

Abstract
Endometrial cancer (EC) is a prevalent gynecological cancer worldwide, often associated with poor prognosis after recurrence or metastasis. Oroxylin A (OA) is an active flavonoid compound with a strong anti-tumor function. However, the effects of OA on EC remain unknown. In this study, we planned to investigate the anti-EC effects of OA and explore its mechanisms. Five cell lines were used for in vitro experiments, and female BALB/c nude mice were applied for xenograft experiments. The cytotoxicity and experimental concentration of OA were detected by CCK-8. Wound healing, transwell, and colony formation assays were used to evaluate the anti-metastatic and anti-proliferative activities of OA on EC cells. TUNEL assay and flow cytometry were applied for the evaluation of apoptosis. Network pharmacology was used to explore potential targets, and molecular dynamics simulations and dockings were applied for the quantification of binding energy, and stability of OA. RT-qPCR, WB, and immunofluorescence were applied for the detection of localization and expression of correlated markers. The results showed that OA notably inhibited the proliferation, migration, and invasion of Ishikawa cells. Meanwhile, in vivo Ishikawa xenograft assays demonstrated that OA notably inhibited growth and promoted apoptosis of EC. Mechanistically, after treatment with OA, the expressions of Cleaved Caspase-3, BAX, E-cadherin, and ERβ were increased, while the expressions of Bcl-2, Vimentin, N-cadherin, MMP2, MMP9, PI3K and phospho-AKT (Ser473) were decreased. Therefore, OA may exhibit significant anti-EC effects by regulating the ERβ/PI3K/AKT pathway to promote apoptosis and inhibit epithelial-mesenchymal transition (EMT).
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-97122-z.
Keywords: Oroxylin A, Endometrial cancer, ERβ, Apoptosis, EMT
Subject terms: Cancer therapy, Metastasis
Introduction
EC, a typical gynecological malignancy, originates from the endometrium and ranks as the sixth most prevalent cancer among the female population. Approximately 75-85% of EC patients are adenocarcinomas (endometrial cancer type I)1,2. Prevalence and incidence of EC have presented to be elevating worldwide, with the onset age of EC patients getting younger and younger3. Within EC’s early stages, the operators typically choose surgical resection as general therapy. Within stages of middle or late, only chemotherapy, surgery, radiotherapy, and immunotherapy can be applied4,5. Despite the effectiveness of surgery in the initial stages, the prognosis remains poor for recurrent, metastatic, and advanced EC cases1,6,7. Consequently, exploring the underlying mechanisms and identifying novel therapeutic targets are crucial for more effective treatment options.
Estrogen is the main female steroid hormone and plays a key role in the occurrence and development of EC. It mainly acts through two receptors: estrogen receptor α (ERα/ESR1) and estrogen receptor β (ERβ/ESR2), among which 17β-estradiol (E2) mainly acts through ERβ in the body8. ERβ generally acts as a cancer suppressor with antiproliferative properties in specific tissues, and ERβ has been shown to be decreased in a variety of cancers, comprising colon, brain, breast, endometrium, prostate, and ovary9,10. Therefore, ERβ may be considered as a potential effective molecular target for the treatment of EC. Mechanistically, some studies have revealed that ERβ exerts its anti-tumor effect by regulating the PI3K/AKT signaling pathway11,12. In addition, increased ERβ expression was associated with sensitivity to doxorubicin via inhibition of the PI3K/AKT/mTOR pathway13. Studies have shown that ERβ acts as an upstream regulator of the PI3K/AKT signaling pathway14, which regulates important pathways relating to cell proliferation, metabolism, metastasis, and angiogenesis15. Meanwhile, the PI3K/AKT pathway has been shown to be frequently dysregulated and overactive in EC16,17. Based on the above studies, the ERβ/PI3K/AKT signaling pathway may play an important role in the treatment of EC.
OA, O-methylated flavone (As shown in Fig. 1A), is a bioactive compound derived from Scutellaria baicalensis roots18, as well as in Aster himalaicus, Anchietea pyrifolia, Oroxylum indicum, and S. lateriflora, all of which hold significant therapeutic value in traditional Chinese medicinal19–21. Existing studies have demonstrated that OA possesses a diverse array of properties, including anti-thrombotic, anti-infective, anti-inflammatory, anti-oxidant, anti-depressant, anti-fibrotic, neuro-protective, liver-protective, heart-protective, bone-protective, and kidney-protective effects. These findings support the considerable potential of OA in disease treatment18,22. OA also exhibits various anti-cancer effects, such as inducing apoptosis, causing cell cycle arrest, suppressing angiogenesis, promoting cellular differentiation, inhibiting migration and invasion, and reversing drug resistance in various cancer cell types18,23–25. However, OA’s effect on EC remains unclear and needs to be further investigated. OA has been shown to regulate the PI3K/AKT signaling pathway in various diseases26,27. In addition, studies have shown that OA, as a phytoestrogen, can reduce vasoconstriction in rat aortic rings through the estrogen receptor signaling pathway28. Therefore, we hypothesized that OA might exert an anti-EC effect, which might be through regulating the ERβ/PI3K/AKT signaling pathway. In this study, we validated this hypothesis through a series of experiments, hoping to provide new therapeutic targets and possible drug options for the clinical treatment of EC.
Fig. 1.

Chemical structure of OA and its effects on cell viability in a range of cell lines. (A) Chemical structure of OA. (B) Cell viability of Ishikawa, RL95-2, HEC-1 A, HEC-1B, and HEK293 cells after treatment with OA for 48 h. (C) Viability of Ishikawa cells after exposure to OA for 24 h, 48 h, and 72 h. (D) Viability of Ishikawa cells after 5-FU treatment for 48 h.
