

Abstract
Background
Breast cancer is one of the most frequent causes of death in Mexican women over 35 years of age. At molecular level, changes in many genetic networks have been reported as associated with this neoplasia. To analyze these changes, we determined gene expression profiles of tumors from Mexican women with breast cancer at different stages and compared these with those of normal breast tissue samples.
Methods
32P-radiolabeled cDNA was synthesized by reverse transcription of mRNA from fresh sporadic breast tumor biopsies, as well as normal breast tissue. cDNA probes were hybridized to microarrays and expression levels registered using a phosphorimager. Expression levels of some genes were validated by real time RT-PCR and immunohistochemical assays.
Results
We identified two subgroups of tumors according to their expression profiles, probably related with cancer progression. Ten genes, unexpressed in normal tissue, were turned on in some tumors. We found consistent high expression of Bik gene in 14/15 tumors with predominant cytoplasmic distribution.
Conclusion
Recently, the product of the Bik gene has been associated with tumoral reversion in different neoplasic cell lines, and was proposed as therapy to induce apoptosis in cancers, including breast tumors. Even though a relationship among genes, for example those from a particular pathway, can be observed through microarrays, this relationship might not be sufficient to assign a definitive role to Bik in development and progression of the neoplasia. The findings herein reported deserve further investigation.
Background
Breast cancer is one of the most frequent causes in Mexican women over 35 years of age and mortality shows a tendency to increase over time [1]. The origin and course of sporadic breast cancer are not clear. At the molecular level, some alterations have been reported as associated with this neoplasia, such as changes in DNA quantity [2], cytogenetic alterations [3], amplification of some proto-oncogenes [4,5], loss of heterozygosity in some chromosomal regions [6,7], and mutations in at least four different susceptibility genes in hereditary forms [8-11].
The traditional way of classifying breast tumors is based on tumor size, degree of dissemination and Histopathology. Alterations in many genetic networks are involved in development of breast cancer; therefore, analysis of isolated genes is not sufficient to adequately understand this neoplasia. Transcriptional analysis of multiple genes expressed by breast tumors should provide a means to define a signature or molecular fingerprint of the disease and might progressively replace conventional diagnostic and prognostic parameters. Likewise, the capability to analyze simultaneous expression levels of thousands of genes offers better possibilities to understand and characterize the complete molecular mechanisms underlying cancer progression. Technologies such as DNA chips permit integral study of the advance of the disease, with the advantage of identifying marker genes for diagnosis, prognosis, and therapy.
In several studies found correlations among expression profiles and clinical characteristics, estrogen receptors, lymphatic nodes or treatment response [12-16]. However, this results can not be generalized to other populations because development of this heterogeneous disease have influence from multiple factors, including age, diet, genetics, environment, geographic location, no pregnancy and race [17]. To generate portraits for each population may help in the sub-classification of tumours, prognosis, and general understanding of breast cancer [18,19] and will allow us to identify characteristic genes from the Mexican population.
Tumor growth rates can be <5% of those predicted by proliferation measurements alone. Several types of human cancers such as colorectal [20], ovarian [21], endometrial [22], and cervical [23], also showed an increase in apoptotic index (by TUNEL assay) during tumorigenesis, which questions if cancer research fields related with inhibition of cell death might be a critical step in cancer development. Thus, in these cases a high rate of cellular proliferation must be responsible for tumor growth.
Human gene expression patterns derived from cDNA microarrays have been increasingly used to identify genes associated with human cancers [12,14,24]. On the basis of these studies, it appears that cDNA microarray based gene expression analysis of breast cancers tissues would reveal molecular characteristics associated with tumorigenesis.
Bcl-2 family proteins contain both, anti-apoptotic and pro-apoptotic, members and are essential to maintain organ systems. For many, but not for all apoptotic signals, balance between these two Bcl-2 subfamilies determine cell fate. The pro-apoptotic Bik protein is a member of one sub-class of the Bcl-2 family designated BH3-alone [25]. The human Bik gene is located on 22q13.3 and codifies a 160 amino acid protein. Its mRNA has ubiquitous distribution with elevated levels in heart and skeletal muscle [26].
Recently, the product of Bik gene has been associated with tumoral reversion in different cell lines and was proposed as therapy to induce apoptosis in cancer including breast tumors [27,28]. Using cDNA microarrays, we obtained gene expression profiles of 15 breast cancers and 5 normal breast tissues. Complete pairwise comparison of selected genes with real-time RT-PCR revealed consistent overexpression of Bik/NBK gene in tumor samples.
