

Abstract
Large-scale studies of linkage disequilibrium (LD) have shown considerable variation in the extent and distribution of pairwise LD within and between populations. Taken at face value, these results suggest that genomewide LD maps for one population may not be generalizable to other populations. However, at least part of this diversity is due to some undesirable features of pairwise LD measures, which are well documented for the D′ and r2 measures. In this report, we compare patterns of LD derived from pairwise measures with statistical estimates of population recombination rates (ρ) along a 10-Mb stretch of chromosome 20 in four population samples, comprising East Asians, African Americans, and U.K. and U.S. individuals of western European descent. The results reveal the expected variability of D′ within and between populations but show better concordance in estimates of r2 for the same markers across the population samples. Estimates of ρ correlate well across populations, but there is still evidence of population-specific spikes and troughs in ρ values. We conclude that it is unlikely that a single haplotype map will provide a definitive guide for association studies of many populations; rather, multiple maps will need to be constructed to provide the best-possible guides for gene mapping.
The search for the genetic basis of complex traits and diseases through genomewide linkage approaches has been, by and large, disappointing (Freimer and Sabatti 2004). This has led to the idea that a more promising strategy would be to perform genomewide association analysis (Risch and Merikangas 1996; Kruglyak 1999; Risch 2000). Although it is currently unfeasible to type every polymorphism in the human genome, it should be possible—because of the presence of linkage disequilibrium (LD)—to genotype a relatively small set of variants that capture most of the common patterns of variation in the genome (Johnson et al. 2001). This is the basic premise of the International Haplotype Mapping Project (HapMap), which aims to characterize the distribution and extent of LD across the entire human genome (Gibbs et al. 2003). It is hoped that HapMap will provide enough information about LD to facilitate association analysis of candidate genes and regions previously identified by linkage analysis, as well as to enable genomewide association analysis.
One of the critical unknowns in association mapping—and for HapMap, in general—is the extent to which patterns of LD are conserved across populations. This question is important because its answer will determine whether one or a few haplotype maps can provide useful information for additional populations or whether separate maps need to be constructed for different populations. Allele frequencies at individual markers have long been known to vary widely across populations (Cavalli-Sforza et al. 1994), and variation in LD between specific pairs of markers can be extremely high (reviewed by Weiss and Clark [2002]). Recent large-scale SNP studies have further highlighted this enormous variability, but they have also shown that some generalizations are possible (Abecasis et al. 2001; Daly et al. 2001; Patil et al. 2001; Dawson et al. 2002; Gabriel et al. 2002; Phillips et al. 2003). For example, the decay of LD with increasing physical distance tends to be faster in samples from Africa than in those from Asia or Europe (Tishkoff et al. 1996, 1998, 2000; Kidd et al. 1998, 2000; Frisse et al. 2001; Reich et al. 2001; Ke et al. 2004). Similarly, broad views of LD tend to be stable across populations, as is apparent in sliding-window plots of pairwise measures of LD (Ke et al. 2004). However, these coarse measures of LD are only useful for obtaining an overall impression of the amount of LD in a region; they do not provide direct guidance for fine-scale association mapping. Of greater practical relevance is the comparison of fine-scale measures of LD across different populations.
We examined genotypes for four population samples along a 10-Mb stretch of chromosome 20q12-20q13.13. The population samples consisted of (1) 96 unrelated white individuals of western European ancestry (U.K. unrelateds), (2) 97 unrelated African American individuals, (3) 42 Asian individuals (32 Japanese and 10 Chinese), and (4) 46 founder individuals from 12 Centre d'Etude du Polymorphisme Humain (CEPH) pedigrees. Comparisons between the unrelated U.K., Asian, and African American samples were based on 4,107 SNPs common to all three populations. Comparisons between the CEPH and unrelated U.K. samples were based on 4,456 common SNPs. Full details of the genotyping have been provided elsewhere (Ke et al. 2004).
