

ABSTRACT
At different stages of life, from embryonic to postnatal, varying oxygen concentrations modulate cellular gene expression by enhancing or repressing hypoxia‐inducible transcription factors. During embryonic/fetal life, these genes encode proteins involved in adapting to a low‐oxygen environment, including the induction of specific enzymes related to glycolytic metabolism, erythropoiesis, angiogenesis, and vasculogenesis. However, oxygen concentrations fluctuate during intrauterine life, enabling the induction of tissue‐specific differentiation processes. Fetal well‐being is thus closely linked to the physiological benefits of a dynamically hypoxic environment. Premature birth entails the precocious exposure of the immature fetus to a more oxygen‐rich environment compared to the womb. As a result, preterm newborns face a condition of relative hyperoxia, which alters the postnatal development of organs and contributes to prematurity‐related diseases. However, until recently, the molecular mechanism by which high oxygen tension alters normal fetal differentiation remained unclear. In this review, we discuss the research trajectory followed by our research group, which suggests that early exposure to a relatively hyperoxic environment may impair preterm neonates due to reduced expression of the β3‐adrenoceptor. Additionally, we explore how these impairments could be prevented through the pharmacological stimulation of the remaining β3‐adrenoceptors. Recent preclinical studies demonstrate that pharmacological stimulation of the β3‐adrenoceptor can decouple exposure to hyperoxia from its harmful effects, offering a glimpse of the possibility to recreating the conditions typical of intrauterine life, even after premature birth.
Keywords: differentiation, fetus, hyperoxia, intrauterine hypoxia, prematurity
Abbreviations
- ATP
adenine triphosphate
- BE
base excess
- BPD
bronchopulmonary dysplasia
- CNS
central nervous system
- CTG
cardiotocography
- Hb
hemoglobin
- HCO3 −
bicarbonate
- HIE
hypoxic‐ischemic encephalopathy
- HIFs
hypoxia‐inducible transcription factors
- IUFD
intrauterine fetal death
- IUGR
intrauterine growth restriction
- NEC
necrotizing enterocolitis
- NIRS
near‐infrared spectroscopy
- O2
oxygen
- OXPHOS
oxidative phosphorylation
- pCO2
partial pressure of carbon dioxide
- pO2
partial pressure of oxygen
- PVL
periventricular leukomalacia
- ROP
retinopathy of prematurity
- VEGF
vascular endothelial growth factor
- β‐ARs
β‐adrenoceptors
1. Introduction
Oxygen (O2) is closely associated with the origin and evolution of life because aerobic organisms require molecular O2 to produce chemical energy in the form of adenine triphosphate (ATP), which is essential for survival [1]. In addition to being a substrate for energy production and acting as the primary electron acceptor, O2 also plays a crucial role as a signaling factor. Indeed, whether O2 levels are very low, as in the early stages of embryonic life, or high, as after birth, O2 modulates cellular gene expression by enhancing or repressing hypoxia‐inducible transcription factors (HIFs) [2]. Under hypoxic conditions, the HIF‐1α protein accumulates due to the inhibition of its proteasome‐mediated degradation, translocates into the nucleus, dimerizes with the HIF‐1β protein, and binds to specific DNA sequences called hypoxia‐responsive elements, inducing the transcription of hundreds of target genes [3, 4]. These genes encode proteins involved in adapting to a low‐O2 environment, including the induction of enzymes involved in glycolytic metabolism, erythropoiesis, angiogenesis, and vasculogenesis [5]. In addition to HIF activation, low O2 levels are also known to promote catecholamine release and adrenergic stimulation [6], which in turn induce the activation of pro‐angiogenic and proinflammatory factors [7].
Through these pathways—some of which have been well studied and others still under investigation—O2 levels and their fluctuations influence the proliferation and differentiation of various stem/progenitor cell populations, either by preserving their stemness or inducing their tissue‐specific differentiation [3]. For instance, high O2 concentrations promote the differentiation of megakaryocytes into platelets [8]; on the other hand, hypoxia drivers the differentiation of specific progenitors into neurons with distinct neurotransmitter phenotypes [9] and human embryonic stem cells toward a cardiomyocyte phenotype [10], as confirmed by the impaired neurogenesis or cardiomyogenesis observed in HIF‐1α knockout models [11, 12]. Therefore, a vast array of biological processes is governed by simple fluctuations in ambient O2 concentrations.
O2 fluctuations are critical for the processes of embryogenesis and morphogenesis. Indeed, embryos develop in a low‐O2 environment, often referred as “Everest in utero” [13], which supports stem cell self‐renewal and proliferation [3, 14]. However, under low O2 conditions, cytotrophoblast proliferation establishes connections with the maternal vasculature, leading to an increase in placental O2 levels [15]. This rise in O2 levels triggers the differentiation of cytotrophoblast cells into multinucleated syncytiotrophoblasts, which adopt a more invasive phenotype, facilitating the formation of a well‐established maternalfetal circulation [16]. Additionally, hypoxia‐induced proangiogenic factors lay the foundation for the embryonic vascular and placental framework, further elevating O2 levels. The role played by O2 in further differentiation of the vascular network is well‐documented in animal models, where vascular structures are completed after birth when tissues are exposed to higher O2 pressures. For instance, proper gliovascular structure in rodent brains forms after birth [17, 18], as well as the vascularization of the mouse retina develops postnatally [19, 20].
1.1. Early Exposure to an O2‐Rich Environment: A Trap for Preterm Infants
This delicate balance of O2 fluctuations during intrauterine life explains why a series of pathologies arise when premature birth exposes precociously an immature newborn to a relatively hyperoxic environment. A well‐documented example is retinopathy of prematurity (ROP), a potentially blinding ocular disorder whose pathogenesis starts and evolves as a function of O2 levels [21]. The biphasic vascular course of ROP—first ischemic, then proliferative—results from initial hyperoxic exposure followed by hypoxia due to vascular regression triggered by excessive O2. Indeed, the primum movens of the disease is represented by the premature exposure of a minimally vascularized retina to a relatively hyperoxic environment, which suppresses proangiogenic factors [21]. This event halts or regresses initial vascularization, a phenomenon ophthalmologists describe as “incomplete vascularization” [22]. As a result, the peripheral retina remains avascular until an approximate postmenstrual age of 32 weeks. Persistent ischemia, coupled with the developing retina's increasing metabolic demands, leads to growing hypoxia in the peripheral areas of the retina, farthest from existing vessels. ROP, therefore, reverses, and the progressive hypoxia developing in the outermost areas of the retina triggers the upregulation of a series of proangiogenic factors, including vascular endothelial growth factor (VEGF), leading to disorganized neovascularization and blood‐retinal barrier breakdown [23].
However, in preterm newborns, particularly those of very low gestational age, the retina is not the only region with immature vascularity where early exposure to high O2 levels triggers alterations typical of prematurity. Premature lungs, for example, are vulnerable to high O2 exposure, which induces alveolar hypoplasia and abnormal vascular organization, which are the typical hallmarks of bronchopulmonary dysplasia (BPD) [24]. Autopsies of infants who die from BPD, in fact, reveal abnormal alveolar microvessels and impaired expression of angiogenic growth factors and their receptors in the lungs [25]. Additionally, very low tracheal VEGF levels within the first week of life are associated with an increased risk of severe BPD, underscoring the strong relationship between impaired lung vascularization and BPD [26]. Hypoxia is also crucial for maintaining physiological homeostasis of the intestine [27], while hyperoxia can lead to intestinal damage, predisposing the newborn to necrotizing enterocolitis (NEC) [28]. Even in NEC, as in many other prematurity‐related conditions, reduced intestinal vascularization and resulting ischemia play a key role in pathogenesis [29]. In addition, the central nervous system (CNS) requires a physiologically hypoxic environment rich in HIF‐1 to support normal vascular development [30]. Hyperoxia has been demonstrated to damage the immature CNS in neonatal animals [31]. In rat pups, exposure to 80% O2 results in reduced brain mass and significant microvascular degeneration, which is more pronounced in the cortex [32]. Similarly, neonatal mice exposed to hyperoxia in the early postnatal period develop vascular structural impairments in the cerebral cortex, such as reduced vessel length and fewer branch points [33]. The close relationship between vascular damage and exposure to high O2 concentrations is further evidenced by human autoptic studies, which show that the vascularity of the white matter between 16 and 32 weeks of gestational age is rudimentary [34]. Therefore, vascular immaturity at birth, combined with vascular regression due to relative hyperoxia exposure, and the reduced viability of preoligodendrocytes exposed to high O2 levels [35], might explain the development of typical white matter lesions seen in developing brains, such as periventricular leukomalacia (PVL) [36].
1.2. Premature Birth and Oxidative Stress
In preterm newborns, exposure to a hyperoxic environment induces oxidative stress, damaging neonatal tissues [37]. This damage results from the excessive production of mitochondrial reactive oxygen species (ROS), such as superoxide anion, hydrogen peroxide, singlet O2, and hydroxyl radicals, which overwhelm the neutralizing capacity of antioxidant defense systems. Preterm newborns are particularly vulnerable to oxidative stress due to their low antioxidant capacity [38]. The formation of reactive nitrogen species (RNS) from the reaction of ROS with nitric oxide, along with lipid peroxidation—damaging cell membranes—leads to cellular and mitochondrial damage, contributing to systemic disorders [39]. Oxidative stress is widely linked to brain damage in preterm infants [40], as well as respiratory damage [41]. Similar mechanisms occur during ischemia/reperfusion injury, where the restoration of O2 to ischemic tissues causes further brain damage [42]. This suggests that both prolonged exposure to high O2 and sudden increases in oxygenation—even when not excessively high—can be harmful.
In addition to hyperoxia, exposure to intermittent [43, 44] or chronic hypoxia [45, 46] also induces oxidative stress in preterm infants. Although counterintuitive, studies have shown increased production of superoxide anion and hydrogen peroxide during hypoxia [45], with ROS production often occurring outside the mitochondria [45]. Thus, hyperoxia, intermittent hypoxia, and chronic hypoxia all contribute to the development of pathologies in preterm newborns via oxidative stress.
Several animal models are used to mimic O2‐induced damage related to prematurity [47, 48, 49]. Among the various models available, newborn rodents exposed to high O2 concentrations are often used due to their anatomical similarity to preterm neonates [47, 48]. Rodent hyperoxia models typically involve 80% O2 exposure, leading to a 2.3‐fold increase on average O2 partial pressure compared to controls [50], similar to what occurs during premature birth [51]. These models, which involve exposure only to hyperoxia rather than mixed models where hyperoxia alternates with periods of hypoxia, allow researchers to explore the effects of hyperoxia without ambiguity. The adoption of the hyperoxia‐based model is also based on the observation that the abrupt transition from an intrauterine hypoxic environment to extrauterine hyperoxia is the primary event that disrupts normal fetal maturation, serving as the primum movens for subsequent hypoxia in the newborn. This is what happens in animal models of BPD, where hyperoxia exposure (at variable concentrations and for variable times) impairs alveolarization, induces fibrosis, and disrupts vascular growth [47, 48]. The same process occurs in the CNS, where hyperoxia leads to vascular regression and, therefore, tissue hypoxia [52]. Intermittent hypoxia following apneas—common in preterm newborns [43]—may also result from premature exposure to hyperoxia. In the rodent model, in fact, perinatal hyperoxia affects carotid body development, impairing the developing respiratory control system, which may lead to hypoventilation and reduced responses to acute hypoxia [53]. Thus, the abrupt and premature transition from a comfortable hypoxic fetal environment to a hyperoxic postnatal environment likely triggers the damage seen in preterm infants, disrupting their physiological growth and differentiation.
Overall, research demonstrates that embryonic and fetal well‐being are closely linked to maintaining a hypoxic environment, essential for proper differentiation and adequate vascularization via HIF‐1‐mediated production of angiogenic factors like VEGF, basic fibroblast growth factor, platelet‐derived growth factor, angiopoietin‐1, angiopoietin‐2, Tie2, and monocyte chemoattractant protein‐1 [54]. However, premature newborns are vulnerable to damage from excessive O2 exposure, which emphasizes the need for cautious O2 administration.
However, if, on the one hand, the extremely relevant role played by physiological hypoxia during intrauterine life is evident, it is surprising how little is known about the embryonic/fetal O2 levels during a healthy pregnancy, their potential fluctuations as gestational age progresses, and the role that these putative fluctuations may have in modulating biological processes of differentiation.
Recent studies on cord blood gas analysis across different gestational ages, from extremely preterm to term infants, revealed that fetal hypoxia is dynamic, allowing us to speculate that the fluctuations of fetal hypoxia may be key to healthy fetal development [55, 56]. Understanding these O2 trends during pregnancy would enhance our knowledge of mechanisms that ensure fetal well‐being. This knowledge will be extraordinarily valuable for researchers who, figuratively speaking, envision bringing the preterm newborn back into the maternal uterus. This ambitious goal could be achieved either through artificial placenta technology [57, 58] or through pharmacological interventions that mimic the biological effects of intrauterine hypoxia [56, 59], which we refer to in this article as a “pharmacological artificial placenta.”
The aim of this review is to describe the pathway that led us to hypothesize that early exposure to a relatively hyperoxic environment may impair preterm neonates due to reduced expression of β3‐Adrenoceptor (β3‐ARs) and that the use of β3‐AR agonist drugs in these newborns may, at least partially, simulate the benefits of the maternal womb.
2. Intrauterine Fluctuations in O2 Levels
Fecundation and embryo development occur under anaerobic conditions, and the fetus also thrives in a very low‐O2 environment [60]. However, the literature suggests that low O2 levels play an important role even before sexual fecundation. In fact, both spermatozoa and oocytes live in a physiologically hypoxic environment. On the male side, the testis is naturally an O2‐deprived organ, where interstitial O2 tension in different animals has been reported to be very low, ranging between 11 and 15 mmHg (approximately 2%) [61, 62, 63]. Hypoxia affects not only germ cells but also the seminiferous tubules [64], which are poorly vascularized. Therefore, O2 reaches the lumen only through diffusion, ensuring that luminal pO2 levels remain very low [65]. On the female side, numerous studies have demonstrated that the O2 concentration in follicular fluid is low, and oxygenation decreases as the ovarian follicle develops [66]. Moreover, O2 levels are particularly low throughout the entire course of the oviduct, approximately 5%, although they fluctuate during different phases of the menstrual cycle [67, 68]. Inside the nonpregnant uterus, O2 levels drop even further, reaching about 2% [69].
The close complementarity between the male and female genital organs during sexual mating suggests that low O2 levels are essential for successful reproduction. On one hand, such anatomical complementarity facilitates the entry of spermatozoa into the female vagina and cervix. On the other hand, the particular adherence between the two genitals suggests a biological interest in maintaining the integrity of this hypoxic environment, avoiding contaminations from air entry. Thus, this mode of sexual reproduction in mammals can be interpreted as a biological strategy aimed at preserving the hypoxia chain, from the testis to the oviduct. In this respect, the importance of low environmental oxygenation in ensuring successful fecundation is highlighted by improved live birth rates of preimplantation embryos cultured under low O2 tension, compared to those cultured in normoxic conditions [70, 71].
However, if the hermetic seal between males and females is necessary to ensure successful sexual fecundation, this means that even temporary exposure to a more oxygenated environment can represent a risk. Hypoxia, in effect, appears to favor the vitality of the spermatozoa, while their exposure to higher O2 concentrations can have detrimental effects [72].
In humans, conception occurs when a sperm cell successfully fertilizes an egg cell in the fallopian tube, where the O2 concentration is approximately 5% [67]. A hypoxic environment remains necessary even after fecundation, as low O2 levels have been identified as key regulators of the harmonious processes of embryonic and placental development [14, 60]. However, immediately after conception, the environmental O2 levels change, and this modulation seems to act as a signal that favors specific developmental processes. For instance, when the morula reaches the uterine cavity, the O2 concentration decreases to around 2% [67, 73, 74]. Interestingly, this transition to a more hypoxic condition is crucial during the first 2−3 weeks of life, allowing the development of mammalian blastocysts. In this environment, the blastocysts form an inner cell mass of embryonic stem cells that are able to proliferate [75, 76] while maintaining their undifferentiated state [77, 78]. In addition to promoting proliferation, increasing hypoxia facilitates the implantation, anchoring, and invasion of the blastocyst into the maternal uterus [15]. Furthermore, exposure to a decreasing O2 gradient induces a metabolic shift in cytotrophoblast cells from oxidative phosphorylation (OXPHOS) to glycolytic metabolism (the so‐called Warburg effect) [79, 80, 81]. This metabolic adjustment offers a proliferative advantage, as it stimulates the production of many intermediates in the pentose phosphate pathway, such as ribose sugars, which are essential for nucleic acid synthesis. It also results in elevated lactic acid levels, which aid in promoting nesting in surrounding tissues [82]. In this respect, the consistent production and extrusion of lactate induce disaggregation of uterine tissues, facilitating trophoblast invasion [83].
The increase in intrauterine hypoxia during the first weeks of life sets the stage for a series of events that provide the foundation for embryonic vascular and placental development, which in turn leads to a further increase in O2 availability [84]. Hypoxia promotes cytotrophoblast proliferation, migration across the uterine epithelium, aggregation into cell columns, and differentiation into extravillous trophoblasts [85, 86]. These phenomena support cytotrophoblast infiltration into both the uterine interstitium and the lumen of spiral arterioles. This infiltration not only anchors the fetus to the mother but also progressively remodels the spiral arteries. The smooth muscle and elastic lamina of the vessel are replaced by cytotrophoblasts [16], transforming the high‐resistance, low‐flow original spiral arteries into low‐resistance, high‐flow vessels. This ensures a gradual increase in O2 delivery in the following weeks of pregnancy [15].
Up until approximately the 10th week of human gestation, intrauterine O2 levels remain very low, less than 20 mmHg (approximately 2% O2), similar to those found within the nonpregnant uterus [84, 87]. However, starting from this point and continuing through the early weeks of the second trimester of pregnancy, placental development promotes a significant increase in O2 availability, as evidenced by a threefold rise in placental oxygenation to around 60 mmHg (approximately 8% O2) [87]. This increased microenvironmental oxygenation promotes the differentiation of cytotrophoblasts into multinucleated syncytiotrophoblasts [16], which are essential for the secretion of various hormones that maintain pregnancy [88].
The tripling of placental O2 content enhances O2 availability to the fetoplacental unit despite several structural factors that tend to reduce the placenta's O2‐diffusing capacity. The immature structure of placental villi and their rudimentary vascularity, which result in a reduced exchange surface area, along with the considerable diffusion distance between maternal−fetal circulations, limit O2 transfer from the mother to the fetus. This explains why the partial pressure of O2 (pO2) in umbilical venous blood, which carries O2 from the placenta to the fetus, is significantly lower than that of both placental [87] and maternal arterial pO2 [89] blood.
