

Abstract
Background
Covariance models (CMs) are probabilistic models of RNA secondary structure, analogous to profile hidden Markov models of linear sequence. The dynamic programming algorithm for aligning a CM to an RNA sequence of length N is O(N3) in memory. This is only practical for small RNAs.
Results
I describe a divide and conquer variant of the alignment algorithm that is analogous to memory-efficient Myers/Miller dynamic programming algorithms for linear sequence alignment. The new algorithm has an O(N2 log N) memory complexity, at the expense of a small constant factor in time.
Conclusions
Optimal ribosomal RNA structural alignments that previously required up to 150 GB of memory now require less than 270 MB.
Background
There are a growing number of RNA gene families and RNA motifs [1,2]. Many (though not all) RNAs conserve a base-paired RNA secondary structure. Computational analyses of RNA sequence families are more powerful if they take into account both primary sequence and secondary structure consensus [3,4].
Some excellent approaches have been developed for database searching with RNA secondary structure consensus patterns. Exact- and approximate-match pattern searches (analogous to PROSITE patterns for proteins) have been extended to allow patterns to specify long-range base pairing constraints [5,6]. In several cases, specialized programs have been developed to recognize specific RNA structures [4] – for example, programs exist for detecting transfer RNA genes [7-9], group I catalytic introns [10], and small nucleolar RNAs [11,12]. All of these approaches, though powerful, lack generality, and they require expert knowledge about each particular RNA family of interest.
In primary sequence analysis, the most useful analysis techniques are general primary sequence alignment algorithms with probabilistically based scoring systems – for example, the BLAST [13], FASTA [14], or CLUSTALW [15] algorithms, and the PAM [16] or BLOSUM [17] score matrices. Unlike specialized programs, a general alignment algorithm can be applied to find homologs of any query sequence(s). Unlike pattern searches, which give yes/no answers for whether a candidate sequence is a match, a scoring system gives a meaningful score that allows ranking candidate hits by their statistical significance. It is of interest to develop general alignment algorithms for RNA secondary structures.
The problem I consider here is as follows. I am given a multiple alignment of an RNA sequence family for which I know the consensus secondary structure. I want to search a sequence database for homologs that significantly match the sequence and structure of my query. The sequence analysis analogue is the use of profile hidden Markov models (profile HMMs) to model multiple alignments of conserved protein domains, and to discover new homologues in sequence databases [18,19]. For instance, if we had an RNA structure equivalent of the HMMER profile HMM program suite http://hmmer.wustl.edu/ it would be possible to develop and efficiently maintain databases of conserved RNA structures and multiple alignments, analogous to the Pfam or SMART databases of conserved protein domains [20,21].
Stochastic context free grammar (SCFG) algorithms provide a general approach to RNA structure alignment [22-24]. SCFGs allow the strong pairwise residue correlations in non-pseudoknotted RNA secondary structure to be taken into account in RNA alignments. SCFGs can be aligned to sequences using a dynamic programming algorithm that guarantees finding a mathematically optimal solution in polynomial time. SCFG alignment algorithms can be thought of as an extension of sequence alignment algorithms (particularly those with fully probabilistic, hidden Markov model formulations) into an additional dimension necessary to deal with 2D RNA secondary structure.
While SCFGs provide a natural mathematical framework for RNA secondary structure alignment problems, SCFG algorithms have high computational complexity that has impeded their practical application. Optimal SCFG-based structural alignment of an RNA structure to a sequence costs O(N3) memory and O(N4) time for a sequence of length N, compared to O(N2) memory and time for sequence alignment algorithms. (Corpet and Michot described a program that implements a different general dynamic programming algorithm for RNA alignment; their algorithm solves the same problem but even less efficiently, requiring O(N4) memory and O(N5) time [25].) SCFG-based alignments of small structural RNAs are feasible. Using my COVE software http://www.genetics.wustl.edu/eddy/software#cove, transfer RNA alignments (~75 nucleotides) take about 0.2 cpu second and 3 Mb of memory. Most genome centers now use an COVE-based search program, tRNAscan-SE, for annotating transfer RNA genes [9]. However, many larger RNAs of interest are OUTSIDE the capabilities of the standard SCFG alignment algorithm. Alignment of a small subunit (SSU) ribosomal RNA sequence to the SSU rRNA consensus structure would take about 23 GB of RAM and an hour of CPU time. Applying SCFG methods to RNAs this large has required clever heuristics, such as using a precalculation of confidently predicted regions of primary sequence alignment to strongly constrain which parts of the SCFG dynamic programming matrix need to be calculated [26]. The steep memory requirement remains a significant barrier to the practicality of SCFG algorithms.
Notredame et al. pointed specifically to this problem [27]. They described RAGA, a program that uses a genetic algorithm (GA) to optimize a pairwise RNA alignment using an objective function that includes base pairing terms. Because GAs have an O(N) memory requirement, RAGA can find reasonable solutions for large RNA alignment problems, including ribosomal RNA alignments. A different memory-efficient approach has also been described [28,29]. However, both approaches are approximate and cannot guarantee a mathematically optimal solution, in contrast to the (mathematically) optimal but more expensive dynamic programming approaches.
Here, I introduce a dynamic programming solution to the problem of structural alignment of large RNAs. The central idea is a divide and conquer strategy. For linear sequence alignment, a divide and conquer algorithm was introduced by Hirschberg [30], an algorithm known in the computational biology community as the Myers/Miller algorithm [31]. (Ironically, at the time, dynamic programming methods for optimal sequence alignment were well known, but were considered impractical on 1970's era computers because of the "extreme" O(N2) memory requirement.) Myers/Miller reduces the memory complexity of a dynamic programming sequence alignment algorithm from O(N2) to O(N), at the cost of a roughly two-fold increase in CPU time. Here I show that a divide and conquer strategy can also be applied to the RNA structural alignment problem, greatly reducing the memory requirement of SCFG alignments and making optimal structural alignment of large RNAs possible.
I will strictly be dealing with the problem of aligning a target sequence of unknown secondary structure to a query of known RNA structure. By "secondary structure" I mean nested (nonpseudoknotted) pairwise RNA secondary structure interactions, primarily Watson-Crick base pairs but also permitting noncanonical base pairs. This RNA structural alignment problem is different from the problem of aligning two known RNA secondary structures together [32], and from the problem of aligning two RNA sequences of unknown structure together under a secondary structure-aware scoring system [33-37].
Algorithm
Prelude: the simpler case of sequence alignment
The essential concepts of a divide and conquer alignment algorithm are most easily understood for the case of linear sequence alignment [30,31].
Dynamic programming (DP) algorithms for sequence alignment fill in an N × M DP matrix of scores F(i,j) for two sequences of lengths N and M (N <M) [38,39]. Each score F(i,j) is the score of the optimal alignment of prefix x1..xi of one sequence to prefix y1..yj of the other. These scores are calculated iteratively, e.g. for global (Needleman/Wunsch) alignment: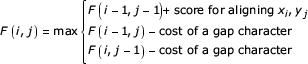
At the end, F(N, M) contains the score of the optimal alignment. The alignment itself is recovered by tracing the individual optimal steps backwards through the matrix, starting from cell (N,M). The algorithm is O(NM) in both time and memory.
If we are only interested in the score, not the alignment itself, the whole F matrix does not have to be kept in memory. The iterative calculation only depends on the current and previous row of the matrix. Keeping two rows in memory suffices (in fact, a compulsively efficient implementation can get away with N + 1 cells). A score-only calculation can be done in O(N) space.
The fill stage of DP alignment algorithms may be run either forwards and backwards. We can just as easily calculate the optimal score B(i, j) of the best alignment of the suffix i + 1..N of sequence 1 to the suffix j + 1..M of sequence 2, until one obtains B(0,0), the overall optimal score – the same number as F(N,M).
