

Abstract
Ease of access to the cornea makes antimicrobial peptides (AMPs) ideal candidates for topical drug application. However, before bringing them to the clinic, it is fundamental to evaluate in vitro: (1) the ability of AMPs to kill bacteria in the presence of human tears, by counting the number of surviving bacteria on agar plates; (2) the potential cytotoxicity of AMPs to mammalian cells by a colorimetric method based on the production of a colored formazan crystals by metabolically active cells; and (3) the ability of AMPs to neutralize the toxic effect of the bacterial cell wall component, lipopolysaccharide (LPS), by measuring the level of the pro-inflammatory cytokine, TNF-α, released from LPS-activated macrophages, using a sandwich enzyme-linked immunosorbent assay.
Keywords: Colony counts, Tear collection, Colorimetric MTT assay, Sandwich immunosorbent assay, LPS detoxification
1. Introduction
Microbial keratitis is a serious vision threatening disease characterized by deterioration of the cornea (the transparent part covering the pupil and iris) with rapid progression to inflammation, necrosis, and potentially vision loss, if not treated [1, 2]. One of the major risk factors for bacterial keratitis is contact lens wear, particularly overnight use of disposable lenses and/or extended wear modalities [3, 4]. Pseudomonas aeruginosa is the most common bacterial pathogen causing microbial keratitis related to contact lens wear [5]. The increasing emergence of microorganisms resistant to the available ophthalmic agents has led to a significant need for novel approaches to prevent and treat ocular infections. Antimicrobial peptides (AMPs) hold promise for the development of new antibiotics. Importantly, in order to be effective at the ocular surface, AMPs must be active at high salt concentration, as well as in the presence of tear film components. The tear film is the liquid layer bathing the cornea and conjunctiva and that provides lubrication, protection, and nutrients to the cornea. It contains a lipid layer in contact with the air that prevents tears from evaporating, a mucus layer that coats the corneal surface and an aqueous layer rich in salts and proteins [6]. Importantly, to be used as new therapeutics, AMPs have to be harmless to corneal epithelial cells. In addition, it is worthwhile mentioning that during bacterial division, but mostly upon antibiotic treatment, lipopolysaccharide (LPS) molecules (the most abundant components of the outer membrane in Gram-negative bacteria) are released in the surroundings [7, 8]. LPS can activate immune cells, e.g., macrophages, triggering the intracellular signaling pathway which controls the secretion of pro-inflammatory cytokines, i.e., TNF-α, whose high levels can induce serious tissue damage [9]. Ease of access to the cornea makes it an ideal candidate for topical drug application, and there have been a small number of studies investigating the efficacy of AMPs in animal models of microbial keratitis (see Chap. 30). However, before bringing AMPs to the clinic, it is fundamental to evaluate in vitro: (1) their ability to kill bacteria in the presence of human tears, (2) their potential cytotoxicity to mammalian cells, and (3) their ability to neutralize the toxic effect of LPS.
A brief description of the methods for in vitro studies of a potential anti-keratitis AMP, according to the aforementioned aims, is reported below.
With reference to the antimicrobial effect in tears, counting surviving cells in terms of colony-forming units (CFU) on plates remains the most accurate and reliable standard method [10, 11].
The toxic effect of AMPs on mammalian cells, in this case corneal epithelial cells, is generally investigated by the colorimetric 3(4,5-dimethylthiazol-2yl)2,5-diphenyltetrazolium bromide (MTT) assay in which the intensity of the dye is proportional to the number of viable cells. MTT is a tetrazolium salt which is reduced to a colored formazan product [12] by mitochondrial reductases which are functional only in metabolically active cells. Acidified isopropanol is then added to dissolve the insoluble purple formazan product into a colored solution. The absorbance of this colored solution can be quantified by measuring the sample at a certain wavelength (usually between 500 and 600 nm) by a spectrophotometer. MTT assays are usually performed in the dark since the MTT reagent is light sensitive [13, 14].
LPS detoxification can be studied by evaluating the peptide’s ability to inhibit the extracellular release of TNF-α from LPS-activated immune cells, e.g., macrophages, which are the most abundant circulating immune cells. The level of human TNF-α in samples can be determined by the sandwich enzyme-linked immunosorbent assay (ELISA), which is designed to measure the amount of the target-bound molecule (i.e., TNF-α) between a matched antibody pair. Briefly, a target-specific antibody, called the capture antibody, is pre-coated in the wells of a microplate. Samples, standards, or controls are then added into wells to bind to the immobilized capture antibody (usually a polyclonal antibody). The sandwich is formed by the addition of a second (detector) biotinylated antibody, which will recognize a different epitope of the target molecule from the capture antibody. Avidin horseradish peroxidase (HRP) is subsequently added. After incubation and washing steps to remove unbound substances, a substrate (3,3′,5,5′-tetramethylbenzidine, TMB, solution) that reacts with the enzyme-antibody-target complex is added to produce a blue color in proportion to the concentration of antigen (TNF-α) present in the sample. Finally, the stop solution changes the reaction color from blue to yellow, and absorbance of the well is read with a microplate reader at 450 and 570 nm.
2. Materials
Prepare all materials at room temperature (R.T.), and diligently follow all waste regulations when disposing waste material.
2.1. Antimicrobial Activity in the Presence of Tears
5 μl microcapillary glass tubes.
Eppendorf microcentrifuge tubes (200 μl capacity).
Ice.
2.1.1. Collection of Human Basal Tears
2.1.2. Bacterial Culture
Luria-Bertani (LB) broth: 1 % (w:v) bacto tryptone, 0.5 % (w:v) yeast extract, and 0.5 % (w:v) NaCl in distilled water; the final pH has to be adjusted to 7.4 with 1 N sodium hydroxide; using a graduated cylinder, dispense aliquots into glass bottles, autoclave them at 120 °C for 20 min, and store the sterilized medium at R.T.
