

Abstract
Deviations from Hardy-Weinberg equilibrium (HWE) can indicate inbreeding, population stratification, and even problems in genotyping. In samples of affected individuals, these deviations can also provide evidence for association. Tests of HWE are commonly performed using a simple χ2 goodness-of-fit test. We show that this χ2 test can have inflated type I error rates, even in relatively large samples (e.g., samples of 1,000 individuals that include ∼100 copies of the minor allele). On the basis of previous work, we describe exact tests of HWE together with efficient computational methods for their implementation. Our methods adequately control type I error in large and small samples and are computationally efficient. They have been implemented in freely available code that will be useful for quality assessment of genotype data and for the detection of genetic association or population stratification in very large data sets.
In the absence of migration, mutation, natural selection, and assortative mating, genotype frequencies at any locus are a simple function of allele frequencies. This phenomenon, now termed “Hardy-Weinberg equilibrium” (HWE), was first described in the early part of the twentieth century (Hardy 1908; Weinberg 1908). The original descriptions of HWE are an important landmark in the history of population genetics (Crow 1988), and it is now common practice to check whether observed genotypes conform to Hardy-Weinberg expectations. These expectations appear to hold for most human populations, and deviations from HWE at particular markers may suggest problems with genotyping or population structure or, in samples of affected individuals, an association between the marker and disease susceptibility.
Here, we describe efficient implementations of exact tests for HWE, which are suitable for use in large-scale studies of SNP data, even when hundreds of thousands of markers are examined. The availability of data on patterns of linkage disequilibrium across the genome (International HapMap Consortium 2003), interest in identifying susceptibility alleles for complex diseases (Cardon and Abecasis 2003), and advances in genotyping technology (Kwok 2001; Weber and Broman 2001) suggest that such large studies will be increasingly common. The principles and procedures used for testing HWE are well established (Levene 1949; Haldane 1954; Hernandez and Weir 1989; Wellek 2004), but the lack of a publicly available, efficient, and reliable implementation for exact tests has led many scientists to rely on asymptotic tests that can perform poorly with realistic sample sizes.
Consider a sample of SNP genotypes for N unrelated diploid individuals measured at an autosomal locus. The sample includes 2N alleles, including nA copies of the rarer allele and nB copies of the common allele. Let the number of heterozygous AB genotypes be nAB, and note that the numbers of AA and BB homozygous genotypes are nAA=(nA-nAB)/2 and nBB=(nB-nAB)/2. Note that there are (2N)!/nA!nB! possible arrangements for the alleles in the sample and that 2nABN!/(nAA!nAB!nBB!) of these arrangements correspond to exactly nAB heterozygotes. Thus, under the assumption of HWE, the probability of observing exactly nAB heterozygotes in a sample of N individuals with nA minor alleles is
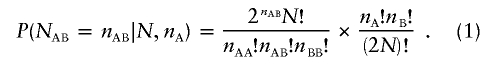 |
This equation holds for each possible number of heterozygotes, nAB. When nA is odd, possible numbers of heterozygotes are 1, 3, 5,…,nA. When nA is even, possible numbers of heterozygotes are 0, 2, 4,…,nA. The expression for P(nAB|N,nA) given in equation (1) leads to natural tests for HWE. For example, one could define one-sided tests that focus on detection of a deficit of heterozygotes, by calculating the statistic Plow=P(NAB⩽nAB|N,nA), or detection of an excess of heterozygotes, by calculating the statistic Phigh=P(NAB⩾nAB|N,nA). In each case, the statistic can be calculated by simply summing over equation (1), to include all possible values of NAB that are lower (for Plow) or higher (for Phigh) than those observed in the actual data. A test for a deficit of heterozygotes in relation to Hardy-Weinberg expectations is appropriate when deviations from HWE due to inbreeding or population stratification are suspected, since both of these increase the proportion of homozygotes in the population. A test for an excess of heterozygotes is appropriate when one suspects problems in genotyping due to the existence of highly homologous regions in the genome, since these low-copy repeats often lead to an increase in the proportion of apparent heterozygotes in the sample. In other settings, it might be appropriate to use both tests. For example, many technologies score genotypes by clustering signals, and misspecified clusters can result in either vast excesses or vast deficits of heterozygotes.