To investigate the variability associated with fine-scale measures of LD across different populations, we calculated pairwise LD measures (r2 and D′) between adjacent markers in all four populations. Note that we examined only adjacent markers, to allow direct comparison of the LD values with point estimates of recombination rates. The correlations between the different populations for D′ and r2 are displayed in figure 1, illustrating the variability in LD between the different populations. For markers at the ∼2-kb density used here, D′ values often reach their maximum value of 1.0 in one or both populations, with the vast majority of points clustering in the high-LD quadrant. This ceiling effect is largely responsible for the low rank correlation between populations for the D′ measure (table 1). In contrast, the correspondence between populations for r2 is more broadly variable, with clusters of both high- and low-LD marker pairs. The r2 measure appears to be less influenced by a ceiling effect and correlates better between the different population samples (table 1). The high variability between the different populations suggests that, by themselves, pairwise measures of LD will be of limited use in association mapping. However, pairwise LD can appear variable even in comparisons of two groups of similar ancestry (fig. 2). For example, comparison of the CEPH and U.K. unrelated samples reveals high correspondence in r2 values but much more variability in D′ values (Spearman rank correlations, ρ[r2]=0.95 and ρ[D′]=0.44).
Figure 1 .

Comparisons of D′ and r2 values between populations. The scatter plots show LD values for all adjacent marker pairs, which are separated by 2.3 kb, on average.
Table 1.
Spearman Rank Correlations between LD Measures and ρ Estimates for Chromosome 20
| ρ |
r2 |
D′ |
||||||
| Measureand Sample | U.K.Unrelated | Asian | AfricanAmerican | U.K.Unrelated | Asian | AfricanAmerican | U.K.Unrelated | Asian |
| ρ: | ||||||||
| Asian | .85 | |||||||
| African American | .82 | .82 | ||||||
| r2: | ||||||||
| U.K. unrelated | −.19 | −.18 | −.18 | |||||
| Asian | −.17 | −.21 | −.19 | .76 | ||||
| African American | −.17 | −.20 | −.22 | .77 | .73 | |||
| D′: | ||||||||
| U.K. unrelated | −.24 | −.21 | −.23 | .54 | .42 | .43 | ||
| Asian | −.15 | −.19 | −.17 | .40 | .55 | .39 | .40 | |
| African American | −.22 | −.22 | −.30 | .39 | .38 | .53 | .42 | .39 |
Figure 2.

Comparison of r2 (left) and D′ (right) values between the CEPH and unrelated U.K. unrelated samples
These results reflect the well-known variability of pairwise LD within populations (Hill and Weir 1994; Watkins et al. 1994; Weiss and Clark 2002) and highlight the possibility that the lack of concordance between the different groups may not be entirely due to their different ancestries but rather may be a consequence of the measures themselves. In particular, pairwise measures of LD have a number of confounding properties for disease-gene mapping—most notably, their dependence on allele frequencies (Hedrick 1987). Since different populations often differ in the allele frequencies of SNPs, pairwise measures of LD may not be the best measures to compare patterns of LD across different populations.
Recently, several groups have developed computational procedures for estimating population recombination rates (ρ) from unphased genotype data: ρ=4Ner, where Ne is the effective population size and r is the recombination rate between a pair of sites (Fearnhead and Donnelly 2001; Hudson 2001; Li and Stephens 2003; McVean et al. 2004). For exploratory association analyses, such as genomewide scans, or for assessments of candidate regions, estimates of ρ have a number of advantages over pairwise measures of LD: they relate the observed patterns of LD directly to the underlying recombination process, they consider all loci simultaneously rather than just pairwise, and they make it possible to compare estimates from studies that have used different types of markers (Pritchard and Przeworski 2001). Preliminary work has also suggested that, compared with pairwise measures of LD, estimates of ρ are more robust to SNP ascertainment strategy, as well as to marker-density and allele-frequency differences (Clark et al. 2003; Li and Stephens 2003; Nielsen and Signorovitch 2003; Crawford et al. 2004; McVean et al. 2004). Estimates of ρ derived from these statistical approaches appear to be similar to estimates derived from simulated data, pedigree studies, and sperm-typing experiments (Li and Stephens 2003; Crawford et al. 2004; McVean et al. 2004).