Methods
Breast tissue samples
The protocol of this work was approved by the Ethical Committee of our hospital with register number 98/718/43 and all patients provided informed consent in signed letter prior to the initiation of any procedure. Samples were taken from non-affected breast tissue and affected tissue from 15 non-related Mexican patients 40 years of age [mean of 52 years and range 40–68 years]. All tumors were sporadic, infiltrating, ductal adenocarcinomas from patients who had not received adjuvant chemotherapy. Tissues were obtained from the Oncology Hospital, at the Centro Medico Nacional Siglo XXI of the Instituto Mexicano del Seguro Social in Mexico City. Each sample was snap-frozen in liquid nitrogen and stored at -70°C until use. Specimens were selected for analysis by two criteria: (i) sufficient material for analysis, and (ii) histological evaluation by two pathologists demonstrating that samples contained at least 70% of tumor cells (in the case of cancer samples). Representative sections from each case were paraffin-embedded for staining with haematoxylin and eosin to assess histopathologic diagnosis using American Cancer Committee criteria [29] and for their use in Immunohistochemistry assays.
RNA extraction and mRNA purification
Total RNA of each sample was obtained with trizol and dissolved in RNase-free water for a final concentration of 1–2 μg/μl. Samples was stored at -70°C. Concentration and purity of each sample was assessed by gel electrophoresis and spectrophotometry (Ultrospec 2000, Pharmacia Biotech). Prior to obtaining poly A+ RNA, total RNA was treated with DNAse I according to Atlas pure total RNA labeling system (Clontech, Palo Alto, CA, USA). Poly A enrichment procedure was used to obtain mRNA (Atlas pure total RNA labeling system®, Clontech, Palo Alto, CA, USA).
Probe labeling and hybridization
cDNA label procedure was used to obtain 32P-labeled cDNA from 1 μg of poly A, with MoMLV reverse transcriptase (Atlas pure total RNA labeling system®, Clontech, Palo Alto, CA, USA). Labeled probes were purified with chroma SPIN-200 columns (Atlas cDNA Expression Arrays®, Clontech, Palo Alto, CA, USA). Incorporation of 32P into the probe was double-checked in a scintillation counter (Beckman, model LS 9000sc).
The hybridization procedure was performed with hybridization solution mixed with the entire pool of labeled cDNA probes with > 1.25 × 106 cpm on Atlas array membrane with 609 genes, double-spotted (Atlas human cancer cDNA expression arrays®; Clontech, Palo Alto, CA, USA). The membrane was hybridized at 68°C for 24 h, and then was washed with high-to-low astringency solutions. Membranes were exposed for 24 h in a phosphor screen (Kodak storage phosphor screen, Molecular Dynamics).
Image analysis and data collection
The digitalized image was obtained in a storm scanner (Storm 680, Molecular Dynamics, Inc.). Expression profiles were obtained with Atlas image 2.0 software (Clontech, Palo Alto, CA, USA) through comparison of normal vs. tumor tissues. We used a normalization coefficient in which average value of all genes was used to normalize the array. This coefficient was determined with the Sum method, which adds values of signal over background for all genes on arrays.
Data analysis
Hybridization profiles were analyzed with J-Express program developed and distributed by Molmine AS and collaborators [30], and Cluster and TreeView software's [31].
Real Time-RT-PCR analysis
The same source of total RNA used to define gene expression profiles was used in real time RT-PCR experiments, following all instructions outlined in the LightCycler-RNA amplification kit SYBR Green I manual (Roche Molecular Biochemicals, Mannheim, Germany). cDNA synthesis was carried out in LightCycler (Roche) in a capillary as follows: 20 μl mix reaction containing 500 ng of DNase I treated total RNA, 4 μl of LightCycler-RT-PCR reaction mix SYBR Green I (final concentration 1X), 5 mM MgCl2, 0.4 μl LightCycler-RT-PCR enzyme mix, and 5.0 pmol forward and reverse primers for both genes. Bik gene primer sequences were designed with OLIGO 4.1 and were as follows: forward 5' GAG ACA TCT TGA TGG AGA CC 3', reverse 5' TCT AAG AAC ATC CCT GAT GT 3'. HPRT gene primers were referred by Pieretti M et al; 1991 [32]. For reverse transcription, the reaction was incubated at 55°C for 30 min and at 95°C for 30 sec.
Amplification was carried out in the same capillary. LightCycler was programmed as follows: 50 three-segment cycles for amplification (10 sec at 95°C, 30 sec at 55°C, and acquisition (single-mode), 20 sec at 72°C) and three-segment cycle of product melting (0 sec at 95°C, 10 sec at 65°C, and 0 sec at 95°C at step-acquisition mode). Temperature transition rate for all segments of amplification cycles and melting curve cycle were set at 20°C/sec except for segment 3 that was set at 0.1°C/sec of product-melting curve analysis. Duplicate reactions were prepared for each sample along with a non-template negative control (H2O control). A standard curve was used with these assays and LightCycler3 data analysis software (Roche Molecular Biochemicals) was used in all processes.
Immunohistochemistry
For identification of Bik (NBK) protein expression, streptavidin-biotin peroxidase complex (DAKO LSAB Kit, Carpinteria, CA, USA) method with diaminobenzidine as chromogen was used. Epitopes were retrieved by autoclaving in 10 mM citric acid buffer, pH 6.1 for 2 min. As primary antibody, polyclonal anti-human NBK/Bik FL-160 (Santa Cruz Biotechnology, work dilution 1:100) in 1% bovine serum albumin-phosphate buffered saline (BSA-PBS) was used. Formalin-fixed, paraffin-embedded sections of normal skin epithelium served as positive controls for Bik (NBK). Negative control slides were processed in parallel, omitting primary antibody.
