

Abstract
Background
CDG2L (MIM#614576) is an autosomal recessive multisystemic disorder due to variants in COG6 gene. Postnatal phenotypes are now well described, while prenatal presentations remain poorly investigated. Only 8 of the 28 published patients have had prenatal ultrasound anomalies reported and no one post‐mortem investigation.
Methods
We used whole‐exome sequencing in a consanguineous Turkish family with four siblings presenting with Pierre Robin sequence, arthrogryposis, heart malformation, splenomegaly, hydrocephaly, corpus callosum dysgenesis, brainstem, and cerebellar hypoplasia.
Results
We identified a novel homozygous pathogenic variant in exon 9 of COG6 (NM_020751.2): c.821del, p.(Arg274Lysfs*32). In this family, our post‐mortem study led us to describe further the prenatal phenotype of CDG2L. In addition, it permits correlating the most relevant anomalies to a maldevelopmental cascade due to a neurodegenerative process of metabolic origin, affecting the entire central nervous system including the spinal cord.
Conclusion
In this context of recurrence of multisystemic disease diagnosed antenatally, exome sequencing is powerful to give a precise diagnosis and allows proposing a molecular prenatal diagnosis at the following pregnancy.
Keywords: arthrogryposis, congenital disorders of glycosylation, corpus callosum, exome sequencing, prenatal ultrasound
1. INTRODUCTION
Congenital disorders of glycosylation (CDGs) are a rare group of metabolic diseases that can affect multiple organ systems (Francisco et al., 2019; Haijes et al., 2020). They are due to deficiencies in the synthesis, attachment, or modification of asparagine (N)‐linked glycans or oligosaccharides on glycoproteins (Péanne et al., 2018). Disorders of N‐linked protein glycosylation can be broadly subdivided into two types. CDG type I, is caused by defects in endoplasmic reticulum‐related glycosylation processes comprising deficiency in the assembly of the dolichol lipid‐linked oligosaccharide chain and its transfer to the nascent protein. CDG type II is due to failing in trimming and processing of the protein bound‐glycans either late in the endoplasmic reticulum or the Golgi compartments.
A sub‐group of CDG type II is due to defects in the conserved oligomeric Golgi complex (COG) (Blackburn et al., 2019), a multiprotein complex involved in intracellular transport and glycoprotein modification composed of eight subunits. CDG2L (MIM#614576) is an autosomal recessive multisystem disorder due to variants in COG6 (OMIM 606977). To date, 28 patients with CDG2L have been described (Alsubhi et al., 2017; Althonaian et al., 2018; Cirnigliaro et al., 2022; Huybrechts et al., 2012; Komlosi et al., 2020; Li et al., 2019; Lübbehusen et al., 2010; Lugli et al., 2021, 2022; Mandel et al., 2020; Rymen et al., 2015; Shaheen et al., 2013; Ververi et al., 2022; Zhao et al., 2021) However, only 8 of these 28 patients had prenatal ultrasound (US) findings (Komlosi et al., 2020; Mandel et al., 2020; Rymen et al., 2015). Here, we report a novel family with recurrence of CDG2L in 4 siblings, including three fetal siblings, diagnosed by exome sequencing. We describe the prenatal features with the clinicopathological findings and compare them to the previously reported patients to better delineate the antenatal presentation.
2. MATERIALS AND METHODS
Ethical Compliance: The genetic analysis and autopsy were performed as part of routine clinical diagnosis, with signed parental consent for both, in accordance with the French law. After genetic counseling, whole‐exome sequencing was performed with parental consent in two affected siblings. The genomic DNA was extracted from amniotic fluid for sibling II.3, fetal tissue for sibling II.4. Briefly, twist libraries were prepared from 5 ng of genomic DNA sheared with a Covaris S2 Ultrasonicator. Exome capture was performed with the Human Core Exome Kit (Twist Bioscience). Sequencing was carried out using NextSeq (Illumina). After demultiplexing, paired‐end sequences were aligned to the reference human genome (NCBI build37/hg19 version) using Burrows‐Wheeler Aligner for Illumina data. The mean depth of coverage obtained for each sample was over 30X with 96% of the exome. Downstream processing was carried out with the Genome Analysis Toolkit (GATK), SAMtools, and Picard (http://www.broadinstitute.org/gatk/guide/topic?name=best‐practices). Variant calls were made with the GATK Unified Genotyper. In each case an in‐house software tool (PolyWeb) was used to annotate (based on Ensembl release 71) and filter variants according to relevant genetic models. We excluded known variants listed >1% or at the homozygous state in gnomAD or in “in‐house” genomic database comprising more than 100,000 individuals (patients with various genetic conditions and unaffected relatives) genotyped by panel or exome sequencing (Polyweb, Imagine genomic platform). Then, we selected variants affecting splice sites or coding regions (non‐synonymous substitutions, insertions, or deletions).