Materials and methods
Chemicals and reagents
OA (MW: 284.263, purity ≥ 99%) was sourced from Shanghai Standard Technology Co., Ltd. (Shanghai, China). PHTPP (HY-103456) was sourced from MedChemExpress (Monmouth Junction, NJ). DMEM medium was purchased from Biosharp (Hefei, China). Primary antibodies against β-Actin (1:10000, AB8227), BAX (1:2000, AB32503), Bcl-2 (1:2000, AB182858), and Cleaved Caspase-3 (1:1000, AB32042) were purchased from Abcam (Shanghai, China). Primary antibodies for GAPDH (1:10000, 60004-1-Ig), Vimentin (1:2000, 10366-1-AP), E-Cadherin (1:2000, 20874-1-AP), N-Cadherin (1:2000, 22018-1-AP), MMP2 (1:2000, 10373-2-AP), MMP9 (1:2000, 10375-2-AP), PI3K (1:1000, 60225-1-Ig) and anti-rabbit secondary antibody (1:20000, SA00001-2) with HRP-conjugation were purchased from Proteintech (Wuhan, China). HRP-conjugated anti-mouse secondary antibody (1:20000, RS0001) and primary antibody for ERβ (1:1000, YT1637) were purchased from Immunoway (Suzhou, China). Primary antibodies for AKT (#4685), and phospho-AKT (Ser473) (#9271) were purchased from Cell Signaling Technology (MA, U.S.A.). Annexin V-FITC/PI apoptosis kit was sourced from MULTI SCIENCES (Hangzhou, China).
Cell culture
Ishikawa, RL95-2, HEC-1 A, HEC-1B and HEK293 cell lines were sourced from Wuhan Pricella Biotechnology Co., Ltd and all of them were preserved within DMEM (Biosharp, Anhui, China) with FBS supplemented (10%, Pricella, Wuhan, China) combined with Penicillin-streptomycin and trypsin (1%, NCM Biotech, Suzhou, China). Cells were kept within the environment of a humid incubator (37 ℃, 5% CO2).
Test of cell viability
Cell Counting Kit-8 (Coolaber, Beijing, China) was applied to quantify cellular viability according to professional instruction. In brief, Ishikawa cells (2000 cells/well) got plated on the plate with 96 wells, starved for 12 h in the medium with serum removed after adherence, and then co-cultured with different concentrations of OA (0-400 µM) at 37 °C and 5% CO2. At 24 h, 48 h, and 72 h, 200 µl of 10% CCK-8 working solution was supplemented in the culture plate, sending the plate for incubation within 37 °C incubator for 2 h. A microplate reader (TECAN, Switzerland) was applied for measuring absorbance (OD) at 450 nm. Quantification of cell viability referred from the equation below:
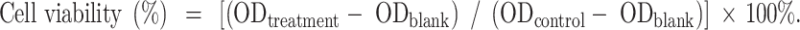 |
Formation of cell colonies
Ishikawa cells were suspended into plates with 6 wells (kept at 37 °C, 5% CO2) at 500 cells/well, starved for 12 h within the medium with serum removed after adherence, then co-cultured with OA (0 µM, 5 µM, 10 µM, and 20 µM) for an exceptional 10 days, with medium changed per three days. At the conclusion of the assay, we dropped the medium, with cells rinsed three times with PBS. The cells were then fixed with 4% paraformaldehyde lasting 20 min. Lastly, the staining of colonies was completed in 0.1% crystal violet lasting 15 min.
Wound healing test
Ishikawa cells (5 × 105 cells/well) were cultured in the plate with 6 wells and grown to approximately 80% confluence. Ishikawa cell monolayer was gently scraped with the tip of a 200 µL pipette. Cell debris was then removed with a rinse using PBS. Ishikawa cells were co-cultured with specified concentrations of OA (0 µM, 2.5 µM, 5 µM, and 10 µM). Gap images of the Ishikawa cells treated by OA were taken under the microscope (0 h and 48 h, respectively). ImageJ software was used for analysis.
Transwell test
The anti-EC cell migrating and evading capabilities of OA were examined by 24-well Transwell® chambers (membrane in 6.5 mm diameter, pore size at 8 μm, LABSELECT,
Hefei, China). The Ishikawa cells were kept starved within the medium with serum removed for 24 h before the assays.
For the migration test, upper chambers got filled with 200 µL Ishikawa cells suspensions (1.5 × 104 cells) with specified OA concentrations (0 µM, 2.5 µM, 5 µM, and 10 µM) in the medium containing 1% FBS, with lower chambers fulfilled with 650 µL corresponding medium and 20% FBS. Following 48 h of incubating operation (5% CO2, 37 °C), upper chambers were wiped by cotton swabs, while cells in lower chambers were sent for fixation in 4% paraformaldehyde lasting 20 min, as well as being stained by 0.1% crystal violet solution for 15 min, and then visualized utilizing microscope. ImageJ software was applied for counting migrated cells selected from 5 fields randomly chosen.
For the test on cell invasion, upper chambers were coated by 100 µL 10% (v: v) Matrigel® (Corning, NY, U.S.), and being solidified (1 h, 37 ℃). 650 µL of the corresponding medium was supplemented within lower chambers, with 20% FBS. 2.5 × 104 Ishikawa cells per well (200 µL) were set in the upper chambers in the corresponding medium with 1% FBS, additionally supplemented by setting certain OA concentrations (0 µM, 2.5 µM, 5 µM, and 10 µM). The remaining steps followed the same protocol as the cell migration test. ImageJ software was utilized to count invaded cells within five random fields.