Results
Expression profiles of normal and malignant breast specimens
We analyzed gene expression patterns in dissected normal or malignant human breast tissue from 20 individuals, 15 infiltrating ductal carcinomas, nine stage II, six stage III, and five normal breast samples. Hierarchical cluster analysis was used to group genes on the basis of similarity in their patterns of expression. We used average cluster linkage, uncentered [31] based on Euclidean distance and removed all genes that were turned on in tumors but that were not expressed in normal tissue (NCK5AI, CDK5 activator, CDC25A, ERK4, K2P, COL11A1, OBCAM, AMPHIREGULIN, BCGF1 and BMP8).
Cluster analysis of tumors and controls is described in Figure 1. Patterns of gene expression among tumors showed great variation (Figure 1A); however, we identified two main groups. The left branch is conformed by four histologic type II and one type III, while the right branch contains five type II and five type III. Left branch shows distinctive underexpression and loss of expression areas with genes such as IFNGR2, interleukin 2, laminin B1, MSH2, MSH6 CASP8 and 10, RB1, PCNA, EGFR and PGS2, among others, and a few overexpressed genes such as MIF, CDK 2, 5, and 7, RPSA, JUP and ITGAE. On the other hand, right branch shows overexpression areas with genes such as CASP 10, CD30-L, and CD-40-L (TNF family), ras-like small, FGF 3 and 5, TDGF1, ERK5, MIF, IFNGR2, MIG, IFN-alpha, beta, gamma, IL-3, -4, -9 and -13, IP-10, HPRT, DCC, TEK, netrin-2, cadherins 4, 8, 12 and 13 [33], MMP3, 8, 10, 12 and 13 (metalloproteinase), COL11A2, CCNG1, CDC-6, ATM, N-MYC, and p53.
Figure 1.
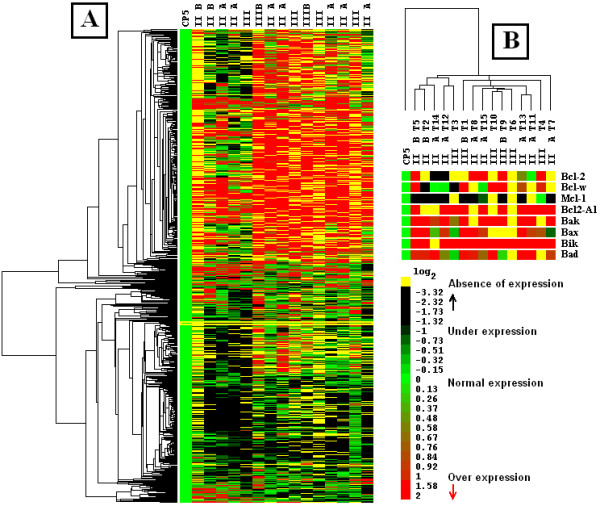
Dendrogram and picture of hierarchical cluster of 15 mammary tumors and normal tissue. Expression profiles from tumor and normal tissue (average from five normal tissues) were obtained through average-linkage, hierarchical, uncentered clustering analysis, as described in Methods. Genes unexpressed in normal tissue were not included in analysis. In dendrograms, each branch corresponds to similar expression profile among genes (A) or samples (B). Green squares in picture correspond to normal expression levels. Yellow squares correspond to absence of expression. Black squares correspond to underexpressed genes. Red squares correspond to overexpressed genes. Picture under dendrogram in panel details behavior of eight apoptosis-related genes along samples.
In Figure 1B, tumors and controls were associated in a dendrogram obtained with expression profile of each sample. With this analysis, tumors were separated in two main branches; left group contained five tumors defined predominantly by histologic type II, while right group was composed by stage II and III tumors, at the same percentage. In the lower part of the same Figure, we showed the gene expression pattern of eight genes related with apoptotic process obtained from each tumor, showing changes across all samples but exposing the possible relation between pro-apoptotic (BAK, BAX, BIK, BAD) and anti-apoptotic genes (BCL2, BCLW, MCL1, BCL2- A1). We showed that pro-apoptotic BCL2-interacting killer gene (BIK/NBK) overexpressed in nearly all samples (14/15), which makes it a possible candidate for further studies on its role in breast cancer. MCL1 was underexpressed or missing, at least 93.3% (14/15), relative to controls.
Due to consistent high expression of Bik gene in breast cancer, we validated its overexpression by real-time PCR and studied distribution of Bik protein in tumor breast samples by Immunohistochemistry.
Real time RT-PCR assay for Bik gene
According to the Methods section, we developed real-time RT-PCR using Bik and HPRT mRNAs as templates. In Figure 2, we showed amplification products of different samples of Bik and HPRT genes. With real-time RT-PCR, results of 9/13 samples (69%) agreed with those obtained through microarray analysis (considering NB and T14) and in 31% of tumors, we were unable to confirm the result. By using melting curves (data not shown), we confirmed that amplification products obtained with real-time RT-PCR were specific. As positive control, HPRT gene (housekeeping) was used (samples numbers 11 and 14) for tumors and normal breast (NB). We used tumor 14 as negative control (tumor that wich show expression for Bik gene in microarray assay).