The sum of F(i,j) and B(i,j) at any cell in the optimal path through the DP matrix is also the optimal overall alignment score. More generally, F(i,j) + B(i,j) at any cell (i,j) is the score of the best alignment that uses that cell. Therefore, since we know the optimal alignment must pass through any given row i somewhere, we can pick some row i in the middle of sequence x, run the forward calculation to i to obtain row F(i), run the backwards calculation back to i to get row B(i), and then find argmaxj F(i,j)+B(i,j). Now I know the optimal alignment passes through cell (i,j). (For clarity, I am leaving out details of how indels and local alignments are handled.)
This divides the alignment into two smaller alignment problems, and these smaller problems can themselves be subdivided by the same trick. Thus, the complete optimal alignment can be found by a recursive series of split point calculations. Although this seems laborious – each calculation is giving us only a single point in the alignment – if we choose our split row i to be in the middle, the size of the two smaller DP problems is decreased by about 4-fold at each split. A complete alignment thus costs only about 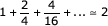 times as much CPU time as doing the alignment in a single DP matrix calculation, but the algorithm is O(N) in memory.
times as much CPU time as doing the alignment in a single DP matrix calculation, but the algorithm is O(N) in memory.
A standard dynamic programming alignment algorithm for SCFGs is the Cocke-Younger-Kasami (CYK) algorithm, which finds an optimal parse tree (e.g. alignment) for a model and a sequence [24,40-42]. (CYK is usually described in the literature as a dynamic programming recognition algorithm for nonstochastic CFGs in Chomsky normal form, rather than as a dynamic programming parsing algorithm for SCFGs in any form. The use of the name "CYK" here is therefore a little imprecise [24].) CYK can be run in a memory-saving "score only" mode. The DP matrix for CYK can also be filled in two opposite directions – either "inside" or "outside", analogous to forward and backward DP matrix fills for linear sequence alignment. I will refer to these algorithms as CYK/inside and CYK/outside (or just inside and outside), but readers familiar with SCFG algorithms should not confuse them with the SCFG Inside and Outside algorithms [43,44] which sum over all possible parse trees rather than finding one optimal parse tree. I am always talking about the CYK algorithm in this paper, and by "inside" and "outside" I am only referring generically to the direction of the CYK DP calculation.
The CYK/inside and CYK/outside algorithms are not as nicely symmetrical as the forward and backward DP fills are in sequence alignment algorithms. The splitting procedure that one obtains does not generate identical types of subproblems, so the divide and conquer procedure for SCFG-based RNA alignment is not as obvious.
Definition and construction of a covariance model
The divide and conquer algorithm I will describe is specific for RNA "covariance models" (CMs). A covariance model is a profile stochastic context free grammar designed to model a consensus RNA secondary structure with position-specific scores [22,24]. My algorithm takes advantage of features of CMs that do not apply to SCFGs in general. Therefore I start with an introduction to what CMs are, how they correspond to a known RNA secondary structure, and how they are built and parameterized.
Definition of a stochastic context free grammar
A stochastic context free grammar (SCFG) consists of the following:
• M different nonterminals (here called states). I will use capital letters to refer to specific nonterminals; V and Y will be used to refer generically to unspecified nonterminals.
• K different terminal symbols (e.g. the observable alphabet, a,c,g,u for RNA). I will use small letters a, b to refer generically to terminal symbols.
• a number of production rules of the form: V → γ, where γ can be any string of nonterminal and/or terminal symbols, including (as a special case) the empty string ε.
• Each production rule is associated with a probability, such that the sum of the production probabilities for any given nonterminal V is equal to 1.
SCFG productions allowed in covariance models
A covariance model is a specific repetitive "profile SCFG" architecture consisting of groups of model states that are associated with base pairs and single-stranded positions in an RNA secondary structure consensus. A covariance model has seven types of states and production rules (Table 1).
Table 1.
| State type | Description | Production | Emission | Transition |
|---|---|---|---|---|
| P | (pair emitting) | P → aYb | ev (a,b) | tv(Y) |
| L | (left emitting) | L → aY | ev(a) | tv(Y) |
| R | (right emitting) | R → Ya | ev(a) | tv(Y) |
| B | (bifurcation) | B → SS | 1 | 1 |
| D | (delete) | D → Y | 1 | tv(Y) |
| S | (start) | S → Y | 1 | tv(Y) |
| E | (end) | E → ε | 1 | 1 |
Each overall production probability is the independent product of an emission probability ev and a transition probability tv, both of which are position-dependent parameters that depend on the state v (analogous to hidden Markov models). For example, a particular pair (P) state v produces two correlated letters a and b (e.g. one of 16 possible base pairs) with probability ev(a, b) and transits to one of several possible new states Y of various types with probability tv(Y). A bifurcation (B) state splits into two new start (S) states with probability 1. The E state is a special case ε production that terminates a derivation.
A CM consists of many states of these seven basic types, each with its own emission and transition probability distributions, and its own set of states that it can transition to. Consensus base pairs will be modeled by P states, consensus single stranded residues by L and R states, insertions relative to the consensus by more L and R states, deletions relative to consensus by D states, and the branching topology of the RNA secondary structure by B, S, and E states. The procedure for starting from an input multiple alignment and determining how many states, what types of states, and how they are interconnected by transition probabilities is described next.
From consensus structural alignment to guide tree
Figure 1 shows an example input file: a multiple sequence alignment of homologous RNAs, with a line describing the consensus RNA secondary structure. The first step of building a CM is to produce a binary guide tree of nodes representing the consensus secondary structure. The guide tree is a parse tree for the consensus structure, with nodes as nonterminals and alignment columns as terminals.
Figure 1.

An example RNA sequence family. Top: a toy multiple alignment of three sequences, with 28 total columns, 24 of which will be modeled as consensus positions. The [structure] line annotates the consensus secondary structure: > and < symbols mark base pairs, x's mark consensus single stranded positions, and .'s mark "insert" columns that will not be considered part of the consensus model. Bottom: the secondary structure of the "human" sequence.
The guide tree has eight types of nodes (Table 2).
Table 2.
| Node | Description | Main state type |
|---|---|---|
| MATP | (pair) | P |
| MATL | (single strand, left) | L |
| MATR | (single strand, right) | R |
| BIF | (bifurcation) | B |
| ROOT | (root) | S |
| BEGL | (begin, left) | S |
| BEGR | (begin, right) | S |
| END | (end) | E |
These consensus node types correspond closely with a CM's final state types. Each node will eventually contain one or more states. The guide tree deals with the consensus structure. For individual sequences, we will need to deal with insertions and deletions with respect to this consensus. The guide tree is the skeleton on which we will organize the CM. For example, a MATP node will contain a P-type state to model a consensus base pair; but it will also contain several other states to model infrequent insertions and deletions at or adjacent to this pair.
The input alignment is first used to construct a consensus secondary structure (Figure 2) that defines which aligned columns will be ignored as non-consensus (and later modeled as insertions relative to the consensus), and which consensus alignment columns are base-paired to each other. Here I assume that both the structural annotation and the labeling of insert versus consensus columns is given in the input file, as shown in the line marked "[structure]" in the alignment in Figure 1. Alternatively, automatic methods might be employed. A consensus structure could be predicted from comparative analysis of the alignment [22,45,46]. The consensus columns could be chosen as those columns with less than a certain fraction of gap symbols, or by a maximum likelihood criterion, as is done for profile HMM construction [18,24].
Figure 2.

The structural alignment is converted to a guide tree. Left: the consensus secondary structure is derived from the annotated alignment in Figure 1. Numbers in the circles indicate alignment column coordinates: e.g. column 4 base pairs with column 14, and so on. Right: the CM guide tree corresponding to this consensus structure. The nodes of the tree are numbered 1..24 in preorder traversal (see text). MATP, MATL, and MATR nodes are associated with the columns they generate: e.g., node 6 is a MATP (pair) node that is associated with the base-paired columns 4 and 14.