Cell apoptosis detection
Annexin V-FITC/PI Apoptosis Kit was applied for apoptosis detection (MULTI SCIENCES, Hangzhou, China). First, Ishikawa cells (per well 2.5 × 104 cells) were positioned into plates with 6 wells then sent for incubation for 12 h, and then co-cultured with OA (0 µM, 5 µM, 10 µM, and 20 µM) for 48 h after being starved for 12 h in the serum-free medium. Harvesting cells by trypsin (NCM Biotech, Suzhou, China) and rinsing them twice with cold PBS. Subsequently, resuspension of cells (1 × 105) with binding buffer 500 µL, then sending for incubation containing Annexin V-FITC 5 µL, PI 10 µL for 15 min (room temperature, light-shading). Consequently, the apoptosis level was immediately evaluated through flow cytometry (BD, USA).
Network pharmacology analysis and molecular docking
In this study, the mechanism of OA in the treatment of EC was studied based on network pharmacology. Initially, 100 genes relevant to OA were integrated from the TCMSP database (https://www.tcmsp-e.com/#/database), while 4978 genes associated with EC were obtained from the GeneCards database (https://www.genecards.org/). To explore the potential targets of OA in EC, an intersection analysis between OA targets and EC targets was performed and a Venn diagram was generated via the Venny 2.1.0 online platform (https://bioinfogp.cnb.csic.es/tools/venny/index.html). Subsequently, the intersecting markers of OA and EC were chosen. Cytoscape 3.7.0 and STRING network database (https://cn.string-db.org/) were applied to establish a network of protein-protein interaction (PPI). The degree value of each target was calculated according to the Network Analysis plugin. Key targets were identified based on their degree values. The KEGG enrichment analysis of potential OA targets for EC treatment was performed in the DAVID database (https://david.ncifcrf.gov/). The Benjamini-Hochberg procedure was used to reduce the bias introduced by multiple testing. The results of the KEGG analysis were visualised using an online tool (https://www.biofoematics.com.cn/).
ERβ 3D structure was obtained in Protein Data Bank (PDB ID: 8BZW), (PDB ID: 7XVY), meanwhile OA structure was sourced from PubChem. PyMoL-2.5.0 was applied to remove molecules of water, as well as other unnecessary ligands (Schrodinger, USA). AutoDockTools-1.5.6 was then employed to transform proteins and ligands into the format of pdbqt file (The Scripps Research Institute, CA, USA), calculate Gasteiger charges, and define the binding pocket for facilitated hydrogenation. Next, AutoDock Vina-1.1.2 was used to process the results of molecular docking (The Scripps Research Institute, CA, USA). Docking conformations were analyzed utilizing PyMOL to visualize the interactions. Binding activity and strength between the key targets and ligands were assessed based on the docking score. Structure with the lowest binding energy was selected as the optimal model, where binding energy below 5 suggested a strong affinity between ligand and receptor.
Molecular dynamics simulations
To better understand the dynamic behavior of OA with ERβ, simulations of molecular dynamics were conducted utilizing the GROMACS package (version 2022.3). For preprocessing the small molecules, AmberTools22 was utilized to apply the GAFF force field, while Gaussian 16 W was employed to introduce hydrogen atoms and calculate the RESP potential. This potential data was incorporated into the topology file of the molecular dynamics system. The simulations were performed under static conditions at a temperature of 300 K and atmospheric pressure (1 Bar). The Amber99sb-ildn force field was used, with water molecules modeled using the TIP3P model, and the total charge of the system was neutralized by adding six Na + ions29. The simulation system utilized the steepest descent method for energy minimization, followed by isothermal-isovolumetric (NVT) and isothermal-isobaric (NPT) equilibrations, each lasting for 100,000 steps with a coupling constant of 0.1 ps and a duration of 100 ps. Subsequently, a free molecular dynamics simulation was conducted over 5,000,000 steps, with a time step of 2 fs, resulting in a total simulation duration of 100 ns. Upon completion of the simulation, the software’s built-in tools were employed to analyze the trajectory, focusing on root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF).
RTqPCR
Ishikawa cells were suspended to plates with 6 wells at 2 × 104 cells/well and sent for incubation for 12 h and then co-cultured with OA (0 µM, 5 µM, 10 µM, and 20 µM) for 48 h after being starved for 12 h in the serum-free medium. Then cells got harvested. TRIzol (Invitrogen) was taken for usage in the extraction of total RNA according to professional instructions. Reactions were repeatedly conducted 3 times, utilizing GAPDH as a reference, and primers utilized in this experiment are detailed in Table 1. RT-qPCR cycling parameters were as follows 95 ℃ for 10 s, followed by 95 ℃ in 40 cycles, each for 30 s, 55 ℃ 30 s, 72 ℃ 30 s. Relative mRNA expressions were determined by the comparative Ct (∆∆Ct) method.
Table 1.
Primers used for RT-qPCR.
| Gene | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| GAPDH-Homo | GGAAGCTTGTCATCAATGGAAATC | TGATGACCCTTTTGGCTCCC |
| ERβ- Homo | GGTCCATCGCCAGTTATCACAT | AGTGAGCATCCCTCTTTGAACC |
Western blot
Ishikawa cells were suspended to 10 cm plates at 8 × 104 cells/well and incubated for 12 h, then co-cultured with OA (0 µM, 5 µM, 10 µM, and 20 µM) for 48 h after being starved for 12 h in the serum-free medium. The harvested cells were lysed utilizing enhanced RIPA buffer (BOSTER, Wuhan, China) with the addition of phosphatase and protease inhibitors (Servicebio, Wuhan, China) in the ice bath for 20 min. After sonication (3 min) and centrifugation (11000 rpm, 10 min) to collect the supernatant. Protein concentration was quantified by bicinchoninic acid (BCA) assay (Coolaber, Beijing, China). Identical protein amounts were then isolated by SDS-PAGE, and transferred to PVDF membranes, which were subsequently blocked by nonfat milk (5%, 2 h, room temperature). Following washing with TBST, PVDF membranes were sent for incubation with specific primary antibodies overnight at 4 °C. Subsequently, PVDF membranes were exposed to secondary antibodies with horseradish peroxidase (HRP)-conjugation (2 h, room temperature) after washing with TBST. Protein bands were visualized via ChemiDoc XRS + imaging system (Bio-Rad Laboratories, USA), and the resulting images were analyzed using ImageJ software.
