

Abstract
Arabidopsis and rice are the only two model plants whose finished phase genome sequence has been completed. Here we report the construction of an oligomer microarray based on the presently known and predicted gene models in the rice genome. This microarray was used to analyze the transcriptional activity of the gene models in representative rice organ types. Expression of 86% of the 41,754 known and predicted gene models was detected. A significant fraction of these expressed gene models are organized into chromosomal regions, about 100 kb in length, that exhibit a coexpression pattern. Compared with similar genome-wide surveys of the Arabidopsis transcriptome, our results indicate that similar proportions of the two genomes are expressed in their corresponding organ types. A large percentage of the rice gene models that lack significant Arabidopsis homologs are expressed. Furthermore, the expression patterns of rice and Arabidopsis best-matched homologous genes in distinct functional groups indicate dramatic differences in their degree of conservation between the two species. Thus, this initial comparative analysis reveals some basic similarities and differences between the Arabidopsis and rice transcriptomes.
Rice is one of the most important crops in the world. With a significantly smaller genome size than other cereals, rice is also an excellent monocot model for genetic, molecular, and genomic studies (Gale and Devos 1998). The availability of the complete sequence of the rice genome (Feng et al. 2002; Goff et al. 2002; Sasaki et al. 2002; Yu et al. 2002, 2005; The Rice Chromosome 10 Sequencing Consortium 2003) makes it possible to estimate the gene number in the genome, to approach gene function on a genomic scale, and to identify candidate genes predicted to regulate traits of interest. Different approaches used to annotate the Oryza sativa L. ssp indica and Oryza sativa L. ssp japonica draft sequences suggested that there are 46,022-55,615 gene models for the former and 32,000-50,000 gene models for the latter (Goff et al. 2002; Yu et al. 2002). Extrapolation from the finished sequences of Oryza sativa L. ssp japonica chromosomes 1, 4, and 10 estimated 62,500, 57,000, and 60,000 gene models for the rice genome, respectively (Feng et al. 2002; Sasaki et al. 2002; The Rice Chromosome 10 Sequencing Consortium 2003). In addition, the estimated gene count for the rice genome was at least 38,000-40,000 if the transposable elements were removed from the consideration (Yu et al. 2005). Each of these calculations placed the number of gene models in rice at the top of all organisms for which the genomes have been sequenced. However, only about half of these gene models (or candidates) were supported by either full-length cDNA clones (Kikuchi et al. 2003) or expressed sequence tags (ESTs) (Wu et al. 2002). This leaves the remaining half of rice gene models without experimental support. Thus, a comprehensive transcriptional analysis of the entire rice gene model set would not only provide insight into the genome expression pattern, but would also provide evidence of expression for those gene models previously lacking experimental support (Ashurst and Collins 2003; Yamada et al. 2003).
Arabidopsis and rice are the best-characterized experimental models for dicot and monocot plants, respectively. The rice genome size is more than three times that of Arabidopsis, and is estimated to have significantly more genes (The Arabidopsis Genome Initiative 2000; Feng et al. 2002; Goff et al. 2002; Sasaki et al. 2002; Yu et al. 2002, 2005; The Rice Chromosome 10 Sequencing Consortium 2003). Given that about half of the rice gene models are highly conserved in the plant kingdom (Yu et al. 2002, 2005), it was reasoned that a comprehensive comparison of the transcriptional activities of the conserved and less-conserved gene models between different species at the whole-genome level would provide novel insights into the genesis and evolution of new rice genes (Koonin et al. 2000). The large gene model number and high proportion of less-conserved gene models in the rice genome may be due to an overannotation of the rice genome (Bennetzen et al. 2004). It has been suggested that more than half of those rice gene models annotated as less conserved in early versions of the rice genome might actually be diverged transposons and retrotransposons, or segments of them (Bennetzen et al. 2004; Jiang et al. 2004). In any case, it would be interesting to know the expression properties of the less-conserved gene models in the rice genome. Therefore, a comparison of the transcriptional activity between rice and Arabidopsis at the whole-genome level should provide a rare opportunity to examine the overall impact of evolution on representative monocot and dicot genomes (Bennetzen 2002; Izawa et al. 2003; Schoof and Karlowski 2003).
DNA microarrays can measure the individual transcript level of tens of thousands of genes simultaneously, thus providing a high-throughput means to analyze gene expression levels at the whole-genome scale (Schena et al. 1995; Chu et al. 1998). The availability of the complete sequence of the rice genome provides the information necessary to design a microarray with essentially all known and predicted gene models in the rice genome, which can, in turn, be used to assay the expression of all the gene models at once. We produced a 70-mer oligomer microarray covering essentially all annotated rice gene models, either with or without experimental support, and used it to analyze rice transcriptomes from representative organs. Furthermore, the transcriptomes between rice and Arabidopsis for similar organ types were compared, providing insights into genome expression and evolution.
Results
Transcriptome analysis in representative rice organs
A 70-mer oligo set for the indica rice genome was designed and printed as a two-slide microarray set (see Methods). This microarray was used to evaluate transcription of the rice genome (namely the entire gene model set) at representative developmental stages during the rice life cycle. A sample microarray hybridization image is shown in Supplemental Figure 1. A remapping of this oligo set to the finished indica genome (Zhao et al. 2004; Yu et al. 2005) indicates that it includes 41,122 physically mapped oligos, representing a set of 41,754 annotated, nontransposable element rice gene models with or without experimental support. This gene model set includes 16,504 full-length cDNA-supported gene models (CG), 5968 EST-supported gene models (SG), and 19,282 predicted gene models lacking experimental support (UCG) (Fig. 1A).
Figure 1.

Microarray expression analysis of confirmed and predicted gene models in the rice genome. (A) Summary of full-length cDNA-confirmed, EST-supported, and total-predicted gene models covered by the 70-mer oligo microarray used in this study. The vertical green line separates the relative proportions of gene models whose expressions are confirmed or not in our analysis. (B) Rice organ-sample collection used for microarray analysis. (C,D) GO categories of gene models expressed in at least one organ or in cultured cells (C) and in all collected organs and cultured cells (D). The details for gene models included in C and D can be found in Supplemental Tables 3 and 4, respectively.