The analysis revealed a homozygous frameshift variation in exon 9 of COG6 (NM_020751.2): c.821del, p.(Arg274Lysfs*32). Sanger sequencing confirmed the presence of the variant at the homozygous state in the three affected siblings and heterozygous state in their parents.
3. RESULTS
Clinicopathological anomalies found in the four affected siblings from a first cousins couple of Turkish origin are summarized in Table 1.
TABLE 1.
Clinical and genetic data of the family studied.
| Patients | Case II1 | Present case II3 | Present case II4 | Present case II5 |
|---|---|---|---|---|
| Gender | M | M | F | ? |
| Death (cause) | TOP 28wg | 1 day | TOP 24wg | TOP 14wg (aspiration) |
| Facial dysmorphism, description | NA | NA | Retrognathism, cleft palate, low set ears, large mouth, depressed nasal bridge, bulbous nose, anteverted nostrils | NA |
| Growth retardation | NA | NA | No | NA |
| Microcephaly | NA | NA | No | NA |
| Brain anomalies | NA | NA | Frontal lobe neuronal ectopies, CC dysmorphic, lateral ventricles asymmetry, vermis hypoplasia, closed V4 | NA |
| Heart malformations | NA | NA | Light ventriculo‐arterial asymmetry (left>right) | NA |
| GI tract anomalies | NA | NA | Voluminous spleen, flat gallbladder | NA |
| Liver and biliary tract anomalies | NA | NA | No | NA |
| Skin | NA | NA | No | NA |
| Inverted nipples | NA | NA | Yes | NA |
| Tooth anomalies | NA | NA | No | NA |
| Skeletal anomalies | NA | NA | Diffuse muscular atrophy with arthrogryposis, club feet, bilateral single transverse palmar crease | NA |
| Others clinical findings | NA | NA | 2 right lung lobes, narrow thorax and shoulders, cervical skin excess, 2 right lung lobes | NA |
| Birth weight | 1500 g | NA | 643 g | NA |
| Prematurity (age in WG) | TOP (28) | No (39) | TOP (24) | TOP (14) |
| Prenatal findings leading to prenatal diagnosis | Hydrocephalus, micropenis, club feet, clenched hands | Colpocephaly, ventriculomegaly 11 mm, partial CC, RV hypoplasia with tricuspid atresia, Pierre Robin sequence, IUGR, small penis, club feet, clenched hands | NT 3,6 mm, dedifferentiated cerebellum with no vermis, hypoplastic CC, retrognathism, club feet, clenched hands, rachidian angulation, diminished fetal movements | NT 8.3 mm, edema, cystic posterior fossa, and club hands, only 1 great vessel |
| Prenatal signs that could have led to antenatal diagnosis | Yes | Yes | Yes | Yes |
Fetus II.1 was the first pregnancy of this couple. During the first trimester screening, elevated nuchal translucency was noted at 3.1 mm. In the second trimester, a polymalformative syndrome was diagnosed on US associating hydrocephalus, micropenis, clubfeet, and clenched hands in this male fetus. The karyotype was normal 46, XY. After counseling, parents asked to terminate the pregnancy at 28 weeks of gestation (WG), in accordance with the French law, but denied autopsy.
Patient II.2 was the second child of this family. All US screenings were normal, the pregnancy was uneventful, and a healthy girl was born at 39 WG, weighing 3600 g.