When neither an increase nor a decrease in the proportion of heterozygotes is specifically expected, one could perform two separate one-sided tests or, instead, use a two-sided test statistic (Weir 1996). A natural two-sided test statistic could be defined as P2α=min(1.0,2Phigh,2Plow). This two-sided statistic is appealing because it leads to rejection of HWE at significance level 2α in instances in which the one-sided tests lead to the rejection of HWE at significance level α. However, because of the asymmetric nature of the distribution of heterozygote counts in a sample, the statistic is quite conservative in practice, and we do not recommend its use. Instead, an appealing approach, analogous to Fisher’s exact test for contingency tables (Fisher 1934), is to calculate the probability of observing a sample configuration that is even less likely than the one being evaluated, conditional on the observed allele counts. This can be achieved using a statistic similar to the Monte Carlo statistic proposed by Guo and Thompson (1992) for multiallelic markers:
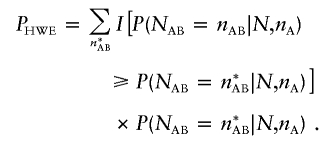 |
In this definition, I[x] is an indicator function that is equal to 1 when the comparison is true and equal to 0 otherwise. The sum should be performed over all heterozygote counts n*AB that are compatible with the observed number of minor alleles, nA.
Most of the computational effort required for performing exact tests of linkage disequilibrium is spent evaluating the factorials in equation (1) for each possible value of nAB. By use of a naive approach, evaluating equation (1) requires 5N–6N multiplications and one division for each possible value of nAB. We simplify calculations by using the recurrence relationships previously recognized by Guo and Thompson (1992) in the implementation of their Markov chain–Monte Carlo sampler:
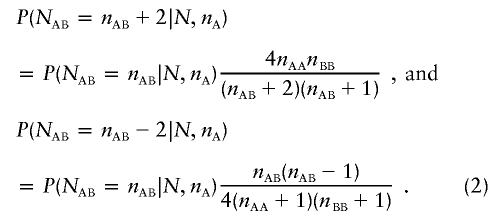 |
In this way, evaluating the probability for each possible number of heterozygotes takes only four multiplications and one division, whatever the sample size N. To avoid underflow, it is best to first calculate the probability of observing the expected number of heterozygotes (in this case, the most likely outcome) and then use the recurrence relationships to calculate probabilities for all other outcomes. A further reduction of computational effort is possible by noting that one need only calculate relative probabilities for each outcome and then scale these to ensure that their sum is 1.0. This means that the probability of observing the expected number of heterozygotes can be replaced with an arbitrary constant when using the recurrence relations in equation (2), provided that the final result is scaled.
Table 1 illustrates the performance of the statistics for a sample of 100 individuals in which 21 copies of the minor allele are present. The observed number of heterozygotes will vary from 1 to 21 and must be odd. Note that only a small number of distinct sample configurations are possible, and each of these is associated with a specific probability for the exact tests. If the desired significance level α does not correspond exactly to one of these discrete outcomes, then the exact test statistics will be conservative (Hernandez and Weir 1989). For example, at the significance level α=0.05, the PHWE and Plow statistics both reject the hypothesis of HWE if ⩽13 heterozygotes are observed in this setting. Since the probability of observing ⩽13 heterozygotes is 0.010, the tests are conservative. In contrast, the asymptotic χ2 test statistic results in rejection of HWE when ⩽15 heterozygotes are observed (for ⩽15 heterozygotes, the χ2 test statistic corresponds to an asymptotic P⩽.045). This results in an inflated type I error rate of 0.070 and therefore is inappropriate. In this sample, it is not possible to reject HWE because of an excess of heterozygous individuals—the probability of observing the maximum of 21 heterozygotes is 0.31, and none of the test statistics gives a P value <.05 for this extreme configuration. Additional examples of the performance of exact test statistics for HWE can be found in the work by Vithayasai (1973).
Table 1.