Extrapolations from animal studies and human fetal studies have shown that by the end of the third trimester of pregnancy, the mean pO2 concentrations in the umbilical vein and umbilical artery are 28 and 19 mmHg, respectively [90]. However, at least in humans, these concentrations do not appear to remain constant throughout the third trimester. Analysis of human umbilical cord arterial and venous blood samples collected via cordocentesis revealed a decrease in fetal pO2 and O2 saturation as gestation advances [91, 92, 93, 94, 95]. While cordocentesis provides insights into the physiological state over a wide gestational age range before the peripartum period, studies using this method did not account for the final weeks of pregnancy. Furthermore, these samples were obtained from pregnancies complicated by maternal infections, fetal structural anomalies, or various prenatal pathologies [91, 92, 93, 94, 95]. Only recently has the analysis of umbilical cord blood gas samples allowed for a clearer understanding of O2 level fluctuations during the last trimester. These studies confirmed that fetal pO2 and O2 saturation decrease as gestation progresses but only up until the 33rd−34th week, after which a significant increase in O2 levels is observed [55, 56, 96].
Essentially, intrauterine life shows that although the embryo/fetus exists in a hypoxic environment, O2 levels fluctuate. These fluctuations likely serve to modulate seemingly contradictory processes, such as the expansion of stemness and differentiation.
Understanding that O2 levels vary according to gestational age has important implications for research on artificial placenta technology, which is currently under development and being tested on animals [57]. This knowledge of the physiological fluctuations in O2 levels during intrauterine life should indicate the oxygenation targets be achieved with extracorporeal membrane oxygenation, which must vary based on the week of gestation to support normal physiological growth and maturation of the fetus through artificial placental technology. Similarly, efforts to mimic the biological effects of increasing intrauterine hypoxia through pharmacologic stimulation [59] will need to consider that fetal hypoxemia increases only until to the 33rd−34th week.
3. Methods for Assessing Fetal O2 Levels
A pathological decrease in the O2 supply from the placenta to the fetus can lead to severe outcomes such as intrauterine fetal death (IUFD) or hypoxic‐ischemic encephalopathy (HIE). As a result, over the years, various methods have been developed to measure O2 levels in fetal blood, with the goal of detecting pathological hypoxia early and thereby preventing IUFD and HIE [97].
The most commonly used method for monitoring fetal oxygenation is electronic fetal heart rate monitoring (EFM), or cardiotocography (CTG), which track fetal heart rate to assess fetal well‐being or detect signs of intrapartum hypoxia [97]. However, the predictive value of CTG for detecting intrapartum fetal hypoxia is only about 30%, and it has a high false‐positive rate of 60% [98]. Therefore, despite clear guidelines for interpretating CTG tracings, the reliability of this technology is limited, providing operators with modest accuracy and, more importantly, only indirect and nonquantitative information about the oxygenation status [98]. Near‐infrared spectroscopy (NIRS) has been investigated as a method for evaluating placental function, particularly in cases of fetal growth restriction [99]. While NIRS offers easy and minimally invasive access to oxygenation data, its low spatial resolution limits its usefulness for fetal monitoring [97]. Additionally, the photoacoustic (PA) technique, which uses multiple light wavelengths to provide real‐time information on blood O2 saturation, has recently been proposed as a promising novel method for fetal oxygenation monitoring. The sensitivity of this technique for detecting tissue hypoxia is comparable to that of NIRS, but PA offers more accurate information due to its higher spatial resolution [97].
Another proposed system for measuring fetal oxygenation during labor is fetal pulse oximetry. Initially, transvaginal fetal pulse oximetry was evaluated. This method consists of placing a device through the birth canal after the rupture of the uterine membrane. The device is positioned on the fetal head to measure O2 saturation [100]. Subsequent studies have sought less invasive but equally reliable methods. Recently, transabdominal fetal pulse oximetry has been developed, utilizing near‐infrared light sources and photodetectors placed on the mother's abdomen to measure fetal O2 saturation in a fully noninvasive manner [101].
In modern obstetric practice, several additional tests have been developed to increase the detection rate of hypoxia. One of these is the analysis of the fetal ECG using ST trait analysis (STAN) [102]. Another method involves assessing fetal scalp blood lactate levels, collected through a small incision in the fetal scalp during labor using a capillary tube [103]. However, this method has been widely questioned due to numerous contraindications, such as maternal infections and fetal coagulation disorders, as well as potential complications like prolonged bleeding, hematoma, or abscess formation at the incision site [104].
These techniques are primarily designed for monitoring oxygenation levels during labor or in the immediate context of complicated childbirth. Generally, these approaches do not allow for prolonged monitoring and have not been used to reconstruct fetal oxygenation status over time.
Umbilical cord blood gas analysis is considered the most accurate and reliable method for quantifying fetal and placental oxygenation and detecting potential fetal hypoxia. This procedure involves isolating a 20 cm segment of the cord between two clamps. Venous and arterial umbilical cord blood is typically collected using blood gas syringes containing spray‐dried calcium‐balanced lithium heparin and is promptly analyzed using an automatic blood gas analyzer [105]. This analysis enables the detection of metabolic acidosis by measuring pH, pO2, partial pressure of carbon dioxide (pCO2), lactates, bicarbonate (HCO3 −), and base excess (BE) [106]. Additionally, measuring hemoglobin (Hb) levels and arterial saturation allows the calculation of O2 content and fetal O2 extraction (the difference between venous and arterial blood O2 contents, divided by venous O2 content). Furthermore, cord blood sampling provides insight into placental and fetal metabolic activity, such as lactate and CO2 production (the difference between arterial lactate or blood CO2 and their respective venous contents, respectively, divided by venous content) [55, 56].
Thus, umbilical cord sampling is the most useful test for evaluating fetal oxygenation at birth and for diagnosing perinatal asphyxia with accuracy [106]. Moreover, the availability of a large number of cord blood gas analyses collected at different gestational ages allows for the reconstruction of fetal oxygenation status as pregnancy progresses [55, 56].
4. Pros and Cons of Umbilical Cord Blood Gas Analysis
Umbilical cord blood gas analysis is one of the most widely used methods in developed countries to monitor fetal well‐being and detect potential fetal hypoxia during delivery. This procedure is simple, painless for the newborn, relatively economical, and provides immediate and valuable feedback to labor ward staff. Additionally, umbilical cord blood analysis often serves as crucial evidence in forensic contexts [106]. Numerous studies over the years have consistently shown that cord blood gas analysis is significantly more accurate and reliable than CTG [107].
However, it is common practice to collect blood from only one cord vessel, especially when the fetus is under stress or compromised. It is generally believed that blood samples from a single vessel are more likely to contain venous blood, as the vein is larger and easier to sample than the artery [107].
The mode of delivery can affect cord blood values, as demonstrated by the lower oxygenation levels observed in newborns delivered by cesarean section compared to those born via vaginal delivery [55, 56]. Several factors may explain this difference, with the impact of delivery type on placental flow likely playing a key role. Neonates delivered by cesarean section experience lower umbilical transfusion force, as they are placed on the maternal abdomen at a higher plane than the placenta, which creates a gravity gradient that opposes the flow of blood from the placenta to the fetus [108]. Furthermore, women undergoing cesarean sections are at a higher risk of developing hypotension [109] or reduced uterine contractility due to uterine incision and anesthesia [110, 111]. Similarly, fetal oxygenation of neonates born through vaginal delivery can be influenced by the engagement of the fetus through the birth canal or by reduced blood flow during uterine contractions [112]. Moreover, neonates delivered by assisted vaginal breech delivery often show lower umbilical cord artery blood pH and pO2, along with increased pCO2, compared to those delivered via cephalic‐vaginal delivery [113].
Another factor that could influence the reliability of umbilical blood gas measurements is the timeliness of sampling. Measurements of pH, pCO2, and pO2, for example, become unreliable if the sample is taken more than 60 min after clamping a double‐segment of the umbilical cord or more than 20 min from an unclamped segment [114]. More controversial, instead, is the effect on the analytical gas status of delaying cord clamping for at least 30−60 s after delivery, which is currently recommended by leading international scientific societies [115]. A recent review showed that the available data are inconclusive on this topic [116]. On the one hand, some studies have suggested that delayed cord clamping is associated with minimal decreases in pH and HCO3 − and increases in pCO2, lactate, and BE [117, 118, 119]. On the other hand, two randomized controlled trials indicated that acid–base balance was not significantly altered by delayed cord clamping, suggesting that placental gas exchange remains effective for at least 1 min after birth [120, 121]. The data related to oxygenation are particularly interesting: no study has shown any significant difference in venous pO2, even with cord clamping delayed up to 45−180 s [116]. In contrast, arterial pO2 values in samples collected after delayed cord clamping were approximately 3−4 mmHg higher, suggesting an influence of the newborn's respiratory activity on umbilical arterial oxygenation [120]. However, these findings were not confirmed in a subsequent study by the same authors [121].
5. Fetal Oxygenation From the 23rd to the Term of Gestation
Recently, fetal oxygenation status was studied through the analysis of umbilical cord blood samples, with results published separately for full‐term [55, 96] and preterm newborns [56]. The decision to report these results separately was made because the sample collection methods were not homogeneous; samples from full‐term newborns are usually collected approximately 60 s after birth (delayed cord clamping), while those from preterm newborns are collected immediately after birth (early cord clamping). The results of these studies demonstrated an apparently biphasic trend in fetal oxygenation, where the level of O2 reaching the fetus from the placenta appears to progressively decrease starting from the 23rd week of pregnancy (the earliest age at which umbilical cord samples could be analyzed) until approximately the 33rd−34th week. After this point, fetal oxygenation reverses this trend, with the placenta seemingly increasing O2 transfer, possibly due to physiological aging [55, 56]. However, this biphasic trend would have been more evident if we had reported the data from preterm and full‐term newborns together, despite the limitations that such a plot entails.
In this review, we combined data from preterm and term neonates by gathering as much information as available. To the previously published data (from full‐term neonates born at the University Hospital of Pisa, Italy, between January 1, 2019, and December 31, 2019 and from preterm neonates born between January 1, 2016, and December 31, 2022) [55, 56], we added data from all umbilical cord blood samples collected from newborns (both full‐term and preterm) born between January 1, 2022, and December 31, 2023. To emphasize the difference in sample collection between full‐term (delayed cord clamping) and preterm (early cord clamping) newborns, the results in the graphs are represented with different colors.
The results were obtained by analyzing 4649 samples, 755 of which were collected from 1450 preterm newborns born between 2016 and 2023, and 3894 of which were collected from 5197 full‐term newborns born between 2019, 2022, and 2023.
Umbilical cord blood samples from pregnancies with intrauterine growth restriction (IUGR) and/or fetal or maternal intrapartum complications (e.g., operative vaginal deliveries involving forceps or vacuum extractor use, abnormal intrapartum CTG requiring emergency cesarean section, meconium‐stained amniotic fluid, placental abruption, cord prolapse, chorioamnionitis, maternal sepsis, convulsions, hemorrhage, uterine rupture, or snapped cord) were excluded from the analysis. Samples suggestive of severe acidosis at birth (pH ≤ 7.00 and/or BE ≤ −12 mmol/L) [107, 122] or deemed unreliable [123] were also excluded. Umbilical parameters with values < or > 3 SD from their respective means were individually evaluated and (i) corrected if likely mid‐entered, (ii) maintained if considered plausible, or (iii) excluded if considered not plausible [97]. The samples were analyzed using an automatic blood gas analyzer (GEM Premier 4000, Instrumentation Laboratory, Lexington, MA, USA). pH, pCO2, pO2, SaO2, lactate, and Hb were measured, while HCO3 − and BE were calculated from the measured pH and pCO2, using the Henderson−Hasselbalch equation: pH = 6.1 + log([HCO3 −]/pCO2 × 0.03) [124] and the formula described by Siggaard−Anderson: (1 − 0.014 × Hb) × (HCO3 − − 24.8 + [1.43 × Hb + 7.7] × [pH ‐7.4]) [95]. The O2 content (mL/dL) was calculated using the formula: (1.36 × Hb [g/dL] × SaO2 [%]/100) + (0.0031 × pO2 [mmHg]], where 1.36 is the volume of O2 (mL) bound by a gram of Hb, and 0.0031 is the solubility coefficient of O2 in blood, representing the volume of O2 (mL) dissolved in 100 mL of plasma for each mmHg of pO2 partial pressure. Fetal O2 extraction was determined as the difference between umbilical venous and arterial blood O2 contents, divided by the umbilical venous O2 content. Fetal CO2 and lactate production were calculated as the difference between arterial blood CO2 or lactate and venous contents, divided by the respective venous content.
The demographic and gas analytical parameters for all newborns are shown in Table 1. Cord blood gas analysis of samples obtained from preterm newborns revealed lower oxygenation (lower venous and arterial pO2, and lower O2 content), lower CO2 and lactate production, and lower glucose extraction compared to samples from full‐term neonates. However, the modes of delivery differed significantly between the two groups, with a predominance of cesarean sections in the preterm newborn group and a similar prevalence of vaginal deliveries in the full‐term group. Since the type of delivery has been shown to significantly influence cord blood gas analysis, particularly oxygenation [55], the data were disaggregated based on delivery mode. Even after this adjustment, preterm fetuses still showed lower oxygenation, CO2 and lactate production, and glucose extraction, although statistical significance was not always achieved for smaller sample sizes (Table 2).
Table 1.
Umbilical cord blood gas analysis in preterm versus term neonates.
|
Preterm newborns (n = 755) |
Term newborns (n = 3894) |
p value | |
|---|---|---|---|
| GA, days, mean (SD) | 235 (23) | 278 (8) | < 0.001 |
| Birth weight, g, mean (SD) | 2030 (673) | 3346 (617) | < 0.001 |
| Male, n (%) | 393 (52.1) | 2003 (51.4) | 0.757 |
| Vaginal delivery, n (%) | 211 (27.9) | 2814 (72.3) | < 0.001 |
| Umbilical venous cord sampling | |||
| pH, mean (SD) | 7.321 (0.08) | 7.322 (0.07) | 0.596 |
| PaCO2, mmHg, mean (SD) | 42.5 (9.2) | 40.1 (8.5) | < 0.001 |
| PaO2, mmHg, mean (SD) | 24.3 (8.4) | 29.1 (8.5) | < 0.001 |
| Bicarbonate, mmol/L, mean (SD) | 21.7 (3.0) | 20.7 (2.7) | < 0.001 |
| BE(B), mmol/L, mean (SD) | −4.3 (3.4) | −5.2 (3.0) | < 0.001 |
| Lactate, mmol/L, mean (SD) | 2.8 (1.4) | 3.8 (1.7) | < 0.001 |
| Hemoglobin, g/dL, mean (SD) | 14.9 (2.3) | 16.2 (1.5) | < 0.001 |
| SaO2, %, mean (SD) | 55.5 (22.0) | 62.3 (18.5) | < 0.001 |
| Umbilical arterial cord sampling | |||
| pH, mean (SD) | 7.268 (0.08) | 7.243 (0.08) | < 0.001 |
| PaCO2, mmHg, mean (SD) | 50.8 (11.4) | 53.4 (11.5) | < 0.001 |
| PaO2, mmHg, mean (SD) | 16.4 (7.3) | 20.6 (9.5) | < 0.001 |
| Bicarbonate, mmol/L, mean (SD) | 22.9 (3.8) | 23.2 (3.1) | 0.034 |
| BE(B), mmol/L, mean (SD) | −4.5 (4.0) | −5.3 (3.6) | < 0.001 |
| Lactate, mmol/L, mean (SD) | 3.1 (1.6) | 4.3 (2.0) | < 0.001 |
| Hemoglobin, g/dL, mean (SD) | 14.7 (2.4) | 16.3 (1.5) | < 0.001 |
| SaO2, %, mean (SD) | 34.8 (19.1) | 38.5 (21.5) | < 0.001 |
| Fetal venous O2 content, mL/100 mL (SD) | 11.3 (4.6) | 13.8 (4.2) | < 0.001 |
| Fetal arterial O2 content, mL/100 mL (SD) | 6.9 (3.9) | 8.6 (4.9) | < 0.001 |
| Fetal O2 extraction, %, mean (SD) | 35.1 (36.4) | 32.8 (41.0) | 0.287 |
| Fetal CO2 production, %, mean (SD) | 22.4 (24.8) | 36.3 (31.8) | < 0.001 |
| Fetal lactate production, %, mean (SD) | 12.6 (20.6) | 16.8 (23.5) | < 0.001 |
| Fetal glucose extraction, %, mean (SD) | 14.1 (13.8) | 16.2 (14.6) | 0.004 |
Abbreviations: BE = base excess, GA = gestational age, PaCO2 = partial pressure of carbon dioxide, PaO2 = partial pressure of oxygen.
Table 2.