Xenograft tumor experiment
6 weeks old 24 female BALB/c nude mice were sourced from Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (Lanzhou, China) and maintained on a 12 h/12 h cycle of light/dark under low stress (50% humidity, low noise, 22 °C). Water/food was freely accessible to the mice. Suspensions of 2 × 106 Ishikawa cells in 100 µL of serum-free PBS were sent for subcutaneous injection to each nude mouse’s right flank. One week later, referring from ranges of body weight and tumor volume, mice loaded with observable tumorous masses were sent for random assignment to four cohorts (n = 6). Mice received treatment of OA (20, 40, 80 mg/kg) intraperitoneal administration every day for 14 days. In contrast, the control group received an equivalent volume of vehicles. Body weight was recorded every 3 days before and after treatment. Tumor sizes were taken measurement every 3 days under a caliper, while volume calculation referred to a standard equation: V = (length × width2)/2. After 14 days, mice were humanely sacrificed after anesthesia with sodium pentobarbital, and the tumors were precisely harvested, measured for weight, and immediately stored at -80 °C for subsequent analysis. Inhibition rate (IR) was computed using the equation: IR (%) = (1 - mean tumorous mass of the treated group/mean tumorous mass of control group) × 100%30. All animal studies received approval from the First Hospital of Lanzhou University Ethical Committee on Experimental Animal Care and Use (No. LDYYLL2024-491) and were conducted in accordance with ARRIVE guidelines. For euthanasia, cervical dislocation was performed after anesthesia with sodium pentobarbital (6 mg/mL) administered intraperitoneally at a dose of 150 µL.
Immunofluorescence (IF)
Ishikawa cells were suspended in plates with 6 wells at 2 × 104 cells/well and incubated for 12 h and then co-cultured with OA (0 µM, 5 µM, 10 µM, and 20 µM) for 48 h after being starved for 12 h in the medium with serum removed. After being washed twice with PBS, cells were immobilized using 4% paraformaldehyde (20 min), being permeabilized in 0.1% Triton X-100 (Servicebio, Wuhan, China) solution for 10 min. The cells were then put in blockage using 5% BSA (2 h, room temperature) and sent for incubation with primary antibody targeted ERβ (1:200 dilution, Immunoway, YT1637, overnight, 4°C). Cells were sent for three times PBS rinses and incubation in the secondary antibody solution, which was conjugated with fluorophores (room temperature, 1 h). After three PBS rinses, products were DAPI-stained (Servicebio, Wuhan, China) for 15 min. Images were captured utilizing an Olympus microscope, and the quantitative analysis was completed utilizing ImageJ.
Tumor tissues were preserved in 4% paraformaldehyde and sent for incubation of the primary antibody targeted ERβ (1:200 dilution, Immunoway, YT1637) at 4 °C overnight. Sections were then exposed to the fluorophore-conjugated secondary antibody. The method for capturing images was performed as previously described.
TUNEL assay
Sections of tissue were 4% paraformaldehyde-fixed for 30 min, following a 0.3% Triton X-100 (Servicebio, Wuhan, China) permeabilization lasting 20 min. After being rinsed by PBS, sections received TUNEL reagent (Servicebio, Wuhan, China) exposure (1 h, 37 °C, light-shading) and DAPI-stained (Servicebio, Wuhan, China). Apoptotic events were counted in five random fields. The method for capturing images was performed as previously described.
Statistical analysis
All experimental results were presented: mean ± standard deviation (SD). The one- or two-way analysis of variance (ANOVA) was applied for statistical comparisons. Statistical analyses were performed in GraphPad Prism 9.5 Software. Significance standard: P<0.05.
Results
OA exhibited a more sensitive cytotoxic effect on Ishikawa cells
As shown in Fig. 1B, the half inhibitory concentration of OA against EC cell Ishikawa was 14.39 µM, which is significantly more sensitive than the other three EC cell lines RL95-2 (51.02 µM), HEC-1 A (179 µM), and HEC-1B (303.1 µM). Simultaneously, it showed lower cytotoxicity in normal human embryonic kidney cells HEK293 (255.8 µM). OA suppressed viability and proliferation of Ishikawa cells time-dependently and dose-dependently (Fig. 1B–C). Additionally, OA showed a stronger suppressive impact on Ishikawa cells compared to 5-FU (32.44 µM) after 48 h of treatment (Fig. 1D). According to the above results, Ishikawa cells were used in subsequent experiments, and the highest concentration of cell co-culture was set to 20 µM, except for the Wound healing and transwell tests, which had a maximum concentration of 10 µM, because it was considered that too high a concentration would directly kill the cells and affect the results.
OA significantly inhibited the proliferation, migration, and invasion of Ishikawa cells
We set the concentration gradient of 0 µM, 5 µM, 10 µM, and 20 µM according to the previous results to conduct colony formation experiments. The results showed that OA had a significant inhibitory effect on the colony formation of Ishikawa cells, and the effect was most obvious at 20 µM (Fig. 2A–B). As Fig. 2C–G showed, we selected 0 µM, 2.5 µM, 5 µM, and 10 µM concentrations for testing to minimize the effects of toxicity on migration and invasion. The results of transwell migration, invasion and wound healing assays showed that OA significantly inhibited the invasion and migration of Ishikawa cells, and the inhibition might be dose-dependent.
Fig. 2.

