., Kim J. S., Yoon J., Chem. Soc. Rev. 2013, 42, 6019–6031. [DOI] [PubMed] [Google Scholar]
- 208. Jia Q., Zhang R., Yan H., Feng Y., Sun F., Yang Z., Qiao C., Mou X., Tian J., Wang Z., Angew. Chem. Int. Ed. 2023, 62, e202313420. [DOI] [PubMed] [Google Scholar]
- 209. Zhou K., Liu H., Zhang S., Huang X., Wang Y., Huang G., Sumer B. D., Gao J., J. Am. Chem. Soc. 2012, 134, 7803–7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Gamcsik M. P., Kasibhatla M. S., Teeter S. D., Colvin O. M., Biomarkers 2012, 17, 671–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Webb B. A., Chimenti M., Jacobson M. P., Barber D. L., Nat. Rev. Cancer 2011, 11, 671–677. [DOI] [PubMed] [Google Scholar]
- 212. Kellum J. A., Song M., Li J., Critical Care 2004, 8, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Bennison L. R., Miller C. N., Summers R. J., Minnis A. M. B., Sussman G., McGuiness W., Wound Practice Res. 2017, 25. [Google Scholar]
- 214. Liu C.-L., Zhang X., Liu J., Wang Y., Sukhova G. K., Wojtkiewicz G. R., Liu T., Tang R., Achilefu S., Nahrendorf M., Libby P., Guo J., Zhang J.-Y., Shi G.-P., Nat. Commun. 2019, 10, 3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Decker Y., Németh E., Schomburg R., Chemla A., Fülöp L., Menger M. D., Liu Y., Fassbender K., Neurobiol. Aging 2021, 101, 40–49. [DOI] [PubMed] [Google Scholar]
- 216. Brooks S. G., Mahmoud R. H., Lin R. R., Fluhr J. W., Yosipovitch G., J. Invest. Dermatol. 2024, 10.1016/j.jid.2024.07.009. [DOI] [PubMed] [Google Scholar]
- 217. Hu Y.-B., Dammer E. B., Ren R.-J., Wang G., Translational Neurodegeneration 2015, 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Pircher T., Wackerhage H., Aszodi A., Kammerlander C., Böcker W., Saller M. M., Front. Plant Physiol. 2021, 12, 684899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Pham K., Parikh K., Heinrich E. C., Front. Plant Physiol. 2021, 12, 10.3389/fphys.2021.676782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Tarbell J., Mahmoud M., Corti A., Cardoso L., Caro C., J. R. Soc. Interface 2020, 17, 20190732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Foresto-Neto O., da Silva A. R. P. A., Cipelli M., Santana-Novelli F. P. R., Camara N. O. S., Kidney Research Clinical Practice 2023, 42, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Nath B., Szabo G., Hepatology (Baltimore, Md.) 2012, 55, 622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Catrina S.-B., Zheng X., Diabetologia 2021, 64, 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Yunus M. H. M., Lee Y., Nordin A., Chua K. H., Idrus R. B. H., Int. J. Mol. Sci. 2022, 23, 5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Morikawa T., Takubo K., Pfluegers Arch. 2016, 468, 13–22. [DOI] [PubMed] [Google Scholar]
- 226. Blikslager A. T., Gastroenterology 2008, 134, 346–348. [DOI] [PubMed] [Google Scholar]
- 227. Weinberg F., Ramnath N., Nagrath D., Cancers 2019, 11, 1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Mittal M., Siddiqui M. R., Tran K., Reddy S. P., Malik A. B., Antioxid. Redox Signaling 2014, 20, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Batty M., Bennett M. R., Yu E., Cells 2022, 11, 3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Lee Y. M., He W., Liou Y.-C., Cell Death Dis. 2021, 12, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Ha H., Hwang I.-A., Park J. H., Lee H. B., Diabetes Res. Clin. Pract. 2008, 82, S42–S45. [DOI] [PubMed] [Google Scholar]