Umbilical cord blood gas analysis in all enrolled preterm and term newborns separately analyzed by the type of delivery.
| Vaginal deliveries | Cesarean deliveries | |||||
|---|---|---|---|---|---|---|
|
Preterm neonates (n = 211) |
Term neonates (n = 2814) |
p value |
Preterm neonates (n = 544) |
Term neonates (n = 1080) |
p value | |
| GA, days, mean (SD) | 233 (28) | 279 (8) | < 0.001 | 236 (22) | 275 (8) | < 0.001 |
| Birth weight, g, mean (SD) | 2084 (732) | 3363 (673) | < 0.001 | 2009 (649) | 3304 (440) | < 0.001 |
| Male, n (%) | 118 (55.9) | 1439 (51.1) | 0.180 | 275 (50.6) | 564 (52.2) | 0.525 |
| Umbilical venous cord sampling | ||||||
| pH, mean (SD) | 7.336 (0.08) | 7.324 (0.07) | 0.026 | 7.315 (0.08) | 7.318 (0.08) | 0.006 |
| pO2, mmHg, mean (SD) | 26.6 (8.9) | 30.3 (8.3) | < 0.001 | 23.4 (8.1) | 26.1 (8.2) | < 0.001 |
| Hemoglobin, g/dL, mean (SD) | 15.6 (1.9) | 16.3 (1.5) | < 0.001 | 14.6 (2.4) | 15.9 (1.7) | < 0.001 |
| SaO2, %, mean (SD) | 58.8 (21.6) | 64.0 (17.8) | < 0.001 | 54.0 (22.1) | 57.2 (19.4) | 0.022 |
| pCO2, mmHg, mean (SD) | 39.6 (9.2) | 38.7 (7.9) | 0.133 | 43.7 (8.9) | 43.5 (8.4) | 0.638 |
| Bicarbonate, mmol/L, mean (SD) | 20.9 (2.7) | 20.2 (2.6) | < 0.001 | 21.9 (3.2) | 22.1 (2.6) | 0.941 |
| BE(B), mmol/L, mean (SD) | −4.6 (3.3) | −5.5 (2.8) | < 0.001 | −4.2 (3.4) | −4.1 (3.2) | 0.509 |
| Lactate, mmol/L, mean (SD) | 3.4 (1.4) | 4.0 (1.6) | < 0.001 | 2.5 (1.3) | 2.9 (1.8) | < 0.001 |
| Glucose, mg/dL, mean (SD) | 94.7 (22.9) | 101.8 (21.8) | < 0.001 | 72.0 (20.5) | 83.2 (21.2) | < 0.001 |
| Umbilical arterial cord sampling | ||||||
| pH, mean (SD) | 7.266 (0.08) | 7.241 (0.08) | < 0.001 | 7.269 (0.08) | 7.250 (0.09) | < 0.001 |
| pO2, mmHg, mean (SD) | 18.8 (8.0) | 22.5 (9.2) | < 0.001 | 15.3 (6.7) | 15.8 (8.3) | 0.338 |
| Hemoglobin, g/dL, mean (SD) | 15.5 (2.1) | 16.4 (1.4) | < 0.001 | 14.3 (2.4) | 15.9 (1.6) | < 0.001 |
| SaO2, %, mean (SD) | 41.1 (20.3) | 41.7 (21.3) | 0.743 | 31.8 (17.8) | 29.0 (19.3) | 0.032 |
| pCO2, mmHg, mean (SD) | 50.4 (11.1) | 52.7 (11.2) | 0.006 | 50.9 (11.5) | 55.4 (12.1) | < 0.001 |
| Bicarbonate, mmol/L, mean (SD) | 22.5 (3.1) | 22.7 (3.0) | 0.322 | 23.1 (4.0) | 24.7 (3.0) | < 0.001 |
| BE, mmol/L, mean (SD) | −5.0 (3.8) | −5.7 (3.2) | 0.005 | −4.3 (4.1) | −4.1 (4.1) | 0.206 |
| Lactate, mmol/L, mean (SD) | 3.8 (1.6) | 4.6 (1.8) | < 0.001 | 2.8 (1.5) | 3.4 (2.0) | < 0.001 |
| Glucose, mg/dL, mean (SD) | 80.7 (21.2) | 84.9 (22.4) | 0.024 | 62.2 (20.0) | 70.9 (21.3) | < 0.001 |
| Fetal venous O2 content, mL/100 mL (SD) | 12.5 (4.6) | 14.2 (4.1) | < 0.001 | 10.7 (4.5) | 12.5 (4.3) | < 0.001 |
| Fetal arterial O2 content, mL/100 mL (SD) | 8.6 (4.3) | 9.4 (4.8) | 0.094 | 6.1 (3.4) | 6.3 (4.2) | 0.444 |
| Fetal O2 extraction, %, mean (SD) | 28.3 (33.5) | 28.6 (40.8) | 0.936 | 38.4 (37.4) | 45.8 (39.2) | 0.012 |
| Fetal CO2 production, %, mean (SD) | 31.7 (31.0) | 39.0 (33.3) | 0.003 | 18.5 (20.5) | 29.2 (26.3) | < 0.001 |
| Fetal lactate production, %, mean (SD) | 13.1 (19.9) | 16.0 (22.9) | 0.095 | 12.7 (20.2) | 19.4 (25.2) | < 0.001 |
| Fetal glucose extraction, %, mean (SD) | 14.5 (13.1) | 16.4 (15.3) | 0.132 | 13.8 (14.2) | 15.5 (12.4) | 0.218 |
Abbreviations: BE = base excess, GA = gestational age, PaCO2 = partial pressure of carbon dioxide, pO2 = partial pressure of oxygen.
To assess whether the different parameters of interest (umbilical venous and arterial O2 levels, fetal O2 extraction, Hb, pH, HCO3 −, BE, CO2, and lactate) varied over the course of pregnancy, gas analysis data were plotted against gestational age, and trends were evaluated using regression models. The best‐fitting model (linear or quadratic) was identified for each parameter, with the equation of the statistically significant model displayed at the top of each figure. If the most representative test (with a lower p‐value) was a linear regression, the parameter was considered to follow a linear trend (either increasing or decreasing) over time. Conversely, if a quadratic regression was more representative, the parameter exhibited a biphasic trend over time: for an increasing quadratic model, the trend reached a maximum value and then decreased, whereas, for a decreasing quadratic model, the trend reached a minimum value and then increased. These tests helped identify the approximate points where the curve reversed.
To assess whether O2 tension varied throughout pregnancy, venous and arterial pO2 data, as well as total venous and arterial O2 content, were analyzed as a function of gestational age (Figure 1A−D). The values from umbilical venous samples demonstrated a biphasic trend in O2 delivery from the placenta, as indicated by a decreasing quadratic regression: pO2 levels progressively decrease until 220−230 days of gestation (approximately the 32nd−34th week of gestation), when pO2 appears to reach its lower point. Then, starting from the 34th week, pO2 increases until delivery, as described in a recent study [55]. This biphasic trend is also observed for arterial pO2, although it is less pronounced compared to venous pO2, total venous and arterial O2 contents (Figure 1A−D).
Figure 1.
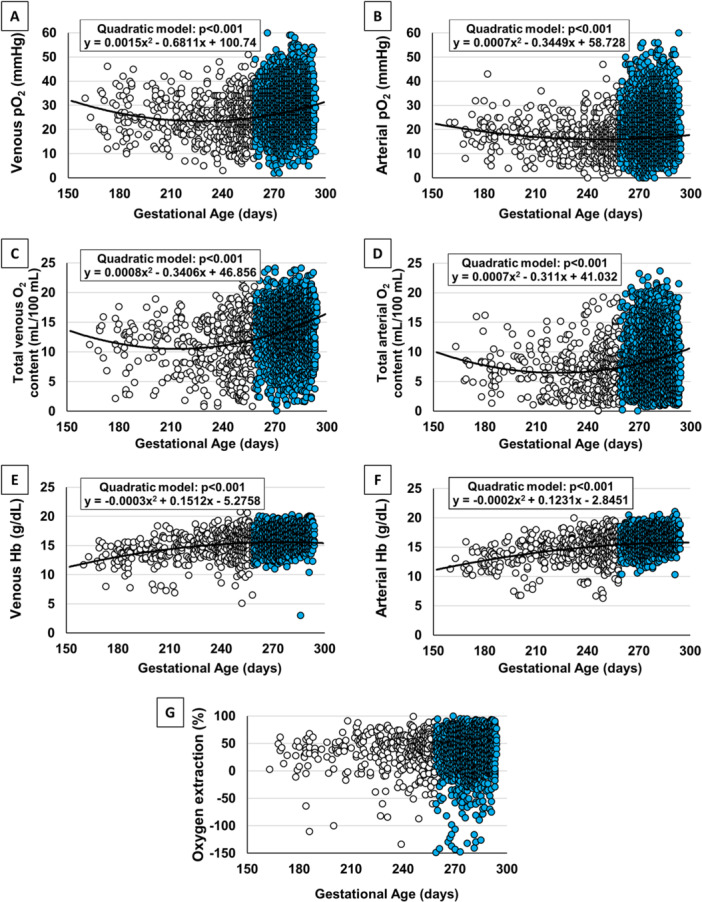
Umbilical cord oxygenation status. Scatter plots with regression lines representing the umbilical cord oxygenation status (venous pO2, A [n = 4286]; arterial pO2, B [n = 4161]; total venous, C [n = 2643] and arterial O2 contents, D [n = 2547]); venous Hb, E (n = 3034); arterial Hb, F (n = 2922); O2 extraction, G (n = 2548) of the whole study population. The equation of the statistically significant model (linear or quadratic) is indicated at the top of each figure. [Color figure can be viewed at wileyonlinelibrary.com]
The biphasic trend in O2 content is accompanied by the inverse trend in Hb levels (Figure 1E,F). Venous and arterial Hb levels progressively increase from the 23rd week onward, reaching a plateau around 250−260 days of gestation (approximately the 36th−38th week of gestation). Overall, O2 extraction does not show significant changes throughout the observed period (Figure 1G).
According to the trend of oxygenation, a biphasic modification in venous and arterial pH, coupled with a mirrored trend in venous HCO3 − and BE, is observed as pregnancy progresses (Figure 2).
Figure 2.

Umbilical cord pH, bicarbonate, and BE status. Scatter plots with regression lines representing the umbilical cord pH, HCO3 − concentration, and BE of the whole study population (venous pH, A [n = 4544]; arterial pH, B [n = 4392]; venous HCO3 −, C [n = 3098]; arterial HCO3 −, D [n = 2975]; venous BE, E [n = 4544]; arterial BE, F [n = 4442]). The equation of the statistically significant model (quadratic) is indicated at the top of each figure. [Color figure can be viewed at wileyonlinelibrary.com]
The trend of umbilical cord pCO2 appears to be opposite to that of pO2. Venous pCO2 shows a progressive increase, following a biphasic trend: starting from the 33rd to 34th week of pregnancy, pCO2 decreases (Figure 3A,B). The trend in arterial pCO2, however, shows a linear increase from the 23rd week until the end of gestation. Overall, CO2 production does not seem to change significantly until the 33rd−34th week but increases during the later stages of pregnancy (Figure 3C). Glucose extraction appears stable throughout the course of pregnancy, although it increases significantly in the final weeks of gestation (Figure 3D).
Figure 3.

Umbilical cord carbon dioxide and lactate status. Scatter plots with regression lines representing umbilical cord carbon dioxide levels (venous pCO2, A [n = 4529]; arterial pCO2, B [n = 4367]; CO2 production, C [n = 4315]); glucose extraction (D [n = 2775]); venous lactate, E ([n = 4119); arterial lactate, F [n = 4051]; fetal lactate production, G [n = 3993]). The equation of the statistically significant model (quadratic) is indicated at the top of each figure. [Color figure can be viewed at wileyonlinelibrary.com]
No significant change in lactate levels is observed in either venous or arterial samples during the early weeks of pregnancy, but an increase in lactate levels becomes evident starting from the 33rd to 34th weeks of gestation. Lactate production also appears to increase in the final weeks of pregnancy (Figure 3E−G).
The type of delivery significantly influences the cord blood gas analysis results, with a notable impact on oxygenation (Table 2). Therefore, umbilical cord venous and arterial O2 contents were analyzed separately according to the type of delivery (Figure S1). However, the results confirm the biphasic trend in the O2 levels that the fetus receives from the placenta, regardless of the type of delivery; this trend is only attenuated in the arterial values of newborns delivered via cesarean section.
The main criticism regarding the reconstruction of these trends concerns the accuracy of plotting blood gas values obtained from cord blood samples taken immediately at birth in preterm newborns, with samples collected from full‐term newborns approximately 60 s after birth. As mentioned above, delayed clamping of 30−60 s does not appear to substantially alter pH, HCO3 −, pCO2, lactate, or BE [117, 118, 119], and no effect on umbilical venous pO2 has ever been observed [116]. However, one study reported that delayed clamping for 2 min increases arterial pO2 by approximately 4 mmHg, likely as a result of the newborn's first breaths while the umbilical cord remains unclamped [120]. To address this issue, all arterial pO2 values of full‐term newborns were deliberately adjusted by subtracting 4 mmHg. Despite this mathematical adjustment, the results still confirm the previously described trends, although the increase in arterial pO2 in the late stages of pregnancy is less pronounced (Figure S2). Essentially, the biphasic trend of fetal oxygenation is confirmed even after correcting the bias due to different sampling methods. Oxygen extraction remains unchanged, stable throughout pregnancy (Figure S3).
In conclusion, the most innovative aspect of these studies is the demonstration that the levels of O2 supplied from the placenta to the fetus follow a biphasic trend, with a gradual reduction in oxygenation up to the 33rd−34th week of gestation, followed by an increase until birth. This insight is particularly valuable for researchers aiming to develop artificial placental technologies, who should take into account the physiological fluctuations in oxygenation levels [57, 58]. However, this awareness also prompts researchers to ask what the functional effects of these fluctuations might be.
6. Putative Functional Effects of Fluctuating O2 Levels
During intrauterine life, the embryo and fetus are supplied with varying levels of O2: immediately after fecundation, during implantation, and in the first weeks of pregnancy, the embryo lives in a highly hypoxic environment. However, the development of the placenta causes a sharp increase in O2 levels. Placentation thus represents a major turning point, with a “before” period characterized by significant hypoxia and an “after” period that is decidedly less hypoxic. Despite this early rise in O2 levels, our data demonstrate that the amount of O2 reaching the fetus from the middle of the second trimester until the first 4 weeks of the third trimester progressively decreases, reaching a minimum around 32−34 weeks. While the O2 level in the maternal uterine vessels remains stable throughout pregnancy [125], the progressive reduction in venous umbilical pO2 suggests that the placenta, as it grows and increases its metabolic activity, progressively extracts more O2 as pregnancy advances, reducing the amount of O2 available to the fetus. This interpretation is supported by samples drawn from the intervillous space of the human placenta, where the mean O2 tension gradually decreases [84, 91, 126], and by evidence that placental O2 consumption increases over time [60]. Therefore, the placenta, through its increase in O2 extraction, is responsible for the progressive reduction in fetal oxygenation observed between the second and third trimesters, while the fetus maintains its own O2 extraction largely unchanged for most of the pregnancy. These findings suggest that the primary function of the placenta is not to ensure maximum O2 supply to the fetus but rather to act as an anatomical barrier that limits O2 availability, providing a progressively more hypoxic environment for the fetus, at least until the 33rd−34th week of pregnancy.
Growing fetal hypoxia until the 33rd−34th week carries several possible consequences:
-
1.
The increasing fetal hypoxia that develops throughout much of pregnancy, until the last 6−8 weeks, upregulates HIF‐1 and induces significant tissue‐specific stem cell recruitment [127], as an O2‐poor environment is essential for preserving naïve stemness potential [128, 129]. The relationship between stemness and O2 levels is particularly evident for endothelial precursor cells (EPCs). Cord blood from preterm infants, in fact, shows a gradual increase in EPCs as gestational age progresses; these EPCs are very low at 24−28 weeks and highly expressed at 28−35 weeks [130] or 34 weeks [131]. This trend suggests that EPC expansion is synchronous with increasing hypoxemia, ceasing around the 33rd−34th week, when O2 levels start to rise. The close link between hypoxia and stemness is also evident during heart development, which is tightly regulated by HIF‐1 expression [10, 132]. Studies in mouse models has shown that hypoxia maintains cardiomyocyte precursors in an undifferentiated state [133, 134], likely allowing them to expand adequately. Similarly, pluripotent stem cells isolated from murine embryoid bodies show impaired differentiation into cardiomyocytes if exposed to hypoxia [135]. Additionally, fetuses made experimentally more hypoxic show an even higher number of immature cardiomyocytes [136], while adult mice gradually exposed to severe systemic hypoxemia show a reactivation of cardiomyocyte mitosis [137].
-
2.
Growing fetal hypoxia, mediated by HIF‐1, induces the differentiation of specific tissues in specific anatomic regions [3]. This is particularly evident in the CNS, where hypoxia regulates various processes, including cell survival, proliferation, catecholaminergic differentiation of isolated neural crest stem cells [138], differentiation of mesencephalic precursor cells into neurons [139], or transition of undifferentiated astrocytes into differentiated astrocytes in the developing retina [140, 141].
-
3.
This hypoxic environment induces a progressive increase in Hb concentration, as its synthesis is stimulated by the activation of the HIF/erythropoietin axis [142, 143]. Despite this increase in Hb, fetal O2 content continues to decrease until approximately the 33rd−34th week, confirming this period as a threshold beyond which umbilical venous oxygenation begins to rise.
-
4.
Increasing fetal hypoxia is associated with a progressive increase in cord venous pCO2 levels. This finding supports the idea that the placental barrier becomes progressively more effective at separating fetal and maternal circulations over time. As the passage of O2 from maternal to fetal circulation is increasingly hindered, the washout of fetal CO2 also becomes less efficient. It is possible that these changes in gas exchange are a consequence of placental growth, which physically better separates the two circulations; however, it is also possible that these effects are secondary to the increased metabolic activity of the placenta, which consumes more O2 and produces more CO2 as it grows [144]. The association of increasing hypoxia and hypercapnia explains the consistent reduction in fetal pH.
-
5.
Despite the increasing hypoxia, fetal metabolism does not appear to be significantly altered, as fetal CO2 production and glucose extraction remain largely unchanged.
However, starting from the 33rd to 34th week of gestation, the gas‐analytical and metabolic trends described in previous weeks abruptly reverse, with fetal oxygenation starting to increase. This gestational age represents the second watershed, beyond which fetal oxygenation rises, likely due to the physiological aging of the placenta, which favors increased diffusion of O2 [145]. The reduction in the distance between fetal vessels and the trophoblastic membrane, detectable in the last weeks of gestation, facilitates the increase in placental O2 transport [146]. The concomitance of other events suggests that the reduced barrier efficiency of the placenta in the final stages of pregnancy can be attributed to its aging. Evidence supporting this include the fact that most immunoglobulin G is acquired by the fetus during the last 4 weeks of pregnancy [147], and that the risk of viral vertical transmission, particularly for cytomegalovirus infections [148], increases with advancing gestational age [149]. These observations suggest that, in the last weeks of pregnancy, the placenta's barrier function between maternal and fetal circulation diminishes. However, this apparent reduction in placental performance observed in the last weeks of gestation may actually benefit the fetus, allowing the acquisition of immunocompetence through the transfer of immunoglobulins and the maturation of various functions in utero. In this regard, it is notable that many animals are born immature, with some structures, such as vascular structures, developing only after birth, likely in response to a more oxygenated environment. For example, rodents' glial−vascular interactions are established postnatally [18, 150], and their retinas, avascular at birth, begin to vascularize during the first postnatal week [20]. Rodent puppies, therefore, at birth are exposed in a condition of fragility, as evidenced by the fact that many mammals are born with their eyelids still sealed. On the contrary, human newborns are born more mature, as evidenced by their open eyes at birth and completed retinal vascularization. It is, therefore, likely that in all mammals, developmental maturation is favored by exposure to a more oxygenated environment, but only in some species (including humans), this maturation occurs during intrauterine life, probably due to the increased availability of O2, at least from the 33rd to 34th week.