Inhibitory effects of OA on the proliferation, migration, and invasion of Ishikawa cells. (A) Cell colony formation and (B) the analysis of colony formation number. (C) Wound healing assay (Scale bar is 100 μm) and (D) the quantification of the wound healing. (E) Transwell migration and invasion assay (Scale bar is 100 μm), and the quantification of (F) migration cells and (G) invasion cells. *p < 0.05, **p < 0.01, ***p < 0.001 vs. OA 0 µM group.
OA can promote apoptosis and inhibit EMT of Ishikawa cells
As illustrated by Fig. 3A, compared with the control group, the Ishikawa cells treated with OA had a shrunken morphology, less adherence to the wall, and a significant decrease in the number of cells, which was most obvious in cells treated with 20 µM. AnnexinV/PI staining results showed that apoptotic cell proportion elevated significantly after OA treatment, which was 6.99%, 17.8%, and 38.8% at 5 µM, 10 µM, and 20 µM, respectively (Fig. 3B and C).
Fig. 3.

Effects of OA on apoptosis induction and EMT inhibition in Ishikawa cells. (A) Morphological changes of Ishikawa cells after OA intervention (Scale bar is 100 μm). (B–C) Evaluation of apoptosis rate and the quantification of apoptosis in Ishikawa cells after treatment with OA for 48 h. (D–G) Effect of OA on the expression of apoptosis-related proteins Bcl2 (E), Bax (F), and Cleaved Caspase3 (G). (H–M) Effects of different concentrations of OA on the expressions of EMT-related proteins N-Cadherin (I), E-Cadherin (J), MMP9 (K), MMP2 (L), and Vimentin (M). *p < 0.05, **p < 0.01, ***p < 0.001 vs. OA 0 μM group. Late apoptosis: #p < 0.05, ##p < 0.01, ###p < 0.001 vs. OA 0 µM group.
Western Blot outcomes demonstrated that following OA treatment, apoptosis-associated proteins Cleaved Caspase-3 and BAX expressions elevated, with Bcl-2 expression dropping (Fig. 3D, E and F, and 3G). In addition, EMT-related protein, E-cadherin expression increased, while Vimentin and N-cadherin expressions decreased as well as MMP2 and MMP9 (Fig. 3H, I, J and K, and 3M). These results suggested that OA may induce apoptosis and inhibite EMT in EC.
ERβ may be a possible target for OA to exert its anti-EC effect
The network pharmacology analysis result stated that the intersection of OA and EC targets comprised 68 factors (Fig. 4A). The PPI network constructed using STRING network and Cytoscape 3.7.0 (Fig. 4B) showed that ESR2 (ERβ) was located in a relatively central position. Combination with KEGG analysis in the DAVID database (Fig. 4F), it was suggested that the estrogen signaling pathway was closely related to OA. Outcomes of molecular docking stated that the binding score of ERβ with OA was 9.1 (Fig. 4C), indicating that OA had a good affinity for ERβ, so we speculated that OA function potentially relied on ERβ regulation. In addition, RMSD of OA and ERβ finally stabilized at 0.24–0.26 nm (Fig. 4D), indicating that the complex was tightly bound and relatively stable. RMSF indicates individual amino acid residue’s flexibility within the protein. The results demonstrated that RMSF values for residues within 305–315, 330–345, and 430-440 ps regions exhibited minimal fluctuations (Fig. 4E). Taken together with the above results, ERβ can be considered as a possible target for OA to exert its anti-EC effect.
Fig. 4.

Target screening of OA for its anti-EC effect. (A) Cross-protein targets of OA and EC. (B) Protein–protein interaction (PPI) network of 68 proteins. (C) Optimal docking mode of OA and ERβ. (D–E) Molecular dynamics simulation between OA and ERβ, RMSD (D), RMSF (E), (chain A: protein, chain B: ligand). (F) KEGG pathway analysis of 68 proteins.
OA may exert its anti-EC effect by regulating the ERβ/PI3K/AKT pathway
As shown in Fig. 5, after OA treatment, mRNA and protein expression levels of ERβ were upregulated dose-dependently (Fig. 5A and B, and 5C), and the same results were also shown by immunofluorescence (Fig. 5J and K). In addition, no significant changes in the expression of AKT were detected, while the expressions of PI3K and p-AKT (Ser473) were downregulated with increasing OA concentrations (Fig. 5B, D and E, and 5F). To further confirm ERβ’s function in response to Ishikawa cells to OA, Ishikawa cells were processed with OA supplemented with PHTPP, a selective ERβ antagonist. The results showed that PHTPP could indeed attenuate the inhibitory effect of OA on Ishikawa cell proliferation and reduce the expression of ERβ. (Figure 5G and H, and 5I).
Fig. 5.

OA exerted anti-EC effects by regulating the ERβ/PI3K/AKT pathway. (A) ERβ mRNA expression level. (B) Protein expression levels of ERβ, AKT, p-AKT(Ser473) and PI3K. (C) Relative protein expression of ERβ. (D) Relative protein expression of AKT. (E) Relative protein expression of p-AKT(Ser473). (F) Relative protein expression of PI3K. (G) CCK-8 used for Ishikawa cells rescue experiment showed that OA inhibited cell proliferation, but the mixture of OA and PHTPP reversed this effect. (H) Protein expression level of ERβ when treated with DMSO, OA (10 µM), PHTPP(1 pM) and OA (10 µM) + PHTPP(1 pM). (I) Relative protein expression of ERβ when treated with DMSO, OA (10 µM), PHTPP(1 pM) and OA (10 µM) + PHTPP(1 pM). (J) Immunofluorescence analysis of subcellular localization and protein expression level of ERβ (red). Cell nuclei were stained with DAPI (blue) (Scale is 20 μm). (K) Average fluorescence of ERβ.
*p < 0.05, **p < 0.01, ***p < 0.001 vs. OA 0 µM group.