Patient II.3 was the third child of the couple. The first trimester US examination was normal with a nuchal translucency (NT) at 0.8 mm for a crown rump length at 65.4 mm. Second trimester maternal serum screening was 1/2500 (alpha‐fetoprotein 0.82, free beta hCG 0.41, and estriol 0.46). US evaluation was unremarkable. At third trimester, US control found growth retardation with major cardiac anomalies. The patient was referred to a multicentric prenatal care center, where additional anomalies were detected mainly cerebral (colpocephaly, ventriculomegaly 11 mm, hypoplastic corpus callosum; Figure 1a,b), cardiac defect with right ventricle hypoplasia with tricuspid atresia (Figure 1c), in association with Pierre Robin sequence, clubfeet and clenched hands, small penis (Figure 1d) and splenomegaly. Parents decided to continue the pregnancy. The CGH array analysis performed after amniocentesis was normal. The baby boy born at 39 WG died 30 min later. Post‐mortem analysis was not performed.
FIGURE 1.

Obstetrical ultrasound images of fetus II.3 at 34 WG. (a) Sagittal view of hypoplastic corpus callosum, white arrow = small and thin corpus callosum; (b) Coronal view of Moose head sign: ascension of lateral ventricles; (c) Cardiac four chamber view showing right ventricle hypoplasia (white arrow). (d) External genitalia: small penis, scrotum with no testis inside.
Fetus II.4 was the fourth pregnancy of the family. The first trimester scanning revealed elevated nuchal translucency 3.6 mm (Figure 2a). CGH array was normal on chorionic villi sampling. At 20 WG, a US examination visualized a female fetus with limb malposition, lumbar angulation (Figure 2b), and hypomotility. At 22 WG, US evaluation disclosed in addition retrognathism and brain anomalies including hypoplastic corpus callosum and abnormal cerebellum (Figure 2c–f). Pregnancy termination and post‐mortem examination were performed at 24 WG on parental request. External and internal anomalies are pictured in Figure 3 and summarized in Table 1. All parameters of fetal biometry were at the 50th centile. The fetus presented with retrognathism, low set ears, large mouth, depressed nasal bridge, bulbous nose with anteverted nares, and cleft palate. Of note, were large and slightly inverted nipples (Figure 3a) with narrow thorax and shoulders, cervical skin excess, diffuse muscular atrophy with distal arthrogryposis (clubfeet, bilateral camptodactyly) and bilateral single transverse palmar crease. On X‐rays, bone age and maturation were in conformity with the fetal age. Visceral evaluation disclosed at the thoracic level ventriculo‐arterial asymmetry with small right ventricle and small pulmonary artery (Figure 3b), and splenomegaly. The placenta was normotrophic with a delay of maturation in histology.
FIGURE 2.
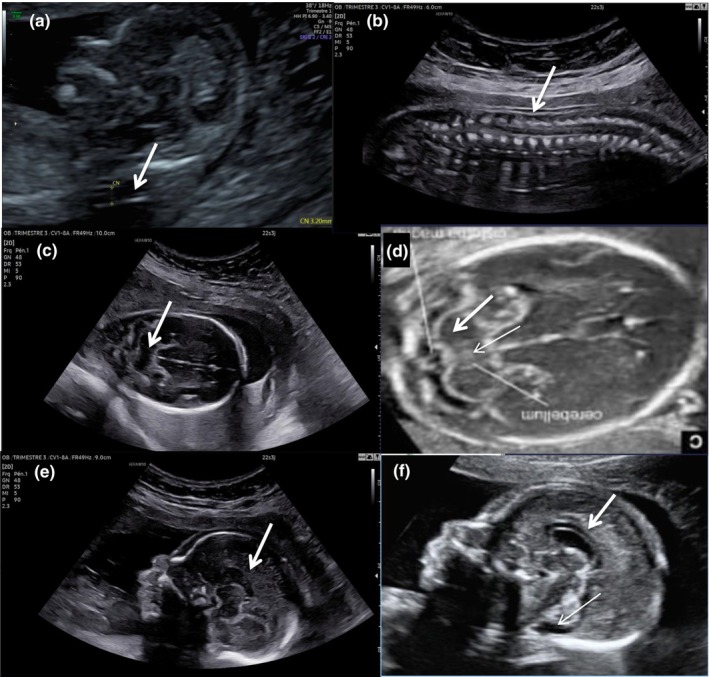
Obstetrical ultrasound images of fetus II.4. (a) Elevated nuchal translucency at first trimester examination; (b) 22 WG, sagittal view of the fetus back showing rachidian angulation, a depression in the lumbar rachis can be noted; (c) 22 WG, abnormal cerebellum with dedifferenciated echogenicity due to the absence of the hyperechogenic ring usually delimiting the cerebellum, no vermis is seen; (d) Normal cerebellum of a 22 WG fetus, note the difference of echogenicity with fetus II.4 and the presence of the vermis in the middle, thick arrow = cerebellum, thin arrow = vermis; (e) 22 WG, sagittal view of face and brain showing retrognathism and hypoplastic corpus callosum, the corpus callosum seems to be normal posteriorly but very thin anteriorly, no vermis is seen; (f) Normal sagittal view of a 22 WG fetus, thick arrow = corpus callosum, thin arrow = vermis.