Possible Sample Configurations and Their Probabilities for a Sample of 100 Individuals and 21 Minor-Allele Copies Are Tabulated[Note]
|
Exact Test P Values |
|||||
| No. ofHeterozygotes(nAB) | Probabilitya | χ2 Test P | PHWE | Phigh | Plow |
| 5 | <.000001 | <.000001b | <.000001b | 1.000000 | <.000001b |
| 7 | .000001 | <.000001b | .000001b | 1.000000 | .000001b |
| 9 | .000047 | <.000001b | .000048b | .999999 | .000048b |
| 11 | .000870 | .000039b | .000919b | .999952 | .000919b |
| 13 | .009375 | .002228b | .010293b | .999081 | .010293b |
| 15 | .059283 | .045180b | .069576 | .989707 | .069576 |
| 17 | .214465 | .342972 | .284042 | .930424 | .284042 |
| 19 | .406355 | .906529 | 1.000000 | .715958 | .690396 |
| 21 | .309604 | .244336 | .593645 | .309604 | 1.000000 |
Note.— The probability of observing each possible outcome is given, together with the corresponding P values for tests of HWE based on the χ2 statistic and on the exact test statistics PHWE, Plow, and Phigh (described in the main text).
P(nAB|N=100,nA=21).
Configurations that would be rejected at the significance level α = 0.05.
In general, the exact test statistics are conservative when a small number of minor-allele copies are present in the sample, but they approximate nominal significance levels as the sample size (and number of minor-allele copies) increases. In contrast, the commonly used χ2 statistic can produce excessively small or large P values for specific outcomes (Hernandez and Weir 1989). To comprehensively evaluate the performance of the χ2 and exact test statistics, we calculated their type I error rates for specified significance levels of α=0.05, 0.01, or 0.001, for sample sizes of N=100 or N=1,000 individuals and varying minor-allele counts. The results are summarized in figure 1 (for samples in which <25% of chromosomes carry the minor allele) and figure 2 (for samples in which >10% of chromosomes carry the minor allele), and it is clear that the statistics exhibit some periodicity in their type I error rates. As expected, both the exact PHWE statistic and the χ2 statistic perform better as the sample size and minor-allele counts increase. Nevertheless, one important difference is that the χ2 statistic can sometimes be extremely anticonservative (e.g., in a sample of 1,000 individuals, when nominal α=0.001, the true type I error rate can exceed 0.06 and is often >0.01 for minor-allele counts <100), whereas the exact statistic never exceeds the nominal significance level. In practical settings, the χ2 statistic could lead to many false rejections of HWE that depend on only the particular count of minor alleles in the sample.
Figure 1.

Type I error rates as a function of minor-allele counts for rare alleles, for samples of either 100 or 1,000 chromosomes and corresponding to a significance threshold of α=0.05, 0.01, or 0.001. Results are plotted as a function of the number of minor alleles in the sample for the exact PHWE statistic (red) and for the asymptotic χ2 test statistic (blue). A gray line denotes the nominal error rate. Note that the Y-axes in figures 1 and 2 differ.
Figure 2.

Type I error rates as a function of minor-allele counts for common alleles, for samples of either 100 or 1,000 chromosomes and corresponding to a significance threshold of α=0.05, 0.01, or 0.001. Results are plotted as a function of the number of minor alleles in the sample for the exact PHWE statistic (red) and for the asymptotic χ2 test statistic (blue). A gray line denotes the nominal error rate. Note that the Y-axes in figures 1 and 2 differ.
To understand the periodicity of the statistics, it is important to consider the discrete nature of the data. For example, for a sample of N=100 individuals including 2–5 copies of the minor allele, we reject HWE at the α=0.05 significance level (fig. 1A) when there is at least one homozygote for the minor allele. The probability of observing more than one homozygote for the minor allele increases gradually from 0.0050 when there are two copies of the allele in the sample up to 0.0499 when there are five copies of the minor allele in the sample. When there are 6–14 copies of the minor allele in the sample, we reject HWE at the α=0.05 significance level (fig. 1A) when at least two homozygotes for the rare allele are observed. Again, the probability of a more extreme event is quite low for small numbers of the rare allele (P=.0011 with six copies of the minor allele in the sample) but gradually increases if there are additional copies of the minor allele in the sample (P=.0482 with 13 copies of the minor allele).