LB agar Petri dishes: add 1.5 % (w:v) agar to freshly prepared LB medium, autoclave at 120 °C for 20 min, and cool the medium to between 45 and 50 °C prior to pouring the plates to minimize the amount of condensation that forms; the thickness of the agar medium in the plates should be around 0.5 cm, which can be achieved by pouring 15–20 ml of medium into 100 mm dish plates; store plates at +4 °C.
50 ml BD Falcon polypropylene tubes.
10 μl sterile inoculating loops.
Phosphate buffer, pH 7.4 (PB): stock solution contains 8.2 mM Na2HPO4 and 1.8 mM KH2PO4 in water, sterilize the buffer solution by filtration (0.22 μm nitrocellulose filters) in a biological safety cabinet, and store at R.T.
Spectrophotometer.
37 °C incubator and a centrifuge for 50 ml tubes.
2.1.3. Antimicrobial Assay
LB agar Petri dishes.
Pipette/micropipettes with disposable tips.
200 μl microcentrifuge tubes.
Eppendorf Thermomixer Comfort; 37 °C incubator and a colony counter.
2.2. MTT Assay to Evaluate Toxicity of AMPs on Corneal Epithelial Cells
2.2.1. Cell Culture, Passaging, and Growth Medium
Telomerase immortalized human corneal epithelial cell line (hTCEpi) [15].
Keratinocyte growth medium-2 (KGM-2) bullet kit (CC-3103 and CC-4152). The kit includes 500 ml basal medium, plus supplements and growth factors.
50 mg/ml normocin: use 1 ml/500 ml media, and prepare the growth media by mixing the 500 ml basal medium with the supplies, growth factors, and normocin.
Tissue culture-treated T25 and T75 flasks (25 cm2 and 75 cm2, respectively).
5 % CO2 incubator for cell cultures.
TrypLE Express phenol red (see Note 1).
Phosphate-buffered saline without calcium and magnesium chloride (CMF-PBS): stock solution (10×) is prepared by mixing 1.37 M NaCl, 27 mM KCl, 100 mM Na2HPO4, and 18 mM KH2PO4 in distilled water; adjust pH to 7.4 with HCl if necessary, and autoclave at 120 °C for 20 min before storage at R.T.; working buffer is prepared by diluting one part of the stock solution with nine parts of distilled water; store at R.T.
2.2.2. Cytotoxicity Assay
Hank’s balanced salt solution (HBSS): 136 mM NaCl; 0.34 mM Na2HPO4; 0.44 mM KH2PO4; 5.4 mM KCl; 4.1 mM NaHCO3, pH 7.2, supplemented with 5.5 mM d-glucose (see Note 2).
MTT stock solution: dissolve 5 mg/ml in sterile HBSS, store single-use aliquots at −20 °C in dark or foil-covered bottles (the compound is light sensitive), and prepare fresh working solutions at the desired concentration (0.5 mg/ml) by dilution with HBSS (see Note 3).
Stop solution: acidified isopropanol by adding 0.04 N HCl.
Sterile 96-well polystyrene flat bottom and tissue culture-treated transparent plates.
0.02 % benzalkonium chloride (BAC).
Pipette/micropipettes with disposable tips.
A microplate reader capable of measuring absorbance at 590 and 635 nm.
2.3. TNF-α Assay
2.3.1. LPS and Peptide Solution
Prepare LPS solution by solubilizing LPS powder (e.g., LPS from P. aeruginosa serotype 10), in sterile water at a final concentration of 5 mg/ml. It is recommended to prepare aliquots and store them at −20 °C. Dilute LPS to the desired concentration before usage.
Peptide powder is dissolved in sterile water at 2 mM (stock solution) and stored at −20 °C.
2.3.2. Macrophage Growth and Passaging
RAW 264.7 murine macrophage cell line.
Dulbecco’s modified high glucose Eagle’s medium (DMEM) supplemented with non-essential amino acids, sodium pyruvate (1 mM final concentration); 2 mM glutamine; 10 % heat-inactivated fetal bovine serum (FBS) and antibiotics (0.1 mg/ml of penicillin and streptomycin).
T25 tissue culture-treated flasks.
5 % CO2 incubator.
CMF-PBS (1×).
Cell scrapers, 1.8 cm blade.
Inverted optical microscope.
Neubauer or Burker chamber.
Cover glasses.
Pipette/micropipette with disposable tips.
50 ml BD Falcon polypropylene tubes.
2.3.3. Neutralization of LPS
Sterile 96-well flat bottom and cell culture-treated transparent plates.
Multichannel pipette to measure volumes ranging from 20 to 200 μl.
2.3.4. ELISA Procedure
eBioscience mouse TNF-α enzyme-linked immunosorbent assay kit.
Microplate reader capable of measuring absorbance at 450 and 570 nm.
Adjustable pipettes to measure volumes ranging from 2 μl to 1 ml.
Phosphate-buffered saline, with MgCl2 and CaCl2 (PBS) sterile filtered.
Wash buffer (PBS + 0.05 % Tween-20, pH 7.4).
Stop solution: 2 N H2SO4 or 1 M H3PO4.
Eppendorf microcentrifuge tubes to prepare standard dilutions.
Plate sealer: parafilm.
Absorbent paper.
Multichannel pipette to measure values ranging from 50 to 200 μl.
3. Methods
3.1. In Vitro Antimicrobial Activity in the Presence of Human Tears
The antimicrobial activity of the peptide is studied in the presence of 50 or 70 % (v:v) human basal tears at 37 °C by a rapid, sensitive, and reliable method based on the reduction in the number of CFU compared to that of peptide-untreated samples.