FIGURE 3.

Clinicopathological and neuropathological findings of fetus II.4. (a) Large and slightly inverted nipples, more pronounced on the left nipple. (b) External cardiac examination: ventriculo‐arterial asymmetry with a small right ventricle (white circle) leading to interventricular sulcus pushed on the right; (c) Histological examination of quadriceps muscle: fibers with regular size in an abundant and loose stroma. (d) External appearance of the cerebral hemispheres with fronto‐parietal lobes filled with nodules, corresponding to (e) Neuroglial ectopias into the arachnoid space; (f) Dysmorphic corpus callosum, thick on the anterior part; (g) Asymmetric lateral ventricles; (h) Hypoplastic vermis (white circle), collapsed fourth ventricle; (i) Poor foliation of the cerebellum.
In histology, skeletal muscle fibers were thin and irregular in an abundant and loose stroma (Figure 3c). Neuropathological examination found normal brain biometries and external configuration. Off note, the irregular surface of fronto‐parietal lobes filled with nodules (Figure 3d), corresponding on histology to neuroglial ectopias into the arachnoid space, through gaps in the glia limitans (the outermost limit of the brain; Figure 3g). In these territories, the underlying cortical plate was irregular, roughly laminated and contained numerous altered elements with nuclear residues. Mid‐sagittal and coronal sections of cerebral hemispheres disclosed a dysmorphic corpus callosum thick anteriorly with moderately enlarged and asymmetric lateral ventricles (Figure 3f,g), and hypoplastic brain stem and vermis (Figure 3h). Parenchymal histological screening disclosed multiple spots of microinfarcts in process of calcification in the subcortical and periventricular white matter as well as in the basal ganglia. Ependymal abrasion was observed at all examined levels with macrophagic proliferation in the sub‐ependymal zone, also present in the cystic ganglionic eminences. The choroid plexus cells were swollen and clarified. In the brainstem, longitudinal tracts were hypoplastic. The cerebellar hemispheres and the vermis had poor foliation with swollen Purkinje cells and neuronal damages on histology (Figure 3i). The spinal cord was filled with damaged elements too.
Fetus II.5 was the fifth pregnancy of the family. The ultrasound performed at 11 WG revealed increased NT (measured at 8.3 mm, Figure 4a), generalized edema (Figure 4b), cystic posterior fossa, club hands, and only one heart great vessel (Figure 4c). A termination of pregnancy was requested by the family, performed by vacuum extraction at 14 WG.
FIGURE 4.

Obstetrical ultrasound images of fetus II.4. (a) Increased NT 8.3 mm. (b) Generalized edema. (c) One heart great vessel.
Exome sequencing in II.1 and II.3 revealed a homozygous frameshift variation in exon 9 of COG6 (NM_020751.2): c.821del, p.(Arg274Lysfs*32). Sanger sequencing confirmed the presence of the variant at the homozygous state in the four affected siblings, and heterozygous state in their parents.
Only 28 patients with CDG2L are reported to date in the literature and summarized in Table 2 along with the four present cases. More precise data are on the Table S1.
TABLE 2.