We used the program PHASE 2.0 (Stephens et al. 2001; Li and Stephens 2003; Stephens and Donnelly 2003) to estimate ρ values across the chromosome 20q12-20q13.13 region in all four populations. We divided the chromosome 20q12-20q13.13 region into nonoverlapping bins of 70 SNPs. Since PHASE 2.0 calculates ρ between adjacent SNPs, each bin was associated with 69 intervals. For each interval, we performed 20,000 iterations after an initial burn-in of 100 iterations and took the median of every 10th estimate.
Figure 3 displays ρ values across U.K. unrelated, Asian, and African American groups. In the present data, the average levels of ρ vary substantially between populations (e.g., African American 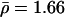 ; Asian
; Asian 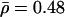 ; and U.K. unrelated
; and U.K. unrelated 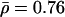 ), but chromosomewide patterns of recombination are highly correlated, in that the low and high values of ρ in one population tend to occur in the same chromosomal regions as the low and high values in other populations (see table 1 for the rank correlations). These results reveal a very close correspondence in rates across populations.
), but chromosomewide patterns of recombination are highly correlated, in that the low and high values of ρ in one population tend to occur in the same chromosomal regions as the low and high values in other populations (see table 1 for the rank correlations). These results reveal a very close correspondence in rates across populations.
Figure 3.

Estimated ρ values along a 10-Mb region of chromosome 20. As a reference point for the basal parameter values, for an average rate of 1 cM/Mb and an effective population size of 10,000, the value of ρ per kb would be 0.4. Each series of points shows the estimates of log10(ρ) per bp. To separate the curves on the Y-axis, arbitrary constants (c) of 6 and 3 were added to individual estimates for the U.K. unrelated and Asian samples, respectively. A, The entire 10-Mb region of chromosome 20. B, Bin of 70 SNPs in which the correlation in ρ between population samples is decreased as a result of population-specific hotspots. C, Bin of 70 SNPs in which the correlation in ρ between population samples is low. Notice that there is little variation in the background levels of ρ. D, Bin of 70 SNPs in which the correlation in ρ between population samples is high. Note both the large variation in ρ across the region and the similar positions of the troughs and spikes in the three population samples. Similar results were obtained using the LDhat program of McVean et al. (2004).
Even though the overall correlations in ρ between the different populations are high, this result conceals substantial heterogeneity, in that some areas of the 10-Mb region exhibit lower correlations than others (fig. 3). In general, the correlation is high in areas in which ρ estimates vary substantially (i.e., they contain many large spikes and troughs) and line up well across populations (fig. 3D). In contrast, the correlation tends to be lower in areas in which there is little variation in the background ρ across the region (fig. 3C). These results are perhaps not surprising, since the model for recombination implemented in PHASE 2.0 (which allows for variation in ρ along the genome) is particularly suited to identifying “hot spots” and “cold spots” of recombination rather than yielding precise estimates of small fluctuations in the background ρ (Li and Stephens 2003).
There are also several population-specific spikes or troughs throughout the region, suggesting that there may be differences in recombination history between different populations (or, alternatively, differences in historical factors affecting the effective population size in local regions of the genome). For example, in figure 3B, there is a large peak at ∼38.45 Mb in the African American and U.K. unrelated samples but not in the Asian sample. Notice also how a spike in the Asian sample is displaced slightly telomeric relative to the peak at ∼38.39 Mb in the African American sample. It is worth noting that, even though these spikes may reflect the same underlying recombination hotspot, they will still lower the correlation in ρ between population samples because they are displaced (inspection of the data revealed that there were many instances in which this was the case).
Similar findings were reported by Crawford et al. (2004), who typed 74 genes in two different populations (an African American and a European American population) and found 16 genes that showed evidence of a recombination hotspot in one sample but not in the other (19 genes showed evidence of a hotspot in both populations, and the remaining 39 lacked hotspots in both populations). Similarly, Clark et al. (2003) estimated ρ in 538 small clusters of SNPs across the genome in Asian, European American, and African American samples. Whereas there was a high correlation in ρ between the different population groups, the authors also noted the existence of several population-specific troughs and spikes.