3.1.1. Collection of Human Basal Tears
Obtain approval for human subject’s research from the relevant regulatory body. When subject has been informed of the study and given consent, collect basal tears as described below and store them on ice during the collection process. If desired reflex tears can be collected by using an appropriate stimulus such as a cotton swab gently inserted in to the nose or onion vapors.
Make sure that the subjects are sitting in a comfortable position with the back of their head against a headrest.
The investigator (i.e., person collecting the tears) can be standing or sitting in order to collect the tears.
Place one capillary tube into the black bulb/holder, and collect tears by holding the collection tube perpendicular to the ocular surface.
Gently pull down the lower lid and collect tears from the fornix (see Note 4 and Fig. 1). This method, when performed properly, rarely induces reflex tearing (see Note 5).
Place your finger over the hole on the back of the plunger, and expel the tears into a sterile 200 μl collection tube (previously put on ice).
Repeat until sufficient tears have been collected. Allow the subjects short breaks in which time they can close their lids and roll their eyes. Multiple samples from the same subject or samples from multiple subjects may be pooled as necessary.
Store the basal tears at −80 °C until usage.
Fig. 1.
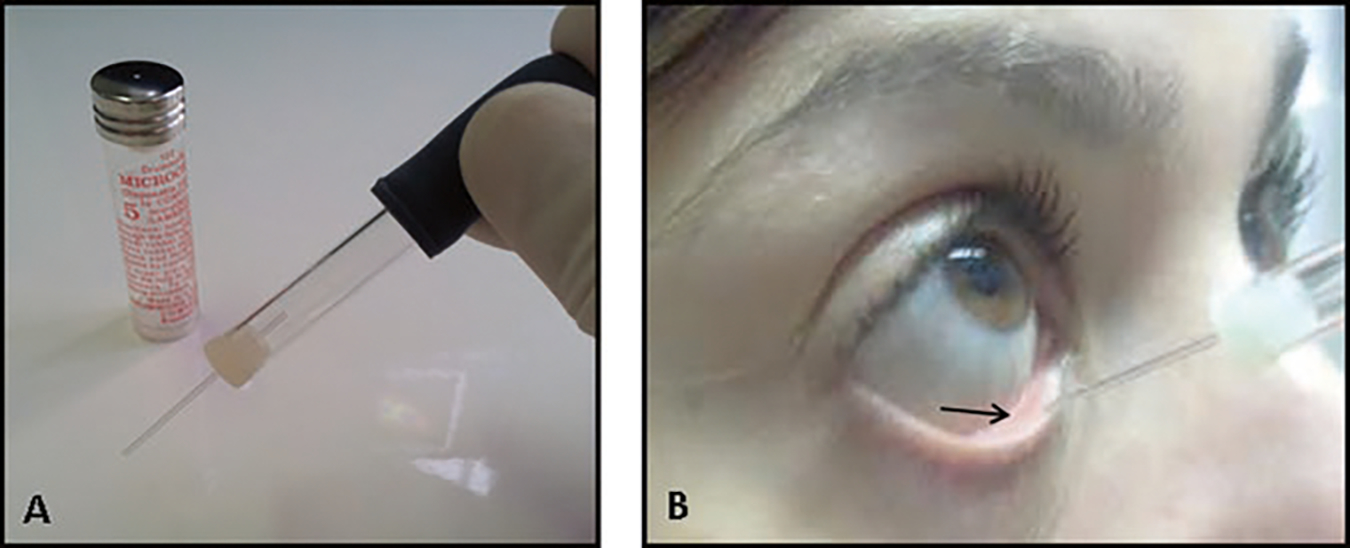
Collection of basal tears. Obtain tears by inserting a 5 μl glass capillary tube (a) into the fornix (indicated by the arrow), in a perpendicular orientation to the ocular surface (b)
3.1.2. Bacterial Culture
Bacterial growth is started from a frozen glycerol stock. Under a biological safety cabinet class II, open the stock tube, scrape off a portion from the top of the frozen glycerol stock with a 10 μl sterile loop, and streak it onto an LB agar plate.
In the case of P. aeruginosa, the plates are incubated at 37 °C for 24 h under aerobic conditions.
A single bacterial colony is picked from the streak plate and then inoculated into 10 ml LB broth, previously put into a 50 ml polypropylene Falcon tube, and grown at 37 °C (use an incubator with shaking at 150 rpm), until an absorbance A590 0.8 is reached (see Note 6).
Bacteria are harvested by centrifugation at 3000 × g for 10 min; the supernatant is discarded, and the pellet is washed twice with PB and resuspended in the same buffer to an optical density of approximately 1 × 107 CFU/ml.
3.1.3. Assay Antimicrobial
Reaction mixtures containing 50 or 70 % (v:v) human basal tears are incubated with 1 × 105 CFU of P. aeruginosa in PB (final volume 50 μl) in microcentrifuge Eppendorf tubes (200 μl capacity), where the peptide is added at the desired concentration. Control samples, without peptide and/or tears, are also included.
Samples are incubated in the thermomixer, with shaking at 600 rpm, at 37 °C for different times (e.g., 30, 90, and 120 min).
At the corresponding time intervals, 5 μl aliquots of peptide-treated samples are withdrawn and plated on LB agar plates using 6 ml of soft agar (see Notes 7 and 8). For the other samples, a 1:10 dilution in PB is made before plating. The plates are incubated at 37 °C overnight, for counting and plotting the data (see Notes 9 and 10).
The results are expressed as percentage of CFU compared to the control (peptide-untreated cells).