The rice organs and tissues that were selected include seedling shoots, tillering-stage shoots and roots, heading and filling-stage panicles (Fig. 1B), and suspension-cultured cells as a common control. Based on a single-color fluorescent dye hybridization analysis (Rinn et al. 2003; see also Methods and Discussion), we estimated that among the 37,132 gene models that correspond to the cross-hybridization-free oligo set in the array, the expression of 32,014 gene models (86.2% of total) can be experimentally detected in at least one of the above-mentioned organs or cultured cells under our experimental conditions (Fig. 1A; Supplemental Table 3). Among them, there were 14,171 (93.5%) CG gene models, 4999 (93.5%) SG gene models, and 12,844 (77.2%) UCG gene models (Fig. 1A; Supplemental Table 3). The other group of 3990 oligos, corresponding to those with possible cross-hybridization effects, matched to 4622 unique gene models, including 1355 CG, 641 SG, and 2626 UCG gene models, though the expression percentage cannot be unambiguously assessed due to possible cross-hybridization. We estimated that 3894 of the 4622 gene models can be experimentally detected in at least one of the above-mentioned organs or cultured cells under our experimental conditions, based on the detection rate of CG, SG, and UCG gene models derived from the above-mentioned unique gene-model match oligo set. The experimental data for those oligos with possible hybridization to more than one unique gene-model match from the above-mentioned organ and cell types are listed in Supplemental Table 2. Together, the expression of about 35,900 gene models was experimentally detected in our experiment (Fig. 1A). For accuracy of the expression analysis, we considered the expression data only from those oligos with a uniquely matched rice gene model in the rice genome in all subsequent analyses.
There were 12,930 (34.8%) gene models whose expression was experimentally detected in all of the above-mentioned organs and cultured cells (common-expressed genes), including 6490 (42.2%) CG gene models, 2548 (47.6%) SG gene models, and 3892 (23.4%) UCG gene models (Supplemental Table 4). The Gene Ontology (GO) functional categories (http://www.geneontology.org; Yu et al. 2005) for those gene models that are either expressed in one or more organ and cultured cells, or in all organs and cultured cells, are shown in Figure 1, C and D, respectively.
We found that the portions of the genome expressed in different organs and in cultured cells was variable, ranging from 49.7% (filling-stage panicle) to 70.2% (heading-stage panicle) (Fig. 2A). Among all selected organs and cultured cells, the percentages of expressed CG and SG gene models were similar and were always higher than those of UCG gene models (Fig. 2A). In general, the average expression level for CG gene models was higher than for SG gene models, with UCG gene models having the lowest average expression level in most selected organs and in cultured cells. An exception was noted in tillering-stage roots, where SG gene models had the highest average expression level (Fig. 2B).
Figure 2.

Rice genome expression in different organs and cultured cells. (A) The percentage of gene models expressed in different organs and cultured cells. (B) The average expression level of all full-length cDNA matched (CG), EST matched (SG), and predicted (UCG) genes in different organs and in cultured cells.
We also examined which gene models were expression enriched in each organ type. Based on the experimental repeats, we identified differentially expressed genes among all of the organs. A gene model was considered to be enriched in a given organ if the expression level of the gene model in that organ was shown to be significantly higher compared with all other organs (see Methods). There were 20 (0.1%), 216 (0.6%), 94 (0.3%), 690 (1.8%), and 387 (1.0%) gene models specifically enriched in seedlings, shoots, roots, heading-stage panicles, and filling-stage panicles, respectively (Fig. 3A). The GO functional categories for these specifically enriched gene models from different organs are shown in Supplemental Figure 2. As expected, the gene models encoding proteins involved in photosynthetic light and dark reactions and in chlorophyll biosynthesis were highly expressed in seedlings, shoots, and panicles, but not in roots, whereas seed-storage proteins were highly expressed in panicles, but not in any other organ. In addition, the orthologs for four well-characterized floral pattern determination genes in Arabidopsis (AP1, AP3, PI, and AG) (Meyerowitz 2002) were highly expressed in panicles, but were either undetected or barely detectable in the other organs.
Figure 3.

Summary of the gene models whose expression levels are enriched in various rice organs. (A) The number of all organ-specific enriched gene models, high-homology (HH) gene models, and low-homology (LH) gene models among all representative organ types. (B) The average expression level of all organ-specific enriched gene models, HH gene models, and LH gene models among all representative organ types.
The corresponding organs from Arabidopsis and rice express similar proportions of their genomes
To facilitate comparison between the Arabidopsis and rice transcriptomes, we divided the gene models in both rice and Arabidopsis into two categories as follows: gene models with significant homologs (high homology, HH) and gene models without significant homologs (low homology, LH) in their counterpart genomes. The purpose of this distinction is to divide both the Arabidopsis and rice genes into two groups of relatively more conserved (HH) or more diverged (LH) gene models, based on their protein sequence homology (see Methods). It should be noted that some of the rice LH gene models may have homologs in Arabidopsis as well, but fall below our cut-off (Jabbari et al. 2004). With this criterion, we calculated that 54.7% of rice gene models have significant homologs in the Arabidopsis genome (HH), whereas 45.3% of the gene models (LH) do not (Fig. 4A). On the other hand, 74.5% of Arabidopsis gene models have significant homologs in the rice genome (HH), while the remaining 25.5% do not (LH) (Fig. 4A).
Figure 4.

Comparative analysis of whole-genome transcription between rice and Arabidopsis. (A) The portion of high-homology (HH) and low-homology (LH) gene models and their expressed percentages in rice and Arabidopsis. (B,C) The average expression levels of HH and LH gene models in rice (B) and Arabidopsis (C). (D) GO categories of the best-matched gene model pair collection between rice and Arabidopsis. The details for gene models included in D can be found in Supplemental Table 5.