Given the consensus structure, consensus base pairs are assigned to MATP nodes and consensus unpaired columns are assigned to MATL or MATR nodes. One ROOT node is used at the head of the tree. Multifurcation loops and/or multiple stems are dealt with by assigning one or more BIF nodes that branch to subtrees starting with BEGL or BEGR head nodes. (ROOT, BEGL, and BEGR start nodes are labeled differently because they will be expanded to different groups of states; this has to do with avoiding ambiguous parse trees for individual sequences, as described below.) Alignment columns that are considered to be insertions relative to the consensus structure are ignored at this stage.
In general there will be more than one possible guide tree for any given consensus structure. Almost all of this ambiguity is eliminated by three conventions: (1) MATL nodes are always used instead of MATR nodes where possible, for instance in hairpin loops; (2) in describing interior loops, MATL nodes are used before MATR nodes; and (3) BIF nodes are only invoked where necessary to explain branching secondary structure stems (as opposed to unnecessarily bifurcating in single stranded sequence). One source of ambiguity remains. In invoking a bifurcation to explain alignment columns i..j by two substructures on columns i..k and k + 1..j, there will be more than one possible choice of k if i..j is a multifurcation loop containing three or more stems. The choice of k impacts the performance of the divide and conquer algorithm; for optimal time performance, we will want bifurcations to split into roughly equal sized alignment problems, so I choose the k that makes i..k and k + 1..j as close to the same length as possible.
The result of this procedure is the guide tree. The nodes of the guide tree are numbered in preorder traversal (e.g. a recursion of "number the current node, visit its left child, visit its right child": thus parent nodes always have lower indices than their children). The guide tree corresponding to the input multiple alignment in Figure 1 is shown in Figure 2.
From guide tree to covariance model
A CM must deal with insertions and deletions in individual sequences relative to the consensus structure. For example, for a consensus base pair, either partner may be deleted leaving a single unpaired residue, or the pair may be entirely deleted; additionally, there may be inserted nonconsensus residues between this pair and the next pair in the stem. Accordingly, each node in the master tree is expanded into one or more states in the CM as follows (Table 3)
Table 3.
| Node | States | total # states | # of split states | # of insert states |
|---|---|---|---|---|
| MATP | [MP ML MR D] IL IR | 6 | 4 | 2 |
| MATL | [ML D] IL | 3 | 2 | 1 |
| MATR | [MR D] IR | 3 | 2 | 1 |
| BIF | [B] | 1 | 1 | 0 |
| ROOT | [S] IL IR | 3 | 1 | 2 |
| BEGL | [S] | 1 | 1 | 0 |
| BEGR | [S] IL | 2 | 1 | 1 |
| END | [E] | 1 | 1 | 0 |
Here we distinguish between consensus ("M", for "match") states and insert ("I") states. ML and IL, for example, are both L type states with L type productions, but they will have slightly different properties, as described below.
The states are grouped into a split set of 1–4 states (shown in brackets above) and an insert set of 0–2 insert states. The split set includes the main consensus state, which by convention is first. One and only one of the states in the split set must be visited in every parse tree (and this fact will be exploited by the divide and conquer algorithm). The insert state(s) are not obligately visited, and they have self-transitions, so they will be visited zero or more times in any given parse tree.
State transitions are then assigned as follows. For bifurcation nodes, the B state makes obligate transitions to the S states of the child BEGL and BEGR nodes. For other nodes, each state in a split set has a possible transition to every insert state in the same node, and to every state in the split set of the next node. An IL state makes a transition to itself, to the IR state in the same node (if present), and to every state in the split set of the next node. An IR state makes a transition to itself and to every state in the split set of the next node.
This arrangement of transitions guarantees that (given the guide tree) there is unambiguously one and only one parse tree for any given individual structure. This is important. The algorithm will find a maximum likelihood parse tree for a given sequence, and we wish to interpret this result as a maximum likelihood structure, so there must be a one to one relationship between parse trees and secondary structures [47].
The final CM is an array of M states, connected as a directed graph by transitions tv (y) (or probability 1 transitions v → (y,z) for bifurcations) with the states numbered such that (y,z) ≥ v. There are no cycles in the directed graph other than cycles of length one (e.g. the self-transitions of the insert states). We can think of the CM as an array of states in which all transition dependencies run in one direction; we can do an iterative dynamic programming calculation through the model states starting with the last numbered end state M and ending in the root state 1. An example CM, corresponding to the input alignment of Figure 1, is shown in Figure 3.
Figure 3.

A complete covariance model. Right: the CM corresponding to the alignment in Figure 1. The model has 81 states (boxes, stacked in a vertical array). Each state is associated with one of the 24 nodes of the guide tree (text to the right of the state array). States corresponding to the consensus are in white. States responsible for insertions and deletions are gray. The transitions from bifurcation state B10 to start states S11 and S46 are in bold because they are special: they are an obligate (probability 1) bifurcation. All other transitions (thin arrows) are associated with transition probabilities. Emission probability distributions are not represented in the figure. Left: the states are also arranged according to the guide tree. A blow up of part of the model corresponding to nodes 6, 7, and 8 shows more clearly the logic of the connectivity of transition probabilities (see main text), and also shows why any parse tree must transit through one and only one state in each "split set".
As a convenient side effect of the construction procedure, it is guaranteed that the transitions from any state are to a contiguous set of child states, so the transitions for state v may be kept as an offset and a count. For example, in Figure 3, state 12 (an MP) connects to states 16, 17, 18, 19, 20, and 21. We can store this as an offset of 4 to the first connected state, and a total count of 6 connected states. We know that the offset is the distance to the next non-split state in the current node; we also know that the count is equal to the number of insert states in the current node, plus the number of split set states in the next node. These properties make establishing the connectivity of the CM trivial. Similarly, all the parents of any given state are also contiguously numbered, and can be determined analogously. We are also guaranteed that the states in a split set are numbered contiguously. This contiguity is exploited by the divide and conquer implementation.
Parameterization
Using the guide tree and the final CM, each individual sequence in the input multiple alignment can be converted unambiguously to a CM parse tree, as shown in Figure 4. Counts for observed state transitions and singlet/pair emissions are then collected from these parse trees. The observed counts are converted to transition and emission probabilities by standard procedures. I calculate maximum a posteriori parameters, using Dirichlet priors.
Figure 4.

Example parse trees. Parse trees are shown for the three sequences/structures from Figure 1, given the CM in Figure 3. For each sequence, each residue must be associated with a state in the parse tree. (The sequences can be read off its parse tree by starting at the upper left and reading counterclockwise around the edge of parse tree.) Each parse tree corresponds directly to a secondary structure – base pairs are pairs of residues aligned to MP states. A collection of parse trees also corresponds to a multiple alignment, by aligning residues that are associated with the same state – for example, all three trees have a residue aligned to state ML4, so these three residues would be aligned together. Insertions and deletions relative to the consensus use nonconsensus states, shown in gray.
Comparison to profile HMMs
The relationship between an SCFG and a covariance model is analogous to the relationship of hidden Markov models (HMMs) and profile HMMs for modeling multiple sequence alignments [18,19,24]. A comparison may be instructive to readers familiar with profile HMMs. A profile HMM is a repetitive HMM architecture that associates each consensus column of a multiple alignment with a single type of model node – a MATL node, in the above notation. Each node contains a "match", "delete", and "insert" HMM state – ML, IL, and D states, in the above notation. The profile HMM also has special begin and end states. Profile HMMs could therefore be thought of as a special case of CMs. An unstructured RNA multiple alignment would be modeled by a guide tree of all MATL nodes, and converted to an unbifurcated CM that would essentially be identical to a profile HMM. (The only difference is trivial; the CM root node includes a IR state, whereas the start node of a profile HMM does not.) All the other node types (especially MATP, MATR, and BIF) and state types (e.g. MP, MR, IR, and B) are SCFG augmentations necessary to extend profile HMMs to deal with RNA secondary structure.