3.2. MTT Assay to Evaluate Toxicity of AMPs on Corneal Epithelial Cells
The colorimetric measurement of viable cells using the chromophore MTT allows an automated quantification of viable cells at low cost without requiring direct reading of the soluble final product.
3.2.1. Cell Culture and Passaging
The hTCEpi cells are cultured in either T25 or T75 tissue culture flasks using KGM-2 medium supplemented with growth factors and normocin (100 μg/ml). They are incubated at 37 °C in 5 % CO2. If they do not reach 80–90 % confluence in 3 days, change medium every day. When cells reach 80–90 % confluence, detach cells from the flask as follows:
Aspirate the medium from the flask and discard it into a bottle under a biological safety cabinet class II. Rinse the cells with CMF-PBS and gently rock the vessel back and forth. Decant CMF-PBS.
Add an appropriate volume of pre-warmed TrypLE Express to the flask (i.e., 1 ml in a T25 flask, 3 ml in a T75 flask).
Gently rock the vessel, allowing the solution to coat the cells completely.
Incubate at 37 °C until the cells are visibly detached (observe at 5–8 min intervals).
Add an appropriate volume of complete growth medium, resuspend cells, and pipette repeatedly to break up any clumps that may be present.
Aspirate 10 μl with a micropipette tip for counting using a Neubauer or Burker chamber (see Note 11).
3.2.2. Cytotoxicity Assay
Seed 10,000 hTCEpi cells/well in a 96-well plate, and incubate the plate at 37 °C and 5 % CO2 for 48 h to allow the cells to attach to the plate and spread.
Stimulate cells with the peptide dissolved in medium at the desired concentration (50 μl final volume), in triplicate. Incubate the plate for 24 h.
Add 50 μl of 0.02 % BAC into three wells for 15 min as a positive control.
Add 50 μl medium into the other wells (now total volume is 100 μl).
Add 10 μl MTT solution (5 mg/ml) into each well.
Incubate the plate at 37 °C for 3 h until purple formazan crystals are visible by light microscopy. An example of MTT assay is reported in Fig. 2.
Stop the reaction by adding 100 μl of acidified isopropanol to solubilize the formazan crystals. Use a multichannel pipette (see Note 12).
Pipette up and down to dissolve the crystals.
Gently pop the bubbles using a 10 μl tip.
Read the plate using a multi-well reader at 590 and 635 nm as reference.
-
Subtract reference absorbance (Abs) value (635 nm) from 590 nm absorbance value and plot graph.
Calculate the percentage of viable cells, according to the formula:
where the blank is given by sample without cells and not treated with the peptide.
Fig. 2.
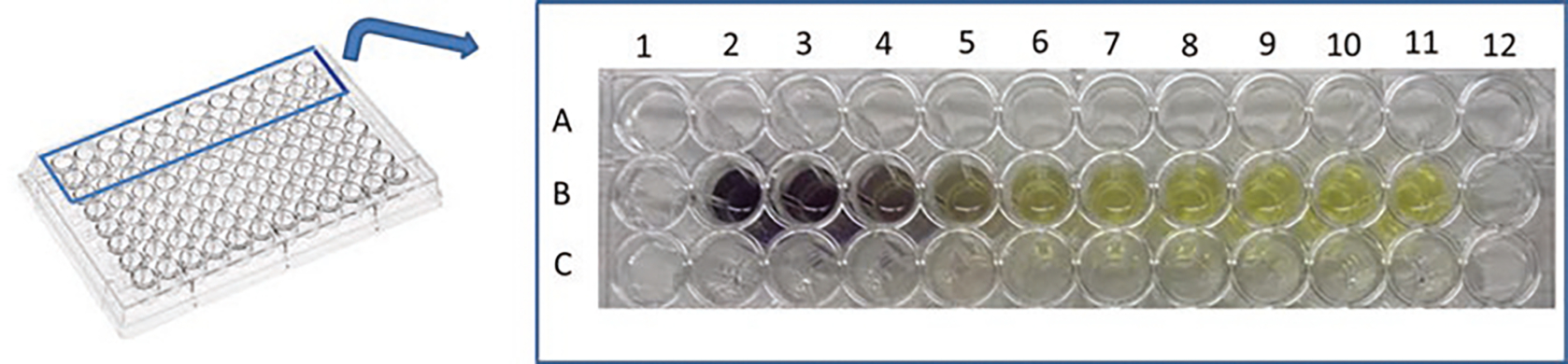
Example of viability assay using the colored indicator MTT. The intensity of the purple dye (formazan) is proportional to the number of viable cells. The transparent yellow color in the B11 well corresponds to MTT solution without cells
3.3. TNF-α Assay
The level of TNF-α released in the extracellular medium upon stimulation of immune cells with LPS can be measured using murine macrophages and a mouse TNF-α enzyme-linked immunosorbent assay kit according to the manufacturer’s protocol. Cells that are stimulated with LPS alone and untreated cells serve as controls. All experiments should be performed in triplicate.
3.3.1. LPS and Peptide Solution
Dilute the peptide (from a stock solution of 2 mM in water) you wish to test in DMEM supplemented with 2 mM glutamine, 1 mM nonessential amino acids, 1 mM sodium pyruvate, and 10 % heat-inactivated FBS without antibiotic (DMEMg).
Since each experiment is performed in triplicate on a 96-well plate, it is recommended to prepare 350 μl mixture containing DMEMg plus peptide (at the desired final concentration) and LPS (10 ng/ml).
In parallel, prepare a mixture without LPS.
3.3.2. Macrophage Growth and Passaging
Grow murine RAW macrophages in their complete culture medium in T25 flasks as described in Subheading 2.3.2.