Figure 2.

Real time RT-PCR validation of expression Bik and HPRT genes. Amplification curves of Bik (Bik T1 to T14), HPRT (T11 and T14) and non-affected breast (NB) mRNAs are shown. Graphic generated by LightCycler-based real time PCR thermocycler. The abscissa shows fluorescence relative units and ordinate cycle number.
Characterization of Bik/NBK expression by immunohistochemistry
After RT-PCR analysis, we checked the distribution and abundance of Bik protein on slides obtained from the same tissue used for histopathologic diagnosis. Only in poorly or well-differentiated tumor areas we found presence of BIK protein with predominant cytoplasmic location (Figure 3A). Figures 3C and 3D (magnification to 400×) show Bik protein in a sample of breast cancer. Presence of BIK protein was specific because, when primary antibody was omitted, we did not observe any signal in neoplasic tissue (Figure 3B).
Figure 3.

Distribution of Bik protein in mammary ducts from adenocarcinomas. Anti-Bik/NBK polyclonal antibody) was reacted with HRP-secondary antibody and revealed with DAB. Haematoxylin was used as counter stain. A, C, and D are different tumor areas. B, negative control from the same patient.
Discussion
Advances in high-density DNA microarray technology have permitted to screen large numbers of genes and to correlate tumoral stages and gene-expression profiles in cancer research. The idea is that tumor behavior is ruled by expression of hundreds of genes; thus microarrays analysis allows those behaviors and clinical features to be predicted. In the present study, 10 genes were turned on in some tumors respect to the normal tissue.
With microarray analysis of samples, we identified two subgroups probably related with cancer progression. In the first group, we identified an underexpression area (lower part of dendrogram in Figure 1) Wich corresponded to four histological stage II tumors, (T2, T5, T12 and T14) and one tumor in histological stage III, (T3). The latter tumor probably corresponded to a stage II tumor at molecular level because its profile expression was similar to others in the same group. We think that this group belongs to an initial stage of development of neoplasia, because it presents fewer alterations than the second group, and because the behavior of some genes related with the beginning of tumor development such as genes related with DNA repairment, apoptosis, TNF, Fas-L route, checkpoint, remodeling, and maintenance of extracellular matrix, were underexpressed or lost their expression while other genes were overexpressed, such as human macrophage migration inhibitory factor and some cyclin-dependent kinases and cytoskeletal proteins.
The right branch (Figure 1) contains stage II or III tumors and probably correspond to more advanced stages of progression of neoplasia. In this group, we found some overexpressed genes; of these, COL11A2 and netrin-2 had not been previously associated with breast cancer. Other genes, such as DCC and cadherin 13, were found overexpressed in our samples but underexpressed in other reports [34]; these genes probably have mutations that differentially affect their proteins.
Apoptosis is a mechanism to control cell death which occurs during normal development, growth, and maintenance in multicellular organisms. It can be activated by stimulation of a cell surface death receptor or release of cytochrome c from mitochondria. In the cascade of events initiated by this signaling, cysteine aspartyl proteases, or caspases are cleaved from an inactive zymogen to an active heterodimer. These active caspases degrade several components critical for cell survival, such as DNA repair elements, structural proteins, and cell signaling peptides [35]. Caspase-3, the primary death effector, serves as a key target for monitoring apoptosis, for it is activated through both cell surface death receptor and mitochondrial release of cytochrome c [36]. Disruption in signaling pathways that regulate apoptosis can lead to a variety of pathologic conditions, making apoptosis an area of intense focus for research [37].
Eight genes related with apoptosis are found in the lower part of Figure 1B. Only two genes showed differential expression in nearly all tumors, Bik gene was overexpressed in 14/15 samples and MCL-1 gene expression was diminished or absent in the same number of tumors. The behavior of the remaining genes was not constant; in tumor 5, for example, nearly all selected genes were overexpressed, whereas in tumor 6 expression of nearly all selected genes was lost. The heterogeneous behavior of these genes makes it difficult to establish their role in apoptosis or survival of neoplasic cells. However, it was possible to establish which genes, as in the case of the Bik gene, are probably related with cancer progression or are involved in other genetic networks.
BH3-only proteins are structurally distant members of the Bcl-2 protein family which triggers apoptosis. These proteins share with each other and with the remainder of the Bcl-2 family only a nine amino acid BH3 (Bcl-2 Homology) region. This domain is required for its ability to bind to Bcl-2-like, pro-survival proteins and to initiate apoptosis. Mammals have at least 10 BH3-only genes that differ in expression pattern and model of activation (regulated by a diverse range of transcription factors). Certain BH3-only proteins, including Bad, Bik/Nbk, Bid, Bim/Bod, and Bmf, are restrained by post-translational modifications that cause their sequestration from pro-survival Bcl-2 family members [38]. Nbk/Bik (natural-born killer/Bcl-2-interacting killer) is a tissue-specific BH3-only protein which molecular function is largely unknown. Nbk fails to induce apoptosis in the absence of Bax. Nbk interacts with Bcl-xL and Bcl-2 but not with Bax. It was suggested that Nbk acts as an indirect killer which triggers Bax-dependent apoptosis, whereas Bak is not sufficient to confer sensitivity to Nbk [39].