Our microarray analysis indicated that for rice, the expression of 89.5% of the HH gene models can be experimentally detected in at least one of the above-mentioned rice organs or in cultured cells, while the percentage of expressed genes was 82.4% for LH gene models (Fig. 4A). To investigate whether the HH and LH gene models in the Arabidopsis genome have similar expression patterns, ideally, expression in the same organ types should be compared. Because of the distinct anatomy of the dicotyledonous Arabidopsis and the monocotyledonous rice, however, the following corresponding Arabidopsis organs and cell types were used for comparison with the rice genome expression data: seedlings, cauline leaves, roots, inflorescences, flowers, siliques, and suspension cultured cells (as a common control). The genome expression profiles for these organs were obtained by hybridization of the cDNA probes derived from each of the above-mentioned organs and cultured cells to a similar 70-mer oligo microarray covering 25,676 unique Arabidopsis gene models (Ma et al. 2005). A comparative analysis of the transcriptomes of similar organ types from rice and Arabidopsis indicated that the proportion of the genome expressed in at least one of the selected organs or cultured cells (expression-detectable gene models), and in all selected organs and cultured cells (commonly expressed gene models) was very similar between rice and Arabidopsis. Expression was detected for 86.2% of rice gene models and 85.5% of Arabidopsis gene models, while 34.8% of rice gene models and 32.4% of Arabidopsis gene models were found to be commonly expressed gene models. The percentages of the Arabidopsis HH and LH gene models with detectable expression were also very similar to those of rice (Fig. 4A). The average expression level for HH gene models was higher than that of LH gene models in most organs in both rice and Arabidopsis (Fig. 4B,C). However, HH and LH gene models showed similar expression levels in root tissue from both rice and Arabidopsis (Fig. 4B,C). HH and LH gene models also showed similar expression levels in Arabidopsis seedlings and in rice filling-stage panicles (Fig. 4B,C).
The expression patterns of orthologous gene groups are conserved to different degrees between Arabidopsis and rice
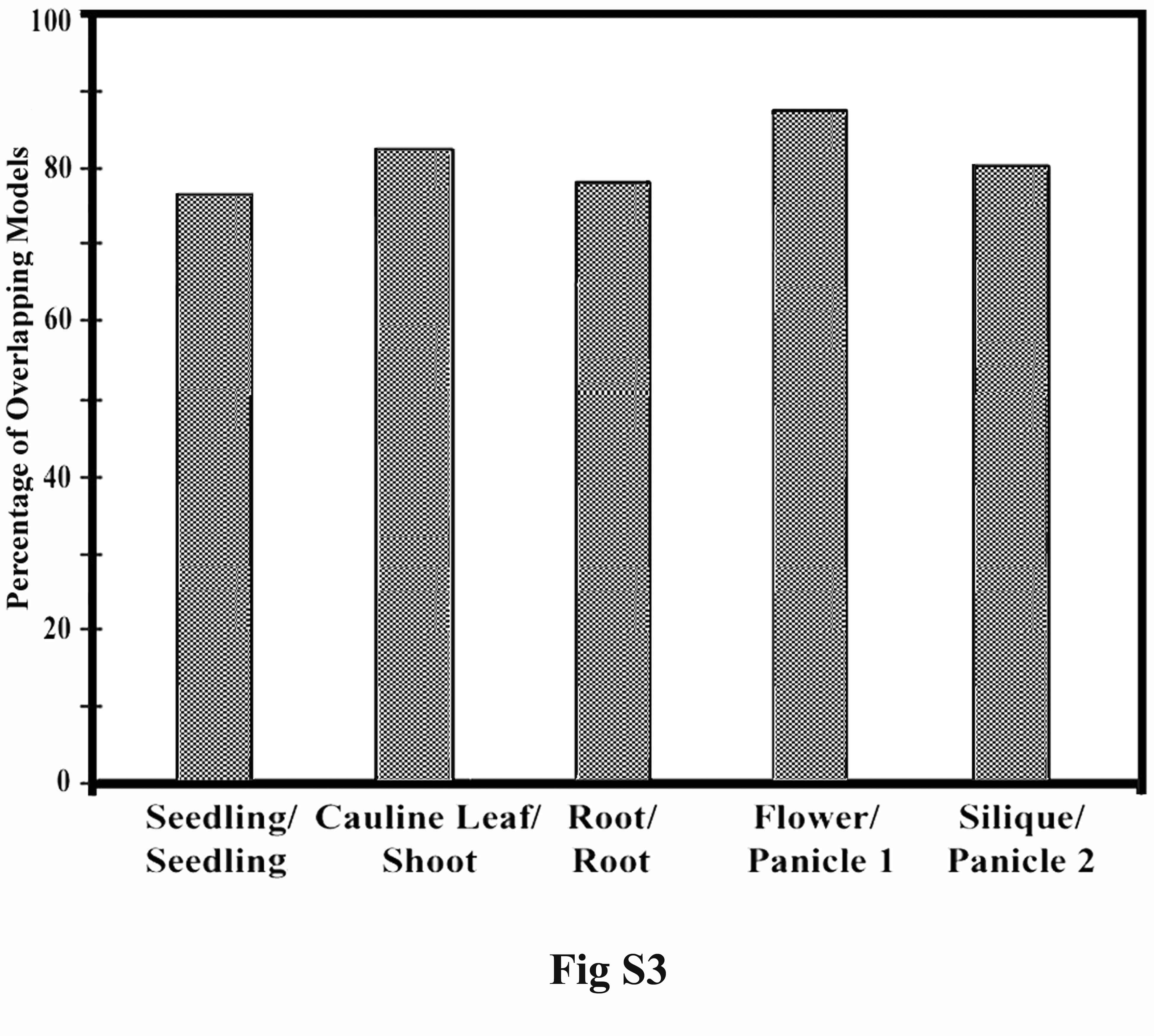
We next examined whether corresponding organs from rice and Arabidopsis express similar sets of gene models and whether those gene models have similar expression levels. For this purpose, the best-matched homologous gene model pairs (potentially enriched for orthologous gene models) between the two plants were compared. The best-matched gene model pairs from the two genomes were basically the closest homologs in the reciprocal homology searches using the above-mentioned homology cut-off criterion. A total of 6314 best-matched pairs of gene models between rice and Arabidopsis were identified (Supplemental Table 5). The GO functional categories for those best-matched gene model pairs are shown in Figure 4D. Among them, there were 415 transcription factor gene models, 321 signal transduction gene models, 270 gene models encoding proteins in the ubiquitin-proteasome pathway, and 198 gene models encoding proteins involved in protein biosynthesis. In addition, gene models involved in plant hormone biosynthesis, including auxin, cytokinin, abscisic acid, gibberellin, ethylene, brassinosteroids, jasmonate, salicylic acid, and polyamine, were all included in the best-matched gene model pairs collection. In all of the corresponding organs of rice and Arabidopsis that were compared, a large overlap (76%-88%) in the expression of the best-matched gene model pairs was observed (Supplemental Figure 3). One potential pitfall for only analyzing the best-matched gene pairs is that there are cases where a gene from one organism can have more than one very closely matched homolog that is potentially functionally indistinguishable. To evaluate the extent of such cases in rice and Arabidopsis, for each rice gene we obtained and examined, the two best homologous Arabidopsis genes (the best and the second best-matched genes). Among 6314 best-matched rice and Arabidopsis gene pairs, there are 1118 or 531 cases where the second best-matched Arabidopsis genes exhibited high-sequence identity (with 70% or 80% identity as the cutoff for the matched sequence in a stretch of not less than 100 amino acids) to the best-matched Arabidopsis genes. The presence of more than one potentially functional redundant ortholog for each rice gene could lead to underestimation of expression conservation in our best-matched pair analysis, however, this impact should be limited due to the relatively small fraction of these in the total population analyzed.