OA can inhibit subcutaneous tumor growth in Ishikawa xenograft mice
Figure 6A shows the subcutaneous tumor implantation and drug administration schedule. Observations revealed no adverse reactions or notable variations in body weight among the OA-treated and vehicle-treated cohorts, suggesting that OA did not exhibit noticeable toxicity to the mice at therapeutic doses (Fig. 6C). Compared with the vehicle, intraperitoneal administration of OA treatment reduced tumor volume and inhibited tumor growth in the mouse xenograft model (Fig. 6B and E). In fact, the subcutaneous tumors of one mouse in the 40 mg/kg group and of two mice in the 80 mg/kg group almost disappeared. The tumor weight IRs of the 20, 40, and 80 mg/kg OA-treated groups were 45.1%, 56.76%, and 72.97%, respectively (Fig. 6D and F). To evaluate in situ cell apoptosis, TUNEL tests were conducted on tumor tissues. Findings indicated that TUNEL-positive Ishikawa cell quantity in tumors from OA-treated mice was markedly greater compared to that in control mice (Fig. 6G). These results indicated that OA had good efficacy against EC in vivo and no apparent toxicity.
Fig. 6.

OA inhibited tumor growth and induced apoptosis in Ishikawa xenograft mice. (A) Schematic diagram of xenograft tumors and drug administration. (B) Photos of Ishikawa xenograft tumors in all mice at the end of the experiment. (C) Changes in mouse body weight during the experimental period. (D) Tumor weight of mice after OA treatment. (E) Tumor volume of mice after OA treatment. (F) Tumor growth inhibition rate after OA treatment. (G) Representative images of ERβ and TUNEL immunofluorescence staining in tumor tissues of Ishikawa xenograft mice after OA treatment (Scale bar is 10 μm). *p < 0.05, **p < 0.01, ***p < 0.001 vs. OA 0 µM group.
Discussion
EC is a heterogeneous disease comprising both ER-positive (ER+) and ER-negative (ER-) cancer cells. In this study, ER+ cell lines Ishikawa, RL95-2, and ER- cell lines HEC-1 A and HEC-1B were used to study the cytotoxic effects of OA on different subtypes of EC cells. As 5-FU is one of the most commonly used chemotherapeutic agents for EC, we chose 5-FU as a positive control drug. The results showed that the in vitro cytotoxic effects of OA on ER+ cell lines Ishikawa and RL95-2 were stronger than those on ER- cell lines HEC-1 A and HEC-1B, and the effect was better than 5-FU at 48 h. At the same time, OA had less toxicity to normal HEK293 cell lines. Therefore, we speculate that OA may be a valuable inhibitor for the prevention and treatment of ER + EC.
The proliferation, migration, and invasion abilities of cancer cells are believed to be crucial for their metastatic and developmental potentials. Compounds that can effectively inhibit these processes would be promising drug candidates for cancer treatment. The results of clone formation test, wound healing test, and transwell test showed that OA could significantly inhibit the proliferation, migration, and invasion ability of Ishikawa cells, and this inhibitory effect was enhanced in a dose-dependent manner.
To further verify the ability of OA to inhibit the proliferation, migration, and metastasis of cancer cells, we performed apoptosis- and EMT-related assays. The results showed that OA significantly increased the apoptosis rate of EC cells in a dose-dependent manner. Since BAX and Bcl-2 play crucial roles in apoptosis modulation, where BAX promotes apoptosis and Bcl-2 inhibits apoptosis31, we assessed the impact of OA on the expression levels of BAX and Bcl-2 using Western Blot analysis. The finding demonstrated that OA significantly increased BAX expression while reducing Bcl-2 levels. It has been established that caspases are crucial mediators in cell apoptosis31. Consequently, we examined the impact of OA on Cleaved Caspase-3, the executor of apoptosis, and found that OA significantly elevated its expression. These results indicated that OA indeed induced apoptosis in EC cells. EMT is a biological process that allows cancer cells to gain invasive and metastatic capabilities32. Our results revealed that OA could elevate E-cadherin, and decrease Vimentin and N-cadherin expressions, which may demonstrate that OA blocked the in vitro metastatic capacity of EC cells by inhibiting EMT. Additionally, we also measured down-regulated MMP2 and MMP9 expressions, the crucial enzymes involved in type IV collagen degradation, and served as potential biomarkers for migrating and invading aggressive tumors33. These results demonstrated that OA suppressed the migration and metastasis ability of EC cells by inhibiting EMT.
To analyze the possible targets of OA’s anti-EC effects, we constructed a "component-target-pathway" model using network pharmacology methods. Combined with the KEGG analysis results in the DAVID database, it was suggested that the estrogen signaling pathway was closely related to OA. Among them, ERβ was identified as the most core target. Our multiple experiments also confirmed that OA indeed exerted its anti-EC effect through ERβ. As mentioned in the introduction, the PI3K/AKT pathway is frequently dysregulated and overactive in EC16,17, and previous studies have shown that ERβ exerted its anticancer effects by regulating the PI3K/AKT pathway11,34,35. Thus, we investigated if OA regulated EC progression via the ERβ/PI3K/AKT pathway. Our findings indicated that OA treatment significantly increased ERβ expression while markedly decreasing the expression of PI3K and phospho-AKT (Ser473). At the same time, after the application of ERβ inhibitors, the above changes were weakened, further confirming that OA exerted its anti-EC effects through the regulation of the ERβ/PI3K/AKT pathway. Furthermore, we performed xenograft tumors to verify the effects of OA on EC in vivo. As expected, OA inhibited EC tumor growth, and apoptosis of tumor cells was also observed. Regarding the possible mechanisms of the OA-induced increase in ERβ expression, we speculated as follows: First, OA may increase the production of ERβ mRNA by binding to specific sequences in the ERβ gene promoter region, thereby initiating gene transcription and providing more templates for protein synthesis, and second, OA may inhibit the degradation of ERβ molecules by binding to ERβ, thus increasing its expression. In the future, more experiments may be needed to investigate potential mechanisms.