The SCFG inside and outside algorithms are analogous to the Forward and Backward algorithms for HMMs [24,48]. The CYK/inside parsing algorithm is analogous to the Viterbi HMM alignment algorithm run in the forward direction. CYK/outside is analogous to a Viterbi DP algorithm run in the backwards direction.
Divide and conquer algorithm
Notation
I use r, v, w, y, and z as indices of states in the model, where r ≤ (v,w,y) ≤ z. These indices will range from 1..M, for a CM G that contains M states. 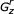 refers to a subgraph of the model, rooted at state r and ending at state z, for a contiguous set of states r..z. Gr, without a subscript, refers to a subgraph of the model rooted at state r and ending at the highest numbered E state descendant from state r. The complete model is
refers to a subgraph of the model, rooted at state r and ending at state z, for a contiguous set of states r..z. Gr, without a subscript, refers to a subgraph of the model rooted at state r and ending at the highest numbered E state descendant from state r. The complete model is 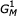 , or G1, or just G.
, or G1, or just G.
Sv refers to the type of state v; it will be one of seven types {D,P,L,R,S,E,B}. Cv is a list of children for state v (e.g. the states that v can transit to); it will contain up to six contiguous indices y with v ≤ y ≤ M. Pv is a list of parents for state v (states that could have transited to state v); it will contain up to six contiguous indices y with 1 ≤ y ≤ v. (Pv parent lists should not be confused with P state types.)
I use g, h, i, j, k, p, and q as indices referring to positions in a sequence x, where g ≤ h ≤ p ≤ q and i ≤ j for all subsequences of nonzero length. These indices range from 1..L, for a sequence of length L. Some algorithms will also use d to refer to a subsequence length, where d = j - i + 1 for a subsequence xi..xj.
The algorithms will have to account for subsequences of zero length (because of deletions). By convention, these will be in the off-diagonal where j = i - 1 or i = j + 1. This special case (usually an initialization condition) is the reason for the qualification that i ≤ j for subsequences of nonzero length.
The CYK/inside algorithm calculates a three-dimensional matrix of numbers αv(i,j), and CYK/outside calculates numbers βv(i,j). I will refer to v (state indices) as deck coordinates in the three-dimensional matrices, whereas j and i (sequence positions) are row and column coordinates within each deck. αv and βv refer to whole two-dimensional decks containing scores αv (i,j) and βv (i,j) for a particular state v. The dividing and conquering will be done in the v dimension, by choosing particular decks as split points.
The CYK/inside algorithm
The CYK/inside algorithm iteratively calculates αv(i,j) – the log probability of the most likely CM parse subtree rooted at state v that generates subsequence xi..xj of sequence x. The calculation initializes at the smallest subgraphs and subsequences (e.g. subgraphs rooted at E states, generating subsequences of length 0), and iterates outwards to progessively longer subsequences and larger CM subgraphs.
For example, if we're calculating αv(i,j) and Sv = P (that is, v is a pair state), v will generate the pair xi, xj and transit to a new state y (one of its possible transitions Cv) which then will have to account for the smaller subsequence xi+1..xj-1. The log probability for a particular choice of next state y is the sum of three terms: an emission term log ev(xi,xj), a transition term log tv(y), and an already calculated solution for the smaller optimal parse tree rooted at y, αy (i + 1, j - 1). The answer for αv (i,j) is the maximum over all possible choices of child states y that v can transit to.
The algorithm INSIDE is as follows:
Input: A CM subgraph and subsequence xg..xq.
Output: Scoring matrix decks αr..αz.
INSIDE(r,z; g,q)
for v ← z down to r
for j ← g - 1 to q
for i ← j + 1 down to g
d ← j - i + 1
if Sv = D or S:
αv (i, j) = 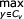 [αy (i, j) + log tv (y)]
[αy (i, j) + log tv (y)]
else if Sv = P and d ≥ 2:
αv (i, j) = log ev (xi, xj) + [αy (i + 1, j - 1) + log tv (y)]
else if Sv = L and d ≥ 1:
αv (i, j) = log ev (xi) + [αy (i + 1, j) + log tv (y)]
else if Sv = R and d ≥ 1:
αv (i, j) = log ev (xj) + [αy (i, j - 1) + log tv (y)]
else if Sv = B:
(y, z) ← left and right S children of state v
αv (i, j) = 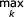 [αy (i, k) + αz (k + 1, j)]
[αy (i, k) + αz (k + 1, j)]
else if Sv = E and d = 0:
αv(i, j) = 0 (initializations)
else
αv(i, j) = -∞ (initializations)
Given a sequence x of length L and a CM G of length M, we could call INSIDE (1, M; 1, L) to align the whole model (states 1..M) to the whole sequence (x1..xL). When INSIDE returns, α1(1, L) would contain the log probability of the best parse of the complete sequence with the complete model.
We do not have to keep the entire α three-dimensional matrix in memory to calculate these scores. As we reach higher decks αv in the three dimensional dynamic programming matrix, our calculations no longer depend on certain lower decks. A lower deck y can be deallocated whenever all the parent decks Py that depend on it have been calculated. (The implementation goes even further and recycles decks when possible, saving some initialization steps and many memory allocation calls; for example, since values in all E decks are identical, only one E deck needs to be calculated and that precalculated deck can be reused whenever Sv= E.)
This deallocation rule has an important property that the divide and conquer algorithm takes advantage of when solving smaller subproblems for CM subgraphs rooted at some state w. When the root state w is an S state, the α matrix returned by INSIDE contains only one active deck αw. (No lower state >w can be reached from any state <w without going through w, so all lower decks are deallocated once deck w is completed.) When the root state w is the first state in a split set w..y (see below for more explanation), all (and only) the decks αw..αy are active when INSIDE returns.
In some cases we want to recover the optimal parse tree itself, not just its score. The INSIDEτ routine is a modified version of INSIDE. It keeps an additional "shadow matrix" τv(i,j). A τv(i,j) traceback pointer either records the index y that maximized αv (i,j) (for state types D,S,P,L,R) or records the split point k that maximized αv(i,j) for a bifurcation (B) state. The τ shadow matrix does not use the deallocation rules – INSIDEτ can only be called for problems small enough that they can be solved within our available memory space. Thus the INSIDEτ routine works by calling INSIDE in a mode that also keeps a shadow matrix τ, and then calls a recursive traceback starting with v, i, j:
Input: A shadow matrix τ for CM subgraph Gv rooted at state v, and subsequence xi..xj.
Output: An optimal parse tree T.
TRACEBACK(v,i,j)
if Sv = E:
attach v
else if Sv = S or D:
attach v
TRACEBACK(τv (i,j), i, j))
else if Sv = P:
attach xi, v, xj
TRACEBACK(τv(i, j), i + 1, j - 1)
else if Sv = L:
attach xi, v
TRACEBACK(τv(i, j), i + 1, j)
else if Sv = R:
attach v, xj
TRACEBACK(τv(i, j), i, j - 1)
else if Sv = B:
(y, z) ← left and right S children of state v
attach v
TRACEBACK(y, i, τv(i, j))
TRACEBACK(z, τv(i, j) + 1, j)
The CYK/outside algorithm
The CYK/outside algorithm iteratively calculates βv(i, j), the log probability of the most likely CM parse tree for a CM generating a sequence x1..xL excluding the optimal parse subtree rooted at state v that accounts for the subsequence xi..xj. The calculation initializes with the entire sequence excluded (e.g. β1(1, L) = 0), and iterates inward to progressively shorter and shorter excluded subsequences and smaller CM subgraphs.