A possible effect of this increased fetal oxygenation in the last weeks of gestation could be to prepare the human fetus for extrauterine life while still in utero, by regulating a series of functions:
-
1.
Increasing fetal O2 levels in the last weeks of gestation, through the downregulation of HIF‐1, induces the differentiation of specific tissues in specific anatomic districts [3]. The surge of O2 promotes precursor cell differentiation in neural retinal tissue [151], pancreatic cells [152], keratinocytes [153], hepatocyte cell lines [154], and, finally, endothelial cells [155]. The effect of O2 on the promotion of endothelial cell differentiation allows us to trace the stages of retinal vascularization in the human fetus: from mid‐gestation to near‐term, the intrauterine environment, which physiologically becomes more hypoxic, promotes the complete development of the astrocytic scaffold and EPC recruitment; during this period, only the EPCs closest to the central retinal artery are affected by an increase in O2 and differentiate into endothelial cells, ensuring centrifugal vascularization. However, the increased availability of O2 in the last weeks of gestation accelerates the complete vascularization of the retina. The example of retinal vascularization is emblematic of the importance of O2 level fluctuations during intrauterine life: these fluctuations are the driver that ensures the synchronization of the different phases of retinal development [156]. O2 tension, in addition to other stimuli, is an environmental cue that modulates also cardiomyocyte maturation [157]. Exposure to a more oxygenated environment, in fact, induces postnatal cardiomyocyte cell‐cycle arrest, likely through ROS upregulation [158]. Moreover, HIF‐1 downregulation by increased O2 tension has been suggested to promote the maturation of cardiomyocytes, inducing a metabolic switch from anaerobic glycolysis to OXPHOS [133].
-
2.
Rising O2 levels may favor the switch from fetal to adult Hb. In this regard, recent discoveries suggest that the production of γ‐globin and thus the production of fetal Hb (HbF) is regulated by HIF‐1 [159], indicating that the increase in O2 and the subsequent downregulation of HIF‐1 may represent a signal for the switch toward adult Hb (HbA) production. Indirect support for this hypothesis comes from the observation that the Hb switch from HbF to HbA begins before birth [160].
-
3.
Increased fetal oxygenation may favor a metabolic shift from a predominantly glycolytic metabolism, which requires minimal O2, to a predominantly oxidative metabolism that instead requires greater O2 supply. The indicators of this metabolic reprogramming are represented by the increase in CO2 production, glucose extraction, and lactate production in the final weeks of pregnancy.
Cord venous and arterial pCO2 levels in the last weeks of pregnancy show a peculiar trend: cord venous pCO2 decreases, and this trend is in line with placental aging, which results in a less efficient barrier effect and therefore greater placental washout of CO2 [145]. In contrast, arterial pCO2 continues to increase linearly from the 23rd week until the end of gestation. The persistent rise in arterial pCO2 even in the last weeks of pregnancy, despite greater placental washout, is likely due to the increased fetal CO2 production, which follows the fetus's increased metabolic activity. This observation is consistent with the simultaneous increase in glucose extraction and lactate production.
In this context, the rise in metabolism in the final stages of pregnancy suggests a possible metabolic shift toward greater mitochondrial activation. A recent study supports this hypothesis, showing that term human placentas exhibit significantly greater mitochondrial content and mitochondrial respiratory activity compared to first‐trimester placentas [161]. The availability of different concentrations of O2 influences the exploitation of different energy sources: to maintain sufficient ATP production to meet bioenergetic needs, anaerobic glycolysis predominates when O2 availability is low, while the mitochondrial OXPHOS pathway is favored when O2 is more abundant [162]. It is well known that HIF‐1 reduces mitochondrial function by inhibiting succinate dehydrogenase, that is, chain complex II [163], and, through the intermediation of nitric oxide, by inhibiting mitochondrial cytochrome c oxidase, that is, electron transport chain complex IV [164]. Additionally, HIF‐1 reduces mitochondrial content by decreasing their biogenesis [165] and increased autophagy [166]. Hypoxia not only reduces mitochondrial function but also diverts placental glucose metabolism toward glycolysis. In fact, HIF‐1 upregulates the transcription of glucose transporters [167, 168] as well as enzymes of the glycolytic pathway, such as aldolase A, phosphoglycerate kinase 1, enolase, and lactate dehydrogenase‐A [169].
Studies on pregnancies with IUGR have confirmed the effects of O2 levels on metabolic reprogramming and the shift between glycolytic and OXPHOS pathways. Particularly hypoxic placentas, such as those in pregnancies with IUGR, reduce their reliance on O2‐dependent mitochondrial OXPHOS and preferentially utilize the O2‐independent glycolytic pathway [170]. Even in this context, the main driver of placental metabolic reprogramming is HIF‐1, which is capable of inducing the Pasteur effect [171], that is, the diversion of carbohydrate metabolism from the mitochondrial oxidative pathway to anaerobic glycolysis [172]. Accordingly, the mitochondrial respiratory chain is less active in the placentas of IUGR pregnancies compared to those in uncomplicated pregnancies [173, 174, 175, 176]. Some authors interpret this reduced mitochondrial activation as a sign of mitochondrial dysfunction secondary to a non‐physiological pregnancy, rather than a compensatory mechanism [176]. However, in placentas from growth restriction pregnancies, ATP production remains unchanged compared to physiological pregnancies [173], suggesting that the switch from mitochondrial OXPHOS to the glycolytic pathway preserves ATP supply while reducing O2 consumption [172]. Consequently, some genes involved in mitochondrial activity and OXPHOS are downregulated in the placentas of growth‐restricted pregnancies [175, 176].
In conclusion, what occurs in the last weeks of an uncomplicated pregnancy appears to be the opposite, a mirror−image scenario to IUGR, equally capable of promoting metabolic reprogramming. In physiological conditions, reprogramming is driven by higher O2 levels and favors a shift toward predominant OXPHOS metabolism. These final weeks of pregnancy therefore seem to serve as a preparatory period for birth and the transition to a more O2‐rich environment.
These considerations help researchers address one of the most fascinating and mysterious questions for perinatologists: why is the fetus capable of modulating its metabolic options (prevalent glycolysis vs. prevalent OXPHOS), thereby demonstrating a notable ability to adapt to a wide range of hypoxic conditions, whereas after birth, the newborn loses this capacity to reprogram its metabolism and no longer tolerates life in a hypoxic environment? In fact, even a minimal reduction in neonatal oxygenation after birth—such as lowering the saturation target from 91%−95% to 85%−89%—leads to a marked increase in infant mortality [177, 178]. So, what are the biological “tricks” that allow the fetus this remarkable metabolic versatility, which the newborn irreversibly loses after birth?
Identifying these “tricks,” that is, the biological mechanisms that enable the fetus to thrive in a hypoxic environment, would be a major scientific breakthrough, opening up previously unexplored possibilities. In fact, if even a small remnant of these biological mechanisms, which allow fetal tolerance to hypoxia, was preserved, their pharmacological modulation might enable the re‐emergence of fetal capabilities and increase hypoxia tolerance in newborns. Figuratively, such modulation could be seen as a way to “bring the premature newborn back into the womb.” Essentially, this pharmacological approach could replicate in premature newborns the same beneficial effects they would have experienced had they remained in utero, benefiting from the gradual increase in hypoxia generated by placental growth. For this reason, we have provocatively referred to this hypothetical pharmacological approach as an attempt to create a “pharmacological artificial placenta.”
For several years, our research has been entirely focused on identifying the biological tricks employed by the fetus and placenta. There is substantial evidence suggesting we are on the right path.
7. The Role of β3‐ARs in Adaptation to Variable O2 Concentrations: A Significant Step Toward a Pharmacological Artificial Placenta
7.1. O2 Dependence of β3‐AR Expression
We previously discussed how HIF‐1 acts as the primary biological sensor responsible for coupling between low intrauterine O2 levels with vascular and metabolic adaptive mechanisms. Thus, HIF‐1 is undoubtedly one of the most crucial transcription factors that equips the fetus with the specific capabilities needed to thrive in a hypoxic environment. However, even though postnatal exposure to a hypoxic environment also leads to the upregulation of HIF‐1, this does not enable the newborn (nor the adult) to tolerate prolonged hypoxia, which is otherwise well tolerated during fetal life. This suggests that, during intrauterine life, other factors or biological mechanisms are essential to fully benefit from the hypoxic environment.
A growing body of research has recently shed new light on the involvement of the adrenergic system in the adaptation to prenatal intrauterine life. Since the late 1990s, the essential role played by the adrenergic system in ensuring embryonic development and fetal survival has become evident, as indicated by the high embryonic lethality observed in mice lacking tyrosine hydroxylase, the rate‐limiting enzyme in catecholamine biosynthesis [179, 180, 181]. Adrenoceptors are well‐known as key mediators of the sympathetic nervous system. By binding catecholamines (adrenaline and noradrenaline), adrenoceptors regulate the function of organs and tissues, maintaining homeostasis and responding to stress. The nine known adrenoceptor subtypes are divided into three families (α1, α2, β). Early studies demonstrated that the effects of catecholamines on fetal cardiac reactivity and survival were mediated through β‐adrenoceptors (β‐ARs), without identifying which specific receptor was most involved [182]. However, later studies identified β1‐AR as the β‐AR subtype most responsible for maintaining fetal heart rate in hypoxic conditions [183], while β2‐ARs have been shown to provide growth and differentiation signals in various tissues [184, 185].
The β3‐AR, the last β‐AR subtype identified in 1989 [186], is a strong candidate for playing an important and early role in embryonic development, even before the enzymatic system that produces catecholamines is fully mature, due to its constitutive activity, which allows it to be potentially active even without ligand stimulation [187]. β3‐ARs are likely involved in conception, as they are expressed in mammalian oocytes [188, 189] and spermatozoa, where they induce motility [190]. β3‐ARs are also expressed in preimplantation embryos [188, 189], in embryos during the early stages of embryogenesis [191], in embryonic tissues and in the placenta [192, 193]. Moreover, β3‐ARs are detected in the mouse uterus [194] and are significantly upregulated in the human pregnant myometrium, where they represent the predominant β‐AR subtype and participate in suppressing spontaneous contractions [195, 196]. Because of this, they are considered ideal targets for tocolytic drugs [197]. Finally, β3‐ARs agonism induces vasodilation of the human umbilical artery ring, suggesting that β3‐ARs can regulate fetal−placental circulation [198]. These data suggest a potential role for β3‐ARs in fecundation, embryo implantation, embryo vascularization, and embryonic/fetal well‐being. The recent observation that repeated administration of high doses of β3‐ARs antagonists to pregnant animals induced preterm delivery or fetal death [199] further supports the active involvement of β3‐ARs in embryonic/fetal adaptation mechanisms.
The consensus in the literature is unanimous that β3‐ARs in various tissues are upregulated under hypoxic conditions [200, 201, 202, 203], and recent evidence suggests direct regulation by HIF‐1 [204]. The close relationship between hypoxia and β3‐AR expression is particularly evident in the mouse retina, where both HIF‐1 and β3‐ARs are strongly expressed during intrauterine life. However, their expression rapidly decreases after birth due to exposure to a more oxygenated environment [205]. This observation is consistent with findings in lymphocytes, where β3‐ARs were upregulated under hypoxic conditions but quickly downregulated after re‐exposure to O2 [206]. Therefore, β3‐ARs appear to be particularly sensitive to and modulated by O2 levels, being upregulated by hypoxia and downregulated by higher O2 concentrations.
At the same time, a growing body of evidence suggests that β3‐ARs are involved in many biological processes that ensure embryo/fetal well‐being and are, unfortunately, reactivated in cancer, allowing both the embryo/fetus and cancer to thrive in an otherwise inhospitable environment [59]. In fact, in addition to their role in vascularization processes induced by hypoxia [207, 208], β3‐ARs, which are intensely expressed in the placenta, promote the development of immune tolerance toward the fetus [209], similarly to how β3‐ARs expressed in cells of the tumor microenvironment promote tolerance toward cancer [206]. Moreover, β3‐ARs seem to participate in the mechanisms by which hypoxia induces the maintenance of stemness traits, as demonstrated by in vitro experiments [203] and confirmed by the observation that pharmacological blockade of β3‐ARs induces the differentiation of tumor stem cells [210, 211]. Finally, β3‐ARs are actively involved in hypoxia‐induced metabolic reprogramming. In embryonic and cancer stem cells, in fact, β3‐ARs induce a distinctive metabolic rearrangement characterized by increased glucose uptake and accelerated glycolytic metabolism, which, at least partially, replaces mitochondrial OXPHOS [212]. This reprogramming occurs through the β3‐ARs‐mediated upregulation of key enzymes involved in glycolytic pathways, such as glucose transporter‐1 (Glut‐1) and hexokinase 2 (HK‐II), as well as transmembrane proteins like monocarboxylate transporter‐4 (MCT‐4), responsible for increased lactate export. At the same time, β3‐ARs induce the upregulation of uncoupling protein 2 (UCP2), which is responsible for mitochondrial dormancy and the reduction of the OXPHOS pathway [212].
Therefore, the knowledge acquired in recent years suggests that β3‐ARs, significantly upregulated by hypoxia during intrauterine life, not only manage various functions that ensure embryonic/fetal well‐being but also shift energy metabolism toward the glycolytic pathway, which is less dependent on O2, rather than the OXPHOS pathway. β3‐ARs thus possess all the characteristics to represent the biological “trick” (or, more likely, one of the biological tricks) that allows the embryo and fetus to thrive in an environment with extremely low O2 levels.
However, as demonstrated by our findings, in a physiological pregnancy, starting from the 33rd to 34th week of gestation, the availability of O2 increases, and O2 levels rise further after birth. It is highly likely that exposure to increasing O2 concentrations leads to a reduction in β3‐ARs expression, causing the upregulation of glycolytic enzymes to cease and the mitochondria to “awaken.” The combination of these phenomena may result in a metabolic shift from the glycolytic pathway to mitochondrial OXPHOS. From this point onward, due to the virtual disappearance of the β3‐ARs, the dependence on O2 would become irreversible, and for this reason, the energy needs of the newborn would no longer be met in an O2‐poor environment.
The involvement of β3‐ARs in numerous processes that ensure fetal well‐being (primarily vascularization) and their modulation by O2 supports the hypothesis that the main damages observable in premature newborns may be caused by the premature downregulation of β3‐ARs following exposure to a hyperoxic environment. Indeed, just as the upregulation of β3‐ARs seems to couple hypoxia with the induction of vascularization [202, 207], the downregulation of β3‐ARs has been hypothesized to be responsible for the coupling between hyperoxia and vascularization arrest [213]. In this regard, it is no coincidence that all pathologies specifically related to prematurity (ROP, BPD, NEC, and PVL) are characterized by an early arrest of vascularization [59].
7.2. β3‐AR Agonism to Prevent Hyperoxia Damages
The recognition of the beneficial role played by β3‐ARs during intrauterine life and the possibility that their downregulation after a preterm birth may contribute to neonatal damage opens up promising new scenarios. In fact, if β3‐ARs do not disappear completely after birth, it may theoretically be possible to reactivate some of their functions using currently available drugs that selectively stimulate them. The data available so far suggest that β3‐ARs contribute to fetal well‐being as intermediaries of certain biological functions promoted by HIF‐1. Thus, pharmacological stimulation of β3‐ARs could maintain these functions even in the absence of hypoxia, HIF‐1, or even in a hyperoxic environment. In other words, the pharmacological activation of these receptors could reproduce the effects normally promoted by hypoxia, even in a high‐O2 environment, thereby counteracting the effects of hyperoxia. Figuratively, this approach could resemble returning the preterm infant to the mother's womb.
Such a perspective is not as far‐fetched as it might initially seem. Until a few years ago, inducing Hb production in anemic patients or athletes required moving to the high altitudes to exploit the effect that hypoxia on red blood cells production [214]. However, once it was demonstrated that hypoxia induces red blood cell production through the mediation of erythropoietin [215], its administration became sufficient to treat anemia or enhance athletic performance without the need for altitude [216, 217]. Figuratively, treatment with erythropoietin is equivalent to moving a patient or athlete to the mountains. Similarly, once it is demonstrated that the hypoxic uterine environment promotes fetal well‐being through β3‐ARs, it is legitimate to hypothesize that pharmacological stimulation of these receptors in preterm newborns could replicate the effects of the uterine environment. This hypothesis of “returning the preterm infant to the womb” effectively paves the way for a pharmacological artificial placenta.
These considerations explain why our current research focuses on evaluating, in newborn animals, whether β3‐AR agonist administration can mitigate the harmful effects of hyperoxia on various tissues. Initial results have recently shown that pharmacological stimulation of β3‐ARs neutralizes the damage induced by hyperoxia in the colon [218] and ileum [219] of neonatal rats.
It is well‐known that neonatal exposure to hyperoxia alters the structure of the colon mucosa in newborn rats [28, 220, 221] and disrupts the balance of the gut microflora [222] due to the generation of ROS, which compromise the integrity of the intestinal barrier [220, 222]. A first study performed in neonatal rats demonstrated that administration of a β3‐AR agonist prevented the shortening of colonic length caused by hyperoxia and ensured the production of mucins by goblet cells within the proximal colon's lining epithelium [218]. These findings provide the first evidence that targeting β3‐ARs pharmacologically could represent a therapeutic option for diseases characterized by impaired development due to hyperoxia. A second study has recently shown similar protective effects of β3‐AR agonism in the ileum. In this anatomical region, hyperoxia also caused damage, evidenced by increased mucosal thickness [221], disruption of the epithelial barrier through effects on tight junctions, and the dilation of capillaries and lacteals [220]. However, the administration of a β3‐AR agonist preserved the integrity of the gut surface epithelium despite hyperoxic exposure [219]. Notably, the significant reduction in vascularization observed in the ileal submucosa and muscle coat of hyperoxic control rats was prevented by β3‐AR agonist treatment [219]. These findings suggest that β3‐AR agonism can neutralize the effects of hyperoxia, restore conditions similar to those in utero, and pave the way for the prevention of NEC.
Similar results have recently been observed in the retina of neonatal mice. In this case, exposure to a hyperoxic environment typically induces the cessation of vascularization in the central part of the retina [223], which is associated with a reduced density of retinal astrocytes [224]. This vascular regression, in turn, leads to hypoxic stress in the peripheral retina, triggering subsequent tumultuous and pathological vascularization [223]. A recent study using the oxygen‐induced retinopathy (OIR) experimental model demonstrated that treatment with β3‐AR agonists administered during the hypoxic phase promoted physiological vascularization in the central retina, restored blood‐retinal barrier integrity, neutralized the effects of hyperoxia, and prevented hypoxic suffering and the induction of pathological peripheral retina vascularization [225]. Although in this third study, the administration of β3‐AR agonists was not synchronous with hyperoxia exposure, also in this experimental setting the activation of β3‐ARs still demonstrated an ability to restore physiological conditions experimentally altered by hyperoxia. These promising results highlight the potential pharmacological prevention of the initial phase of ROP.