Finally, in figure 4, we display estimates of ρ along the first 750 kb of the 10-Mb region for the two groups of similar background (i.e., the CEPH and U.K. unrelated samples). There is high similarity not only in the location of the troughs and spikes but also in the background ρ (Spearman rank correlation 0.90). The results in figure 4 are representative of the entire 10-Mb region and strongly suggest that a single fine-scale recombination map may provide useful information on samples drawn from populations of close ancestry.
Figure 4.

Estimated ρ values along the first 750 kb of the 10-Mb region in CEPH and U.K. unrelated individuals. The unbroken line represents estimates of ρ for CEPH individuals, and the broken line represents estimates of ρ for U.K. unrelated individuals.
One potential weakness of our method is the existence of a possible ascertainment bias against rare SNPs, which might influence estimates of ρ (Clark et al. 2003; Nielsen and Signorovitch 2003). Most of the SNPs genotyped here were initially identified via shotgun sequencing of flow-sorted chromosome 20 DNA from four individuals (one U.K. unrelated individual, one East Asian, one African American, and one African pygmy), augmented by a small proportion of SNPs from an early version of dbSNP (release 114) (see the study by Ke et al. [2004]). Because the SNPs typed in the present study were first identified in a small population of individuals, it is likely that these SNPs predominantly represent common polymorphisms. Since common SNPs tend to be older and have had more chances to recombine, it is likely that the estimates of ρ in our study are inflated. This ascertainment may also be partially responsible for the high correlation between different populations in estimates of ρ. Clark et al. (2003) and Nielsen and Signorovitch (2003) have developed methods to formally assess such effects, which will form the basis of a subsequent investigation.
In summary, we have examined three different statistics—two pairwise measures of LD and one estimate of ρ—and their correlations across four different population samples. Our results have highlighted the well-known variability in pairwise estimates of LD across the genome (Hill and Weir 1994; Watkins et al. 1994; Weiss and Clark 2002). Our results show that D′ correlates poorly between populations, suggesting that individual estimates of pairwise D′ are likely to be of limited use in guiding association mapping in different populations. In contrast, pairwise r2 correlates much better between the population samples (despite its dependence on allele frequency), suggesting that this simple measure may carry useful information for transpopulation disease-gene mapping.
Our results also show that estimates of ρ correlate well across populations. Unlike pairwise measures of LD, estimates of ρ directly relate observed patterns of LD to the underlying recombination process (Pritchard and Przeworski 2001). Thus, a fine-scale recombination map of the genome may be more useful for guiding association studies of multiple populations than maps based on pairwise LD patterns. For example, it is often the case that a relatively large genomic region is identified by linkage and association analysis. Since areas of high recombination may delineate boundaries of genes and may indicate how far to extend the search for functional variants (Cardon and Abecasis 2003), a fine-scale recombination map may be of some use in guiding fine-scale mapping in different population samples. Given the high degree of similarity between estimates of ρ for the CEPH and U.K. unrelated samples, a haplotype map may be particularly useful for guiding association mapping in groups of similar ancestry. However, we emphasize that, even though the overall correlation between estimates of ρ in different populations was quite high, there was still considerable evidence for population-specific spikes and troughs in the ρ estimates (Clark et al. 2003; Crawford et al. 2004). Thus, it is unlikely that a single haplotype map will provide a definitive guide for association studies in many populations; rather, multiple maps will need to be constructed to provide the best-possible guides for gene mapping. Of course, we have only reported the results for one small region of chromosome 20. It remains to be seen whether these results will hold true for the rest of the genome.
Acknowledgments
This work was supported by the Wellcome Trust, the SNP Consortium, the National Institutes of Health (grant EY-126562 [to L.R.C]), and the Medical Research Council (grant G9801327). We thank Panos Deloukas and David Bentley, at the Wellcome Sanger Institute, for the chromosome 20 data. We also thank Andy Clark and James Signorovitch, for useful discussions, and two anonymous reviewers, for their helpful comments.