A complete implementation of the CYK/outside algorithm requires first calculating the CYK/inside matrix α because it is needed to calculate βv when the parent of v is a bifurcation [24,43,44]. However, the divide and conquer algorithm described here only calls OUTSIDE on unbifurcated, linear CM subgraphs (only the final state z may be a B state; there are no internal bifurcations that lead to branches in the model). Thus the parent of a state v is never a bifurcation, and the implementation can therefore be streamlined as follows:
Input: An unbifurcated CM subgraph and subsequence xg..xq.
Output: Scoring matrix decks βr..βz.
OUTSIDE(r,z; g,q)
βv(i, j) ← - ∞ ∀ v, i, j
βr(g, q) ← 0
for v ← r + 1 to z
for j ← q down to g - 1
for i ← g to j + 1
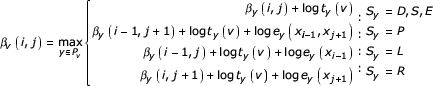
As with INSIDE, we do not keep the entire β matrix in memory. A deck βv can be deallocated when all child decks Cv that depend on the values in βv have been calculated. This means that if the last deck z is a bifurcation or end state, βz will be the only active allocated deck when OUTSIDE returns. If z is the last state in a split set w..z, all (and only) the split set decks βw..βz will be active when OUTSIDE returns.
Using CYK/inside and CYK/outside to divide and conquer
Now, for any chosen state v, argmaxi,j [αv (i, j) + βv(,i,j)] tells us which cell v, i, j the optimal parse tree passes through, conditional on using state v in the parse. We know that any parse tree must include all the bifurcation and start states of the CM, so we know that the optimal alignment must use any chosen bifurcation state v and its child start states w and y. Thus, we are guaranteed that (when Sv = B and Cv = w, y):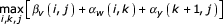
is the optimal overall alignment score, and we also know that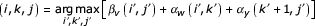
gives us a triplet that identifies three cells that must be in the optimal alignment – (v, i, j), (w, i, k), and (y, k + 1, j). This splits the remaining problem into three smaller subproblems – an alignment of the sequence xi..xk to a CM subgraph w..y - 1, an alignment of the sequence xk+1..xj to a CM subgraph y..M, and an alignment of the two-piece sequence x1..zi-1//xj+1..xL to a CM subgraph 1..v.
The subproblems are then themselves split, and this splitting can continue recursively until all the bifurcation triplets on the optimal parse tree have been determined.
At this point the remaining alignment subproblems might be small enough to be solved by straightforward application of the standard CYK/inside algorithm (e.g. INSIDEτ). However, this is not guaranteed to be the case. A more general division strategy is needed that does not depend on splitting at bifurcations.
For the more general strategy we take advantage of the fact that we know that the optimal parse tree must also include one and only one state from the split set of each node (e.g. the non-insert states in the node). Let w..y be the indices of a split set of states in the middle of the current model subgraph. (w..y can be at most 4 states.) We know that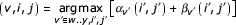
gives us a new cell (v, i, j) in the optimal parse tree, and splits the problem into two smaller problems. This strategy can be applied recursively all the way down to single nodes, if necessary. We can therefore guarantee that we will never need to carry out a full CYK/inside alignment algorithm on any subproblem. The most memory-intensive alignment problem that needs to be solved is the very first split. The properties of the first split determine the memory complexity of the algorithm.
The bifurcation-dependent strategy is a special case of this more general splitting strategy, where the B state is the only member of its split set, and where we also take advantage of the fact that αv(i,j) = maxk αw(i, k) + αy(k + 1,j). By carrying out the maxk operation during the split, rather than before, we can split the current problem into three optimal pieces instead of just two.
If we look at the consequences of these splitting strategies, we see we will have to deal with three types of problems (Figure 5):
Figure 5.

The three types of problems that need to be split. The sequence axis (e.g. xg..xq) is horizontal. The model subgraph axis for a contiguous set of states (e.g. states r..z) is vertical, where a solid lines means an unbifurcated model subgraph, and a dashed line means a model subgraph that may contain bifurcations. Closed circles indicate "inclusive of", and open circles indicate "exclusive of".
• A generic problem means finding the optimal alignment of a CM subgraph to a contiguous subsequence xg..xq. The subgraph corresponds to a complete subtree of the CM's guide tree – e.g. state r is a start (S), and state z is an end (E). may contain bifurcations. The problem is solved in one of two ways. If contains no bifurcations, it is solved as a wedge problem (see below). Else, the problem is subdivided by the bifurcation-dependent strategy: an optimal triple (i, k, j) is found for a bifurcation state v and its children w, y, splitting the problem into a V problem and two generic problems.
• A wedge problem means finding the optimal alignment of an unbifurcated CM subgraph to a contiguous subsequence xg..xq. State z does not have to be a start state (S); it may be a state in a split set (MP, ML, MR, or D). State z is an end (E). A wedge problem is solved by the split set-dependent strategy: an optimal (v, i, j) is found, splitting the problem into a V problem and a smaller wedge problem.
• A V problem consists of finding the optimal alignment of an unbifurcated CM subgraph to a noncontiguous, two-piece sequence xg..xh//xp..xg, exclusive of the residues xh and xp (open circles in Figure 5). State r can be a start state or any state in a split set; the same is true for z. A V problem is solved by a split set-dependent strategy: an optimal (v, i, j) is found, splitting the problem into two V problems.
The three recursive splitting algorithms to solve these problems are as follows:
The generic_splitter routine
Input: A generic problem, for CM subgraph and subsequence xg..q.
Output: An optimal parse subtree T.
GENERIC_SPLITTER(r, z; g,q)
if no bifurcation in :
return WEDGE_SPLITTER(r,z; g,q)
else
v ← lowest numbered bifurcation state in subgraph .
w,y ← left and right S children of v.
βv ← OUTSIDE(r,w; g,q)
αw ← INSIDE(w,y-1; g,q)
αy ← INSIDE(y,z; g,q)
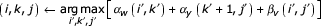
T1 ← V_SPLITTER(r,v; g,i; j,q)
T2 ← GENERIC_SPLITTER(w,y-1; i,k)
T3 ← GENERIC_SPLITTER(y,z; k+l,j)
Attach S state w of T2 as left child of B state v in T1.
Attach S state y of T3 as right child of B state v in T2.
return T1.
The wedge_splitter routine
Input: A wedge problem, for unbifurcated CM subgraph and subsequence xg..q.
Output: An optimal parse subtree T.
WEDGE_SPLITTER(r,z; g,q)
(w..y) ← a split set chosen from middle of
(αw..αy) ← INSIDE(w,z; g,q)
(βw..βy) ← OUTSIDE(r,y; g,q)
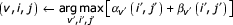
T1 ← V_SPLITTER(r,v; g,i; j,q)
T2 ← WEDGE_SPLITTER(v,z; i,j)
Attach T2 to T1 by merging at state v.
return T1.
The V_splitter routine
Input: A V problem, for unbifurcated CM subgraph and two-part subsequence xg..h//xp..q.
Output: An optimal parse subtree T.
V_SPLITTER(r,z; g,h; p,q)
(w..y) ← a split set chosen from middle of
(αw..αy) ← VINSIDE(w,z; g,h; p,q)
(βw..βy) ← VOUTSlDE(r,y; g,h; p,q)
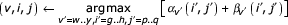
T1 ← V_SPLITTER(r,v; g,i; j,q)
T2 ← V_SPLITTER(v,z; i,h; p,j)
Attach T2 to T1 by merging at state v.
return T1.