In conclusion, our research suggested that OA could serve as a promising new therapeutic option for ER-positive EC, potentially exerting its anti-tumor effects through the modulation of the ERβ/PI3K/AKT pathway (Fig. 7). Moreover, consistent with OA’s reputation for low toxicity36,37, no significant systemic or tissue toxicity was observed in the dose range applied in this study. These findings may pave the way for the development of innovative hormonal and molecular targeted therapies for EC. Additionally, our results will enhance the clinical application of phytoestrogens in medical practice. However, this study did not explore the effects and mechanisms of OA on ERα, which may require further investigation.
Fig. 7.

Potential mechanisms of action of OA in EC (BioRender.com is used for the creation of Fig. 7).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Author contributions
X.F.: Conceptualization, Methodology, Investigation, Writing-original draft. L.W.: Methodology, Investigation, Writing—review & editing. T.C.: Investigation. J.T.: Investigation. J.T.: Investigation. S.T.: Investigation. W.L.: Supervision. F.T.: Funding acquisition. Y.W.: Conceptualization, Funding acquisition, Project administration, Writing—review & editing.
Funding
This work was supported by the Science and Technology Program of Gansu Province (grant 24JRRA910, 24JRRA699, 24JRRL003).
Data availability
Data will be available from the corresponding author on reasonable request.
Declarations
Competing interests
The authors declare no competing interests.
Ethical approval
We promise that all animal studies received approval from First Hospital of Lanzhou University Ethical Committee on Experimental Animal Care and Use (No. LDYYLL2024-491) and were conducted in accordance with ARRIVE guidelines. The mice were humanely sacrificed after anesthesia with sodium pentobarbital.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xue Fan and Luming Wu authors contributed equally to this work.
References
- 1.Crosbie, E. J. et al. Endometrial cancer. Lancet399, 1412–1428. 10.1016/s0140-6736(22)00323-3 (2022). [DOI] [PubMed] [Google Scholar]
- 2.Emons, G. & Gründker, C. The role of gonadotropin-releasing hormone (GnRH) in endometrial cancer. Cells1010.3390/cells10020292 (2021). [DOI] [PMC free article] [PubMed]
- 3.He, S., Fang, X., Xia, X., Hou, T. & Zhang, T. Targeting CDK9: A novel biomarker in the treatment of endometrial cancer. Oncol. Rep.10.3892/or.2020.7746 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu, M. et al. Adjuvant chemotherapy versus radiotherapy in high-risk, early-stage endometrioid endometrial carcinoma. Curr. Med. Sci.42, 185–191. 10.1007/s11596-021-2437-8 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Yao, L., Chen, S. & Li, W. Fatostatin inhibits the development of endometrial carcinoma in endometrial carcinoma cells and a xenograft model by targeting lipid metabolism. Arch. Biochem. Biophys.684, 108327. 10.1016/j.abb.2020.108327 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Mitamura, T., Dong, P., Ihira, K., Kudo, M. & Watari, H. Molecular-targeted therapies and precision medicine for endometrial cancer. Jpn J. Clin. Oncol.49, 108–120. 10.1093/jjco/hyy159 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Wang, J., Song, T., Zhou, S. & Kong, X. YAP promotes the malignancy of endometrial cancer cells via regulation of IL-6 and IL-11. Mol. Med.2510.1186/s10020-019-0103-4 (2019). [DOI] [PMC free article] [PubMed]
- 8.Qiao, D. et al. Estradiol mediates the interaction of LINC01541 and miR-429 to promote angiogenesis of G1/G2 endometrioid adenocarcinoma in-vitro: A pilot study. Front. Oncol.1210.3389/fonc.2022.951573 (2022). [DOI] [PMC free article] [PubMed]
- 9.GY, R. et al. Pharmacological Inhibition of KDM1A LSD1 enhances Estrogen receptor beta-mediated tumor suppression in ovarian cancer. Cancer Lett.57510.1016/j.canlet.2023.216383 (2023). [DOI] [PMC free article] [PubMed]
- 10.Hojnik, M. et al. The Co-Expression of Estrogen receptors ERα, ERβ, and GPER in endometrial cancer. Int. J. Mol. Sci.2410.3390/ijms24033009 (2023). [DOI] [PMC free article] [PubMed]
- 11.Yang, M., Liu, B., Jin, L., Tao, H. & Yang, Z. Estrogen receptor Β exhibited anti-tumor effects on osteosarcoma cells by regulating integrin, IAP, NF-kB/BCL-2 and PI3K/Akt signal pathway. J. Bone Oncol.9, 15–20. 10.1016/j.jbo.2017.09.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marzagalli, M. et al. Estrogen receptor Β in melanoma: From molecular insights to potential clinical utility. Front. Endocrinol.7, 140. 10.3389/fendo.2016.00140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lei, S. et al. Elevated Estrogen receptor Β expression in triple negative breast cancer cells is associated with sensitivity to doxorubicin by inhibiting the PI3K/AKT/mTOR signaling pathway. Exp. Ther. Med.20, 1630–1636. 10.3892/etm.2020.8809 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu, Y. et al. Cisplatin-activated ERβ/DCAF8 positive feedback loop induces chemoresistance in non-small cell lung cancer via PTEN/Akt axis. Drug Resist. Updates7110.1016/j.drup.2023.101014 (2023). [DOI] [PubMed]
- 15.He, Y. et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal. Transduct. Target. Therapy610.1038/s41392-021-00828-5 (2021).