Clinical and genetic data of 28 patients reported in the literature and the four present cases.
| Patient | Reference | Consanguinity | Facial | Brain | Heart | Skeletal | Prenatal findings | Possible prenatal signs |
|---|---|---|---|---|---|---|---|---|
| P1 | Rymen et al. (2015) | No | Yes | Yes | Yes | Yes | Yes | Yes |
| P2 | Rymen et al. (2015) | Yes | Yes | Yes | NA | NA | NA | ? |
| P3 | Rymen et al. (2015) | Yes | Yes | Yes | Yes | Yes | NA | Yes |
| P4.1 | Rymen et al. (2015) | No | Yes | NA | NA | No | NA | No |
| P4.2 | Rymen et al. (2015) | No | No | NA | NA | No | NA | No |
| P5.1 | Rymen et al. (2015) | No | NA | No | Yes | NA | NA | No |
| P6.1 | Rymen et al. (2015) | No | Yes | Yes | Yes | Yes | NA | Yes |
| P5.2 | Lübbehusen et al. (2010) | No | NA | NA | NA | NA | NA | No |
| P7 | Huybrechts et al. (2012) | Yes | Yes | No | NA | Yes | NA | Yes |
| P8 | Shaheen et al. (2013) | Yes | Yes | NA | NA | NA | NA | No |
| P9 | Alsubhi et al. (2017) | NA | Yes | Yes | Yes | Yes | NA | Yes |
| P10 | Albushi et al. (2017) | NA | Yes | Yes | No | No | NA | No |
| P11 | Alsubhi et al. (2017) | NA | Yes | Yes | Yes | No | Yes | Yes |
| P12 | Alsubhi et al. (2017) | NA | Yes | NA | Noss | Yes | NA | Yes |
| P13 | Alsubhi et al. (2017) | NA | Yes | Yes | No | No | NA | No |
| P14 | Alsubhi et al. (2017) | NA | Yes | Yes | No | No | NA | No |
| P15 | Alsubhi et al. (2017) | NA | Yes | Yes | No | No | NA | No |
| P16 | Althonaian et al. (2018) | Yes | Yes | Yes | NA | Yes | Yes | Yes |
| P17 | Li et al. (2019) | No | No | No | Yes | Yes | NA | Yes |
| P18.1 | Komlosi et al. (2020) | No | NA | NA | NA | Yes | NA | No |
| P18.2 | Komlosi et al. (2020) | No | Yes | NA | Yes | Yes | Yes | Yes |
| P19.1 | Mandel et al. (2020) | Yes | Yes | Yes | NA | Yes | Yes | Yes |
| P19.2 | Mandel et al. (2020) | Yes | Yes | Yes | NA | Yes | Yes | Yes |
| P20 | Zhao et al. (2021) | No | Yes | Yes | Yes | Yes | Yes | No |
| P21.1 | Lugli et al. (2021) | Yes | Yes | Yes | NA | Yes | Yes | Yes |
| P21.2 | Lugli et al. (2021) | Yes | Yes | Yes | Yes | Yes | Yes | No |
| P22 | Cirnigliaro et al. 2022 | No | Yes | Yes | No | Yes | NA | Yes |
| P23 | Ververi et al. 2022 | No | Yes | NA | NA | Yes | NA | Yes |
| Total | 28 | 9 (32%) | 23 (82%) | 17 (61%) | 10 (36%) | 17 (61%) | 9 (32%) | 15 (54%) |
Only eight patients are described to have prenatal findings on US examination namely P1, P16, P18.2, P19.1, P19.2, P20, P21.1, and P21.2 (Table 2). However, clinical signs are often present at birth, and some could be accessible to US examination during pregnancy, in particular hydrocephalus, abnormal corpus callosum and cerebellum, arthrogryposis and postaxial polydactyly, retrognathism, and ambiguous genitalia. Here, we add a family with four siblings and describe the US pathological findings. Arthrogryposis is the most consistent finding with clubfeet and clenched hands (leading to diminished fetal movements) and was observed in 8 of 12 cases. Intrauterine growth restriction was detected in seven cases, and was the only characteristic seen in three patients. Polyhydramnios was detected in three cases. Structural brain anomalies were seen in six cases, three being anomalies of the corpus callosum. Abnormal genitalia were described in four patients, and only for male fetuses. Cardiac defects were present in four cases (ventricular septal defect and atrial septal defect are mostly described), and facial dysmorphism in three cases (including retrognathism in two cases). Increased nuchal translucency was present in three cases during first trimester screening.
4. DISCUSSION
CDG2L is a multisystem disorder caused by deficiency of subunit 6 of COG, and characterized by growth retardation, global developmental delay, muscular hypotonia, microcephaly, liver and gastrointestinal disease, thrombocytopenia, recurrent infections, episodic fever, congenital heart defects, generalized joint contractures, and early lethality (Rymen et al., 2015). It has been proposed that a distinctive feature of CDG2L compared to other CDG can be additional ectodermal signs comprising hyperhidrosis with dry skin, hyperthermia, thickened skin, hyperkeratosis, and tooth anomalies (Komlosi et al., 2020).