In table 2, the overall type I error rates for each statistic are summarized for sample sizes of 100 or 1,000 individuals and various ranges of minor-allele counts. It is clear that, on average, the χ2 test approximates nominal significance levels as the number of minor alleles in the sample increases. Nevertheless, as illustrated in figure 1, this is achieved at the cost of inflated error rates for samples with specific numbers of minor alleles. Even in a sample of 1,000 individuals, the type I error rate at α = 0.001 for the χ2 test is inflated when there are <200 copies of the minor allele (corresponding to an allele frequency of ∼10%). The exact tests approximate nominal significance levels with increasing sample size but remain conservative because of the discrete nature of the data.
Table 2.
Actual Error Rates for the χ2 Test Statistic and the PHWE Test Statistic for Nominal Significance Level α = 0.01 or 0.001[Note]
| α = 0.01a |
α = 0.001a |
|||
| Sample andMinor-Allele Count | χ2 | PHWE | χ2 | PHWE |
| N=1,000 | ||||
| 1–100 | .0208b (.0208)b | .0039 (.0039) | .0088b (.0088)b | .0004 (.0004) |
| 101–200 | .0100 (.0154)b | .0065 (.0052) | .0017b (.0053)b | .0006 (.0005) |
| 201–400 | .0097 (.0126)b | .0083 (.0067) | .0010 (.0032)b | .0008 (.0006) |
| 401–1,000 | .0100 (.0110)b | .0090 (.0081) | .0010 (.0018)b | .0009 (.0008) |
| N=100 | ||||
| 1–10 | .0292b (.0292)b | .0024 (.0024) | .0114b (.0114)b | .0001 (.0001) |
| 11–20 | .0191b (.0242)b | .0035 (.0030) | .0035b (.0074)b | .0003 (.0002) |
| 21–40 | .0083 (.0162)b | .0037 (.0033) | .0016b (.0045)b | .0004 (.0003) |
| 41–100 | .0099 (.0124)b | .0072 (.0057) | .0009 (.0023)b | .0006 (.0005) |
Note.— Results are tabulated for samples of 100 and 1,000 individuals and represent simple averages for each range of minor-allele counts.
The error rate for each bin is tabulated, followed by the cumulative error rate in parenthesis. The cumulative error rate is calculated by including each bin and all previous bins. For example, for a sample of size 1,000, when α = 0.001, the type I error rate for the standard χ2 test in a sample with 101–200 copies of the minor allele is 0.0017 and the cumulative error rate, corresponding to samples with 1–200 copies of the minor allele, is 0.0053.
Exceeds nominal significance level.
As a final evaluation of our approach, we applied our method to a subset of the genotypes collected by the International HapMap Consortium (2003). We focused on a set of 18,460 SNP markers genotyped independently by two different centers with no discrepancies between the two sets of experimental results. For each of these markers, we evaluated evidence against HWE by using both the exact PHWE statistic and the asymptotic χ2 statistic. Results were broadly similar for 14,889 markers with minor-allele frequencies ⩾20%. However, we observed noticeable differences for 3,571 markers with minor-allele frequencies <20%. For example, the χ2 test rejected HWE for 71 of these markers at α=0.01 (twice as many as the 35 markers expected to fail this test by chance), whereas the exact test rejected HWE for only 33 markers. At the more stringent α=0.001 significance level, the χ2 test rejected HWE for 28 markers (rejection for 3 markers is expected by chance), whereas the exact PHWE statistic rejected HWE for only 5 markers.