We further examined the expression level of individual best-matched gene model pairs in the corresponding organs of rice and Arabidopsis. We used both one-channel intensity and relative expression ratio (a given organ vs. cultured cells) to calculate the correlation of expression between organs, and obtained similar results using each method. We found that the expression level of best-matched gene model pairs in rice and Arabidopsis was highly correlated, with significant P values. The overall correlation of expression level, calculated based on the one-channel expression level for all the best-matched gene model pairs as a whole, among selected organs between the two species, is summarized in Table 1. The correlation coefficient is relatively low, but it is at a similar level to that from a Caenorhabditis elegans and Drosophila melanogaster comparison (McCarroll et al. 2004). This result suggested that the expression change for most of the best-matched gene pairs is species specific, but that the expression for some of these gene pairs (roughly several hundred) was evidently conserved between the two species. These gene pairs were likely involved in the conserved pathways. We found that there was no significant difference in the correlation coefficient between these different homologous organ pairs between the two species, however, the correlation coefficient values decrease when noncorresponding organ types were compared between rice and Arabidopsis (Table 1).
Table 1.
The correlation of best-matched gene model pairs' expression levels between rice and Arabidopsis organ types
| Rice Arabidopsis | Seedling | Shoot | Root | Heading stage panicle | Filling stage panicle |
|---|---|---|---|---|---|
| Seedling | 0.207*** | 0.221*** | 0.149*** | 0.182*** | 0.116* |
| Cauline leaf | 0.221*** | 0.243*** | 0.150*** | 0.192*** | 0.119** |
| Root | 0.138*** | 0.143*** | 0.212*** | 0.186*** | 0.136*** |
| Flower | 0.199*** | 0.211*** | 0.152*** | 0.195*** | 0.117* |
| Silique | 0.190*** | 0.198*** | 0.139*** | 0.186*** | 0.112* |
The significance of the correlation was classified into the three categories: *P-value of <1.5E-20, **P-value of <2.8E-21, ***P-value of <2.8E-23 by the t-Test.
The correlation of expression levels for different categories of the best-matched gene model pairs between the two species was further examined. As shown in Table 2, in general, the correlation of the expression level for the best-matched gene models encoding proteins involved in the ubiquitin-proteasome pathway was higher than those of the best-matched gene models encoding proteins involved in signal transduction, whereas there is no significant correlation for genes encoding components of the protein biosynthesis pathway and the transcription factor group between the corresponding organs from the two species (Table 2). These results suggest that the expression pattern for gene models encoding ubiquitin-proteasome pathway proteins are the most conserved between the two model plant species.
Table 2.
The correlation of expression for different functional categories of rice and Arabidopsis best-matched gene model pairs from corresponding organs
| Arabidopsis/Rice | Seedling/seedling | Cauline leaf/shoot | Root/root | Flower/heading stage panicle | Silique/filling stage panicle |
|---|---|---|---|---|---|
| Transcription factor | 0.134a | 0.197 | 0.141 | 0.077 | 0.076 |
| (0.006)b | (5.3E-05) | (0.004) | (0.118) | (0.123) | |
| Proteasome pathway | 0.259 | 0.321 | 0.240 | 0.285 | 0.165 |
| (1.7E-5) | (7.3E-8) | (6.8E-5) | (1.97E-6) | (6.0E-4) | |
| Protein synthesis pathway | 0.024 | 0.067 | 0.223 | 0.049 | 0.098 |
| (0.736) | (0.345) | (0.02) | (0.494) | (0.172) | |
| Signal transduction | 0.168 | 0.213 | 0.202 | 0.251 | 0.180 |
| (0.03) | (0.001) | (0.0002) | (5.1E-6) | (0.01) |
aCorrelation coefficient; bt-test significance P-value.
A large proportion of LH genes are expressed in rice
As shown in Figure 4A, expression of about 82.4% of rice LH gene models was detected in the examined organs or cultured cells using our experimental conditions. Among the organ-specific enriched rice gene models, there are both HH and LH gene models in all five organs, and, in general, the HH gene models are more numerous than the LH gene models in most organs (Fig. 3A). However, the organ-specific enriched gene models in filling-stage panicles and tillering-stage roots contain similar numbers of HH and LH gene models (Fig. 3A). Among the gene models specifically enriched in different rice organs, the average expression level of LH gene models is similar to that of the HH gene models in shoots and filling-stage panicles, and is higher than that of HH gene models in other organs (Fig. 3B). A significant fraction of the LH gene models exhibited high expression levels in one or more organs. Because panicles and roots expressed a higher proportion of LH gene models than other organs, we examined the possible function of those highly expressed LH gene models in both panicles and roots. The panicle-specific enriched LH gene models included those encoding seed-storage proteins and protease inhibitors, proteins involved in amino acid biosynthesis and secondary metabolite (e.g., nicotinate, nicotinamide, pyrimidine, and purine etc.) biosynthesis. Root-specific, highly expressed LH gene models included those encoding metal-binding proteins and transporters, presumably functioning in nutrient absorption and transportation. These results indicate that a large proportion of LH gene models are expressed, and imply that some of these highly expressed LH gene models in rice may have developed specific functions that underlie agriculturally and economically important traits.