Once cells reach confluence, remove the medium with a 10 ml pipette, and discard it into a bottle under the biological safety cabinet class II.
Wash three times with 4 ml CMF-PBS. The first two washes are very quick; for the last wash, leave the buffer for 3 min, before removing it.
Add 3 ml of DMEMg and detach cells with an appropriate scraper.
Transfer the cell suspension in to a 50 ml BD Falcon tube, and take out a small amount (10 μl) for counting in a Neubauer or Burker chamber under a microscope (see Note 11).
Dilute the cells to 1 × 105/ml in DMEMg.
3.3.3. Neutralization of LPS
One day before running the assay, seed 1 × 105 RAW 264.7 macrophages (suspended in 100 μl of DMEMg) in a 96-well plate, and incubate the plate at 37 °C and 5 % CO2.
The following day remove the medium using a multichannel pipette, wash each well with 100 μl of DMEMg, and replace the medium with 100 μl of the peptide mixture containing or not LPS. One hundred μl of medium (without LPS or peptide) are added to the other wells as control.
Incubate the plate for 4 h at 37 °C and 5 % CO2.
Aspirate the samples for each treatment and keep at −20 °C before ELISA test (see Note 13).
Perform ELISA test according to the manufacture’s manual.
3.3.4. ELISA Procedure
Coat all wells of a 96-well plate with 100 μl of capture antibody previously diluted in coating buffer provided with the ELISA kit. Seal the plate and incubate it overnight (16–18 h) at 4 °C.
Aspirate and wash the plate for ~1 min with wash buffer (250 μl per well). Repeat three times. Be careful not to touch (with the plastic tip) the adjacent wells during washing and pipetting to avoid contamination.
To remove any residual buffer, turn the plate upside down on absorbent paper.
To block nonspecific binding and reduce background, add 200 μl of Assay Diluent 1× (diluted with 4 parts water) per well, seal the plate, and incubate it at R.T. for 1 h.
While the plate is being blocked, prepare the appropriate samples dilution (if necessary) and standards.
Wash the plate as in steps 2 and 3.
Using Assay Diluent 1×, dilute TNF-α standards and add 100 μl/well (at the highest concentration) in triplicate to wells A1, A2, and A3. Add 100 μl of half TNF-α concentration to the second row (B1, B2, and B3) and so on until row G (see Notes 13 and 14). Do not add TNF-α to H row. Wells H1, H2, and H3 contain only media (Assay Diluent 1×) and are used as blank control. A schematic representation is shown in Fig. 3.
Add 100 μl/well of your samples to the appropriate wells. Seal the plate with parafilm to prevent evaporation and incubate overnight at 4 °C or at R.T. for 2 h.
Wash and blot the plate four times as in steps 2 and 3.
Add 100 μl/well of detection antibody diluted in Assay Diluent (as suggested by the manufacturer’s procedure).
Seal the plate and incubate at R.T. for 1 h.
Wash and blot the plate four times as in steps 2 and 3.
Add 100 μl/well of avidin-HRP diluted in Assay Diluent (as suggested by the manufacturer’s manual). Seal the plate and incubate at R.T. for 30 min.
Wash plate seven times. In this wash step, soak wells in wash buffer for 1–2 min before aspiration to minimize background.
Add 100 μl/well of TMB (substrate solution) to each well in the dark (use a multichannel pipette).
Incubate the plate at R.T. for 15 min in the dark. Positive wells should turn blue in color. It is not necessary to seal the plate during this step.
Add 50 μl of stop solution to each well. Positive wells should turn from blue to yellow.
Read absorbance at 450 and 570 nm (see Notes 15 and 16). A schematic representation is reported in Fig. 4.
Fig. 3.
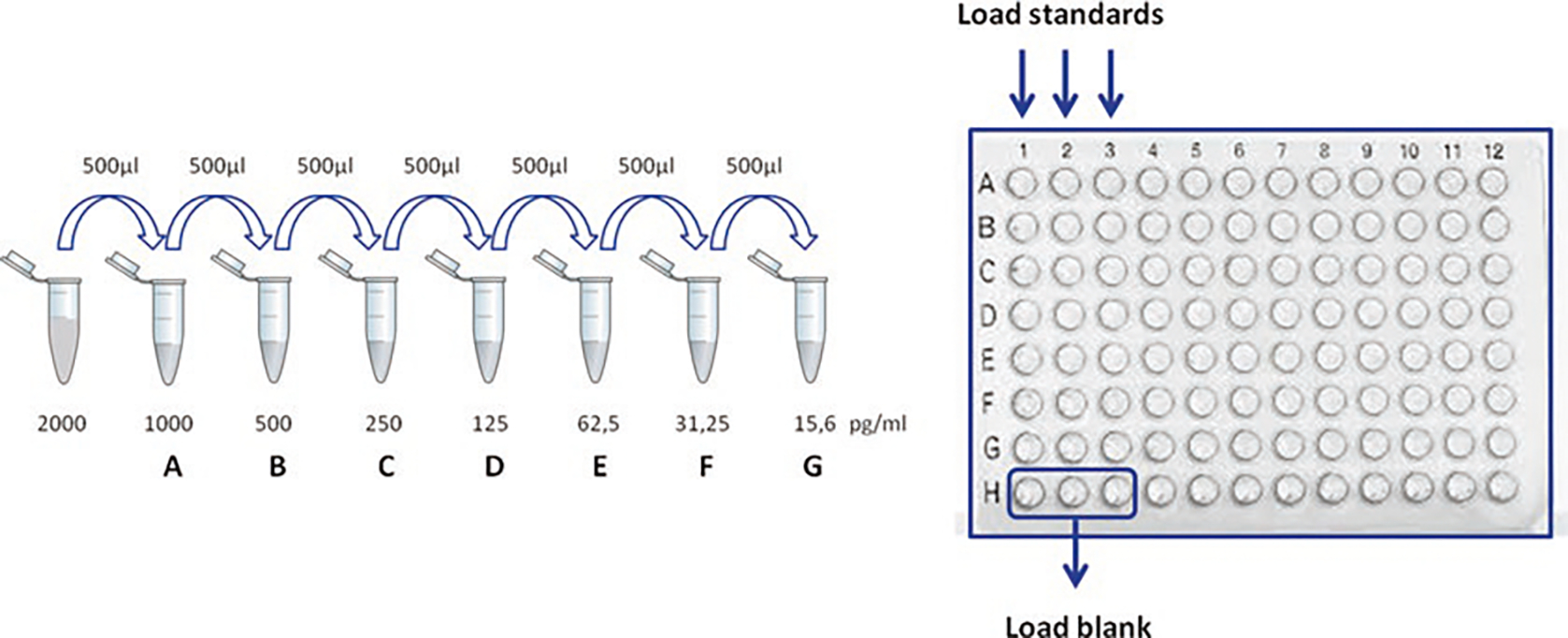
Representation of TNF-α standard dilutions and their loading in a 96-well plate. Put 500 μl of Assay Diluent 1× in seven Eppendorf tubes, from A to G, and perform twofold serial dilutions by transferring 500 μl from the top standard (2000 pg/ml) to tube A, then from A to B, and so on until G. Transfer 100 μl of diluted standards in the corresponding wells (in triplicate) and load Assay Diluent 1× only in three wells H
Fig. 4.