References
- Abecasis GR, Noguchi E, Heinzmann A, Traherne JA, Bhattacharyya S, Leaves NI, Anderson GG, Zhang Y, Lench NJ, Carey A, Cardon LR, Moffatt MF, Cookson WOC (2001) Extent and distribution of linkage disequilibrium in three genomic regions. Am J Hum Genet 68:191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardon LR, Abecasis GR (2003) Using haplotype blocks to map human complex trait loci. Trends Genet 19:135–140 10.1016/S0168-9525(03)00022-2 [DOI] [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Menozzi P, Piazza A (1994) History and geography of human genes. Princeton University Press, Princeton [Google Scholar]
- Clark AG, Nielsen R, Signorovitch J, Matise TC, Glanowski S, Heil J, Winn-Deen ES, Holden AL, Lai E (2003) Linkage disequilibrium and inference of ancestral recombination in 538 single-nucleotide polymorphism clusters across the human genome. Am J Hum Genet 73:285–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Bhangale T, Li N, Hellenthal G, Rieder MJ, Nickerson DA, Stephens M (2004) Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat Genet 36:700–706 10.1038/ng1376 [DOI] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES (2001) High-resolution haplotype structure in the human genome. Nat Genet 29:229–232 10.1038/ng1001-229 [DOI] [PubMed] [Google Scholar]
- Dawson E, Abecasis GR, Bumpstead S, Chen Y, Hunt S, Beare DM, Pabial J, et al (2002) A first-generation linkage disequilibrium map of human chromosome 22. Nature 418:544–548 10.1038/nature00864 [DOI] [PubMed] [Google Scholar]
- Fearnhead P, Donnelly P (2001) Estimating recombination rates from population genetic data. Genetics 159:1299–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freimer N, Sabatti C (2004) The use of pedigree, sib-pair and association studies of common diseases for genetic mapping and epidemiology. Nat Genet 36:1045–1051 10.1038/ng1433 [DOI] [PubMed] [Google Scholar]
- Frisse L, Hudson RR, Bartoszewicz A, Wall JD, Donfack J, Di Rienzo A (2001) Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am J Hum Genet 69:831–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D (2002) The structure of haplotype blocks in the human genome. Science 296:2225–2229 10.1126/science.1069424 [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium (2003) The International HapMap Project. Nature 426:789–796 10.1038/nature02168 [DOI] [PubMed] [Google Scholar]
- Hedrick PW (1987) Gametic disequilibrium measures: proceed with caution. Genetics 117:331–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Weir BS (1994) Maximum-likelihood estimation of gene location by linkage disequilibrium. Am J Hum Genet 54:705–714 [PMC free article] [PubMed] [Google Scholar]
- Hudson RR (2001) Two-locus sampling distributions and their application. Genetics 159:1805–1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GC, Esposito L, Barratt BJ, Smith AN, Heward J, Di Genova G, Ueda H, Cordell HJ, Eaves IA, Dudbridge F, Twells RC, Payne F, Hughes W, Nutland S, Stevens H, Carr P, Tuomilehto-Wolf E, Tuomilehto J, Gough SC, Clayton DG, Todd JA (2001) Haplotype tagging for the identification of common disease genes. Nat Genet 29:233–237 10.1038/ng1001-233 [DOI] [PubMed] [Google Scholar]
- Ke X, Hunt S, Tapper W, Lawrence R, Stavrides G, Ghori J, Whittaker P, Collins A, Morris AP, Bentley D, Cardon LR, Deloukas P (2004) The impact of SNP density on fine-scale patterns of linkage disequilibrium. Hum Mol Genet 13:577–588 10.1093/hmg/ddh060 [DOI] [PubMed] [Google Scholar]