- 232. Allameh A., Niayesh-Mehr R., Aliarab A., Sebastiani G., Pantopoulos K., Antioxidants 2023, 12, 1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Palma F. R., Gantner B. N., Sakiyama M. J., Kayzuka C., Shukla S., Lacchini R., Cunniff B., Bonini M. G., Oncogene 2024, 43, 295–303. [DOI] [PubMed] [Google Scholar]
- 234. Sandalio L. M., Rodríguez-Serrano M., Romero-Puertas M. C., del Río L. A., Subcell. Biochem. 2013, 69, 231–255. [DOI] [PubMed] [Google Scholar]
- 235. Ozgur R., Turkan I., Uzilday B., Sekmen A. H., J. Exp. Bot. 2014, 65, 1377–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Mortimer P. M., Intyre S. A. M., Thomas D. C., Front. Immunol. 2021, 12, 733918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Choi D.-I., Park J.-H., Choi J.-Y., Piao M., Suh M.-S., Lee J.-B., Yun S.-J., Lee S.-C., Annals of Dermatology 2020, 33, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Eguchi N., Vaziri N. D., Dafoe D. C., Ichii H., Int. J. Mol. Sci. 2021, 22, 1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Kos J., Int. J. Mol. Sci. 2022, 23, 4632.35563022 [Google Scholar]
- 240. Goetzke C. C., Ebstein F., Kallinich T., J. Clin. Med. 2021, 10, 1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Slack M. A., Gordon S. M., Arterioscler. Thromb. Vasc. Biol. 2019, 39, e210–e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Rai M., Curley M., Coleman Z., Demontis F., Aging Cell 2022, 21, e13603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Ruiz-Blázquez P., Pistorio V., Fernández-Fernández M., Moles A., J. Hepatol. 2021, 75, 1192–1202. [DOI] [PubMed] [Google Scholar]
- 244. Lucena F., McDougall J. J., Int. J. Mol. Sci. 2021, 22, 9352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Biancheri P., Di Sabatino A., Corazza G. R., MacDonald T. T., Cell Tissue Res. 2013, 351, 269–280. [DOI] [PubMed] [Google Scholar]
- 246. Kammerl I. E., Meiners S., Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L328–L336. [DOI] [PubMed] [Google Scholar]
- 247. Williams M. R., Nakatsuji T., Sanford J. A., Vrbanac A. F., Gallo R. L., J. Invest. Dermatol. 2016, 137, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Antalis T. M., Conway G. D., Peroutka R. J., Buzza M. S., Curr. Opin. Hematol. 2016, 23, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Kiyozumi D., Ikawa M., Front. Endocrinol. 2022, 13, 10.3389/fendo.2022.876370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Boyce B. F., Yao Z., Xing L., Crit. Rev. Eukaryotic Gene Expression 2009, 19, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Cheng K., Qi J., Zhang J., Li H., Ren X., Wei W., Meng L., Li J., Li Q., Zhang H., Deng W., Sun H., Mei L., Biomaterials 2022, 291, 121916. [DOI] [PubMed] [Google Scholar]
- 252. Yin J., Ouyang C., Shen S., Zhou Y., He G., Zhang H., Zhou K., Chen G., Ren L., Mol. Pharmaceutics 2022, 19, 2441–2455. [DOI] [PubMed] [Google Scholar]
- 253. Ha S. Y. Y., Zhou Y., Fong W.-P., Ng D. K. P., J. Med. Chem. 2020, 63, 8512–8523. [DOI] [PubMed] [Google Scholar]
- 254. Tam L. K. B., He L., Ng D. K. P., Cheung P. C. K., Lo P., Chem. A Eur. J. 2022, 28, e202201652. [DOI] [PubMed] [Google Scholar]
- 255. Nestoros E., de Moliner F., Nadal-Bufi F., Seah D., Ortega-Liebana M. C., Cheng Z., Benson S., Adam C., Maierhofer L., Kozoriz K., Lee J.-S., Unciti-Broceta A., Vendrell M., Nat. Commun. 2024, 15, 7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Wei R., Dong Y., Tu Y., Luo S., Pang X., Zhang W., Yao W., Tang W., Yang H., Wei X., Jiang X., Yuan Y., Yang R., ACS Appl. Mater. Interfaces 2021, 13, 14004–14014. [DOI] [PubMed] [Google Scholar]