A significant fraction of neighboring genes show a coexpression pattern in the rice genome
Recent results suggest that the regulation of genome expression in some species involves coordinated regulation of adjacent gene models in chromosomal regions defined as chromatin domains (Hurst et al. 2004). In order to investigate whether this is also the case in rice, we monitored the coexpression patterns for adjacent gene models in the rice genome using all of the expression ratio data sets for each organ vs. cultured cells. We calculated the number of coexpressed adjacent gene models in different window sizes along the chromosomes using the method described by Spellman and Rubin (2002). As the window size increased from two to 11, the net number of coexpressed gene models increased proportionally. The net number of gene models began to plateau at a window size of 11 (Fig. 5A), suggesting that most of the coordinately expressed gene model groups include about 11 gene models. We also found that most of the coordinately expressed adjacent gene model clusters are about 100 kp of genomic sequence in length (Fig. 5B). The adjacent genes that can be organized into a coexpression group are shown in Supplemental Table 6. We considered that the rice genome contains many tandem-repeat gene models as well and excluded them from this calculation. Overall, our result suggests that about 10% of the rice genome shows a coordinated expression pattern (Table 3; Supplemental Table 6) and is, therefore, likely organized into so-called coexpressed chromosomal regions.
Figure 5.

Summary of the chromosomal regions with coexpressed gene models in the rice genome. (A) The total number of coexpressed adjacent gene models obtained using different window sizes for estimation. The number of net gene models is the total number of gene models from the rice genome showing a coexpression pattern. Window size is the average number of gene models that are coexpressed in one group. (B) The length of genomic DNA sequence for the coexpressed adjacent gene models. The x-axis represents the genomic DNA length along the chromosome, and the y-axis represents the distribution of coexpressed gene models within a specific length of chromosome.
Table 3.
The total number of gene models identified as being within a coexpression chromosomal region, with genes ordered in their native chromosomal locations (ordered genes) compared with the situation where all genes are randomized in their relative positions (randomized genes), at various significance levels
| Significance (P-value) | Ordered gene models | Randomized gene models |
|---|---|---|
| 0.001 | 558 | 95 |
| 0.005 | 1981 | 695 |
| 0.01 | 3324 | 1350 |
Calculation of the chromosomal regions is based on a recently reported method (Spellman and Rubin 2002) with a chromosomal region window size of 11 gene models and is detailed in Supplemental Table 6.
Discussion
In addition to providing expression data for many computer-predicted gene models in the rice genome, this study also describes the genome expression pattern for several representative rice organs. A whole-genome comparative analysis of expression between the monocot model rice and the dicot model Arabidopsis provides expression evidence for the majority of LH gene models, and reveals a trend of changes in expression patterns of best-matched gene model pairs during plant evolution. This fundamental knowledge should provide a valuable basis for a more complete description of the rice genome.
It is worth noting that various methods may be used to define whether a given gene model is expressed or not, using microarray analysis. Although no universal criterion for this purpose is available so far, several common approaches have been described. For example, determination of gene-model expression has been based on the reproducibility of a detectable expression level among replicates (Rinn et al. 2003), on the expression signal level compared with the background signal (Kim et al. 2003), or by relying on internal negative controls (Kapranov et al. 2002). In the current study, we considered all of these factors together. We first determined the gene expression status based on the internal negative and positive controls in each replicate, and checked its reproducibility among the replicates. We scored the gene model as expressed only when it reproducibly exhibited a signal higher than both background and negative controls with a 90% confidence in three replicates. This means that our estimation could have an upper limit of a 10% false positive rate. Given that about 85% of rice genes were considered to be expressed in our microarray analysis (Fig. 1A), the expressed portion of the total number of rice gene models is estimated to be at least between 77% and 85% in the organs/tissues examined. Although different microarray systems were used to detect the expressed portion of the genome between our present work and the work from Kim et al. (2003) on Arabidopsis chromosome 2, the number for the portion of expressed genes in the present study is consistent with the number from Arabidopsis chromosome 2 gene models in their report (Kim et al. 2003).
Analysis of rice transcriptome provides support for the expression of the majority of those predicted gene models without prior expression support
About half of the computer-based annotated gene models in the rice genome have full-length cDNA or EST support, while the other half do not. In the present study, expression of 93% of the full-length cDNA or EST-supported genes could be experimentally detected (Fig. 1A), suggesting that our microarray system is sensitive and accurate for detection of transcripts in rice tissue. Thus, the results obtained by microarray analysis can be used to evaluate the accuracy of the rice genome annotation, especially for those gene models lacking experimental support by examining the presence of their transcripts. In this regard, we found that about 86% of the total known and predicted genes in the rice genome had detectable transcripts in the six organ and cell types examined (Fig. 1A). For those purely predicted gene models, 77% were found to be transcribed (Fig. 1A). Therefore, our results indicate that most of the predicted rice gene models are expressed, at least in terms of producing detectable RNA transcripts.
LH gene models have been suggested to be the byproduct of the process of genome evolution by gene duplication (Prince and Pickett 2002; Kellogg 2003; Yu et al. 2005). A further hypothesis is that these duplicated genes may take on different fates—some dying in the process of nonfunctionalization, while others may be fast evolving, resulting in new functions (Prince and Pickett 2002; Domazet-Loso and Tautz 2003). Alternatively, it has been suggested that the majority of LH genes might not be real genes, but instead are derived from highly diverged or truncated transposons (Bennetzen et al. 2004; Jiang et al. 2004). In the present study, we found that 82% of the LH gene models in rice were transcribed, compared with 89% of the HH gene models (Fig. 4A). Furthermore, the average expression level for the LH gene models was similar to the HH gene models in rice roots and panicles (Figs, 3B, 4B). Thus, our results suggest that a large proportion of rice LH gene models are expressed at the transcript level. It may be true then that at least a portion of these LH group gene models are potentially functional genes in rice. Further analysis of the expressed LH genes might provide insights into the rice genome.
The ubiquitin-proteasome pathway has fundamental roles in plant development and evolution
Regulated proteolysis of individual proteins plays an essential role in the development of all organisms. In eukaryotes, this is achieved by the tagging of proteins with ubiquitin and their subsequent recognition and degradation by the 26S proteasome (Pickart and Cohen 2004). In plants, the ubiquitin-proteasome pathway is involved in degrading a wide range of proteins (Sullivan et al. 2003; Vierstra 2003). Recent studies have indicated that the ubiquitin-proteasome pathway is involved in the control of diverse developmental processes, including floral development, responses to plant hormones and pathogens, and the regulation of photomorphogenesis (Hellmann and Estelle 2002; Serino and Deng 2003; Sullivan et al. 2003).