- Kidd JR, Pakstis AJ, Zhao H, Lu R-B, Okonofua FE, Odunsi A, Grigorenko E, Bonne-Tamir B, Friedlaender J, Schulz LO, Parnas J, Kidd KK (2000) Haplotypes and linkage disequilibrium at the phenylalanine hydroxylase locus, PAH, in a global representation of populations. Am J Hum Genet 66:1882–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd KK, Morar B, Castiglione CM, Zhao H, Pakstis AJ, Speed WC, Bonne-Tamir B, Lu R-B, Goldman D, Lee C, Nam YS, Grandy DK, Jenkins T, Kidd JR (1998) A global survey of haplotype frequencies and linkage disequilibrium at the DRD2 locus. Hum Genet 103:211–227 10.1007/s004390050809 [DOI] [PubMed] [Google Scholar]
- Kruglyak L (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 22:139–144 10.1038/9642 [DOI] [PubMed] [Google Scholar]
- Li N, Stephens M (2003) Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics 165:2213–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GA, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P (2004) The fine-scale structure of recombination rate variation in the human genome. Science 304:581–584 10.1126/science.1092500 [DOI] [PubMed] [Google Scholar]
- Nielsen R, Signorovitch J (2003) Correcting for ascertainment biases when analyzing SNP data: application to the estimation of linkage disequilibrium. Theor Popul Biol 63:245–255 10.1016/S0040-5809(03)00005-4 [DOI] [PubMed] [Google Scholar]
- Patil N, Berno AJ, Hinds DA, Barrett WA, Doshi JM, Hacker CR, Kautzer CR, Lee DH, Marjoribanks C, McDonough DP, Nguyen BT, Norris MC, Sheehan JB, Shen N, Stern D, Stokowski RP, Thomas DJ, Trulson MO, Vyas KR, Frazer KA, Fodor SP, Cox DR (2001) Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 294:1719–1723 10.1126/science.1065573 [DOI] [PubMed] [Google Scholar]
- Phillips MS, Lawrence R, Sachidanandam R, Morris AP, Balding DJ, Donaldson MA, Studebaker JF, et al (2003) Chromosome-wide distribution of haplotype blocks and the role of recombination hot spots. Nat Genet 33:382–387 10.1038/ng1100 [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Przeworski M (2001) Linkage disequilibrium in humans: models and data. Am J Hum Genet 69:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, Lander ES (2001) Linkage disequilibrium in the human genome. Nature 411:199–204 10.1038/35075590 [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 [DOI] [PubMed] [Google Scholar]
- Risch NJ (2000) Searching for genetic determinants in the new millennium. Nature 405:847–856 10.1038/35015718 [DOI] [PubMed] [Google Scholar]
- Stephens M, Donnelly P (2003) A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73:1162–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Dietzsch E, Speed W, Pakstis AJ, Kidd JR, Cheung K, Bonne-Tamir B, Santachiara-Benerecetti AS, Moral P, Krings M (1996) Global patterns of linkage disequilibrium at the CD4 locus and modern human origins. Science 271:1380–1387 [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Goldman A, Calafell F, Speed WC, Deinard AS, Bonne-Tamir B, Kidd JR, Pakstis AJ, Jenkins T, Kidd KK (1998) A global haplotype analysis of the myotonic dystrophy locus: implications for the evolution of modern humans and for the origin of myotonic dystrophy mutations. Am J Hum Genet 62:1389–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Pakstis AJ, Stoneking M, Kidd JR, Destro-Bisol G, Sanjantila A, Lu R-b, Deinard AS, Sirugo G, Jenkins T, Kidd KK, Clark AG (2000) Short tandem-repeat polymorphism/Alu haplotype variation at the PLAT locus: implications for modern human origins. Am J Hum Genet 67:901–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins WS, Zenger R, O’Brien E, Nyman D, Eriksson AW, Renlund M, Jorde LB (1994) Linkage disequilibrium patterns vary with chromosomal location: a case study from the von Willebrand factor region. Am J Hum Genet 55:348–355 [PMC free article] [PubMed] [Google Scholar]
- Weiss KM, Clark AG (2002) Linkage disequilibrium and the mapping of complex human traits. Trends Genet 18:19–24 10.1016/S0168-9525(01)02550-1 [DOI] [PubMed] [Google Scholar]