The vinside and voutside routines
The VINSIDE and VOUTSIDE routines are just INSIDE and OUTSIDE, modified to deal with a two-piece subsequence xg..xh//xp..xg instead of a contiguous sequence xg..xq. These modifications are fairly obvious. The range of i, j is restricted so that i ≤ h and j ≥ p. Also, VINSIDE (w, z; g, h; p, q) initializes αz (h, p) = 0: that is, we know that sequence xh..xp has already been accounted for by a CM parse tree rooted at z.
Implementation
In the description of the algorithms above, some technical detail has been omitted – in particular, a detailed description of efficient initialization steps, and details of how the the dynamic programming matrices are laid out in memory. These details are not necessary for a high level understanding of the divide and conquer algorithm. However, they may be necessary for reproducing a working implementation. Commented ANSI/C source code for a reference implementation is therefore freely available at http://www.genetics.wustl.edu/eddy/infernal/ under a GNU General Public License. This code has been tested on GNU/Linux platforms.
In this codebase, the CM data structure is defined in structs.h. The CM construction procedure is in modelmaker. c:Handmodelmaker(). The guide tree is constructed in HandModelmaker(). A CM is constructed from the guide tree by cm_from_master(). Individual parse trees are constructed using the guide tree by transmogrify(). The divide and conquer algorithm is implemented in smallcyk. c:CYKDivideAndConquer(), which will recursively call a set of functions: the three splitting routines GENERIC_SPLITTER(), wedge_splitter(), and v_splitter(); the four alignment engines INSIDE(), OUTSIDE(), VINSIDE(), and VOUTSIDE(); and the two traceback routines INSIDET() and VINSIDET().
Results and discussion
Memory complexity analysis
The memory complexity of normal CYK/inside is O(N2M), for a model of M states and a query sequence of N residues, since the full 3D dynamic programming matrix is indexed N × N × M (and since N ∝ M, we can alternatively state the upper bound as O(N3)). The memory complexity of the divide and conquer algorithm is O(N2 log M). The analysis that leads to this conclusion is as follows.
For a model with no bifurcations, the divide and conquer algorithm will never require more than 10 decks in memory at once. In the case of two adjacent MATP nodes, we will need six decks to store the scores for the current node we're calculating, and four decks for the split set of the adjacent node that we're connecting to (and dependent upon) (Figure 3).
Bifurcations will require some number of additional decks for start states (BEGL_S and BEGR_S) to be kept. In INSIDE, whenever we reach a deck for a start state, we will keep that deck in memory until we reach the parent bifurcation state. Half the time, that will mean waiting until another complete subgraph of the model is calculated (e.g. the subgraph rooted at the other start child of that bifurcation); that is, to calculate deck αv for a bifurcation v, we need both decks αw and αy for its child start states w and y, so we have to hold on to αy until we reach αw. In turn, the subgraph rooted at w might contain bifurcations, so our calculation of αw might require additional decks to be kept. Each start deck we reach in the INSIDE iteration means holding one extra deck in memory, and each bifurcation we reach means deallocating the two start decks it depends on; therefore we can iteratively calculate the maximum number of extra decks we will require:
xM ← -1
for v ← M -1 to 1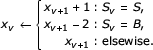
return maxv xv
This number depends on the topology and order of evaluation of the states in the CM. Think of the bifurcating structure of the CM as a binary tree numbered in preorder traversal (e.g. left children are visited first, and have lower indices than right children). If this is a complete balanced tree with B bifurcations, we will need log2B extra decks. If it is a maximally unbalanced tree in which bifurcations only occur in left children, we will need B extra decks (all the right children). If it is a maximally unbalanced tree in which bifurcations only occur in right children, we will only ever need 1 extra deck. A left-unbalanced binary tree can be converted to a right-unbalanced binary tree just by swapping branches. For a CM, we can't swap branches without affecting the order of the sequence that's generated. We can, however, get the same effect by renumbering the CM states in a modified preorder traversal. Instead of always visiting the left subtree first, we visit the best subtree first, where "best" means the choice that will optimize memory usage. This reordering is readily calculated in O(M) time (not shown; see cm. c:CMRebalance() in the implementation). This way, we can never do worse than the balanced case, and we will often do better. We never need more than log2B extra decks. Since B <M, we can guarantee a O(N2 log M) bound on memory complexity.
Time complexity analysis
The time complexity of the standard algorithm is O(MN2 + BN3), for a model of M states (B of which are bifurcations) aligned to a sequence of N residues. Since B <M, and M ∝ N, we can also state the upper bound as O(MN3) or O(N4).
The time complexity of the divide and conquer algorithm depends on how close each split is to dividing a problem into equal sized subproblems. In the most ideal case, each call to GENERIC_SPLITTER could split into three subproblems that each contained 1/3 of the states and residues: splitting those three subproblems would only cost:
3 × 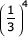 × MN3
× MN3
in time, e.g. only about 1/27 the time it took to split the first problem. Thus in an ideal case the time requirement is almost completely dominated by the first split, and the extra time required to do the complete divide and conquer algorithm could be negligible. In pathological cases, optimal splits might lead to a series of very unequally sized problems. We never need to do more splits than there are states in the model, so we cannot do worse than O(M2N3) in time.
An example of a pathological case is an RNA structure composed of a series of multifurcation loops such that each bifurcation leads to a small stem on one side, and the rest of the structure on the other. In such a case, every call to GENERIC_SPLITTER will split into a small subproblem containing a small stem (e.g. only removing a constant number of states and residues per split) and a large subproblem containing all the remaining states and sequence. This case can be avoided. It only arises because of the decision to implement a simplified CYK/Outside algorithm and always split at the highest bifurcations. Better time performance could be guaranteed if a complete CYK/Outside algorithm were implemented (at the cost of complexity in the description and implementation of the algorithm). This would allow us to choose a split point in a generic problem at any state in the CM (for instance, in the middle) regardless of its bifurcating structure.
In practice, empirical results on a variety of real RNAs (see below) indicate that the extra time required to do the divide and conquer is a small constant factor. A more complex implementation does not seem to be necessary.
Empirical results
Six structural RNA families were chosen for empirical evaluations of the algorithm, using available RNA database resources – tRNA [49], 5S ribosomal RNA [50], signal recognition particle (SRP) RNA [51], RNase P [52], small subunit (SSU) ribosomal RNA [53], and large subunit (LSU) ribosomal RNA [54]. For each family, a secondary structure annotated multiple alignment of four or five example sequences was extracted, and used to construct a CM.
The top of Table 4 shows some statistics about these alignments and CMs. The number of consensus columns in the alignments ranges from 72 (tRNA) to 2898 (LSU rRNA); about 55–60% are involved in consensus base pairs. The number of CM states is consistently about 3-fold more than the consensus alignment length, ranging from 230 states (tRNA) to 9023 (LSU). About 1/150 of the states in each model are bifurcations. After optimal reordering of the model states, the number of extra decks required by the alignment algorithm is small, ranging up to 3 for SSU and 5 for LSU rRNA. Therefore the minimum constant of 10 decks required in iterations across unbifurcated model segments dominates the memory requirement. The memory required for extra decks does not have much impact even for the largest structural RNAs. (Even without optimal reordering, the number of extra decks required for SSU and LSU are only 7 and 9, respectively. State reordering was only needed to assure a O(N2 log M) memory complexity bound.)
Table 4.