Overall, the preliminary data suggest that treatment with β3‐AR agonists neutralizes much of the damage caused by hyperoxia, which is critical in the development of major prematurity‐related diseases (Figure 4). The most promising effect appears to be the restoration of normal vascularization, which would otherwise be impaired by hyperoxic exposure. However, the benefits of β3‐AR agonist treatment do not seem to be limited to vascularization. In fact, preliminary data from animal studies suggest that β3‐AR agonism can also reverse certain hyperoxia‐induced alterations even in the nervous system. The study conducted on the OIR mouse model showed that the restoration of normal vascularization induced by β3‐AR agonism was accompanied by a recovery in astrocyte density in the central avascular retina. The exact mechanism by which β3‐AR stimulation restores the retinal astrocyte population is not yet clear. However, the induction of specific transcription factors suggests that β3‐AR activation may promote astrocyte migration or maturation [225]. Similarly, a study conducted on neonatal rats exposed to prolonged hyperoxia demonstrated significant effects of β3‐AR agonist treatment on the myenteric neuronal population. Hyperoxia exposure caused a notable increase in neurons expressing choline acetyltransferase and a marked reduction in neurons expressing neuronal nitric oxide synthase (implicated in the production of two opposing neurotransmitters, the excitatory acetylcholine and the inhibitory nitric oxide, respectively). However, these hyperoxia‐induced changes in neuronal chemical coding were prevented by β3‐AR agonism [218], restoring conditions typically seen under physiological normoxia.
Figure 4.

Putative involvement of β3‐ARs in the pathogenesis of prematurity related diseases. β3‐ARs are actively involved in prenatal vascularization, participating in the coupling between hypoxia and the promotion of vasculogenesis (left). Premature birth involves early exposure to a relatively hyperoxic environment that downregulates HIF‐1 and β3‐ARs. If the downregulation of β3‐ARs is responsible for the arrest of vascularization observed in preterm newborns (center), this could be reactivated with pharmacological stimulation of the remaining β3‐ARs (right). Thus, it could be possible to reactivate typically fetal functions promoted by hypoxia (e.g., vascularization) even after birth, even without the need to reduce the O2 supply, through pharmacological stimulation. This strategy aims to prevent all pathologies related to prematurity that are triggered by an arrest of vascularization. [Color figure can be viewed at wileyonlinelibrary.com]
It is, therefore, likely that the effects of stimulating β3‐ARs during the neonatal period could be even more numerous, warranting further investigation in the future. In particular, we expect that the pharmacological stimulation of β3‐ARs will have significant effects on the metabolic programming of the newborn. Since the activation of β3‐ARs has already been shown to promote the glycolytic pathway at the expense of mitochondrial activity [212], we can anticipate that preterm newborns treated with β3‐AR agonists may experience a metabolic shift toward greater reliance of glycolysis and reduced OXPHOS activity. This shift would likely result in an increased need for glucose, while simultaneously reducing mitochondrial activity. Theoretically, reduced mitochondrial activation should lead to lower production of mitochondrial free radicals, decreased dependence on O2, and possibly greater tolerance to lower oxygenation levels. All of these conditions could provide theoretical benefits to preterm newborns, but these effects will need to be verified and thoroughly analyzed in future studies.
8. Limitations and Potential Risks
The strategy of reactivating fetal competencies in preterm newborns through the administration of β3‐AR agonist drugs does not aim to completely nullify the effects of early interruption of intrauterine life and premature exposure to a hyperoxic environment. It is evident that the physiological hypoxia experienced by the fetus in the maternal womb regulates, via HIF‐1, a myriad of functions, only a portion of which is likely mediated by β3‐ARs. Therefore, our goal is not to replace the extreme complexity of the maternal uterus with drugs, but rather to help preterm newborns maintain at least the biological effects that would have been ensured by the increased expression of β3‐ARs. The objective we reasonably set is to mitigate the consequences of prematurity and limit the damage caused by hyperoxia.
One of the current limitations of this strategy is the lack of studies demonstrating the safety of β3‐AR agonist drugs during the neonatal period, particularly in preterm newborns, although these drugs have been used in pediatric patients for different indications [226]. At present, there are no reports on the use of β3‐AR agonist drugs in preterm newborns, and thus no specific adverse events have been described. Theoretically, one of the potential risks in preterm infants could be the induction of persistent patent ductus arteriosus. In recent years, our research group has demonstrated the presence of β3‐ARs in endothelial and smooth muscle cells of the murine fetal ductus arteriosus and their involvement in maintaining its patency. Indeed, treating pregnant mice with β3‐AR antagonists led to partial closure of the ductus arteriosus in fetuses [199]. Therefore, it cannot be ruled out that treating preterm newborns with a β3‐AR agonist might contribute to the persistence of a patent ductus arteriosus. Fortunately, evidence showing that β3‐ARs agonism administered to neonatal mice immediately after birth did not prevent the physiological ductus arteriosus closure, raises hope that the role of β3‐ARs in maintaining duct patency is marginal [199].
9. Alternative Strategies
It is reasonable to imagine that treatment with β3‐ARs agonists is not the only possible path to achieving a pharmacological reconstruction of the uterine environment, thus creating a virtual artificial placenta. If the fetus indeed develops in utero through the combined action of HIF‐1 and β3‐ARs, pharmacological upregulation of HIF‐1 could be a valid alternative approach to mimic the uterine environment. In this context, prolyl hydroxylase domain (PHD) inhibitors represent a novel class of drugs capable of preventing the hydroxylation and degradation of HIF‐1, even in the presence of O2. Therefore, these drugs can stabilize and maintain elevated levels of HIF‐1 even in the absence of hypoxia. Preclinical studies have suggested that these drugs may be effective in counteracting vascular regression induced by hyperoxia. For instance, the systemic administration of competitive PHD inhibitors, such as dimethyloxalylglycine or Roxadustat, to neonatal animal exposed to hyperoxia prevented retinal vascular regression by increasing levels of HIF‐1α and pro‐angiogenic factors such as VEGF and erythropoietin [227, 228]. A number of PHD inhibitors are already in clinical use to mimic the effects of hypoxia for systemic conditions such as anemia [229], acute and chronic kidney disease, or cardiovascular diseases [230]. However, despite the promise of this approach, concerns exist regarding the safety of HIF‐1 modulation, especially in immature infants who are still undergoing key developmental processes. The upregulation of a transcription factor like HIF‐1, which controls hundreds of genes involved in the regulation of delicate biological functions, may lead to severe adverse effects, such as cancer, thromboembolism, pulmonary hypertension, or hyperkaliemia [231].
10. Conclusion
The preclinical results available thus far suggest that pharmacological treatments stimulating β3‐ARs—currently available, inexpensive, and safe for use in pediatric populations—can effectively counteract the effects of early exposure to a highly oxygenated environment on neonatal tissues. The goal of this strategy is to restore in premature newborns the same fetal capabilities that were induced by hypoxia in utero, even in the presence of an O2‐rich environment. This approach aims to replicate the physiological benefits typically provided by the placenta using a drug, without relying on sophisticated and complex technology. However, further steps are required before advancing to human trials.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supplementary Fig. 1. Umbilical cord oxygenation status according to the type of delivery. Scatter plots with regression lines representing umbilical cord oxygenation status [total venous O2 content, panel A (n = 1788) and arterial O2 content, panel B (n = 1740)] in neonates born by vaginal delivery and oxygenation status [total venous O2 content, panel C (n = 855) and arterial O2 content, panel D (n = 808)] in neonates born by cesarean delivery. The equation of the statistically significant model (linear or quadratic) is indicated at the top of each figure.
Supplementary Fig. 2. Umbilical cord oxygenation status after subtracting 4 mmHg from the arterial pO 2 values. Scatter plots with regression lines representing the umbilical cord oxygenation status [venous pO2, panel A (n = 4286); arterial pO2, panel B (n = 4161); total venous, panel C (n = 2643) and arterial O2 contents, panel D (n = 2547)] of the whole study population after artificial correction of arterial pO2 values, subtracting 4 mmHg from the original values. The equation of the statistically significant model (linear or quadratic) is indicated at the top of each figure.
Supplementary Fig. 3. Umbilical cord O 2 extraction after subtracting 4 mmHg from the arterial pO 2 values. Scatter plot representing umbilical cord O2 extraction of the whole study population after artificial correction of arterial pO2 values minus 4 mmHg to the original values (n = 2548).
Acknowledgments
We are most grateful to Jean‐Luc Baroni and Cristina Ranzato for their valuable support in conducting this study. This research was funded by Ministero dell'Università e della Ricerca, PRIN 2022 D.D. 104 del 02/02/2022 funded by Europe Union–NextGenerationEU‐OBERON, 2022FYBMEX, Italy. Open access publishing facilitated by Universita degli Studi di Pisa, as part of the Wiley ‐ CRUI‐CARE agreement.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Thannickal V. J., “Oxygen in the Evolution of Complex Life and the Price We Pay,” American Journal of Respiratory Cell and Molecular Biology 40, no. 5 (May 2009): 507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Semenza G. L., “Hypoxia‐Inducible Factor 1: Control of Oxygen Homeostasis in Health and Disease,” Pediatric Research 49, no. 5 (May 2001): 614–617. [DOI] [PubMed] [Google Scholar]
- 3. Simon M. C. and Keith B., “The Role of Oxygen Availability in Embryonic Development and Stem Cell Function,” Nature Reviews Molecular Cell Biology 9, no. 4 (April 2008): 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pringle K. G., Kind K. L., Sferruzzi‐Perri A. N., Thompson J. G., and Roberts C. T., “Beyond Oxygen: Complex Regulation and Activity of Hypoxia Inducible Factors in Pregnancy,” Human Reproduction Update 16, no. 4 (July/August 2010): 415–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dengler V. L., Galbraith M. D., and Espinosa J. M., “Transcriptional Regulation by Hypoxia Inducible Factors,” Critical Reviews in Biochemistry and Molecular Biology 49, no. 1 (January/February 2014): 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Borovsky V., Herman M., Dunphy G., Caplea A., and Ely D., “CO2 Asphyxia Increases Plasma Norepinephrine in Rats via Sympathetic Nerves,” American Journal of Physiology 274, no. 1 (January 1998): 19–22. [DOI] [PubMed] [Google Scholar]
- 7. Park S. Y., Kang J. H., Jeong K. J., et al., “Retracted: Norepinephrine Induces VEGF Expression and Angiogenesis by a Hypoxia‐Inducible Factor‐1α Protein‐Dependent Mechanism,” International Journal of Cancer 128, no. 10 (May 2011): 2306–2316. [DOI] [PubMed] [Google Scholar]
- 8. Mostafa S. S., Miller W. M., and Papoutsakis E. T., “Oxygen Tension Influences the Differentiation, Maturation and Apoptosis of Human Megakaryocytes,” British Journal of Haematology 111, no. 3 (December 2000): 879–889. [PubMed] [Google Scholar]
- 9. Francis K. R. and Wei L., “Human Embryonic Stem Cell Neural Differentiation and Enhanced Cell Survival Promoted by Hypoxic Preconditioning,” Cell Death & Disease 1, no. 2 (2010): e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Niebruegge S., Bauwens C. L., Peerani R., et al., “Generation of Human Embryonic Stem Cell‐Derived Mesoderm and Cardiac Cells Using Size‐Specified Aggregates in an Oxygen‐Controlled Bioreactor,” Biotechnology and Bioengineering 102, no. 2 (February 2009): 493–507. [DOI] [PubMed] [Google Scholar]
- 11. Tomita S., Ueno M., Sakamoto M., et al., “Defective Brain Development in Mice Lacking the Hif‐1α Gene in Neural Cells,” Molecular and Cellular Biology 23, no. 19 (October 2003): 6739–6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ateghang B., Wartenberg M., Gassmann M., and Sauer H., “Regulation of Cardiotrophin‐1 Expression in Mouse Embryonic Stem Cells by HIF‐1α and Intracellular Reactive Oxygen Species,” Journal of Cell Science 119, no. pt. 6 (March 2006): 1043–1052. [DOI] [PubMed] [Google Scholar]
- 13. Eastman N. J., “Mount Everest in Utero,” American Journal of Obstetrics and Gynecology 67, no. 4 (April 1954): 701–711. [DOI] [PubMed] [Google Scholar]
- 14. Fathollahipour S., Patil P. S., and Leipzig N. D., “Oxygen Regulation in Development: Lessons From Embryogenesis Towards Tissue Engineering,” Cells Tissues Organs 205, no. 5–6 (2018): 350–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao H., Wong R. J., and Stevenson D. K., “The Impact of Hypoxia in Early Pregnancy on Placental Cells,” International Journal of Molecular Sciences 22, no. 18 (September 2021): 9675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Robins J. C., Heizer A., Hardiman A., Hubert M., and Handwerger S., “Oxygen Tension Directs the Differentiation Pathway of Human Cytotrophoblast Cells,” Placenta 28, no. 11–12 (November/December 2007): 1141–1146. [DOI] [PubMed] [Google Scholar]
- 17. Coelho‐Santos V., Cruz A. J. N., and Shih A. Y., “Does Perinatal Intermittent Hypoxia Affect Cerebrovascular Network Development?,” Developmental Neuroscience 46, no. 1 (2024): 44–54. [DOI] [PubMed] [Google Scholar]
- 18. Semple B. D., Blomgren K., Gimlin K., Ferriero D. M., and Noble‐Haeusslein L. J., “Brain Development in Rodents and Humans: Identifying Benchmarks of Maturation and Vulnerability to Injury Across Species,” Progress in Neurobiology 106–107 (July/August 2013): 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aguilar E., Dorrell M. I., Friedlander D., et al., “Chapter 6. Ocular Models of Angiogenesis,” Methods in Enzymology 444 (2008): 115–158. [DOI] [PubMed] [Google Scholar]
- 20. Stahl A., Connor K. M., Sapieha P., et al., “The Mouse Retina as an Angiogenesis Model,” Investigative Opthalmology & Visual Science 51, no. 6 (June 2010): 2813–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hartnett M. E. and Lane R. H., “Effects of Oxygen on the Development and Severity of Retinopathy of Prematurity,” Journal of American Association for Pediatric Ophthalmology and Strabismus 17, no. 3 (June 2013): 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chiang M. F., Quinn G. E., Fielder A. R., et al., “International Classification of Retinopathy of Prematurity, Third Edition,” Ophthalmology 128, no. 10 (October 2021): e51–e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hellström A., Smith L. E., and Dammann O., “Retinopathy of Prematurity,” Lancet 382, no. 9902 (October 2013): 1445–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Buczynski B. W., Maduekwe E. T., and O'Reilly M. A., “The Role of Hyperoxia in the Pathogenesis of Experimental BPD,” Seminars in Perinatology 37, no. 2 (April 2013): 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bhatt A. J., Pryhuber G. S., Huyck H., Watkins R. H., Metlay L. A., and Maniscalco W. M., “Disrupted Pulmonary Vasculature and Decreased Vascular Endothelial Growth Factor, Flt‐1, and TIE‐2 in Human Infants Dying With Bronchopulmonary Dysplasia,” American Journal of Respiratory and Critical Care Medicine 164, no. 10 pt. 1 (November 2001): 1971–1980. [DOI] [PubMed] [Google Scholar]
- 26. Mariduena J., Ramagopal M., Hiatt M., Chandra S., Laumbach R., and Hegyi T., “Vascular Endothelial Growth Factor Levels and Bronchopulmonary Dysplasia in Preterm Infants,” Journal of Maternal‐Fetal & Neonatal Medicine 35, no. 8 (April 2022): 1517–1522. [DOI] [PubMed] [Google Scholar]
- 27. Liu W., Fan X., Jian B., et al., “The Signaling Pathway of Hypoxia Inducible Factor in Regulating Gut Homeostasis,” Frontiers in Microbiology 14 (October 2023): 1289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang H. C., Chou H. C., and Chen C. M., “Molecular Mechanisms of Hyperoxia‐Induced Neonatal Intestinal Injury,” International Journal of Molecular Sciences 24, no. 5 (February 2023): 4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen Y., Chang K. T. E., Lian D. W. Q., et al., “The Role of Ischemia in Necrotizing Enterocolitis,” Journal of Pediatric Surgery 51, no. 8 (August 2016): 1255–1261. [DOI] [PubMed] [Google Scholar]