In this consanguineous family, the recurrence of a multisystemic disease and the post‐mortem findings were concordant with a metabolic disorder causing neuronal death and glia limitans fragility. Off note, the abnormal nipples often observed in CDG type I were evocative of a CDG deficit (Magalhães et al., 2020). When looking for « inverted nipples » on HPO and reviews of published cases, only CDG type I is described with this sign. Thus, this fetal case shows that anomalies of nipples can be a CDG type II sign as well. Among neuropathological findings, neuroglial ectopia into the arachnoid space results in the obliteration of the arachnoid space leading to ventricular dilatation as in cobblestone lissencephaly (or lissencephaly type II) (Devisme et al., 2012). Overmigration into the arachnoid space occurs through gaps in the glia limitans, also described in abnormal O'glycosylation (Ververi et al., 2022). Etiopathogenical approach permits us in addition, to correlate the Pierre Robin sequence, as well as the fetal hypomobility and the micropenis, to the diffuse neurodegeneration affecting the entire nervous system including the spinal cord.
Thus, we could say that there is no specific prenatal manifestation that clearly hint toward CDG2L but the association of arthrogryposis with another US finding, such as intrauterine growth restriction, abnormal corpus callosum, abnormal male genitalia or cardiac defect should indicate that CDG2L variants have to be evaluated.
Since the first report of COG6 variants, only 16 pathogenic variants have been described. The c.1646G>T variant is reported in four patients. Two are homozygous for c.1646G>T variation and are not related: one is from Morocco and one from Turkey. The two others are siblings and carried compound heterozygous variants along with the c.785A>G variant in trans. The c.511C>T non‐sense variant is reported in three unrelated patients. The c.821del, p.(Arg274Lysfs*32) we report here is a novel variant. Due to the limited number of CDG2L patients reported to date, no clear genotype–phenotype correlation has been established. Only the deep intronic splice site variant c.1167‐24A>G described in seven patients (including three siblings), all from Saudi Arabia, is correlated with Shaheen syndrome (MIM#615328) (Shaheen et al., 2013), with milder clinical features.
In conclusion, we report the first pathological findings of CDG2L and review the prenatal manifestations. An accurate, early diagnosis is essential for genetic counseling and management. Suspecting CDG2L is challenging in the prenatal setting and at birth, with early death preventing an exhaustive biochemical screening for metabolic disorders. In these contexts, exome sequencing is a powerful strategy to make the diagnosis. In the family we report, it allowed us to propose a molecular prenatal diagnosis during the following pregnancy that hopefully showed that the fetus was not affected with CDG2L. It also allows us to discuss the possibility of preimplantation genetic diagnosis for further pregnancies.
AUTHOR CONTRIBUTIONS
Conception, design and writing: Sarah Guterman, Agnese Feresin and Tania Attié‐Bitach. Acquisition of data: Stanislas Lyonnet, Jean‐Pierre Bernard, Claire Colmant, Philippe Roth, Nicolas Bourgon, Pierre Mace, Ferechté Encha‐Razavi, Bettina Bessières and Joana Bengoa. Technical support: Zaina Ait Arkoub, Cécile Fourrage, Alice Thoreau. Analysis and interpretation of data: Sarah Guterman, Tania Attié‐Bitach Lucile Boutaud. Review & editing: all authors.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest to declare.
ETHICS STATEMENT
The study was carried out in accordance with all institutional and national ethical guidelines. The genetic analysis and autopsy were performed as part of routine clinical diagnosis, with signed parental consent for both, in accordance with the French law.
Supporting information
Table S1.
ACKNOWLEDGMENTS
None.