- 16.Dong, L., Du, M. & Lv, Q. Picropodophyllin inhibits type I endometrial cancer cell proliferation via disruption of the PI3K/Akt pathway. Acta Biochim. Biophys. Sin.51, 753–760. 10.1093/abbs/gmz055 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Lee, I. I. & Kim, J. J. Influence of AKT on progesterone action in endometrial diseases. Biol. Reprod.9110.1095/biolreprod.114.119255 (2014). [DOI] [PMC free article] [PubMed]
- 18.Tuli, H. S. et al. Anticancer potential of oroxylin A: from mechanistic insight to synergistic perspectives. Naunyn. Schmiedebergs Arch. Pharmacol.396, 191–212. 10.1007/s00210-022-02298-0 (2022). [DOI] [PubMed] [Google Scholar]
- 19.Shah, R. C., Mehta, C. R. & Wheeler, T. S. 131. The constitution of oroxylin-A, a yellow colouring matter from the root-bark of Oroxylum indicum, vent. J. Chem. Soc. (Resumed) (1936). 10.1039/jr9360000591
- 20.Li, H. B. & Chen, F. Isolation and purification of Baicalein, Wogonin and oroxylin A from the medicinal plant scutellaria baicalensis by high-speed counter-current chromatography. J. Chromatogr. A. 1074, 107–110. 10.1016/j.chroma.2005.03.088 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Jongen, V. et al. Expression of estrogen receptor-alpha and -beta and progesterone receptor-A and -B in a large cohort of patients with endometrioid endometrial cancer. Gynecol. Oncol.112, 537–542. 10.1016/j.ygyno.2008.10.032 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Sajeev, A. et al. Oroxylin A: A promising flavonoid for prevention and treatment of chronic diseases. Biomolecules1210.3390/biom12091185 (2022). [DOI] [PMC free article] [PubMed]
- 23.Hui, H. et al. Oroxylin A has therapeutic potential in acute myelogenous leukemia by dual effects targeting PPARγ and RXRα. Int. J. Cancer. 134, 1195–1206. 10.1002/ijc.28435 (2013). [DOI] [PubMed] [Google Scholar]
- 24.H, W. et al. An Estrogen receptor dependent mechanism of oroxylin A in the repression of inflammatory response. PLoS One810.1371/journal.pone.0069555 (2013). [DOI] [PMC free article] [PubMed]
- 25.Qu, J., Liu, F., Zhang, X., Wang, J. & Oroxylin A reduces vasoconstriction in rat aortic rings through promoting NO production and NOS protein expression via estrogen receptor signal pathway. Evid.-Based Complement. Altern. Med. 1–6 (2020). (2020). 10.1155/2020/9257950 [DOI] [PMC free article] [PubMed]
- 26.Shen, M. et al. ROS-dependent Inhibition of the PI3K/Akt/mTOR signaling is required for oroxylin A to exert anti-inflammatory activity in liver fibrosis. Int. Immunopharmacol.8510.1016/j.intimp.2020.106637 (2020). [DOI] [PubMed]
- 27.Cao, H. J. et al. A mixture of baicalein, wogonin, and oroxylin-A inhibits EMT in the A549 cell line via the PI3K/AKT-TWIST1-glycolysis pathway. Front. Pharmacol.12, 821485. 10.3389/fphar.2021.821485 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qu, J. et al. A Reduces vasoconstriction in rat aortic rings through promoting NO production and NOS protein expression via estrogen receptor signal pathway. Evid.-Based Complement. Altern. Med.10.1155/2020/9257950 (2020). [DOI] [PMC free article] [PubMed]
- 29.Wu, T. et al. Identification of novel PD-1/PD-L1 small molecule inhibitors: virtual screening, synthesis and in vitro characterisation. J. Enzyme Inhib. Med. Chem.3910.1080/14756366.2024.2353711 (2024). [DOI] [PMC free article] [PubMed]
- 30.Ruan, G. Y. et al. An integrated approach of network pharmacology, molecular docking, and experimental verification uncovers Kaempferol as the effective modulator of HSD17B1 for treatment of endometrial cancer. J. Transl. Med.20410.1186/s12967-023-04048-z (2023). [DOI] [PMC free article] [PubMed]
- 31.D, T. The molecular machinery of regulated cell death. Cell Res.2910.1038/s41422-019-0164-5 (2019). [DOI] [PMC free article] [PubMed]
- 32.TX, H. et al. Oroxylin A inhibits the migration of hepatocellular carcinoma cells by inducing NAG-1 expression. Acta Pharmacol. Sin.4310.1038/s41401-021-00695-4 (2022). [DOI] [PMC free article] [PubMed]
- 33.Tang, T. T. et al. Saikosaponin D exerts cytotoxicity on human endometrial cancer Ishikawa cells by inducing apoptosis and inhibiting metastasis through MAPK pathways. Food Chem. Toxicol.17710.1016/j.fct.2023.113815 (2023). [DOI] [PubMed]
- 34.Lindberg, K., Helguero, L. A., Omoto, Y., Gustafsson, J. Å. & Haldosén, L. A. Estrogen receptor Β represses Akt signaling in breast cancer cells via downregulation of HER2/HER3 and upregulation of PTEN: Implications for tamoxifen sensitivity. Breast Cancer Res.1310.1186/bcr2865 (2011). [DOI] [PMC free article] [PubMed]
- 35.Li, W. et al. Involvement of estrogen receptor Β5 in the progression of glioma. Brain Res.1503, 97–107. 10.1016/j.brainres.2013.02.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao, J. Y. et al. Novel CDK9 inhibitor oroxylin A promotes wild-type P53 stability and prevents hepatocellular carcinoma progression by disrupting both MDM2 and SIRT1 signaling. Acta Pharmacol. Sin.43, 1033–1045. 10.1038/s41401-021-00708-2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei, L. et al. Oroxylin A activates PKM1/HNF4 alpha to induce hepatoma differentiation and block cancer progression. Cell Death Dis.8, e2944. 10.1038/cddis.2017.335 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be available from the corresponding author on reasonable request.