- 30. Trollmann R. and Gassmann M., “The Role of Hypoxia‐Inducible Transcription Factors in the Hypoxic Neonatal Brain,” Brain and Development 31, no. 7 (August 2009): 503–509. [DOI] [PubMed] [Google Scholar]
- 31. Xue‐Jiao H. and Jian‐Hua F., “A Review of the Effects of Early Postnatal Hyperoxia Exposure on the Immature Brain,” Experimental Neurology 370 (December 2023): 114550. [DOI] [PubMed] [Google Scholar]
- 32. Sirinyan M., Sennlaub F., Dorfman A., et al., “Hyperoxic Exposure Leads to Nitrative Stress and Ensuing Microvascular Degeneration and Diminished Brain Mass and Function in the Immature Subject,” Stroke 37, no. 11 (November 2006): 2807–2815. [DOI] [PubMed] [Google Scholar]
- 33. Lithopoulos M. A., Toussay X., Zhong S., et al., “Neonatal Hyperoxia in Mice Triggers Long‐Term Cognitive Deficits via Impairments in Cerebrovascular Function and Neurogenesis,” Journal of Clinical Investigation 132, no. 22 (November 2022): e146095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ballabh P., Braun A., and Nedergaard M., “Anatomic Analysis of Blood Vessels in Germinal Matrix, Cerebral Cortex, and White Matter in Developing Infants,” Pediatric Research 56, no. 1 (July 2004): 117–124. [DOI] [PubMed] [Google Scholar]
- 35. Vottier G., Pham H., Pansiot J., et al., “Deleterious Effect of Hyperoxia at Birth on White Matter Damage in the Newborn Rat,” Developmental Neuroscience 33, no. 3–4 (2011): 261–269. [DOI] [PubMed] [Google Scholar]
- 36. Gerstner B., DeSilva T. M., Genz K., et al., “Hyperoxia Causes Maturation‐Dependent Cell Death in the Developing White Matter,” Journal of Neuroscience 28, no. 5 (January 2008): 1236–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Torres‐Cuevas I., Parra‐Llorca A., Sánchez‐Illana A., et al., “Oxygen and Oxidative Stress in the Perinatal Period,” Redox Biology 12 (2017): 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rogers S., Witz G., Anwar M., Hiatt M., and Hegyi T., “Antioxidant Capacity and Oxygen Radical Diseases in the Preterm Newborn,” Archives of Pediatrics & Adolescent Medicine 154, no. 6 (2000): 544–548. [DOI] [PubMed] [Google Scholar]
- 39. Houldsworth A., “Role of Oxidative Stress in Neurodegenerative Disorders: A Review of Reactive Oxygen Species and Prevention by Antioxidants,” Brain Communications 6, no. 1 (2024): fcad356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Panfoli I., Candiano G., Malova M., et al., “Oxidative Stress as a Primary Risk Factor for Brain Damage in Preterm Newborns,” Frontiers in Pediatrics 6 (2018): 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cannavò L., Perrone S., Viola V., Marseglia L., Di Rosa G., and Gitto E., “Oxidative Stress and Respiratory Diseases in Preterm Newborns,” International Journal of Molecular Sciences 22, no. 22 (2021): 12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Torres‐Cuevas I., Corral‐Debrinski M., and Gressens P., “Brain Oxidative Damage in Murine Models of Neonatal Hypoxia/Ischemia and Reoxygenation,” Free Radical Biology and Medicine 142 (2019): 3–15. [DOI] [PubMed] [Google Scholar]
- 43. Di Fiore J. M. and Raffay T. M., “The Relationship Between Intermittent Hypoxemia Events and Neural Outcomes in Neonates,” Experimental Neurology 342 (2021): 113753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Di Fiore J. M. and Vento M., “Intermittent Hypoxemia and Oxidative Stress in Preterm Infants,” Respiratory Physiology & Neurobiology 266 (2019): 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zorov D. B., Juhaszova M., and Sollott S. J., “Mitochondrial Reactive Oxygen Species (ROS) and ROS‐Induced ROS Release,” Physiological Reviews 94, no. 3 (2014): 909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tong Y., Zhang S., Riddle S., Zhang L., Song R., and Yue D., “Intrauterine Hypoxia and Epigenetic Programming in Lung Development and Disease,” Biomedicines 9, no. 8 (2021): 944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Berger J. and Bhandari V., “Animal Models of Bronchopulmonary Dysplasia. The Term Mouse Models,” American Journal of Physiology‐Lung Cellular and Molecular Physiology 307, no. 12 (2014): L936–L947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leary S. and Bhandari V., “Chapter 3: Animal Models of Bronchopulmonary Dysplasia,” in Updates on Neonatal Chronic Lung Disease, eds. Kallapur S. G. and Pryhuber G. S. (Elsevier, 2020), 33–44. [Google Scholar]
- 49. Endesfelder S., “Caffeine: The Story Beyond Oxygen‐Induced Lung and Brain Injury in Neonatal Animal Models: A Narrative Review,” Antioxidants 13, no. 9 (2024): 1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schmitz T., Ritter J., Mueller S., Felderhoff‐Mueser U., Chew L. J., and Gallo V., “Cellular Changes Underlying Hyperoxia‐Induced Delay of White Matter Development,” Journal of Neuroscience 31, no. 11 (2011): 4327–4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sotiropoulos J. X., Saugstad O. D., and Oei J. L., “Aspects on Oxygenation in Preterm Infants Before, Immediately After Birth, and Beyond,” Neonatology 121, no. 5 (2024): 562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Song W., Hoppe G., Hanna D., DeSilva T. M., and Sears J. E., “Hyperoxia Induced Hypomyelination,” Biomedicines 11, no. 1 (2022): 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bavis R. W., “Effects of Perinatal Hyperoxia on Breathing,” Comprehensive Physiology 10, no. 2 (2020): 597–636. [DOI] [PubMed] [Google Scholar]
- 54. Krock B. L., Skuli N., and Simon M. C., “Hypoxia‐Induced Angiogenesis: Good and Evil,” Genes & Cancer 2, no. 12 (December 2011): 1117–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Filippi L., Scaramuzzo R. T., Pascarella F., et al., “Fetal Oxygenation in the Last Weeks of Pregnancy Evaluated Through the Umbilical Cord Blood Gas Analysis,” Frontiers in Pediatrics 11 (April 2023): 1140021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Filippi L., Pascarella F., Pini A., et al., “Fetal Oxygenation From the 23rd to the 36th Week of Gestation Evaluated Through the Umbilical Cord Blood Gas Analysis,” International Journal of Molecular Sciences 24, no. 15 (August 2023): 12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Spencer B. L. and Mychaliska G. B., “Milestones for Clinical Translation of the Artificial Placenta,” Seminars in Fetal and Neonatal Medicine 27, no. 6 (December 2022): 101408. [DOI] [PubMed] [Google Scholar]
- 58. Usuda H., Watanabe S., T H., et al., “Artificial Placenta Technology: History, Potential and Perception,” Placenta 141 (September 2023): 10–17. [DOI] [PubMed] [Google Scholar]
- 59. Filippi L., Pini A., Cammalleri M., Bagnoli P., and Dal Monte M., “β3‐Adrenoceptor, a Novel Player in the Round‐Trip From Neonatal Diseases to Cancer: Suggestive Clues From Embryo,” Medicinal Research Reviews 42, no. 3 (May 2022): 1179–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Carter A. M., “Placental Gas Exchange and the Oxygen Supply to the Fetus,” Comprehensive Physiology 5, no. 3 (July 2015): 1381–1403. [DOI] [PubMed] [Google Scholar]
- 61. Cross B. A. and Silver I. A., “Neurovascular Control of Oxygen Tension in the Testis and Epididymis,” Reproduction (Cambridge, England) 3 (1962): 377–395. [DOI] [PubMed] [Google Scholar]
- 62. Free M. J., Schluntz G. A., and Jaffe R. A., “Respiratory Gas Tensions in Tissues and Fluids of the Male Rat Reproductive Tract,” Biology of Reproduction 14 (1976): 481–488. [DOI] [PubMed] [Google Scholar]
- 63. Lysiak J. J., Nguyen Q. A., and Turner T. T., “Fluctuations in Rat Testicular Interstitial Oxygen Tensions Are Linked to Testicular Vasomotion,” Biology of Reproduction 63, no. 5 (November 2000): 1383–1389. [DOI] [PubMed] [Google Scholar]
- 64. Li Z., Wang S., Gong C., et al., “Effects of Environmental and Pathological Hypoxia on Male Fertility,” Frontiers in Cell and Developmental Biology 9 (September 2021): 725933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Giaccia A. J., Simon M. C., and Johnson R., “The Biology of Hypoxia: The Role of Oxygen Sensing in Development, Normal Function, and Disease,” Genes & Development 18, no. 18 (September 2004): 2183–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lim M., Thompson J. G., and Dunning K. R., “Hypoxia and Reproductive Health: Hypoxia and Ovarian Function: Follicle Development, Ovulation, Oocyte Maturation,” Reproduction (Cambridge, England) 161, no. 1 (January 2021): F33–F40. [DOI] [PubMed] [Google Scholar]
- 67. Ottosen L. D., Hindkjær J., Husth M., Petersen D. E., Kirk J., and Ingerslev H. J., “Observations on Intrauterine Oxygen Tension Measured by Fibre‐Optic Microsensors,” Reproductive Biomedicine Online 13, no. 3 (September 2006): 380–385. [DOI] [PubMed] [Google Scholar]
- 68. Ng K. Y. B., Mingels R., Morgan H., Macklon N., and Cheong Y., “In Vivo Oxygen, Temperature and pH Dynamics in the Female Reproductive Tract and Their Importance in Human Conception: A Systematic Review,” Human Reproduction Update 24, no. 1 (January 2018): 15–34. [DOI] [PubMed] [Google Scholar]
- 69. Kaufman D. L. and Mitchell J. A., “Intrauterine Oxygen Tension During the Oestrous Cycle in the Hamster: Patterns of Change,” Comparative Biochemistry and Physiology Part A: Physiology 107, no. 4 (April 1994): 673–678. [DOI] [PubMed] [Google Scholar]
- 70. Nastri C. O., Nóbrega B. N., Teixeira D. M., et al., “Low Versus Atmospheric Oxygen Tension for Embryo Culture in Assisted Reproduction: A Systematic Review and Meta‐Analysis,” Fertility and Sterility 106, no. 1 (July 2016): 95–104.e17. [DOI] [PubMed] [Google Scholar]
- 71. Houghton F. D., “Hypoxia and Reproductive Health: Hypoxic Regulation of Preimplantation Embryos: Lessons From Human Embryonic Stem Cells,” Reproduction (Cambridge, England) 161, no. 1 (January 2021): F41–F51. [DOI] [PubMed] [Google Scholar]
- 72. Bar‐Sagie D., Mayevsky A., and Bartoov B., “Effects of Hyperbaric Oxygenation on Spermatozoan Motility Driven by Mitochondrial Respiration,” Journal of Applied Physiology 50, no. 3 (March 1981): 531–537. [DOI] [PubMed] [Google Scholar]
- 73. Yedwab G. A., Paz G., Homonnai T. Z., David M. P., and Kraicer P. F., “The Temperature, pH, and Partial Pressure of Oxygen in the Cervix and Uterus of Women and Uterus of Rats During the Cycle,” Fertility and Sterility 27, no. 3 (March 1976): 304–309. [DOI] [PubMed] [Google Scholar]
- 74. Fischer B. and Bavister B. D., “Oxygen Tension in the Oviduct and Uterus of Rhesus Monkeys, Hamsters and Rabbits,” Reproduction (Cambridge, England) 99, no. 2 (1993): 673–679. [DOI] [PubMed] [Google Scholar]
- 75. Mohyeldin A., Garzón‐Muvdi T., and Quiñones‐Hinojosa A., “Oxygen in Stem Cell Biology: A Critical Component of the Stem Cell Niche,” Cell Stem Cell 7, no. 2 (August 2010): 150–161. [DOI] [PubMed] [Google Scholar]
- 76. Abdollahi H., Harris L. J., Zhang P., et al., “The Role of Hypoxia in Stem Cell Differentiation and Therapeutics,” Journal of Surgical Research 165, no. 1 (January 2011): 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Stamati K., Mudera V., and Cheema U., “Evolution of Oxygen Utilization in Multicellular Organisms and Implications for Cell Signalling in Tissue Engineering,” Journal of Tissue Engineering 2, no. 1 (2011): 2041731411432365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Närvä E., Pursiheimo J. P., Laiho A., et al., “Continuous Hypoxic Culturing of Human Embryonic Stem Cells Enhances SSEA‐3 and MYC Levels,” PLoS One 8, no. 11 (November 2013): e78847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Leese H., “Metabolic Control During Preimplantation Mammalian Development,” Human Reproduction Update 1, no. 1 (January 1995): 63–72. [DOI] [PubMed] [Google Scholar]
- 80. Thompson J. G., Partridge R. J., Houghton F. D., Cox C. I., and Leese H. J., “Oxygen Uptake and Carbohydrate Metabolism by In Vitro Derived Bovine Embryos,” Reproduction (Cambridge, England) 106, no. 2 (March 1996): 299–306. [DOI] [PubMed] [Google Scholar]
- 81. Vaupel P. and Multhoff G., “Revisiting the Warburg Effect: Historical Dogma Versus Current Understanding,” Journal of Physiology 599, no. 6 (March 2021): 1745–1757. [DOI] [PubMed] [Google Scholar]
- 82. Krisher R. L. and Prather R. S., “A Role for the Warburg Effect in Preimplantation Embryo Development: Metabolic Modification to Support Rapid Cell Proliferation,” Molecular Reproduction and Development 79, no. 5 (May 2012): 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ma L. N., Huang X. B., Muyayalo K. P., Mor G., and Liao A. H., “Lactic Acid: A Novel Signaling Molecule in Early Pregnancy?,” Frontiers in Immunology 11 (February 2020): 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Burton G. J., Cindrova‐Davies T., Yung H., and Jauniaux E., “Hypoxia and Reproductive Health: Oxygen and Development of the Human Placenta,” Reproduction (Cambridge, England) 161, no. 1 (January 2021): F53–F65. [DOI] [PubMed] [Google Scholar]
- 85. Cowden Dahl K. D., Fryer B. H., Mack F. A., et al., “Hypoxia‐Inducible Factors 1α and 2α Regulate Trophoblast Differentiation,” Molecular and Cellular Biology 25, no. 23 (December 2005): 10479–10491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Adelman D. M., Gertsenstein M., Nagy A., Simon M. C., and Maltepe E., “Placental Cell Fates Are Regulated In Vivo by HIF‐Mediated Hypoxia Responses,” Genes & Development 14, no. 24 (December 2000): 3191–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jauniaux E., Watson A., and Burton G., “Evaluation of Respiratory Gases and Acid‐Base Gradients in Human Fetal Fluids and Uteroplacental Tissue Between 7 and 16 Weeks' Gestation,” American Journal of Obstetrics and Gynecology 184, no. 5 (April 2001): 998–1003. [DOI] [PubMed] [Google Scholar]
- 88. Chang C. W., Wakeland A. K., and Parast M. M., “Trophoblast Lineage Specification, Differentiation and Their Regulation by Oxygen Tension,” Journal of Endocrinology 236, no. 1 (January 2018): R43–R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wilkening R. B. and Meschia G., “Current Topic: Comparative Physiology of Placental Oxygen Transport,” Placenta 13, no. 1 (January/February 1992): 1–15. [DOI] [PubMed] [Google Scholar]
- 90. Meschia G., “Placental Respiratory Gas and Exchange and Fetal Oxygenation,” in Creasy and Resnik's Maternal‐Fetal Medicine: Principles and Practice, eds. Resnik R., Lockwood C. J., Moore T. R., Greene M. F., Copel J. A., and Silver R. M. (Philadephia, PA: Elsevier, 2014), 210–222. [Google Scholar]
- 91. Soothill P. W., Nicolaides K. H., Rodeck C. H., and Campbell S., “Effect of Gestational Age on Fetal and Intervillous Blood Gas and Acid‐Base Values in Human Pregnancy,” Fetal Therapy 1 4 (1986): 168–175. [DOI] [PubMed] [Google Scholar]
- 92. Weiner C. P., Sipes S. L., and Wenstrom K., “The Effect of Fetal Age Upon Normal Fetal Laboratory Values and Venous Pressure,” Obstetrics and Gynecology 79, no. 5 pt. 1 (May 1992): 713–718. [PubMed] [Google Scholar]
- 93. Pardi G., Cetin I., Marconi A. M., et al., “Diagnostic Value of Blood Sampling in Fetuses With Growth Retardation,” New England Journal of Medicine 328, no. 10 (March 1993): 692–696. [DOI] [PubMed] [Google Scholar]
- 94. Nava S., Bocconi L., Zuliani G., Kustermann A., and Nicolini U., “Aspects of Fetal Physiology From 18 to 37 Weeks' Gestation as Assessed by Blood Sampling,” Obstetrics & Gynecology 87, no. 6 (June 1996): 975–980. [DOI] [PubMed] [Google Scholar]
- 95. Siggaard‐Andersen O. and Huch R., “The Oxygen Status of Fetal Blood,” Acta Anaesthesiologica Scandinavica Supplementum 107 (1995): 129–135. [DOI] [PubMed] [Google Scholar]
- 96. Richardson B. S., de Vrijer B., Brown H. K., Stitt L., Choo S., and Regnault T. R. H., “Gestational Age Impacts Birth to Placental Weight Ratio and Umbilical Cord Oxygen Values With Implications for the Fetal Oxygen Margin of Safety,” Early Human Development 164 (June 2022): 105511. [DOI] [PubMed] [Google Scholar]
- 97. Okawa K. S., Hirasawa T., Okawa S., Fujita M., and Ishihara M., “Real‐Time Fetal Monitoring Using Photoacoustic Measurement of Placental Oxygen Saturation in a Rabbit Hypoxia Model,” Placenta 146 (February 2024): 110–119. [DOI] [PubMed] [Google Scholar]
- 98. Pinas A. and Chandraharan E., “Continuous Cardiotocography During Labour: Analysis, Classification and Management,” Best Practice & Research Clinical Obstetrics & Gynaecology 30 (January 2016): 33–47. [DOI] [PubMed] [Google Scholar]
- 99. Hasegawa J., Nakamura M., Matsuoka R., et al., “Evaluation of Placental Function Using Near Infrared Spectroscopy During Fetal Growth Restriction,” Journal of Perinatal Medicine 38, no. 1 (2010): 29–32. [DOI] [PubMed] [Google Scholar]
- 100. Garite T. J., “The Search for an Adequate Back‐Up Test for Intrapartum Fetal Heart Rate Monitoring,” American Journal of Obstetrics and Gynecology 208, no. 3 (March 2013): 163–164. [DOI] [PubMed] [Google Scholar]