- 257. Linden G., Zhang L., Pieck F., Linne U., Kosenkov D., Tonner R., Vázquez O., Angew. Chem. Int. Ed. 2019, 58, 12868–12873. [DOI] [PubMed] [Google Scholar]
- 258. Wang X., Liew S. S., Huang J., Hu Y., Wei X., Pu K., J. Am. Chem. Soc. 2024, 146, 22689–22698. [DOI] [PubMed] [Google Scholar]
- 259. Chen D., Xu Q., Wang W., Shao J., Huang W., Dong X., Small 2021, 17, 2006742. [DOI] [PubMed] [Google Scholar]
- 260. Lu B., Wang L., Tang H., Cao D., J. Mater. Chem. B 2023, 11, 4600–4618. [DOI] [PubMed] [Google Scholar]
- 261. Li X., Kwon N., Guo T., Liu Z., Yoon J., Angew. Chem. Int. Ed. 2018, 57, 11522–11531. [DOI] [PubMed] [Google Scholar]
- 262. Mattila H., Khorobrykh S., Havurinne V., Tyystjärvi E., J. Photochem. Photobiol. B 2015, 152, 176–214. [DOI] [PubMed] [Google Scholar]
- 263. Walsh C., Rajora M. A., Ding L., Nakamura S., Endisha H., Rockel J., Chen J., Kapoor M., Zheng G., Chem. Biomed. Imaging 2023, 1, 66–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Benson S., Kiang A., Lochenie C., Lal N., Mohanan S. M. P. C., Williams G. O. S., Dhaliwal K., Mills B., Vendrell M., Theranostics 2023, 13, 3814–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Lochenie C., Duncan S., Zhou Y., Fingerhut L., Kiang A., Benson S., Jiang G., Liu X., Mills B., Vendrell M., Adv. Mater. 2024, 36, 2404107. [DOI] [PubMed] [Google Scholar]
- 266. Wang H., Cheng K., Sun S., Wang P., Zhou Y., Sun H., Wang X., Shen H., Li S., Lin H., Adv. Healthcare Mater. 2024, 13, 2302481. [DOI] [PubMed] [Google Scholar]
- 267. Li B., Zhao M., Lai W., Zhang X., Yang B., Chen X., Ni Q., Angew. Chem. Int. Ed. 2023, 62, e202302676. [DOI] [PubMed] [Google Scholar]
- 268. Wu G., Liu F., Li N., Fu Q., Wang C., Yang S., Xiao H., Tang L., Wang F., Zhou W., Wang W., Kang Q., Li Z., Lin N., Wu Y., Chen G., Tan X., Yang Q., Adv. Sci. 2023, 10, 2304104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Yuan M., Fang X., Liu J., Yang K., Xiao S., Yang S., Du W., Song J., Small 2023, 19, 2206666. [DOI] [PubMed] [Google Scholar]
- 270. An H., Guo C., Li D., Liu R., Xu X., Guo J., Ding J., Li J., Chen W., Zhang J., ACS Appl. Mater. Interfaces 2020, 12, 17230–17243. [DOI] [PubMed] [Google Scholar]
- 271. Zhao H., Li Y., Zhang X., Wu K., Lv J., Chen C., Liu H., Shi Z., Ju H., Liu Y., Biomaterials 2022, 291, 121873. [DOI] [PubMed] [Google Scholar]
- 272. Choi V., Rajora M. A., Zheng G., Bioconjugate Chem. 2020, 31, 967–989. [DOI] [PubMed] [Google Scholar]
- 273. Xu C., Huang J., Jiang Y., He S., Zhang C., Pu K., Nat. Biomed. Eng. 2023, 7, 298–312. [DOI] [PubMed] [Google Scholar]
- 274. Xu C., Qin X., Wei X., Yu J., Zhang Y., Zhang Y., Ding D., Song J., Pu K., Nat. Nanotechnol. 2025, 20, 286–295. [DOI] [PubMed] [Google Scholar]
- 275.M. A. Rajora, Supramolecular Structure-Enabled Delivery of Porphyrin to Glioblastomas Beyond the Blood-Brain Barrier. 2024. University of Toronto. Canada.
- 276. Wang G. D., Nguyen H. T., Chen H., Cox P. B., Wang L., Nagata K., Hao Z., Wang A., Li Z., Xie J., Theranostics 2016, 6, 2295–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. He L., Yu X., Li W., ACS Nano 2022, 16, 19691–19721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.