BH3-only proteins require multidomain pro-apoptotic members Bax and Bak to release cytochrome c from mitochondria and kill cells. Short peptides representing alpha-helical BH3 domains of Bid or Bim are capable of inducing oligomerization of Bak and Bax to release cytochrome c. BH3 peptides from Bad and Bik cannot directly activate Bax or Bak, but instead bind anti-apoptotic members of BCL-2, resulting in displacement of Bid-like BH3 domains which initiate mitochondrial dysfunction. These data support two types of BH3 domains: Bid-like domains which activate Bax, Bak, and Bad-like domains which sensitize by occupying the pocket of anti-apoptotic members [40].
Bik, a BH3-alone protein (Bcl-2 homology domain), is a pro-apoptotic member of the BCL2 family. Missense Bik gene mutations and sequence alterations in intronic regions were observed in cell lymphomas; these data indicate that mutation of the Bik gene is relatively frequent [41,42]. The importance of phosphorylation and desphosphorylation reactions in intracellular signaling pathways has long been accepted. The importance of serine/threonine protein phosphatases in many processes including apoptosis is recognized. The phosphorylation state of anti-apoptotic (Bcl-2 and Bcl-xL) and pro-apoptotic (Bad, Bid and Bik) Bcl-2 proteins regulates their cellular activity and, therefore, cell survival and cell death. For instance, desphosphorylation of Bad allows it to interact with Bcl-xL and initiate cell death [43]. However, mutation of the Bik phosphorylation sites, in which the Thr and Ser residues were changed to alanine residues, reduced the apoptotic activity of Bik protein without significantly affecting its ability to heterodimerize with BCL-2 [44]
Of note, a significant fraction of either ectopic or endogenous BIK was found associated with the endoplasmic reticulum, suggesting that this organelle, in addition to mitochondria, may be a target of BIK function [45]. Collectively, the results identify BIK as an initiator of cytochrome c release from mitochondria operating from a location at the ER [46]. These results indicate that any function of Bik in programmed cell death and stress-induced apoptosis must overlap that of other BH3-only proteins [47].
Because the product of Bik gene is pro-apoptotic, this protein has been used to induce apoptosis in several cancer cell lines (PC-3, HT-29, MCF-7, MDA-MB-231, 435, 468 and A540) [27,28,48]. Fay M et al; in 2003 [49] reported underexpression of the CUL-5 gene in breast tumor tissue, whereas expression levels in several cancer cell lines (MCF7, MDA-MB-231) are essentially identical to those of normal breast. Probably breast tumor tissues (i.e., Bik gene) have a different behavior vs. breast cancer cell lines at a molecular level. Moreover we found some reports with different expression levels of Bik gene among multiple human tissues: Daniel T et al; 1999 [48] reported absence of Bik expression in heart, skeletal muscle, and brain, while Verma et al; 2000 [26] reported higher expression in heart and skeletal muscle. It is difficult to reconcile these reports.
Conclusion
It is not known whether Bik gene works as an apoptosis activator or only sensitizes the cell for death. In any event, it could be a prognostic factor and a possible therapy target in breast cancer. The findings reported in this paper deserve further investigation.