In the Arabidopsis genome, ∼5% (1350) of the genome encodes for components of the ubiquitin-proteasome pathway (The Arabidopsis Genome Initiative 2000). Among these 1350 gene models, 270 have best-matched pairs in rice (this proportion is very similar to the proportion of best-matched gene model pairs versus the number of total gene models in Arabidopsis) (Supplemental Table 3). Of the four functional groups of gene models we considered (those coding for transcription factors, proteins involved in protein biosynthesis, proteins involved in signal transduction, and those involved in the ubiquitin-proteasome pathway), the best-matched gene model pairs for the ubiquitin-proteasome pathway had the highest correlation coefficients in general, meaning that their expression patterns are the most similar between the two species (Table 2).
In an attempt to test whether the highly correlated gene model expression pattern in this pathway between the two species is due to evolutionary conservation, we examined the expression pattern for all gene models involved in the above-mentioned four pathways among all organs from Arabidopsis and rice. We found that a high proportion of the gene models involved in the ubiquitin-proteasome pathway also showed differential expression between light- and dark-grown organs, or between different organs and cell-type pairs (organ vs. cultured cell) in the same species. Furthermore, the proportion and the average fold change of differentially expressed gene models are similar to those of gene models involved in the remaining three pathways (data not shown). Thus, the change in expression level for gene models encoding proteins involved in the ubiquitin-proteasome pathway is at a level similar to that of the other pathway genes in response to environmental or developmental signals. Strikingly, we find that the variation in expression level among best-matched gene model pairs encoding proteins involved in the ubiquitin-proteasome pathway in each organ or cell type is the smallest. This suggests that the expression patterns for the gene models encoding proteins involved in the ubiquitin-proteasome pathway might be more conserved during plant evolution. Thus, our genomic evidence indicates that proteolysis has a crucial regulatory role throughout both the individual plant life cycle and plant evolution.
The possible mechanism of coexpression in neighboring gene models
Recent results from human (Caron et al. 2001; Lercher et al. 2002), Drosophila (Spellman and Rubin 2002), Arabidopsis (Birnbaum et al. 2003; Ma et al. 2005), and yeast (Cohen et al. 2000) studies suggest that the regulation of genome expression involves coordinated regulation of adjacent genes or gene models in so-called chromatin domains. However, the physical size of these coexpression clusters varies from species to species, ranging from a few kilobases (kb) in yeast to several megabases in mammals, and the size seems to be correlated to the organismal complexity and evolutionary scale (Cohen et al. 2000; Lercher et al. 2002; Hurst et al. 2004). Our result suggests that ∼10% of the rice genome shows a coexpression pattern (Table 3; Supplemental Table 6), and that most of these coexpression clusters include 11 gene models and are about 100 kb of genomic sequence in length (Fig. 5A,B; Supplemental Table 6). Given that the structure of chromosomes is generally organized into loops of roughly 50-100 kb of genomic sequence (Alberts et al. 2002), each coexpression chromosomal region in rice covers about one to two loops of DNA in a given chromosome.
Still, the mechanism for this coexpression pattern in the genome is not clear. One reasonable possibility is the involvement of a chromatin-level modification mechanism in the coexpressed gene model clusters. When core histones in the nucleosomes around one gene model are covalently modified (e.g., acetylation) by chromatin remodeling mediators, according to a given signal, chromatin opening is initiated, and this modification spreads along a chromosome until it reaches a boundary element (Labrador and Corces 2002). In this way, all of the genes within the whole chromatin domain within the boundary may be expressed in a similar manner. To understand the mechanism involved, further genome-wide analysis will be necessary to sort out the possible effects of histone acetylation or methylation states, regulatory protein/chromosome-binding patterns from representative rice organs, or responses to a given signal.
Methods
Plant materials
The rice subspecies used in this study was the cultivar of Oryza sativa L. ssp indica 93-11. The seeds were grown in soil in a green house until the seedling stage. The seedlings were then transferred to the field. The upper part of the seedlings was collected from 7-d-old plants, the shoot was collected from fourth tillering-stage plants, and panicles were collected from both heading- and filling-stage plants. Roots were collected from fourth tillering-stage plants.
Arabidopsis tissue was collected from plants of the ecotype Columbia. Seedlings were grown on growth medium (GM) agar plates. The seedlings were grown in a plant growth chamber under continuous white light for 6 d. The white light intensity used was 150 mol m-2sec-1. Adult Arabidopsis plants were grown in soil in a walk-in Environmental Growth Chamber under continuous white light (250 mol m-2sec-1). Siliques were collected 3 d post-pollination.
The suspension rice culture cells were prepared in a liquid medium containing 2 mg/mL 2,4-D and 0.2 mg/mL 6-BA (Nojiri et al. 1996). Suspension Arabidopsis culture cells were prepared as described by Martinez-Zapater and Salinas (1998). The cultured cells used for RNA isolation were collected at the logarithmic growth phase.
Oligo microarray design and production
Based on a phase II rice genome assembly version available in October, 2002 (http://rise.genomics.org.cn), which was significantly improved from the initial draft version (Yu et al. 2002), and on the available full-length cDNAs (Kikuchi et al. 2003) and all available EST information, we chose 61,123 unique known and predicted gene models to be included in our microarray design. A 70-mer oligo corresponding to the sequence within the coding region of each of those 61,123 gene models was designed. After correcting for such factors as oligo cross-hybridization, uniform TM value, GC content, and hairpin/stem nucleotide number (Sengupta and Tompa 2002), a total of 58,404 70-mer oligos were retained. After the release of the complete gene centric map (Zhao et al. 2004; Yu et al. 2005), we remapped these oligos to the new version of the complete Oryza sativa L. ssp indica genome. A total of 41,122 oligos were matched to 41,754 known and predicted nontransposable element gene models (Zhao et al. 2004; Yu et al. 2005). Therefore, our current oligo set includes 41,122 physically mapped oligos, representing a set of 41,754 annotated nontransposable element rice gene models with or without experimental support. This gene model set includes 16,504 full-length cDNA-supported gene models (CG), 5968 EST-supported gene models (SG), and 19,282 predicted gene models lacking experimental support (UCG) (Fig. 1A). Among this current oligo set, there were 37,132 oligos, for which each oligo matched to one unique gene model with a 70% or higher identity (Supplemental Table 1), while the remaining 3990 oligos might have potential cross-hybridization to more than one gene model in the rice genome at a 70% or higher identity (Supplemental Table 2). We did all analyses in the study with gene models represented by the 37,132 cross-hybridization-free set of oligos. Only for calculating the experimentally detectable gene model number shown in Figure 1A did we include the gene expression numbers from the 3990 oligos with possible cross-hybridization based on the gene model composition covered by this subset of oligos. The oligo set was synthesized by Qiagen/Operon, and all oligos were randomized with respect to their genome location before printing onto polylysine-coated microscope slides in the DNA microarray laboratory at Yale University (http://info.med.yale.edu/wmkeck/dna_arrays.htm) and at the Institute of Human Genetics, University of Aarhus. For more details for the rice and Arabidopsis slides used in this study, please check the Supplemental Methods.