Results of empirical tests of memory and CPU time required by CM structural alignment algorithms on six known structural RNAs of various sizes.
| tRNA | 5S rRNA | SRP RNA | RNase P | SSU rRNA | LSU rRNA | |
|---|---|---|---|---|---|---|
| # of consensus columns | 72 | 116 | 301 | 379 | 1545 | 2898 |
| # of consensus base pairs | 21 | 35 | 89 | 113 | 462 | 794 |
| # of consensus unpaired | 30 | 46 | 123 | 153 | 621 | 1310 |
| CM states (M) | 230 | 357 | 927 | 1176 | 4789 | 9023 |
| bifurcations (B) | 2 | 1 | 4 | 7 | 30 | 65 |
| Maximum extra decks needed | 1 | 1 | 2 | 2 | 3 | 5 |
| Example sequence length (N) | 73 | 120 | 300 | 377 | 1542 | 2904 |
| Full CYK RAM (MB) | 2.6 | 10.7 | 168.9 | 336.7 | 22705.0 | 151349.7 |
| Divide & conquer RAM (MB) | 0.1 | 0.4 | 2.4 | 3.7 | 66.8 | 270.9 |
| Full CYK CPU time (sec) | 0.2 | 0.7 | 12.7 | 28.6 | n.d. | n.d. |
| CYK CPU time, no trace (sec) | 0.1 | 0.6 | 10.4 | 23.8 | 2614.8 | 25151.2 |
| Divide & conquer time (sec) | 0.2 | 0.9 | 22.8 | 37.8 | 3594.4 | 31649.4 |
To determine the memory and CPU time requirements for a structural alignment, one example sequence from each family was aligned to the CM. CPU time was measured and memory requirements were calculated for three algorithms: (1) the full CYK/inside algorithm, but in memory-saving score-only mode (e.g. INSIDE(1,M; 1,L); (2) the full CYK/inside algorithm, with shadow matrix and traceback to recover the optimal alignment (e.g. INSIDEτ (1,M; 1,L)); and (3) the divide and conquer algorithm to recover an optimal alignment (e.g. GENERIC_SPLITTER (1,M; 1,L)). The most important comparison is between the full CYK/inside algorithm and the divide and conquer algorithm. The score-only CYK/inside algorithm was included, because a complete CYK alignment couldn't be done on SSU and LSU rRNA (because of the steep memory requirement). In all cases where comparison could be done, the scores and alignments produced by these algorithms were verified to be identical.
The results of these tests are shown in the bottom half of Table 1. The memory required by divide and conquer alignment ranges up to 271 MB for LSU rRNA, compared to a prohibitive 151 GB for the standard CYK algorithm. The extra CPU time required by the divide and conquer is small; usually about 20% more, with a maximum of about two-fold more for SRP-RNA.
The same results are plotted in Figure 6. Memory requirements scale as expected: N3 for standard CYK alignment, and better than N2 log N for the divide and conquer algorithm. Empirical CPU time requirements scale similarly for the two algorithms (N3.24 – N3.29). The observed performance is better than the theoretical worst case of O (N4). The proportion of extra time required by divide and conquer is roughly constant over a wide range of RNAs. The difference shown in Figure 6 is exaggerated because times are plotted for score-only CYK, not complete CYK alignment, in order to include CPU times for SSU and LSU rRNA. Because score-only CYK does not keep a shadow traceback matrix nor perform the traceback, it is about 20% faster than CYK alignment, as seen in the data in Table 4.
Figure 6.

Empirical time and memory requirements for structural alignment. Plots of data from Table 1. Filled circles: divide and conquer algorithm; open circles: standard CYK algorithm. Left: Memory use in megabytes on a log-log scale. Lines represent weighted least-squares regression fits to the theoretically expected memory scaling: aN2 log N for divide and conquer (solid line) and aN3 for standard CYK (dashed line). Right: CPU times in seconds on a log-log scale. Lines represent least-squares regression fits to a power law (aNb). According to this fit, divide and conquer time (solid line) empirically scales as N3.24, and standard CYK without traceback (dashed line) scales as N3.29. A line representing O(N4) scaling (the theoretical upper bound on performance) is shown for comparison.
Conclusions
The divide and conquer algorithm described here makes it possible to align even the largest structural RNAs to secondary structure consensus models, without exceeding the available memory on current computational hardware. Optimal SSU and LSU rRNA structural alignments can be performed in 70 MB and 270 MB of memory, respectively. Previous structural alignment algorithms had to sacrifice mathematical optimality to achieve ribosomal RNA alignments.
The CPU time requirement of the alignment algorithm is still significant, and even prohibitive for certain important applications. However, CPU time is generally an easier issue to deal with. A variety of simple parallelization strategies are possible. Banded dynamic programming algorithms (e.g. calculating only relevant parts of the matrix) of various forms can also be explored, including not only heuristic schemes, but also optimal algorithms based on branch and bound ideas. (Properly implemented, banded DP algorithms would also save additional memory.)
The algorithm takes advantage of the structure of covariance models (profile SCFGs), splitting in the dimension of the states of the model rather than in the sequence dimensions. The approach does not readily apply, therefore, to unprofiled SCFG applications in RNA secondary structure prediction [34,36,55,56], where the states are fewer and more fully interconnected. For these applications, it would seem to be necessary to divide and conquer in the sequence dimensions to obtain any significant improvement in memory requirements, and it is not immediately apparent how one would do this.
The current implementation of the algorithm is not biologically useful. It is meant only as a testbed for the algorithm. It outputs a raw traceback structure and alignment score, not a standardly formatted alignment file. Most importantly, the probability parameters for models are calculated in a very quick and simple minded fashion, and are far from being reasonable for producing robustly accurate structural alignments. The next step along this line is to produce good prior distributions for estimating better parameters, by estimating mixture Dirichlet priors from known RNA structures [57]. At this stage it would not be meaningful to compare the biological alignment accuracy of this implementation to (for instance) the excellent performance of the RAGA genetic algorithm [27]. A biologically useful implementation with accurate alignment performance is of course the eventual goal of this line of work, but is not the point of the present paper.
Acknowledgements
I gratefully acknowledge financial support from the Howard Hughes Medical Institute, NIH R01 HG01363, and Alvin Goldfarb. I also wish to thank Elena Rivas and Robin Dowell for critical comments on the manuscript.
References
- Eddy SR. Non-coding RNA genes and the modern RNA world. Nat. 2001;2:919–929. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- Erdmann VA, Barciszewska MZ, Symanski M, Hochberg A, de Groot N, Barciszewski J. The non-coding RNAs as riboregulators. Nucl. 2001;29:189–193. doi: 10.1093/nar/29.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S. Computational genomics of noncoding RNA genes. Cell. 2002;109:137–140. doi: 10.1016/s0092-8674(02)00727-4. [DOI] [PubMed] [Google Scholar]
- Dandekar T, Hentze MW. Finding the hairpin in the haystack: Searching for RNA motifs. Trends Genet. 1995;11:45–50. doi: 10.1016/S0168-9525(00)88996-9. [DOI] [PubMed] [Google Scholar]
- Laferriere A, Gautheret D, Cedergren R. An RNA pattern matching program with enhanced performance and portability. Comput. 1994;10:211–212. doi: 10.1093/bioinformatics/10.2.211. [DOI] [PubMed] [Google Scholar]
- Dsouza M, Larsen N, Overbeek R. Searching for patterns in genomic data. Trends Genet. 1997;13:497–498. doi: 10.1016/S0168-9525(97)01347-4. [DOI] [PubMed] [Google Scholar]
- Fichant GA, Burks C. Identifying potential tRNA genes in genomic DNA sequences. J. 1991;220:3659–671. doi: 10.1016/0022-2836(91)90108-i. [DOI] [PubMed] [Google Scholar]
- El Mabrouk N, Lisacek F. Very fast identification of RNA motifs in genomics DNA. Application to tRNA search in the yeast genome. J. 1996. pp. 46–55. [DOI] [PubMed]
- Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucl. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisacek F, Diaz Y, Michel F. Automatic identification of group I intron cores in genomic DNA sequences. J. 1994;235:1206–1217. doi: 10.1006/jmbi.1994.1074. [DOI] [PubMed] [Google Scholar]
- Nicoloso M, Qu LH, Michot B, Bachellerie JP. Intron-encoded, antisense small nucleolar RNAs: The characterization of nine novel species points to their direct role as guides for the 2'-O-ribose methylation of rRNAs. J. 1996;260:178–195. doi: 10.1006/jmbi.1996.0391. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. A computational screen for methylation guide snoRNAs in yeast. Science. 1998;283:1168–1171. doi: 10.1126/science.283.5405.1168. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucl. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties, and weight matrix choice. Nucl. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayhoff MO, Schwartz RM, Orcutt BC. In: Atlas of Protein Sequence and Structure. Dayhoff MO, editor. Washington DC, National Biomedical Research Foundation; 1978. A model of evolutionary change in proteins. pp. 345–352. [Google Scholar]
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Brown M, Mian IS, Sjolander K, Haussler D. Hidden Markov models in computational biology: Applications to protein modeling. J. 1994;235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths Jones S, Howe KL, Marshall M, Sonnhammer EL. The Pfam protein families database. Nucl. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Goodstadt L, Dickens NJ, Doerks T, Schultz J, Mott R, Ciccarelli F, Copley RR, Ponting CP, Bork P. Recent improvements to the SMART domain-based sequence annotation resource. Nucl. 2002;30:242–244. doi: 10.1093/nar/30.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucl. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara Y, Brown M, Hughey R, Mian IS, Sjolander K, Underwood RC, Haussler D. Stochastic context-free grammars for tRNA modeling. Nucl. 1994;22:5112–5120. doi: 10.1093/nar/22.23.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin R, Eddy SR, Krogh A, Mitchison GJ. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge UK, Cambridge University Press. 1998.