Although we focus on testing the agreement of observed genotypes with HWE proportions, computationally efficient exact tests can be constructed for any desired genotype proportions. In brief, let the expected proportion of heterozygotes be pAB and the two homozygote proportions be pAA and pBB. For example, in a population with inbreeding coefficient f, we might expect the proportion of heterozygotes to be 2(1-f)pApB. Define the quantity θ=p2AB/pAApBB so that θ=4 when HWE holds. Then, the probability of observing nAB heterozygotes is
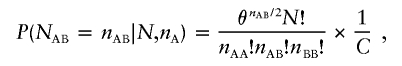 |
where
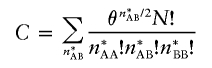 |
(Wellek 2004). It is simple to verify that the recurrence relationships given in equation (2) can be extended to this setting by replacing the number 4 with the quantity θ in each expression.
The exact test statistics for HWE described here are accurate for a variety of allele frequencies and can be computed in an inexpensive manner. We recommend that they be used instead of the standard χ2 test statistic in all situations. For large data sets, rather than fixing an arbitrary threshold for rejecting HWE, we suggest that methods based on the false-discovery rate (Benjamini and Hochberg 1995) be used to identify a subset of markers whose genotypes do not conform to the expected equilibrium distribution.
The PHWE test statistic described here is implemented in the Pedstats software package (see Pedstats Web site), which generates summaries and checks the integrity of genetic data. In addition, code for calculating Plow, Phigh, and PHWE in C/C++, R, and Fortran is available from the authors’ Web site. With appropriate citation, our code is freely available for use and can be incorporated into other programs. The HapMap Project genotype data are freely available at the HapMap Web site.
Acknowledgments
We gratefully acknowledge grant support from the National Human Genome Research Institute and the National Eye Institute. The manuscript was improved by helpful comments from reviewers.
Electronic-Database Information
The URLs for data presented herein are as follows:
- Authors' Web site, http://www.sph.umich.edu/csg/abecasis/
- HapMap, http://www.hapmap.org/
- Pedstats, http://www.sph.umich.edu/csg/abecasis/Pedstats/
References
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300 [Google Scholar]
- Cardon LR, Abecasis GR (2003) Using haplotype blocks to map human complex trait loci. Trends Genet 19:135–140 10.1016/S0168-9525(03)00022-2 [DOI] [PubMed] [Google Scholar]
- Crow JF (1988) Eighty years ago: the beginnings of population genetics. Genetics 119:473–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA (1934) Statistical methods for research workers. Oliver and Boyd, Edinburgh [Google Scholar]
- Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48:361–372 [PubMed] [Google Scholar]
- Haldane JBS (1954) An exact test for randomness of mating. J Genet 52:631–635 [Google Scholar]
- Hardy HG (1908) Mendelian proportions in a mixed population. Science 28:49–50 [DOI] [PubMed] [Google Scholar]
- Hernandez JL, Weir BS (1989) A disequilibrium coefficient approach to Hardy-Weinberg equilibrium testing. Biometrics 45:53–70 [PubMed] [Google Scholar]
- International HapMap Consortium (2003) The International HapMap Project. Nature 426:789–796 10.1038/nature02168 [DOI] [PubMed] [Google Scholar]
- Kwok PY (2001) Methods for genotyping single nucleotide polymorphisms. Annu Rev Genomics Hum Genet 2:235–258 10.1146/annurev.genom.2.1.235 [DOI] [PubMed] [Google Scholar]
- Levene H (1949) On a matching problem arising in genetics. Ann Math Stat 21:91–94 [Google Scholar]
- Vithayasai C (1973) Exact critical values of the Hardy-Weinberg test statistic for two alleles. Communic Stat 1:229–242 [Google Scholar]
- Weber JL, Broman KW (2001) Genotyping for human whole-genome scans: past, present, and future. Adv Genet 42:77–96 [DOI] [PubMed] [Google Scholar]
- Weinberg W (1908) On the demonstration of heredity in man. In: Boyer SH, trans (1963) Papers on human genetics. Prentice Hall, Englewood Cliffs, NJ [Google Scholar]
- Weir BS (1996) Genetic data analysis II. Sinauer Associates, Sunderland, MA [Google Scholar]
- Wellek S (2004) Tests for establishing compatibility of an observed genotype distribution with Hardy-Weinberg equilibrium in the case of a biallelic locus. Biometrics 60:694–703 10.1111/j.0006-341X.2004.00219.x [DOI] [PubMed] [Google Scholar]