List of abbreviations
ATM Ataxia telangiectasia
BAD BCL2-antagonist of cell death
BAK BCL2-antagonist/killer 1
BAX BCL2-associated × protein
BCGF1 B-cell growth factor
BCL2- A1 BCL2-related protein A1
BCL2 B-cell CLL/lymphoma 2
Bcl-xL BCL2-like 1
CUL-5 Cullin 5
BCLW BCL2-like 2
Bik/Nbk BCL2-interacting killer/ natural-born killer
BMP8 Bone morphogenetic protein 8
CASP10 Apoptotic cysteine protease MCH4
CASP8 Apoptotic cysteine protease MCH5
CCNG1 Cyclin G1
CD30-L CD30 ligand
CD-40-L CD40 ligand
CDC25A Cell division cycle 25a
CDC-6 CDC6-related protein
CDK 2 Cyclin-dependent kinase 2
CDK 5 Cell division protein kinase 5
CDK 7 Cyclin-dependent kinase 7
CDK5 activator Cyclin-dependent kinase 5 activator p35
COL11A1 Collagen type xi alpha-1
COL11A2 Collagen type xi alpha-2
cpm Counts per million
DCC Tumor suppressor protein DCC
EGFR Epidermal growth factor receptor
ERK4 Extracellular signal-regulated kinase 4
ERK5 Extracellular signal-regulated kinase 5
FGF 3 Fibroblast growth factor-3
FGF 5 Fibroblast growth factor-5
HPRT Hypoxanthine-guanine phosphoribosyltransferase
IFN-alpha Interferon alpha-c leukocyte
IFN-beta Interferon beta-1
IFN-gamma Interferon gamma
IFNGR2 Interferon gamma accessory factor-1
IL-13 Interleukin-13
IL-3 Interleukin-3
IL-4 Interleukin-4
IL-9 Interleukin-9
IP-10 IFN-gamma-inducible chemokine IP-10
ITGAE Integrin alphae
JUP Junction plakoglobin
K2P Keratin, type ii cytoskeletal 2 oral
MCL1 Myeloid cell leukemia sequence 1
MIF Macrophage migration inhibitory factor
MIF Macrophage migration inhibitory factor
MIG Gamma interferon induced monokine
MMP10 Matrix metalloproteinase 10
MMP12 Matrix metalloproteinase 12
MMP13 Matrix metalloproteinase 13
MMP3 Matrix metalloproteinase 3
MMP8 Matrix metalloproteinase 8
MSH2 DNA mismatch repair protein MSH2
MSH6 DNA mismatch repair protein MSH6
NB Normal breast
NCK5AI Cyclin-dependent kinase 5 activator isoform p39i
N-MYC N-MYC proto-oncogene
OBCAM Opioid binding cell adhesion molecule
p53 cellular tumor antigen p53
PCNA Proliferating cell nuclear antigen
PGS2 Dermatan sulfate proteoglycan core protein
ras-like small Ras-like small GTPase TTF
RB1 Retinoblastoma susceptibility
RPSA Laminin 37kd receptor
TDGF1 Teratocarcinoma-derived growth factor
TEK TYROSINE-PROTEIN KINASE RECEPTOR
TNF Tumor necrosis factor
BH3 Bcl-2 Homology
Bid BH3 interacting domain death agonist
Bim/Bod Bcl-2 like11 (apoptosis facilitator)
Bmf Bcl-2 modifying factor
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
NG performed microarrays and real-time RT-PCR assays, contributed toward the design of the study and drafted the manuscript. FS participated in the conception and design of the study. HA performed real-time RT-PCR and immunohistochemistry assays. ECQ participated in the conception and design study. IA obtained and performed histopathologic diagnosis of the samples. RP participated in the conception of the study. DA drafted the manuscript, participated in the conception and design of study and its coordination. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
This work was supported by grants No. 34451-M and Salud-2003-C01-074 from Mexican Council of Science and Technology (CONACYT) and by the Medical Research Council of Mexican Social Security Institute (IMSS) FOFOI FP-0038/1248; FP-0038/1247; FP-0038/763 México. The authors would like to thank PhD José Moreno (Specialties Hospital), PhD Guadalupe Rico (Pediatrics Hospital) for technical cooperation, M.A. Maggie Brunner (Managing Editor, Archives of Medical Research) for technical English assess, National Medical Center Century-XXI, IMSS, and PhD Juan Burgueño to assessment on statistical approach. E Curiel-Quesada is a COFAA fellow.
Contributor Information
Normand García, Email: normandgarcia@yahoo.com.
Fabio Salamanca, Email: fasalam@prodigy.net.mx.
Horacio Astudillo-de la Vega, Email: hastud2@aol.com.
Everardo Curiel-Quesada, Email: cuqe520728@hotmail.com.
Isabel Alvarado, Email: isa98@prodigy.net.mx.
Rosenda Peñaloza, Email: rosendaipe@hotmail.com.
Diego Arenas, Email: arenasdi@servidor.unam.mx.