- Corpet F, Michot B. RNAlign program: Alignment of RNA sequences using both primary and secondary structures. Comput. 1994;10:389–399. doi: 10.1093/bioinformatics/10.4.389. [DOI] [PubMed] [Google Scholar]
- Brown MP. Small subunit ribosomal RNA modeling using stochastic context-free grammars. Proc. Int. Conf. on Intelligent Systems in Molecular Biology. 2000;8:57–66. [PubMed] [Google Scholar]
- Notredame C, OBrien EA, Higgins DG. RAGA: RNA sequence alignment by genetic algorithm. Nucl. 1997;25:4570–4580. doi: 10.1093/nar/25.22.4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhof HP, Reinert K, Vingron M. A polyhedral approach to RNA sequence structural alignment. RECOMB 98 ACM Press. 1998. pp. 153–162. [DOI] [PubMed]
- Lenhof HP, Reinert K, Vingron M. A polyhedral approach to RNA sequence structure alignment. J. 1998;5:517–530. doi: 10.1089/cmb.1998.5.517. [DOI] [PubMed] [Google Scholar]
- Hirschberg DS. A linear space algorithm for computing maximal common subsequences. Communications of the ACM. 1975;18:341–343. doi: 10.1145/360825.360861. [DOI] [Google Scholar]
- Myers EW, Miller W. Optimal alignments in linear space. Comput. 1988;4:11–17. doi: 10.1093/bioinformatics/4.1.11. [DOI] [PubMed] [Google Scholar]
- Shapiro BA, Zhang K. Comparing multiple RNA secondary structures using tree comparisons. Comput. 1990;6:309–318. doi: 10.1093/bioinformatics/6.4.309. [DOI] [PubMed] [Google Scholar]
- Sankoff D. Simultaneous solution of the RNA folding, alignment, and protosequence problems. SIAM J. 1985;45:810–825. [Google Scholar]
- Knudsen B, Hein J. RNA secondary structure prediction using stochastic context-free grammars and evolutionary history. Bioinformatics. 1999;15:446–454. doi: 10.1093/bioinformatics/15.6.446. [DOI] [PubMed] [Google Scholar]
- Gorodkin J, Stricklin SL, Stormo GD. Discovering common stem-loop motifs in unaligned RNA sequences. Nucl. 2001;29:2135–2144. doi: 10.1093/nar/29.10.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes I, Rubin GM. Pairwise RNA structure comparison with stochastic context-free grammars. Pac. 2002. pp. 163–174. [DOI] [PubMed]
- Mathews DH, Turner DH. Dynalign: an algorithm for finding the secondary structure common to two RNA sequences. J. 2002;317:191–203. doi: 10.1006/jmbi.2001.5351. [DOI] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Kasami T. An efficient recognition and syntax algorithm for context-free algorithms. Technical Report AFCRL-65-758 Air Force Cambridge Research Lab Bedford, Mass. 1965.
- Younger DH. Recognition and parsing of context-free languages in time n3. Information and Control. 1967;10:189–208. [Google Scholar]
- Hopcroft JE, Ullman JD. Introduction to Automata Theory, Languages, and Computation. Addison-Wesley. 1979.
- Lari K, Young SJ. The estimation of stochastic context-free grammars using the INSIDE-OUTSIDE algorithm. Computer Speech and Language. 1990;4:35–56. [Google Scholar]
- Lari K, Young SJ. Applications of stochastic context-free grammars using the INSIDE-OUTSIDE algorithm. Computer Speech and Language. 1991;5:237–257. [Google Scholar]
- Chiu DKY, Kolodziejczak T. Inferring consensus structure from nucleic acid sequences. Comput. 1991;7:347–352. doi: 10.1093/bioinformatics/7.3.347. [DOI] [PubMed] [Google Scholar]
- Gutell RR, Power A, Hertz GZ, Putz EJ, Stormo GD. Identifying constraints on the higher-order structure of RNA: Continued development and application of comparative sequence analysis methods. Nucl. 1992;20:5785–5795. doi: 10.1093/nar/20.21.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giegerich R. In: Proceedings of the 11th Annual Symposium on Combinatorial Pattern Matching. Giancarlo R, Sankoff D, editor. Vol. 1848. Montreal, Canada, Springer-Verlag, Berlin; 2000. Explaining and controlling ambiguity in dynamic programming. pp. 46–59. [Google Scholar]
- Rabiner LR. A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE. 1989;77:257–286. doi: 10.1109/5.18626. [DOI] [Google Scholar]
- Steinberg S, Misch A, Sprinzl M. Compilation of tRNA sequences and sequences of tRNA genes. Nucl. 1993;21:3011–3015. doi: 10.1093/nar/21.13.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski M, Barciszewska MZ, Erdmann VA, Barciszewski J. 5S ribosomal RNA database. Nucl. 2002;30:176–178. doi: 10.1093/nar/30.1.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen N, Zwieb C. SRP-RNA sequence alignment and secondary structure. Nucl. 1991;19:209–215. doi: 10.1093/nar/19.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JW. The ribonuclease P database. Nucl. 1999;27:314. doi: 10.1093/nar/27.1.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neefs JM, van Peer de Y, De Rijk P, Chapelle S, De Wachter R. Compilation of small ribosomal subunit RNA structures. Nucl. 1993;21:3025–3049. doi: 10.1093/nar/21.13.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rijk P, Van Peer de Y, Chapelle S, De Wachter R. Database on the structure of large ribosomal subunit RNA. Nucl. 1994;22:3495–3501. doi: 10.1093/nar/22.17.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas E, Eddy SR. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J. 1999;285:2053–2068. doi: 10.1006/jmbi.1998.2436. [DOI] [PubMed] [Google Scholar]
- Rivas E, Eddy SR. The language of RNA: A formal grammar that includes pseudoknots. Bioinformatics. 2000;16:326–333. doi: 10.1093/bioinformatics/16.4.326. [DOI] [PubMed] [Google Scholar]
- Sjolander K, Karplus K, Brown M, Hughey R, Krogh A, Mian IS, Haussler D. Dirichlet mixtures: A method for improving detection of weak but significant protein sequence homology. Comput. 1996;12:327–345. doi: 10.1093/bioinformatics/12.4.327. [DOI] [PubMed] [Google Scholar]