Guterman, S. , Feresin, A. , Boutaud, L. , Jacquin, C. , Lyonnet, S. , Bernard, J.‐P. , Colmant, C. , Roth, P. , Bourgon, N. , Mace, P. , Thoreau, A. , Ville, Y. , Bengoa, J. , Ait Arkoub, Z. , Fourrage, C. , Encha‐Razavi, F. , Bessières, B. , & Attié‐Bitach, T. (2025). COG6 ‐related prenatal phenotype (CDG2L): Clinico‐pathological report and review of the literature. Molecular Genetics & Genomic Medicine, 13, e2442. 10.1002/mgg3.2442
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Alsubhi, S. , Alhashem, A. , Faqeih, E. , Alfadhel, M. , Alfaifi, A. , Altuwaijri, W. , Alsahli, S. , Aldhalaan, H. , Alkuraya, F. S. , Hundallah, K. , Mahmoud, A. , Alasmari, A. , Mutairi, F. A. , Abduraouf, H. , AlRasheed, L. , Alshahwan, S. , & Tabarki, B. (2017). Congenital disorders of glycosylation: The Saudi experience. American Journal of Medical Genetics. Part A, 173(10), 2614–2621. [DOI] [PubMed] [Google Scholar]
- Althonaian, N. , Alsultan, A. , Morava, E. , & Alfadhel, M. (2018). Secondary Hemophagocytic syndrome associated with COG6 gene defect. JIMD Reports, 42, 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn, J. B. , D'Souza, Z. , & Lupashin, V. V. (2019). Maintaining order: COG complex controls Golgi trafficking, processing, and sorting. FEBS Letters, 593(17), 2466–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirnigliaro, L. , Bianchi, P. , Sturiale, L. , Garozzo, D. , Mangili, G. , Keldermans, L. , Rizzo, R. , Matthijs, G. , Fiumara, A. , Jaeken, J. , & Barone, R. (2022). COG6‐CDG: Novel variants and novel malformation. Birth Defects Research, 114(5–6), 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devisme, L. , Bouchet, C. , Gonzalès, M. , Alanio, E. , Bazin, A. , Bessières, B. , Bigi, N. , Blanchet, P. , Bonneau, D. , Bonnières, M. , Bucourt, M. , Carles, D. , Clarisse, B. , Delahaye, S. , Fallet‐Bianco, C. , Figarella‐Branger, D. , Gaillard, D. , Gasser, B. , Delezoide, A. L. , … Encha‐Razavi, F. (2012). Cobblestone lissencephaly: Neuropathological subtypes and correlations with genes of dystroglycanopathies. Brain, 135(2), 469–482. [DOI] [PubMed] [Google Scholar]
- Francisco, R. , Marques‐da‐Silva, D. , Brasil, S. , Pascoal, C. , Dos Reis, F. V. , Morava, E. , & Jaeken, J. (2019). The challenge of CDG diagnosis. Molecular Genetics and Metabolism, 126(1), 1–5. [DOI] [PubMed] [Google Scholar]
- Haijes, H. A. , Jaeken, J. , & van Hasselt, P. M. (2020). Hypothesis: Determining phenotypic specificity facilitates understanding of pathophysiology in rare genetic disorders. Journal of Inherited Metabolic Disease, 43(4), 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huybrechts, S. , De Laet, C. , Bontems, P. , Rooze, S. , Souayah, H. , Sznajer, Y. , Sturiale, L. , Garozzo, D. , Matthijs, G. , Ferster, A. , Jaeken, J. , & Goyens, P. (2012). Deficiency of subunit 6 of the conserved oligomeric Golgi complex (COG6‐CDG): Second patient. JIMD Reports, 4, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlosi, K. , Gläser, S. , Kopp, J. , Hotz, A. , Alter, S. , Zimmer, A. D. , Beger, C. , Heinzel, S. , Schmidt, C. , & Fischer, J. (2020). Neonatal presentation of COG6‐CDG with prominent skin phenotype. JIMD Reports, 55(1), 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G. , Xu, Y. , Hu, X. , Li, N. , Yao, R. , Yu, T. , Wang, X. , Guo, W. , & Wang, J. (2019). Compound heterozygous variants of the COG6 gene in a Chinese patient with deficiency of subunit 6 of the conserved oligomeric Golgi complex (COG6‐CDG). European Journal of Medical Genetics, 62(1), 44–46. [DOI] [PubMed] [Google Scholar]