To check whether this Oryza sativa L. ssp indica-derived oligo set can effectively represent the gene models in Oryza sativa L. ssp japonica, we further aligned these 37,132 unique oligos to the Syngenta Oryza sativa L. ssp japonica (Goff et al. 2002) and the IRGSP Oryza sativa L. ssp japonica (Japan Rice Genome Program; http://rgp.dna.affrc.go.jp/index.html) sequences, and found that 92% (34,248) of the oligos matched to the Oryza sativa L. ssp japonica genome sequences (see Methods). This analysis suggests that, as intended, our genome-wide oligo set can be used for examining the transcriptomes of both the Oryza sativa L. ssp indica and japonica subspecies. For more details on the oligo design and coverage, please check the Supplemental Methods.
RNA isolation, probe labeling, and hybridization
RNA preparation, fluorescent labeling of the probe, slide hybridization, washing, and scanning were performed as described previously (Ma et al. 2001, 2002).
Data processing and normalization
Spot intensities were quantified using Axon GenePix Pro 3.0 image analysis software.
To determine the threshold for expression, we followed a commonly used strategy (Kim et al. 2003; Rinn et al. 2003) with minor adjustments. For more details for the data analysis, please check the Supplemental Methods.
Homology search and transcription correlation analysis between rice and Arabidopsis
We searched the sequences of the rice genome (Yu et al. 2005) and the sequences of the Arabidopsis genome (March 20, 2003 version) by means of TBLASTN (Altschul and Gish 1996). The expectation value cutoff was set to 1E-7, and the blast hit length is no less than 100 amino acids or 50% of the full-length protein (for those deduced proteins that are <200 amino acids in length). For the best-matched gene pair search, we used the same criteria to search the rice and Arabidopsis genomes.
Correlation analysis
The correlation for gene expression levels between corresponding organs from rice and Arabidopsis was analyzed using SPSS (version 10.0) software with the function of bivariate Pearson correlations and the two-tailed test of significance analysis.
Calculation of chromosomal regions with coexpressed adjacent gene models
We used the method reported by Spellman and Rubin (2002) to identify coregulated, adjacent gene models.
Supplementary Material
Acknowledgments
We thank Jessica Habashi, Elizabeth Strickland, Lei Li, and Timothy Nelson for reading and commenting on this manuscript, and the Yale DNA microarray laboratory of the Keck Biological Resource Center for the printing of both the rice and Arabidopsis 70-mer oligo microarray slides used in this study (http://info.med.yale.edu/wmkeck/dna_arrays.htm). This research was supported by the 863 rice functional genomics program from the Ministry of Science and Technology of China, a strategic international cooperation project grant (#30221120261) from the National Science Foundation of China, Chinese Academy of Sciences, Commission for Economy Planning, 973 project (#2001CB1088) and, in part, by grants from National Institutes of Health (GM-47850) and National Science Foundation Plant Genome Program (DBI-0325821). L.M. was a long-term post-doctoral fellow of the Human Frontier Science Program.
Footnotes
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GEO under accession no. GSE2691.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3657405.
References
- Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., and Walter, P. 2002. Molecular Biology of the Cell. Garland Publishing, New York.
- Altschul, S.F. and Gish, W. 1996. Local alignment statistics. Methods Enzymol. 266: 460-480. [DOI] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796-815. [DOI] [PubMed] [Google Scholar]
- Ashurst, J.L. and Collins, J.E. 2003. Gene annotation: Prediction and testing. Annu. Rev. Genomics Human Genet. 4: 69-88. [DOI] [PubMed] [Google Scholar]
- Bennetzen, J. 2002. The rice genome: Opening the door to comparative plant biology. Science 296: 60-63. [DOI] [PubMed] [Google Scholar]
- Bennetzen, J., Coleman, C., Liu, R., Na, J., and Ramakrishna, W. 2004. Consistent over-estimation of gene number in complex plant genomes. Curr. Opin. Plant Biol. 7: 732-736. [DOI] [PubMed] [Google Scholar]
- Birnbaum, K., Shasha, D.E., Wang, J.Y., Jung, J.W., Lambert, G.M., Galbraith, D.W., and Benfey P.N. 2003. A gene expression map of the Arabidopsis root. Science 302: 1956-1960. [DOI] [PubMed] [Google Scholar]
- Caron, H., van Schaik, B., van der Mee, M., Baas, F., Riggins, G., van Sluis, P., Hermus, M., van Asperen, R., Boon, K., Voûte, P.A., et al. 2001. The human transcriptome map: Clustering of highly expressed genes in chromosomal domains. Science 291: 1289-1292. [DOI] [PubMed] [Google Scholar]
- Chu, S., DeRisi, J., Eisen, M., Mulholland, J., Botstein, D., Brown, P.O., and Herskowitz, I. 1998. The transcriptional program of sporulation in budding yeast. Science 282: 699-705. [DOI] [PubMed] [Google Scholar]
- Cohen, B.A., Mitra, R.D., Hughes, J.D., and Church, G.M. 2000. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat. Genet. 26: 183-186. [DOI] [PubMed] [Google Scholar]
- Domazet-Loso, T. and Tautz, D. 2003. An evolutionary analysis of orphan genes in Drosophila. Genome Res. 13: 2213-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Q., Zhang, Y., Hao, P., Wang, S., Fu, G., Huang, Y., Li, Y., Zhu, J., Liu, Y., Hu, X., et al. 2002. Sequence and analysis of rice chromosome 4. Nature 420: 316-320. [DOI] [PubMed] [Google Scholar]