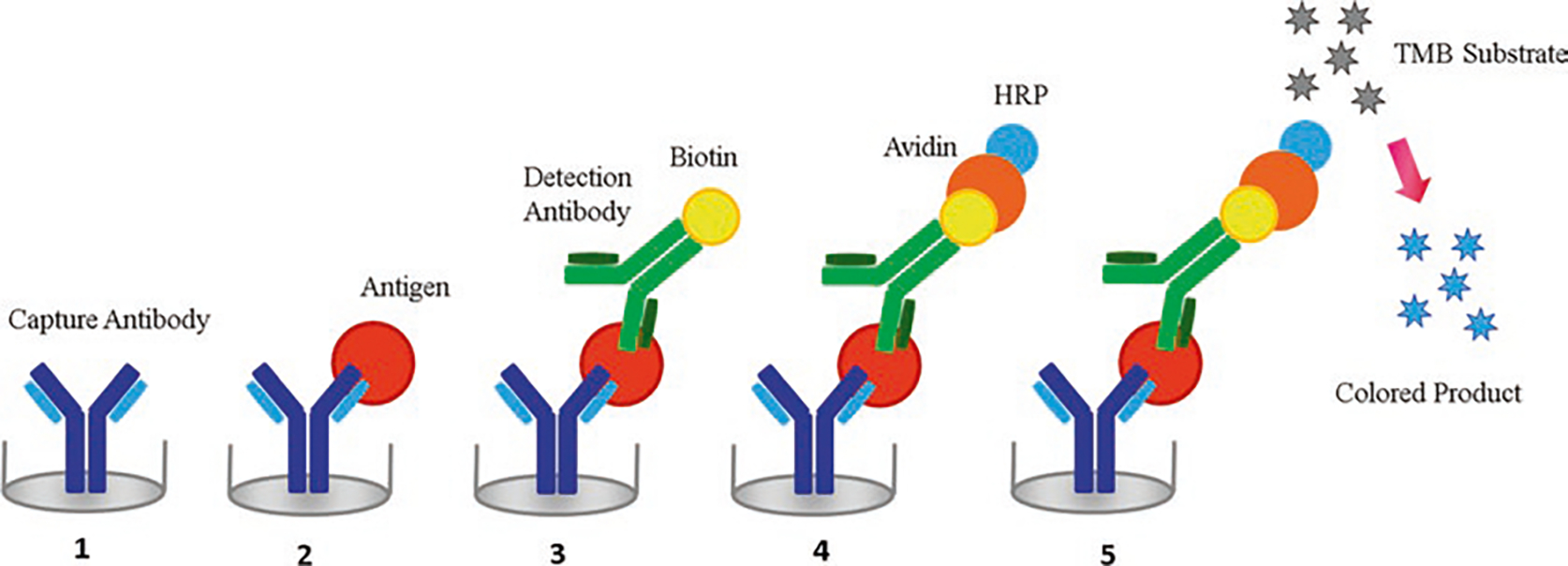
Schematic representation of a sandwich enzyme-linked immunosorbent assay (ELISA). The plate is coated with a suitable capture antibody (1). Then the sample is added and the antigen present is bound to the capture antibody (2). A suitable biotin-labeled detection antibody (BDAb) which binds to the antigen is added (3). Avidin-HRP binds BDAb (4). Finally, TMB substrate is added and converted to a detectable form
4. Notes
TrypLE Express 10× is designed for the detachment of cells with strong adhesive properties to plastic surfaces and can be used at 10× concentration or properly diluted depending on the cell type. TrypLE Express can be substituted by trypsin-EDTA, but make sure to use media containing 10 % FBS to stop the trypsin reaction.
If HBSS is made from scratch, sterilize it in a biological safety cabinet by filtering through a sterile filtration device fitted with 0.22 μm nitrocellulose filter. Store at +4 °C.
Be careful because MTT is toxic. Weigh the compound in the dark in a conical tube and solubilize it in HBSS by shaking with a vortex mixer. Sterilize it in a biological safety cabin by filtering through a sterile filtration device fitted with a 0.22 μm nitrocellulose filter and wrap the tube in aluminum foil. Fresh working solution can be stored at +4 °C for a few days.