References
- SUIVE Sistema Unico de Informacion para la Vigilancia Epidemiologica /DGE/SSA. Incidencia de casos nuevos de enfermedades por grupo de edad Estados Unidos Mexicanos. Registro Histopalógico de Neoplasias Malignas. 2002. http://www.dgepi.salud.gob.mx/diveent/RHNM.htm
- Cornelisse CJ, Kuipers-Dijkshoorn N, van Vliet M, Hermans J, Devilee P. Fractional allelic imbalance in human breast cancer increases with tetraploidization and chromosome loss. Int J Cancer. 1992;50:544–548. doi: 10.1002/ijc.2910500408. [DOI] [PubMed] [Google Scholar]
- Pandis N, Jin Y, Gorunova L, Petersson C, Bardi G, Idvall I, Johansson B, Ingvar C, Mandahl N, Mitelman F. Chromosome analysis of 97 primary breast carcinomas: identification of eight karyotypic subgroups. Genes Chromosomes Cancer. 1995;12:173–185. doi: 10.1002/gcc.2870120304. [DOI] [PubMed] [Google Scholar]
- Borg A, Baldetorp B, Ferno M, Killander D, Olsson H, Sigurdsson H. ERBB2 amplification in breast cancer with a high rate of proliferation. Oncogene. 1991;6:137–143. [PubMed] [Google Scholar]
- Borg A, Baldetorp B, Ferno M, Olsson H, Sigurdsson H. c-myc amplification is an independent prognostic factor in postmenopausal breast cancer. Int J Cancer. 1992;51:687–691. doi: 10.1002/ijc.2910510504. [DOI] [PubMed] [Google Scholar]
- Bieche I, Lidereau R. Genetic alterations in breast cancer. Genes Chromosomes Cancer. 1995;14:227–251. doi: 10.1002/gcc.2870140402. [DOI] [PubMed] [Google Scholar]
- Phelan CM, Borg A, Cuny M, Crichton DN, Baldersson T, Andersen TI, Caligo MA, Lidereau R, Lindblom A, Seitz S, Kelsell D, Hamann U, Rio P, Thorlacius S, Papp J, Olah E, Ponder B, Bignon YJ, Scherneck S, Barkardottir R, Borresen-Dale AL, Eyfjord J, Theillet C, Thompson AM, Larsson C. Consortium study on 1280 breast carcinomas: allelic loss on chromosome 17 targets subregions associated with family history and clinical parameters. Cancer Res. 1998;58:1004–1012. [PubMed] [Google Scholar]
- Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J, Miki Y. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266:120–122. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- Collins N, McManus R, Wooster R, Mangion J, Seal S, Lakhani SR, Ormiston W, Daly PA, Ford D, Easton DF. Consistent loss of the wild type allele in breast cancers from a family linked to the BRCA2 gene on chromosome 13q12-13. Oncogene. 1995;10:1673–1675. [PubMed] [Google Scholar]
- Birch JM, Hartley AL, Tricker KJ, Prosser J, Condie A, Kelsey AM, Harris M, Jones PH, Binchy A, Crowther D. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 1994;54:1298–1304. [PubMed] [Google Scholar]
- Vorechovsky I, Luo L, Lindblom A, Negrini M, Webster AD, Croce CM, Hammarstrom L. ATM mutations in cancer families. Cancer Res. 1996;56:4130–4133. [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de RM, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Bertucci F, Houlgatte R, Benziane A, Granjeaud S, Adelaide J, Tagett R, Loriod B, Jacquemier J, Viens P, Jordan B, Birnbaum D, Nguyen C. Gene expression profiling of primary breast carcinomas using arrays of candidate genes. Hum Mol Genet. 2000;9:2981–2991. doi: 10.1093/hmg/9.20.2981. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de RM, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein LP, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono JS, Tolcher AW, Rowinsky EK. The future of cytotoxic therapy: selective cytotoxicity based on biology is the key. Breast Cancer Res. 2003;5:154–159. doi: 10.1186/bcr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanzer JG, Eberwine JH. Expression profiling of small cellular samples in cancer: less is more. Br J Cancer. 2004;90:1111–1114. doi: 10.1038/sj.bjc.6601668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai KV, Kavanaugh CJ, Calvo A, Green JE. Chipping away at breast cancer: insights from microarray studies of human and mouse mammary cancer. Endocr Relat Cancer. 2002;9:207–220. doi: 10.1677/erc.0.0090207. [DOI] [PubMed] [Google Scholar]
- Forus A, Sorlie T, Borresen-Dale AL, Myklebost O. [Microarray technology--potential in cancer research] Tidsskr Nor Laegeforen. 2001;121:2498–2503. [PubMed] [Google Scholar]
- Liu ET. Classification of cancers by expression profiling. Curr Opin Genet Dev. 2003;13:97–103. doi: 10.1016/S0959-437X(03)00008-X. [DOI] [PubMed] [Google Scholar]
- Watanabe I, Toyoda M, Okuda J, Tenjo T, Tanaka K, Yamamoto T, Kawasaki H, Sugiyama T, Kawarada Y, Tanigawa N. Detection of apoptotic cells in human colorectal cancer by two different in situ methods: antibody against single-stranded DNA and terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling (TUNEL) methods. Jpn J Cancer Res. 1999;90:188–193. doi: 10.1111/j.1349-7006.1999.tb00732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WY, Cheung KK, Schorge JO, Huang LW, Welch WR, Bell DA, Berkowitz RS, Mok SC. Bcl-2 and p53 protein expression, apoptosis, and p53 mutation in human epithelial ovarian cancers. Am J Pathol. 2000;156:409–417. doi: 10.1016/S0002-9440(10)64744-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioffe OB, Papadimitriou JC, Drachenberg CB. Correlation of proliferation indices, apoptosis, and related oncogene expression (bcl-2 and c-erbB-2) and p53 in proliferative, hyperplastic, and malignant endometrium. Hum Pathol. 1998;29:1150–1159. doi: 10.1016/S0046-8177(98)90429-4. [DOI] [PubMed] [Google Scholar]