- 101. Fong D. D., Yamashiro K. J., Vali K., et al., “Design and In Vivo Evaluation of a Non‐Invasive Transabdominal Fetal Pulse Oximeter,” IEEE Transactions on Biomedical Engineering 68, no. 1 (January 2021): 256–266. [DOI] [PubMed] [Google Scholar]
- 102. Amer‐Wahlin I., Arulkumaran S., Hagberg H., Maršál K., and Visser G., “Fetal Electrocardiogram: ST Waveform Analysis in Intrapartum Surveillance,” BJOG: An International Journal of Obstetrics & Gynaecology 114, no. 10 (October 2007): 1191–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. East C. E., Leader L. R., Sheehan P., Henshall N. E., Colditz P. B., and Lau R., “Intrapartum Fetal Scalp Lactate Sampling for Fetal Assessment in the Presence of a Non‐Reassuring Fetal Heart Rate Trace,” Cochrane Database of Systematic Reviews 2015, no. 5 (May 2015): CD006174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Carbonne B., Pons K., and Maisonneuve E., “Foetal Scalp Blood Sampling During Labour for pH and Lactate Measurements,” Best Practice & Research Clinical Obstetrics & Gynaecology 30 (January 2016): 62–67. [DOI] [PubMed] [Google Scholar]
- 105. ACOG Committee on Obstetric Practice ,ACOG Committee Opinion No. 348, November: Umbilical Cord Blood Gas and Acid‐Base Analysis,” Obstetrics & Gynecology 108, no. 5 (2006): 1319–1322. [DOI] [PubMed] [Google Scholar]
- 106. Ayres‐de‐Campos D. and Arulkumaran S., “FIGO Intrapartum Fetal Monitoring Expert Consensus Panel. FIGO Consensus Guidelines on Intrapartum Fetal Monitoring: Physiology of Fetal Oxygenation and the Main Goals of Intrapartum Fetal Monitoring,” International Journal of Gynaecology and Obstetrics 131, no. 1 (2015): 5–8. [DOI] [PubMed] [Google Scholar]
- 107. Olofsson P., “Umbilical Cord pH, Blood Gases, and Lactate at Birth: Normal Values, Interpretation, and Clinical Utility,” supplement, American Journal of Obstetrics and Gynecology 228, no. S5 (May 2023): S1222–S1240. [DOI] [PubMed] [Google Scholar]
- 108. Yao A. and Lind J., “Effect of Gravity on Placental Transfusion,” Lancet 294, no. 7619 (September 1969): 505–508. [DOI] [PubMed] [Google Scholar]
- 109. Kakkilaya V., Pramanik A. K., Ibrahim H., and Hussein S., “Effect of Placental Transfusion on the Blood Volume and Clinical Outcome of Infants Born by Cesarean Section,” Clinics in Perinatology 35, no. 3 (September 2008): 561–570. [DOI] [PubMed] [Google Scholar]
- 110. Yao A., Hirvensalo M., and Lind J., “Placental Transfusion‐Rate and Uterine Contraction,” Lancet 291, no. 7539 (February 1968): 380–383. [DOI] [PubMed] [Google Scholar]
- 111. Aladangady N., McHugh S., Aitchison T. C., Wardrop C. A. J., and Holland B. M., “Infants' Blood Volume in a Controlled Trial of Placental Transfusion at Preterm Delivery,” Pediatrics 117, no. 1 (January 2006): 93–98. [DOI] [PubMed] [Google Scholar]
- 112. Brotanek V., Hendricks C. H., and Yoshida T., “Changes in Uterine Blood Flow During Uterine Contractions,” American Journal of Obstetrics and Gynecology 103, no. 8 (April 1969): 1108–1116. [DOI] [PubMed] [Google Scholar]
- 113. Daniel Y., Fait G., Lessing J., et al., “Umbilical Cord Blood Acid‐Base Values in Uncomplicated Term Vaginal Breech Deliveries,” Acta Obstetricia et Gynecologica Scandinavica 77, no. 2 (February 1998): 182–185. [PubMed] [Google Scholar]
- 114. Armstrong L., “Effect of Delayed Sampling on Umbilical Cord Arterial and Venous Lactate and Blood Gases in Clamped and Unclamped Vessels,” Archives of Disease in Childhood‐Fetal and Neonatal Edition 91, no. 5 (September 2006): F342–F345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. American College of Obstetricians and Gynecologists' Committee on Obstetric Practice ,Delayed Umbilical Cord Clamping After Birth: ACOG Committee Opinion, Number 814,” Obstetrics and Gynecology 136, no. 6 (December 2020): e100–e106. [DOI] [PubMed] [Google Scholar]
- 116. Nudelman M. J. R., Belogolovsky E., Jegatheesan P., Govindaswami B., and Song D., “Effect of Delayed Cord Clamping on Umbilical Blood Gas Values in Term Newborns: A Systematic Review,” Obstetrics & Gynecology 135, no. 3 (March 2020): 576–582. [DOI] [PubMed] [Google Scholar]
- 117. Valero J., Desantes D., Perales‐Puchalt A., Rubio J., Diago Almela V. J., and Perales A., “Effect of Delayed Umbilical Cord Clamping on Blood Gas Analysis,” European Journal of Obstetrics & Gynecology and Reproductive Biology 162, no. 1 (May 2012): 21–23. [DOI] [PubMed] [Google Scholar]
- 118. Wiberg N., Källén K., and Olofsson P., “Delayed Umbilical Cord Clamping at Birth Has Effects on Arterial and Venous Blood Gases and Lactate Concentrations,” BJOG: An International Journal of Obstetrics & Gynaecology 115, no. 6 (May 2008): 697–703. [DOI] [PubMed] [Google Scholar]
- 119. Lievaart M. and de Jong P. A., “Acid‐Base Equilibrium in Umbilical Cord Blood and Time of Cord Clamping,” Obstetrics and Gynecology 63, no. 1 (January 1984): 44–47. [PubMed] [Google Scholar]
- 120. De Paco C., Florido J., Garrido M. C., Prados S., and Navarrete L., “Umbilical Cord Blood Acid‐Base and Gas Analysis After Early Versus Delayed Cord Clamping in Neonates at Term,” Archives of Gynecology and Obstetrics 283, no. 5 (May 2011): 1011–1014. [DOI] [PubMed] [Google Scholar]
- 121. De Paco C., Herrera J., Garcia C., et al., “Effects of Delayed Cord Clamping on the Third Stage of Labour, Maternal Haematological Parameters and Acid‐Base Status in Fetuses at Term,” European Journal of Obstetrics & Gynecology and Reproductive Biology 207 (December 2016): 153–156. [DOI] [PubMed] [Google Scholar]
- 122.Executive Summary: Neonatal Encephalopathy and Neurologic Outcome, Second Edition. Report of the American College of Obstetricians and Gynecologists' Task Force on Neonatal Encephalopathy” Obstetrics and Gynecology 123 no. 4 (April 2014): 896–901. [DOI] [PubMed]
- 123. Westgate J., Garibaldi J. M., and Greene K. R., “Umbilical Cord Blood Gas Analysis at Delivery: A Time for Quality Data,” BJOG: An International Journal of Obstetrics & Gynaecology 101, no. 12 (December 1994): 1054–1063. [DOI] [PubMed] [Google Scholar]
- 124. Ramsay A. G., “Clinical Application of the Henderson‐Hasselbalch Equation,” Applied Therapeutics 7, no. 9 (September 1965): 730–736. [PubMed] [Google Scholar]
- 125. Templeton A. and Kelman G. R., “Maternal Blood‐Gases, PAo2‐‐Pao2), Physiological Shunt and VD/VT in Normal Pregnancy,” British Journal of Anaesthesia 48, no. 10 (October 1976): 1001–1004. [DOI] [PubMed] [Google Scholar]
- 126. Schneider H., “Oxygenation of the Placental‐Fetal Unit in Humans,” Respiratory Physiology & Neurobiology 178, no. 1 (August 2011): 51–58. [DOI] [PubMed] [Google Scholar]
- 127. Shahbazi M. N., Jedrusik A., Vuoristo S., et al., “Self‐Organization of the Human Embryo in the Absence of Maternal Tissues,” Nature Cell Biology 18, no. 6 (June 2016): 700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Di Mattia M., Mauro A., Citeroni M. R., et al., “Insight Into Hypoxia Stemness Control,” Cells 10, no. 8 (August 2021): 2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Semenza G. L., “Dynamic Regulation of Stem Cell Specification and Maintenance by Hypoxia‐Inducible Factors,” Molecular Aspects of Medicine 47–48 (February/March 2016): 15–23. [DOI] [PubMed] [Google Scholar]
- 130. Javed M. J., Mead L. E., Prater D., et al., “Endothelial Colony Forming Cells and Mesenchymal Stem Cells Are Enriched at Different Gestational Ages in Human Umbilical Cord Blood,” Pediatric Research 64, no. 1 (July 2008): 68–73. [DOI] [PubMed] [Google Scholar]
- 131. Borghesi A., Massa M., Campanelli R., et al., “Circulating Endothelial Progenitor Cells in Preterm Infants With Bronchopulmonary Dysplasia,” American Journal of Respiratory and Critical Care Medicine 180, no. 6 (September 2009): 540–546. [DOI] [PubMed] [Google Scholar]
- 132. Compernolle V., “Cardia Bifida, Defective Heart Development and Abnormal Neural Crest Migration in Embryos Lacking Hypoxia‐Inducible Factor‐1α,” Cardiovascular Research 60, no. 3 (2003): 569–579. [DOI] [PubMed] [Google Scholar]
- 133. Menendez‐Montes I., Escobar B., Palacios B., et al., “Myocardial VHL‐HIF Signaling Controls an Embryonic Metabolic Switch Essential for Cardiac Maturation,” Developmental Cell 39, no. 6 (2016): 724–739. [DOI] [PubMed] [Google Scholar]
- 134. Knox C., Camberos V., Ceja L., et al., “Long‐Term Hypoxia Maintains a State of Dedifferentiation and Enhanced Stemness in Fetal Cardiovascular Progenitor Cells,” International Journal of Molecular Sciences 22, no. 17 (2021): 9382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Medley T. L., Furtado M., Lam N. T., et al., “Effect of Oxygen on Cardiac Differentiation in Mouse iPS Cells: Role of Hypoxia Inducible Factor‐1 and Wnt/Beta‐Catenin Signaling,” PLoS One 8, no. 11 (2013): e80280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Meng X., Zhang P., and Zhang L., “Fetal Hypoxia Impacts on Proliferation and Differentiation of Sca‐1+ Cardiac Progenitor Cells and Maturation of Cardiomyocytes: A Role of MicroRNA‐210,” Genes 11, no. 3 (2020): 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Nakada Y., Canseco D. C., Thet S., et al., “Hypoxia Induces Heart Regeneration in Adult Mice,” Nature 541, no. 7636 (2017): 222–227. [DOI] [PubMed] [Google Scholar]
- 138. Morrison S. J., Csete M., Groves A. K., Melega W., Wold B., and Anderson D. J., “Culture in Reduced Levels of Oxygen Promotes Clonogenic Sympathoadrenal Differentiation by Isolated Neural Crest Stem Cells,” Journal of Neuroscience 20, no. 19 (October 2000): 7370–7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Studer L., Csete M., Lee S. H., et al., “Enhanced Proliferation, Survival, and Dopaminergic Differentiation of CNS Precursors in Lowered Oxygen,” Journal of Neuroscience 20, no. 19 (October 2000): 7377–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zhang Y., Porat R. M., Alon T., Keshet E., and Stone J., “Tissue Oxygen Levels Control Astrocyte Movement and Differentiation in Developing Retina,” Developmental Brain Research 118, no. 1–2 (December 1999): 135–145. [DOI] [PubMed] [Google Scholar]
- 141. Nakamura‐Ishizu A., Kurihara T., Okuno Y., et al., “The Formation of an Angiogenic Astrocyte Template Is Regulated by the Neuroretina in a HIF‐1‐Dependent Manner,” Developmental Biology 363, no. 1 (March 2012): 106–114. [DOI] [PubMed] [Google Scholar]
- 142. Jopling J., Henry E., Wiedmeier S. E., and Christensen R. D., “Reference Ranges for Hematocrit and Blood Hemoglobin Concentration During the Neonatal Period: Data From a Multihospital Health Care System,” Pediatrics 123, no. 2 (February 2009): e333–e337. [DOI] [PubMed] [Google Scholar]
- 143. Madan A., Varma S., and Cohen H. J., “Co‐Transactivation of the 3’ Erythropoietin Hypoxia Inducible Enhancer by the HIF‐1 Protein,” Blood Cells, Molecules, and Diseases 23, no. 2 (August 1997): 169–176. [DOI] [PubMed] [Google Scholar]
- 144. Aye I. L. M. H., Aiken C. E., Charnock‐Jones D. S., and Smith G. C. S., “Placental Energy Metabolism in Health and Disease‐Significance of Development and Implications for Preeclampsia,” supplement, American Journal of Obstetrics and Gynecology 226, no. S2 (February 2022): S928–S944. [DOI] [PubMed] [Google Scholar]
- 145. Saini B. S., Morrison J. L., and Seed M., “Gas Exchange Across the Placenta,” in Respiratory Disease in Pregnancy, eds. Lapinsky S. E. and Plante L. A. (Cambridge University Press, 2020), 34–56. [Google Scholar]
- 146. Jackson M. R., Mayhew T. M., and Boyd P. A., “Quantitative Description of the Elaboration and Maturation of Villi From 10 Weeks of Gestation to Term,” Placenta 13, no. 4 (July/August 1992): 357–370. [DOI] [PubMed] [Google Scholar]
- 147. Palmeira P., Quinello C., Silveira‐Lessa A. L., Zago C. A., and Carneiro‐Sampaio M., “IgG Placental Transfer in Healthy and Pathological Pregnancies,” Clinical and Developmental Immunology 2012 (2012): 985646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Gindes L., Teperberg‐Oikawa M., Sherman D., Pardo J., and Rahav G., “Congenital Cytomegalovirus Infection Following Primary Maternal Infection in the Third Trimester,” BJOG: An International Journal of Obstetrics & Gynaecology 115, no. 7 (June 2008): 830–835. [DOI] [PubMed] [Google Scholar]
- 149. Khalil A., Sotiriadis A., Chaoui R., et al., “ISUOG Practice Guidelines: Role of Ultrasound in Congenital Infection,” Ultrasound in Obstetrics & Gynecology 56, no. 1 (July 2020): 128–151. [DOI] [PubMed] [Google Scholar]
- 150. Coelho‐Santos V. and Shih A. Y., “Postnatal Development of Cerebrovascular Structure and the Neurogliovascular Unit,” Wiley Interdisciplinary Reviews: Developmental Biology 9, no. 2 (March 2020): e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Gao L., Chen X., Zeng Y., et al., “Intermittent High Oxygen Influences the Formation of Neural Retinal Tissue From Human Embryonic Stem Cells,” Scientific Reports 6 (July 2016): 29944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Heinis M., Simon M. T., Ilc K., et al., “Oxygen Tension Regulates Pancreatic β‐Cell Differentiation Through Hypoxia‐Inducible Factor 1α,” Diabetes 59, no. 3 (March 2010): 662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ngo M. A., Sinitsyna N. N., Qin Q., and Rice R. H., “Oxygen‐Dependent Differentiation of Human Keratinocytes,” Journal of Investigative Dermatology 127, no. 2 (February 2007): 354–361. [DOI] [PubMed] [Google Scholar]
- 154. van Wenum M., Adam A. A. A., vad der Mark V. A., et al., “Oxygen Drives Hepatocyte Differentiation and Phenotype Stability in Liver Cell Lines,” Journal of Cell Communication and Signaling 12, no. 3 (September 2018): 575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Berthelemy N., Kerdjoudj H., Schaaf P., et al., “O2 Level Controls Hematopoietic Circulating Progenitor Cells Differentiation Into Endothelial or Smooth Muscle Cells,” PLoS One 4, no. 5 (2009): e5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Puebla M., Tapia P. J., and Espinoza H., “Key Role of Astrocytes in Postnatal Brain and Retinal Angiogenesis,” International Journal of Molecular Sciences 23, no. 5 (February 2022): 2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Guo Y. and Pu W. T., “Cardiomyocyte Maturation: New Phase in Development,” Circulation Research 126, no. 8 (2020): 1086–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Puente B. N., Kimura W., Muralidhar S. A., et al., “The Oxygen‐Rich Postnatal Environment Induces Cardiomyocyte Cell‐Cycle Arrest Through DNA Damage Response,” Cell 157, no. 3 (2014): 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Feng R., Mayuranathan T., Huang P., et al., “Activation of γ‐Globin Expression by Hypoxia‐Inducible Factor 1α,” Nature 610, no. 7933 (October 2022): 783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Amoyal I. and Fibach E., “Hemoglobin Switch in the Newborn: A Flow Cytometry Analysis,” Neonatology 91, no. 1 (2007): 61–68. [DOI] [PubMed] [Google Scholar]
- 161. Holland O. J., Hickey A. J. R., Alvsaker A., et al., “Changes in Mitochondrial Respiration in the Human Placenta Over Gestation,” Placenta 57 (September 2017): 102–112. [DOI] [PubMed] [Google Scholar]
- 162. Nakazawa M. S., Keith B., and Simon M. C., “Oxygen Availability and Metabolic Adaptations,” Nature Reviews Cancer 16, no. 10 (September 2016): 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Dahia P. L. M., Ross K. N., Wright M. E., et al., “A HIF1α Regulatory Loop Links Hypoxia and Mitochondrial Signals in Pheochromocytomas,” PLoS Genetics 1, no. 1 (July 2005): e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Erusalimsky J. D. and Moncada S., “Nitric Oxide and Mitochondrial Signaling: From Physiology to Pathophysiology,” Arteriosclerosis, Thrombosis, and Vascular Biology 27, no. 12 (December 2007): 2524–2531. [DOI] [PubMed] [Google Scholar]
- 165. Zhang H., Gao P., Fukuda R., et al., “HIF‐1 Inhibits Mitochondrial Biogenesis and Cellular Respiration in VHL‐Deficient Renal Cell Carcinoma by Repression of C‐MYC Activity,” Cancer Cell 11, no. 5 (May 2007): 407–420. [DOI] [PubMed] [Google Scholar]
- 166. Zhang H., Bosch‐Marce M., Shimoda L. A., et al., “Mitochondrial Autophagy Is an HIF‐1‐Dependent Adaptive Metabolic Response to Hypoxia,” Journal of Biological Chemistry 283, no. 16 (April 2008): 10892–10903. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 167. Hayashi M., Sakata M., Takeda T., et al., “Induction of Glucose Transporter 1 Expression Through Hypoxia‐Inducible Factor 1α Under Hypoxic Conditions in Trophoblast‐Derived Cells,” Journal of Endocrinology 183, no. 1 (October 2004): 145–154. [DOI] [PubMed] [Google Scholar]
- 168. Lauer V., Grampp S., Platt J., et al., “Hypoxia Drives Glucose Transporter 3 Expression Through Hypoxia‐Inducible Transcription Factor (HIF)‐Mediated Induction of the Long Noncoding RNA NICI,” Journal of Biological Chemistry 295, no. 13 (March 2020): 4065–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Semenza G. L., Roth P. H., Fang H. M., and Wang G. L., “Transcriptional Regulation of Genes Encoding Glycolytic Enzymes by Hypoxia‐Inducible Factor 1,” Journal of Biological Chemistry 269, no. 38 (September 1994): 23757–23763. [PubMed] [Google Scholar]