The tear film can be gently collected also from the lateral canthus, while the patient can blink normally. The subject needs to look superior nasal (away from the collection site) and tilt the head toward the investigator.
Reflex tears are produced in response to stimulation and irritation of the cornea and conjunctiva or in response to emotions. Reflex tears are not simply a more diluted version of basal tears because the concentration of some components such as lysozyme is higher, whereas some interleukins but mainly mucins are more concentrated in basal tears than reflex.
It is important to optimize the assay with your particular strain. For example, in the case of P. aeruginosa, an optical density of 0.8 at 590 nm corresponds to 4 × 108 CFU/ml.
Soft agar differs from LB agar by having 0.75 % (w/v) agar concentration.
5 μl aliquots can be added into 6 ml of soft agar (previously put into 15 ml centrifuge tubes) and then spread on top of the agar plate. Leave the plate drying at R.T. for 10–20 min; then, invert the plate and incubate as desired.
When enumerating CFU, a proper number of countable bacterial cells are between 50 and 300 CFU per plate. Typically, a tenfold dilution series is prepared from the original sample, using a suitable diluent, such as PBS.
Incubation in a closed humidified incubator will help to avoid problems with plates drying out especially when working with slow-growing colonies.
Place the pipette tip close to the glass cover edge, previously put above the Neubauer or Burker chamber (see Fig. 5); release the plunger slowly watching how the liquid enters the chamber uniformly, being absorbed by capillarity. In case of bubble formation, rinse the chamber and repeat the loading process. If you have a very high concentration of cells, make a dilution. Start counting the cells in two big squares (each one contains 16 small squares, bounded by triple lines in Burker chamber or multiple lines in Neubauer chamber) (see Fig. 5). Repeat the operation for the other side of the chamber (Fig. 5). The equation used when counting cells in the big squares will be: concentration (cells/ml) = number of cells × 10,000/number of squares. In the case of a dilution, the concentration will be divided by the dilution applied.
Note that full solubilization may take a few hours. Incubation of the microplate at 37 °C and soft agitation for 1 h speed up the process.
If necessary, centrifuge cell culture supernatants to remove debris prior to analysis. After reconstitution of the lyophilized standard with Assay Diluent 1X, aliquot it into propylene tubes and store at −70 °C for up to 1 month. Avoid repeated freeze/thaw cycles.
Prepare TNF-α solution at a concentration of 2000 pg/ml, by dissolving the standard TNF-α provided by the kit in 10 ml of Assay Diluent 1X. Six twofold serial dilutions are recommended.
Read the plate within several minutes after adding the stop solution, not days/hours later. Once you have determined measurements at both 450 and 570 nm, subtract the values of 570 nm from those of 450 nm and plot the data (it reduces your background).
Plot the standard curve on log-log axis graph paper with TNF-α concentration on the x-axis and absorbance on the y-axis. Draw the best fit line through the standard points. Once you have determined the correct curve-fitting algorithm, you can transform the absorbance values to pg/ml. For example, if the standard curve is a linear line with the following equation y = 0.98x + 0.032 where y is the OD and x is the concentration of TNF-α, the unknown x values of your samples can be calculated from the corresponding OD measurements as follows: x = (OD value − 0.0325)/0.98. If the sample’s absorbance is outside the standard curve range, the sample needs to be reanalyzed at a higher or lower dilution, as appropriate.