- Lübbehusen, J. , Thiel, C. , Rind, N. , Ungar, D. , Prinsen, B. H. C. M. T. , de Koning, T. J. , van Hasselt, P. M. , & Körner, C. (2010). Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Human Molecular Genetics, 19(18), 3623–3633. [DOI] [PubMed] [Google Scholar]
- Lugli, L. , Bariola, M. C. , Ferri, L. , Lucaccioni, L. , Bertucci, E. , Cattini, U. , Torcetta, F. , Morrone, A. , Iughetti, L. , & Berardi, A. (2021). Disorder of sex development associated with a novel homozygous nonsense mutation in COG6 expands the phenotypic spectrum of COG6‐CDG. American Journal of Medical Genetics, 185(4), 1187–1194. [DOI] [PubMed] [Google Scholar]
- Lugli, L. , Pollazzon, M. , Bigoni, S. , Caraffi, S. G. , Ferlini, A. , Ferri, L. , Morrone, A. , Calabrese, O. , Iughetti, L. , Garavelli, L. , & Berardi, A. (2022). Correspondence on “disorder of sex development associated with a novel homozygous nonsense mutation in COG6 expands the phenotypic spectrum of COG6‐CDG”. American Journal of Medical Genetics, 188(1), 382–383. [DOI] [PubMed] [Google Scholar]
- Magalhães, A. P. P. S. , Burin, M. G. , Souza, C. F. M. , de Bitencourt, F. H. , Sebastião, F. M. , Silva, T. O. , Vairo, F. P. E. , & Schwartz, I. V. D. (2020). Transferrin isoelectric focusing for the investigation of congenital disorders of glycosylation: Analysis of a ten‐year experience in a Brazilian center. The Journal of Pediatrics, 96(6), 710–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel, H. , Cohen Kfir, N. , Fedida, A. , Shuster Biton, E. , Odeh, M. , Kalfon, L. , Ben‐Harouch, S. , Fleischer Sheffer, V. , Hoffman, Y. , Goldberg, Y. , Dinwiddie, A. , Dumin, E. , Eran, A. , Apel‐Sarid, L. , Tiosano, D. , & Falik‐Zaccai, T. C. (2020). COG6‐CDG: Expanding the phenotype with emphasis on glycosylation defects involved in the causation of male disorders of sex development. Clinical Genetics, 98, 402–407. [DOI] [PubMed] [Google Scholar]
- Péanne, R. , de Lonlay, P. , Foulquier, F. , Kornak, U. , Lefeber, D. J. , Morava, E. , Pérez, B. , Seta, N. , Thiel, C. , van Schaftingen, E. , Matthijs, G. , & Jaeken, J. (2018). Congenital disorders of glycosylation (CDG): Quo vadis? European Journal of Medical Genetics, 61(11), 643–663. [DOI] [PubMed] [Google Scholar]
- Rymen, D. , Winter, J. , Van Hasselt, P. M. , Jaeken, J. , Kasapkara, C. , Gokçay, G. , Haijes, H. , Goyens, P. , Tokatli, A. , Thiel, C. , Bartsch, O. , Hecht, J. , Krawitz, P. , Prinsen, H. C. , Mildenberger, E. , Matthijs, G. , & Kornak, U. (2015). Key features and clinical variability of COG6‐CDG. Molecular Genetics and Metabolism, 116(3), 163–170. [DOI] [PubMed] [Google Scholar]
- Shaheen, R. , Ansari, S. , Alshammari, M. J. , Alkhalidi, H. , Alrukban, H. , Eyaid, W. , & Alkuraya, F. S. (2013). A novel syndrome of hypohidrosis and intellectual disability is linked to COG6 deficiency. Journal of Medical Genetics, 50(7), 431–436. [DOI] [PubMed] [Google Scholar]
- Ververi, A. , Stathopoulou, T. , Kontou, A. , Farini, M. , Vlahou, G. , Demiris, N. , & Sarafidis, K. (2022). Lethal COG6‐CDG in neonatal patient with arachnodactyly, joint contractures, and skin manifestations: Founder mutation in the southeastern European population? Pediatric Dermatology, 39(2), 314–315. [DOI] [PubMed] [Google Scholar]
- Zhao, P. , Zhang, L. , Tan, L. , Luo, S. , Huang, Y. , Peng, H. , Cao, J. , & He, X. (2021). Genetic analysis and prenatal diagnosis in a Chinese with growth retardation, abnormal liver function, and microcephaly. Molecular Genetics & Genomic Medicine, 9(9), e1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.