- 170. Cetin I., Taricco E., Mandò C., et al., “Fetal Oxygen and Glucose Consumption in Human Pregnancy Complicated by Fetal Growth Restriction,” Hypertension 75, no. 3 (March 2020): 748–754. [DOI] [PubMed] [Google Scholar]
- 171. Kierans S. J. and Taylor C. T., “Regulation of Glycolysis by the Hypoxia‐Inducible Factor (HIF): Implications for Cellular Physiology,” Journal of Physiology 599, no. 1 (June 2021): 23–37. [DOI] [PubMed] [Google Scholar]
- 172. Illsley N. P., Caniggia I., and Zamudio S., “Placental Metabolic Reprogramming: Do Changes in the Mix of Energy‐Generating Substrates Modulate Fetal Growth?,” International Journal of Developmental Biology 54, no. 2–3 (2010): 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Guitart‐Mampel M., Juarez‐Flores D. L., Youssef L., et al., “Mitochondrial Implications in Human Pregnancies With Intrauterine Growth Restriction and Associated Cardiac Remodelling,” Journal of Cellular and Molecular Medicine 23, no. 6 (June 2019): 3962–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Mandò C., De Palma C., Stampalija T., et al., “Placental Mitochondrial Content and Function in Intrauterine Growth Restriction and Preeclampsia,” American Journal of Physiology‐Endocrinology and Metabolism 306, no. 4 (February 2014): E404–E413. [DOI] [PubMed] [Google Scholar]
- 175. Madeleneau D., Buffat C., Mondon F., et al., “Transcriptomic Analysis of Human Placenta in Intrauterine Growth Restriction,” Pediatric Research 77, no. 6 (June 2015): 799–807. [DOI] [PubMed] [Google Scholar]
- 176. Beyramzadeh M., Dikmen Z. G., Erturk N. K., Tuncer Z. S., and Akbiyik F., “Placental Respiratory Chain Complex Activities in High Risk Pregnancies,” Journal of Maternal‐Fetal & Neonatal Medicine 30, no. 24 (December 2017): 2911–2917. [DOI] [PubMed] [Google Scholar]
- 177. Carlo W. A., Finer N. N., Walsh M. C., et al., “Target Ranges of Oxygen Saturation in Extremely Preterm Infants,” New England Journal of Medicine 362, no. 21 (May 2010): 1959–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Tarnow‐Mordi W., Stenson B., Kirby A., et al., “Outcomes of Two Trials of Oxygen‐Saturation Targets in Preterm Infants,” New England Journal of Medicine 374, no. 8 (February 2016): 749–760. [DOI] [PubMed] [Google Scholar]
- 179. Zhou Q. Y., Quaife C. J., and Palmiter R. D., “Targeted Disruption of the Tyrosine Hydroxylase Gene Reveals That Catecholamines Are Required for Mouse Fetal Development,” Nature 374, no. 6523 (April 1995): 640–643. [DOI] [PubMed] [Google Scholar]
- 180. Thomas S. A., Matsumoto A. M., and Palmiter R. D., “Noradrenaline Is Essential for Mouse Fetal Development,” Nature 374, no. 6523 (April 1995): 643–646. [DOI] [PubMed] [Google Scholar]
- 181. Slotkin T. A., Auman J. T., and Seidler F. J., “Ontogenesis of β‐Adrenoceptor Signaling: Implications for Perinatal Physiology and for Fetal Effects of Tocolytic Drugs,” Journal of Pharmacology and Experimental Therapeutics 306, no. 1 (July 2003): 1–7. [DOI] [PubMed] [Google Scholar]
- 182. Portbury A. L., Chandra R., Groelle M., et al., “Catecholamines Act via a β‐Adrenergic Receptor to Maintain Fetal Heart Rate and Survival,” American Journal of Physiology‐Heart and Circulatory Physiology 284, no. 6 (June 2003): H2069–H2077. [DOI] [PubMed] [Google Scholar]
- 183. Chandra R., Portbury A. L., Ray A., Ream M., Groelle M., and Chikaraishi D. M., “β1‐Adrenergic Receptors Maintain Fetal Heart Rate and Survival,” Neonatology 89, no. 3 (2006): 147–158. [DOI] [PubMed] [Google Scholar]
- 184. Kwon J. H., Eves E. M., Farrell S., et al., “β‐Adrenergic Receptor Activation Promotes Process Outgrowth in an Embryonic Rat Basal Forebrain Cell Line and in Primary Neurons,” European Journal of Neuroscience 8, no. 10 (1996): 2042–2055. [DOI] [PubMed] [Google Scholar]
- 185. Gharami K. and Das S., “Thyroid Hormone‐Induced Morphological Differentiation and Maturation of Astrocytes Are Mediated Through the β‐Adrenergic Receptor,” Journal of Neurochemistry 75, no. 5 (2000): 1962–1969. [DOI] [PubMed] [Google Scholar]
- 186. Emorine L. J., Marullo S., Briend‐Sutren M. M., et al., “Molecular Characterization of the Human β3‐Adrenergic Receptor,” Science 245, no. 4922 (September 1989): 1118–1121. [DOI] [PubMed] [Google Scholar]
- 187. Perrone M. G. and Scilimati A., “β(3)‐Adrenoceptor Agonists and (Antagonists as) Inverse Agonists History, Perspective, Constitutive Activity, and Stereospecific Binding,” Methods in Enzymology 484 (2010): 197–230. [DOI] [PubMed] [Google Scholar]
- 188. Čikoš Š., Veselá J., Il'ková G., Rehák P., Czikková S., and Koppel J., “Expression of Beta Adrenergic Receptors in Mouse Oocytes and Preimplantation Embryos,” Molecular Reproduction and Development 71, no. 2 (June 2005): 145–153. [DOI] [PubMed] [Google Scholar]
- 189. Čikoš Š., Czikková S., Chrenek P., et al., “Expression of Adrenergic Receptors in Bovine and Rabbit Oocytes and Preimplantation Embryos,” Reproduction in Domestic Animals 49, no. 1 (February 2014): 92–100. [DOI] [PubMed] [Google Scholar]
- 190. Adeoya‐Osiguwa S. A., Gibbons R., and Fraser L. R., “Identification of Functional α2‐ and β‐Adrenergic Receptors in Mammalian Spermatozoa,” Human Reproduction 21, no. 6 (June 2006): 1555–1563. [DOI] [PubMed] [Google Scholar]
- 191. Fujinaga M. and Scott J. C., “Gene Expression of Catecholamine Synthesizing Enzymes and β Adrenoceptor Subtypes During Rat Embryogenesis,” Neuroscience Letters 231, no. 2 (August 1997): 108–112. [DOI] [PubMed] [Google Scholar]
- 192. Resch B. E., Ducza E., Gáspár R., and Falkay G., “Role of Adrenergic Receptor Subtypes in the Control of Human Placental Blood Vessels,” Molecular Reproduction and Development 66, no. 2 (October 2003): 166–171. [DOI] [PubMed] [Google Scholar]
- 193. Hynes P. G., Friel A. M., Smith T. J., and Morrison J. J., “β‐Adrenoceptor Subtype Expression in Human Placenta and Umbilical Arteries in Normal and Preeclamptic Pregnancies,” Hypertension in Pregnancy 27, no. 2 (2008): 169–181. [DOI] [PubMed] [Google Scholar]
- 194. Hakak Y., Shrestha D., Goegel M. C., Behan D. P., and Chalmers D. T., “Global Analysis of G‐Protein‐Coupled Receptor Signaling in Human Tissues,” FEBS Letters 550, no. 1–3 (August 2003): 11–17. [DOI] [PubMed] [Google Scholar]
- 195. Rouget C., Bardou M., Breuiller‐Fouché M., et al., “β3‐Adrenoceptor Is the Predominant β‐Adrenoceptor Subtype in Human Myometrium and Its Expression Is Up‐Regulated in Pregnancy,” Journal of Clinical Endocrinology & Metabolism 90, no. 3 (March 2005): 1644–1650. [DOI] [PubMed] [Google Scholar]
- 196. Bardou M., Rouget C., Breuiller‐Fouché M., et al., “Is the Beta3‐Adrenoceptor (ADRB3) a Potential Target for Uterorelaxant Drugs?,” supplement, BMC Pregnancy and Childbirth 7, no. S1 (June 2007): S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Buxton I. L. O., Asif H., and Barnett S. D., “β3 Receptor Signaling in Pregnant Human Myometrium Suggests a Role for β3 Agonists as Tocolytics,” Biomolecules 13, no. 6 (June 2023): 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Dennedy M. C., Houlihan D. D., McMillan H., and Morrison J. J., “β2‐ and β3‐Adrenoreceptor Agonists: Human Myometrial Selectivity and Effects on Umbilical Artery Tone,” American Journal of Obstetrics and Gynecology 187, no. 3 (September 2002): 641–647. [DOI] [PubMed] [Google Scholar]
- 199. Pini A., Fazi C., Nardini P., et al., “Effect of Beta 3 Adrenoreceptor Modulation on Patency of the Ductus Arteriosus,” Cells 9, no. 12 (December 2020): 2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Moniotte S., Kobzik L., Feron O., Trochu J., Gauthier C., and Balligand J. L., “Upregulation of β3‐Adrenoceptors and Altered Contractile Response to Inotropic Amines in Human Failing Myocardium,” Circulation 103, no. 12 (March 2001): 1649–1655. [DOI] [PubMed] [Google Scholar]
- 201. Cheng H. J., Zhang Z. S., Onishi K., Ukai T., Sane D. C., and Cheng C. P., “Upregulation of Functional β3‐Adrenergic Receptor in the Failing Canine Myocardium,” Circulation Research 89, no. 7 (September 2001): 599–606. [DOI] [PubMed] [Google Scholar]
- 202. Ristori C., Filippi L., Dal Monte M., et al., “Role of the Adrenergic System in a Mouse Model of Oxygen‐Induced Retinopathy: Antiangiogenic Effects of β‐Adrenoreceptor Blockade,” Investigative Opthalmology & Visual Science 52, no. 1 (January 2011): 155–170. [DOI] [PubMed] [Google Scholar]
- 203. Calvani M., Pelon F., Comito G., et al., “Norepinephrine Promotes Tumor Microenvironment Reactivity Through β3‐Adrenoreceptors During Melanoma Progression,” Oncotarget 6, no. 7 (2015): 4615–4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Amato R., Pisani F., Laudadio E., et al., “HIF‐1‐Dependent Induction of β3 Adrenoceptor: Evidence From the Mouse Retina,” Cells 11, no. 8 (April 2022): 1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Cammalleri M., Amato R., Dal Monte M., Filippi L., and Bagnoli P., “The β3 Adrenoceptor in Proliferative Retinopathies: “Cinderella” Steps Out of Its Family Shadow,” Pharmacological Research 190 (April 2023): 106713. [DOI] [PubMed] [Google Scholar]
- 206. Calvani M., Bruno G., Dal Monte M., et al., “β3‐Adrenoceptor as a Potential Immuno‐Suppressor Agent in Melanoma,” British Journal of Pharmacology 176, no. 14 (July 2019): 2509–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Dal Monte M., Filippi L., and Bagnoli P., “Beta3‐Adrenergic Receptors Modulate Vascular Endothelial Growth Factor Release in Response to Hypoxia Through the Nitric Oxide Pathway in Mouse Retinal Explants,” Naunyn‐Schmiedeberg's Archives of Pharmacology 386, no. 4 (April 2013): 269–278. [DOI] [PubMed] [Google Scholar]
- 208. Dal Monte M., Fornaciari I., Nicchia G. P., Svelto M., Casini G., and Bagnoli P., “β3‐Adrenergic Receptor Activity Modulates Melanoma Cell Proliferation and Survival Through Nitric Oxide Signaling,” Naunyn‐Schmiedeberg's Archives of Pharmacology 387, no. 6 (June 2014): 533–543. [DOI] [PubMed] [Google Scholar]
- 209. Calvani M., Dabraio A., Subbiani A., et al., “β3‐Adrenoceptors as Putative Regulator of Immune Tolerance in Cancer and Pregnancy,” Frontiers in Immunology 11 (September 2020): 2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Bruno G., Cencetti F., Pini A., et al., “β3‐Adrenoreceptor Blockade Reduces Tumor Growth and Increases Neuronal Differentiation in Neuroblastoma via SK2/S1P2 Modulation,” Oncogene 39, no. 2 (January 2020): 368–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Calvani M., Bruno G., Dabraio A., et al., “β3‐Adrenoreceptor Blockade Induces Stem Cells Differentiation in Melanoma Microenvironment,” International Journal of Molecular Sciences 21, no. 4 (February 2020): 1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Calvani M., Cavallini L., Tondo A., et al., “β3‐Adrenoreceptors Control Mitochondrial Dormancy in Melanoma and Embryonic Stem Cells,” Oxidative Medicine and Cellular Longevity 2018 (November 2018): 6816508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Filippi L., Cammalleri M., Amato R., et al., “Decoupling Oxygen Tension From Retinal Vascularization as a New Perspective for Management of Retinopathy of Prematurity. New Opportunities From Β‐Adrenoceptors,” Frontiers in Pharmacology 13 (January 2022): 835771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Lobigs L. M., Sharpe K., Garvican‐Lewis L. A., et al., “The Athlete's Hematological Response to Hypoxia: A Meta‐Analysis on the Influence of Altitude Exposure on Key Biomarkers of Erythropoiesis,” American Journal of Hematology 93, no. 1 (January 2018): 74–83. [DOI] [PubMed] [Google Scholar]
- 215. Kietzmann T., “Hypoxia‐Inducible Erythropoietin Expression: Details Matter,” Haematologica 105, no. 12 (December 2020): 2704–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Tong E. M. and Nissenson A. R., “Erythropoietin and Anemia,” Seminars in Nephrology 21, no. 2 (March 2001): 190–203. [DOI] [PubMed] [Google Scholar]
- 217. Robinson N., Giraud S., Saudan C., et al., “Erythropoietin and Blood Doping,” supplement, British Journal of Sports Medicine 40, no. S1 (July 2006): i30–i34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Filippi L., Nardini P., Zizi V., et al., “β3 Adrenoceptor Agonism Prevents Hyperoxia‐Induced Colonic Alterations,” Biomolecules 13, no. 12 (December 2023): 1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Nardini P., Zizi V., Molino M., et al., “Protective Effects of Beta‐3 Adrenoceptor Agonism on Mucosal Integrity in Hyperoxia‐Induced Ileal Alterations,” Antioxidants 13, no. 7 (2024): 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Chen C. M. and Chou H. C., “Hyperoxia Disrupts the Intestinal Barrier in Newborn Rats,” Experimental and Molecular Pathology 101, no. 1 (2016): 44–49. [DOI] [PubMed] [Google Scholar]
- 221. Giannone P., Bauer J., Schanbacher B., and Reber K., “Effects of Hyperoxia on Postnatal Intestinal Development,” Biotechnic & Histochemistry 82, no. 1 (2007): 17–22. [DOI] [PubMed] [Google Scholar]
- 222. Liu D. Y., Lou W. J., Zhang D. Y., and Sun S. Y., “ROS Plays a Role in the Neonatal Rat Intestinal Barrier Damages Induced by Hyperoxia,” BioMed Research International 2020 (2020): 8819195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Smith L. E., Wesolowski E., McLellan A., et al., “Oxygen‐Induced Retinopathy in the Mouse,” Investigative Ophthalmology & Visual Science 35, no. 1 (1994): 101–111. [PubMed] [Google Scholar]
- 224. Bucher F., Stahl A., Agostini H. T., and Martin G., “Hyperoxia Causes Reduced Density of Retinal Astrocytes in the Central Avascular Zone in the Mouse Model of Oxygen‐Induced Retinopathy,” Molecular and Cellular Neuroscience 56 (2013): 225–233. [DOI] [PubMed] [Google Scholar]
- 225. Melecchi A., Canovai A., Amato R., et al., “Agonism of β3‐Adrenoceptors Inhibits Pathological Retinal Angiogenesis in the Model of Oxygen‐Induced Retinopathy,” Investigative Ophthalmology & Visual Science 65, no. 10 (2024): 34. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 226. Kim J. K., De Jesus M. J., Lee M. J., et al., “β3‐Adrenoceptor Agonist for the Treatment of Bladder Dysfunction in Children: A Systematic Review and Meta‐Analysis,” Journal of Urology 207, no. 3 (2022): 524–533. [DOI] [PubMed] [Google Scholar]
- 227. Sears J. E., Hoppe G., Ebrahem Q., and Anand‐Apte B., “Prolyl Hydroxylase Inhibition During Hyperoxia Prevents Oxygen‐Induced Retinopathy,” Proceedings of the National Academy of Sciences of the United States of America 105, no. 50 (December 2008): 19898–19903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Hoppe G., Yoon S., Gopalan B., et al., “Comparative Systems Pharmacology of HIF Stabilization in the Prevention of Retinopathy of Prematurity,” Proceedings of the National Academy of Sciences of the United States of America 113, no. 18 (May 2016): E2516–E2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Chen H., Cheng Q., Wang J., Zhao X., and Zhu S., “Long‐Term Efficacy and Safety of Hypoxia‐Inducible Factor Prolyl Hydroxylase Inhibitors in Anaemia of Chronic Kidney Disease: A Meta‐Analysis Including 13,146 Patients,” Journal of Clinical Pharmacy and Therapeutics 46, no. 4 (August 2021): 999–1009. [DOI] [PubMed] [Google Scholar]
- 230. Miao M., Wu M., Li Y., et al., “Clinical Potential of Hypoxia Inducible Factors Prolyl Hydroxylase Inhibitors in Treating Nonanemic Diseases,” Frontiers in Pharmacology 13 (February 2022): 837249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Hirota K., “HIF‐α Prolyl Hydroxylase Inhibitors and Their Implications for Biomedicine: A Comprehensive Review,” Biomedicines 9, no. 5 (April 2021): 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. Umbilical cord oxygenation status according to the type of delivery. Scatter plots with regression lines representing umbilical cord oxygenation status [total venous O2 content, panel A (n = 1788) and arterial O2 content, panel B (n = 1740)] in neonates born by vaginal delivery and oxygenation status [total venous O2 content, panel C (n = 855) and arterial O2 content, panel D (n = 808)] in neonates born by cesarean delivery. The equation of the statistically significant model (linear or quadratic) is indicated at the top of each figure.
Supplementary Fig. 2. Umbilical cord oxygenation status after subtracting 4 mmHg from the arterial pO 2 values. Scatter plots with regression lines representing the umbilical cord oxygenation status [venous pO2, panel A (n = 4286); arterial pO2, panel B (n = 4161); total venous, panel C (n = 2643) and arterial O2 contents, panel D (n = 2547)] of the whole study population after artificial correction of arterial pO2 values, subtracting 4 mmHg from the original values. The equation of the statistically significant model (linear or quadratic) is indicated at the top of each figure.
Supplementary Fig. 3. Umbilical cord O 2 extraction after subtracting 4 mmHg from the arterial pO 2 values. Scatter plot representing umbilical cord O2 extraction of the whole study population after artificial correction of arterial pO2 values minus 4 mmHg to the original values (n = 2548).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.