Fig. 5.
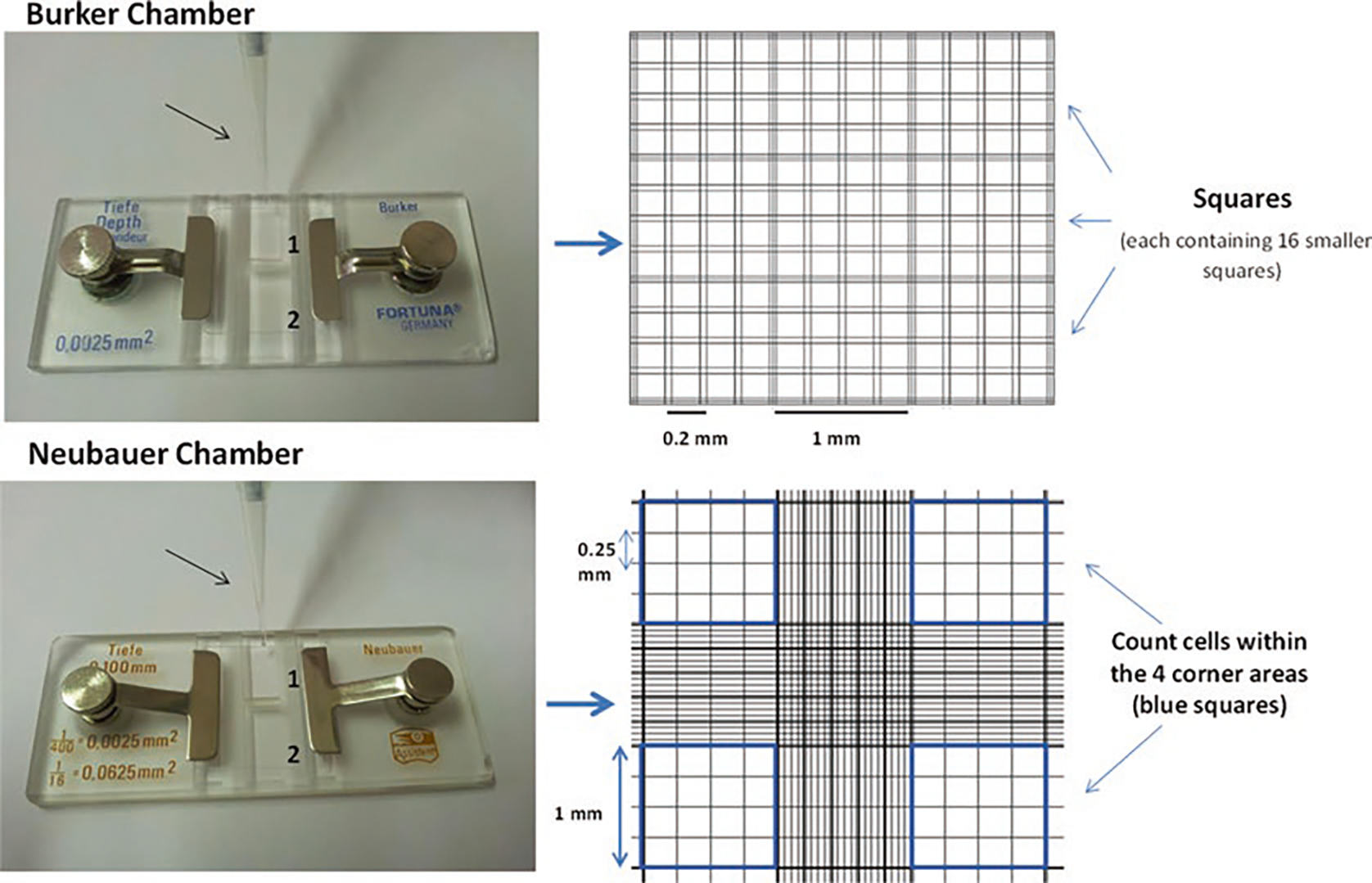
Representation of a Burker and a Neubauer chamber. Left side: using a micropipette tip (indicated by the arrow), inject 10 μl of cell suspension under the cover glass (previously put over the chamber). It is recommended to count both sides (1 and 2) of the chamber. Right side: typical grids of Burker and Neubauer chambers, showing squares, each of which is 1 mm long and contains 16 smaller squares
Acknowledgments
This work was supported grants from Sapienza Università di Roma (MLM) and NIH grant EY13175 (AMM). Part of the content of this work is object of a US patent application No. 14/506,383.
References
- 1.Keay L, Edwards K, Naduvilath T, Taylor HR, Snibson GR, Forde K, Stapleton F (2006) Microbial keratitis predisposing factors and morbidity. Ophthalmology 113:109–116 [DOI] [PubMed] [Google Scholar]
- 2.Thomas PA, Geraldine P (2007) Infectious keratitis. Curr Opin Infect Dis 20:129–141 [DOI] [PubMed] [Google Scholar]
- 3.Hsu HY, Nacke R, Song JC, Yoo SH, Alfonso EC, Israel HA (2010) Community opinions in the management of corneal ulcers and ophthalmic antibiotics: a survey of 4 states. Eye Contact Lens 36:195–200 [DOI] [PubMed] [Google Scholar]
- 4.Matthews TD, Frazer DG, Minassian DC, Radford CF, Dart JK (1992) Risks of keratitis and patterns of use with disposable contact lenses. Arch Ophthalmol 110:1559–1562 [DOI] [PubMed] [Google Scholar]
- 5.Evans DJ, Fleiszig SM (2013) Why does the healthy cornea resist Pseudomonas aeruginosa infection? Am J Ophthalmol 155:961–970.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butovich IA (2013) Tear film lipids. Exp Eye Res 117:4–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H et al. (1994) Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J 8:217–225 [DOI] [PubMed] [Google Scholar]
- 8.Trent MS, Stead CM, Tran AX, Hankins JV (2006) Diversity of endotoxin and its impact on pathogenesis. J Endotoxin Res 12:205–223 [DOI] [PubMed] [Google Scholar]
- 9.Rosenfeld Y, Shai Y (2006) Lipopolysaccharide (Endotoxin)-host defense antibacterial peptides interactions: role in bacterial resistance and prevention of sepsis. Biochim Biophys Acta 1758:1513–1522 [DOI] [PubMed] [Google Scholar]
- 10.Jett BD, Hatter KL, Huycke MM, Gilmore MS (1997) Simplified agar plate method for quantifying viable bacteria. Biotechniques 23:648–650 [DOI] [PubMed] [Google Scholar]
- 11.Koch AC (1994) Growth measurement. In: Gerhardt P, Murray RGE, Wood WA, Krieg NR (eds) Methods for general and molecular bacteriology. ASM Press, Washington, DC, pp 254–257 [Google Scholar]
- 12.Kiderlen AF, Kaye PM (1990) A modified colorimetric assay of macrophage activation for intracellular cytotoxicity against Leishmania parasites. J Immunol Methods 127:11–18 [DOI] [PubMed] [Google Scholar]
- 13.van de Loosdrecht AA, Beelen RH, Ossenkoppele GJ, Broekhoven MG, Langenhuijsen MM (1994) A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J Immunol Methods 174:311–320 [DOI] [PubMed] [Google Scholar]
- 14.Gerlier D, Thomasset N (1986) Use of MTT colorimetric assay to measure cell activation. J Immunol Methods 94:57–63 [DOI] [PubMed] [Google Scholar]
- 15.Robertson DM, Li L, Fisher S, Pearce VP, Shay JW, Wright WE, Cavanagh HD, Jester JV (2005) Characterization of growth and differentiation in a telomerase-immortalized human corneal epithelial cell line. Invest Ophthalmol Vis Sci 46:470–478 [DOI] [PubMed] [Google Scholar]