- Astudillo H, Lopez T, Castillo S, Gariglio P, Benitez L. p53, Bcl-2, PCNA expression, and apoptotic rates during cervical tumorigenesis. Ann N Y Acad Sci. 2003;1010:771–774. doi: 10.1196/annals.1299.138. [DOI] [PubMed] [Google Scholar]
- Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Borresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci U S A. 2002;99:12963–12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- Verma S, Budarf ML, Emanuel BS, Chinnadurai G. Structural analysis of the human pro-apoptotic gene Bik: chromosomal localization, genomic organization and localization of promoter sequences. Gene. 2000;254:157–162. doi: 10.1016/S0378-1119(00)00276-6. [DOI] [PubMed] [Google Scholar]
- Tong Y, Yang Q, Vater C, Venkatesh LK, Custeau D, Chittenden T, Chinnadurai G, Gourdeau H. The pro-apoptotic protein, Bik, exhibits potent antitumor activity that is dependent on its BH3 domain. Mol Cancer Ther. 2001;1:95–102. [PubMed] [Google Scholar]
- Zou Y, Peng H, Zhou B, Wen Y, Wang SC, Tsai EM, Hung MC. Systemic tumor suppression by the proapoptotic gene bik. Cancer Res. 2002;62:8–12. [PubMed] [Google Scholar]
- Cotran R, Kumar V, Robins S, Schoen F. Pathologic basis of disease. Fifth edition. Saunders; 1994. [Google Scholar]
- Bioinformatics software solutions. J-Express. 2002. http://www.molmine.com
- Statsoft TreeView & Cluster. 2000. http://www.statsoft.com/textbook/stcluan.html
- Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-I. [DOI] [PubMed] [Google Scholar]
- Lee SW. H-cadherin, a novel cadherin with growth inhibitory functions and diminished expression in human breast cancer. Nat Med. 1996;2:776–782. doi: 10.1038/nm0796-776. [DOI] [PubMed] [Google Scholar]
- O'Reilly PE, Raab SS, Niemann TH, Rodgers JR, Robinson RA. p53, proliferating cell nuclear antigen, and Ki-67 expression in extrauterine leiomyosarcomas. Mod Pathol. 1997;10:91–97. [PubMed] [Google Scholar]
- Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Duan W, Pedersen WA, Culmsee C. Neurodegenerative disorders and ischemic brain diseases. Apoptosis. 2001;6:69–81. doi: 10.1023/A:1009676112184. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Strasser A. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 2002;9:505–512. doi: 10.1038/sj.cdd.4400998. [DOI] [PubMed] [Google Scholar]
- Gillissen B, Essmann F, Graupner V, Starck L, Radetzki S, Dorken B, Schulze-Osthoff K, Daniel PT. Induction of cell death by the BH3-only Bcl-2 homolog Nbk/Bik is mediated by an entirely Bax-dependent mitochondrial pathway. EMBO J. 2003;22:3580–3590. doi: 10.1093/emboj/cdg343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/S1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Arena V, Martini M, Luongo M, Capelli A, Larocca LM. Mutations of the BIK gene in human peripheral B-cell lymphomas. Genes Chromosomes Cancer. 2003;38:91–96. doi: 10.1002/gcc.10245. [DOI] [PubMed] [Google Scholar]
- Klumpp S, Krieglstein J. Serine/threonine protein phosphatases in apoptosis. Curr Opin Pharmacol. 2002;2:458–462. doi: 10.1016/S1471-4892(02)00176-5. [DOI] [PubMed] [Google Scholar]
- Verma S, Zhao LJ, Chinnadurai G. Phosphorylation of the pro-apoptotic protein BIK: mapping of phosphorylation sites and effect on apoptosis. J Biol Chem. 2001;276:4671–4676. doi: 10.1074/jbc.M008983200. [DOI] [PubMed] [Google Scholar]
- Mathai JP, Germain M, Marcellus RC, Shore GC. Induction and endoplasmic reticulum location of BIK/NBK in response to apoptotic signaling by E1A and p53. Oncogene. 2002;21:2534–2544. doi: 10.1038/sj.onc.1205340. [DOI] [PubMed] [Google Scholar]
- Germain M, Mathai JP, Shore GC. BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J Biol Chem. 2002;277:18053–18060. doi: 10.1074/jbc.M201235200. [DOI] [PubMed] [Google Scholar]
- Coultas L, Bouillet P, Stanley EG, Brodnicki TC, Adams JM, Strasser A. Proapoptotic BH3-only Bcl-2 family member Bik/Blk/Nbk is expressed in hemopoietic and endothelial cells but is redundant for their programmed death. Mol Cell Biol. 2004;24:1570–1581. doi: 10.1128/MCB.24.4.1570-1581.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel PT, Pun KT, Ritschel S, Sturm I, Holler J, Dorken B, Brown R. Expression of the death gene Bik/Nbk promotes sensitivity to drug-induced apoptosis in corticosteroid-resistant T-cell lymphoma and prevents tumor growth in severe combined immunodeficient mice. Blood. 1999;94:1100–1107. [PubMed] [Google Scholar]
- Fay MJ, Longo KA, Karathanasis GA, Shope DM, Mandernach CJ, Leong JR, Hicks A, Pherson K, Husain A. Analysis of CUL-5 expression in breast epithelial cells, breast cancer cell lines, normal tissues and tumor tissues. Mol Cancer. 2003;2:40. doi: 10.1186/1476-4598-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]