- Gale, M.D. and Devos, K.M. 1998. Plant comparative genetics after 10 years. Science 282: 656-659. [DOI] [PubMed] [Google Scholar]
- Goff, S.A., Ricke, D., Lan, T., Presting, G., Wang, R., Dunn, M., Glazebrook, J., Sessions, A., Oeller, P., Varma, H., et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296: 92-100. [DOI] [PubMed] [Google Scholar]
- Hellmann, H. and Estelle, M. 2002. Plant development: Regulation by protein degradation. Science 297: 793-797. [DOI] [PubMed] [Google Scholar]
- Hurst, L.D., Pal, C., and Lercher, M.J. 2004. The dynamics of eukaryotic gene order. Nat. Rev. Genet. 5: 299-310. [DOI] [PubMed] [Google Scholar]
- Izawa, T., Takahashi, Y., and Yano, M. 2003. Comparative biology comes into bloom: Genomic and genetic comparison of flowering pathways in rice and Arabidopsis. Curr. Opin Plant Biol. 6: 113-120. [DOI] [PubMed] [Google Scholar]
- Jabbari, K., Cruveiller, S., Clay, O., Saux, J.L., and Bernardi, G. 2004. The new genes of rice: A closer look. Trends Plant Sci. 9: 281-285. [DOI] [PubMed] [Google Scholar]
- Jiang, N., Bao, Z., Zhang, X., Eddy, S., and Wessler, S.R. 2004. Pack-MULES: Transposon-mediated gene evolution in plants. Nature 431: 569-573. [DOI] [PubMed] [Google Scholar]
- Kapranov, P., Cawley, S.E., Drenkow, J., Bekiranov, S., Strausberg, R.L., Fodor, S.P., and Gingeras, T.R. 2002. Large-scale transcriptional activity in chromosome 21 and 22. Science 296: 916-919. [DOI] [PubMed] [Google Scholar]
- Kellogg, E.A. 2003. What happens to genes in duplicated genomes. Proc. Natl. Acad. Sci. 100: 4369-4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi, S., Satoh, K., Nagata, T., Kawagashira, N., Doi, K., Kishimoto, N., Yazaki, J., Ishikawa, M., Yamada, H., Ooka, H., et al. 2003. Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science 301: 376-379. [DOI] [PubMed] [Google Scholar]
- Kim, H., Snesrud, E.C., Haas, B., Cheung, F., Town, C.D., and Quackenbush, J. 2003. Gene expression analysis of Arabidopsis chromosome 2 using a genomic DNA amplicon microarray. Genome Res. 13: 327-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin, E.V., Aravind, L., and Kondrashov, A.S. 2000. The impact of comparative genomics on our understanding of evolution. Cell 101: 573-576. [DOI] [PubMed] [Google Scholar]
- Labrador, M. and Corces, V.G. 2002. Setting the boundaries of chromatin domains and nuclear organization. Cell 111: 151-154. [DOI] [PubMed] [Google Scholar]
- Lercher, M.J., Urrutia, A.O., and Hurst, L.D. 2002. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat. Genet. 31: 180-183. [DOI] [PubMed] [Google Scholar]
- Ma, L., Li, J., Qu, L., Hager, J., Chen, Z., Zhao, H., and Deng, X.W. 2001. Light control of Arabidopsis development entails coordinated regulation of genome expression and cellular pathways. Plant Cell 13: 2589-2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L., Gao, Y., Li, J., Chen, Z., Li, J., Zhao, H., and Deng, X.W. 2002. Genomic evidence for COP1 as a repressor of light-regulated gene expression and development in Arabidopsis. Plant Cell 14: 2383-2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L., Sun, N., Liu, X., Jiao, Y., Zhao, H., and Deng, X.W. 2005. Organ-specific genome expression atlas during Arabidopsis development. Plant Physiol. 138: 80-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Zapater, J.M. and Salinas, J. 1998. Arabidopsis protocols, pp. 27-30. Humana Press, Totowa, NJ.
- McCarroll, S.A., Murphy, C.T., Zou, S., Pletcher, S.D., Chin, C.S., Jan, Y.N., Kenyon, C., Bargmann, C.I., and Li, H. 2004. Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat. Genet. 36: 197-204. [DOI] [PubMed] [Google Scholar]
- Meyerowitz, E.M. 2002. Plants compared to animals: The broadest comparative study of development. Science 295: 1482-1485. [DOI] [PubMed] [Google Scholar]
- Nojiri, H., Sugimori, M., Yamane, H., Nishimura, Y., Yamada, A., Shibuya, N., Kodama, O., Murofushi, N., and Omori, T. 1996. Involvement of jasmonic acid in elicitor-induced phytoalexin production in suspension-cultured rice cells. Plant Physiol. 110: 387-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart, C.M. and Cohen, R.E. 2004. Proteasomes and their kin: Proteases in the machine age. Nat. Rev. Mol. Cell. Biol. 5: 177-187. [DOI] [PubMed] [Google Scholar]
- Prince, V.E. and Pickett, F.B. 2002. Splitting pairs: The diverging fates of duplicated genes. Nat. Rev. Genet. 3: 827-837. [DOI] [PubMed] [Google Scholar]
- The Rice Chromosome 10 Sequencing Consortium. 2003. In-depth view of structure, activity, and evolution of rice chromosome 10. Science 300: 1566-1569. [DOI] [PubMed] [Google Scholar]
- Rinn, J.L., Euskirchen, G., Bertone, P., Martone, R., Luscombe, N.M., Hartman, S., Harrison, P.M., Nelson, F.K., Miller, P., Gerstein, M., et al. 2003. The transcriptional activity of human Chromosome 22. Genes & Dev. 17: 529-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki, T., Matsumoto, T., Yamamoto, K., Sakata, K., Baba, T., Katayose, Y., Wu, J., Niimura, Y., Cheng, Z., Nagamura, Y., et al. 2002. The genome sequence and structure of rice chromosome 1. Nature 420: 312-316. [DOI] [PubMed] [Google Scholar]
- Schena, M., Shalon, D., Davis, R.W., and Brown, P.O. 1995. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270: 467-470. [DOI] [PubMed] [Google Scholar]
- Schoof, H. and Karlowski, W.M. 2003. Comparison of rice and Arabidopsis annotation. Curr. Opin. Plant Biol. 6: 106-112. [DOI] [PubMed] [Google Scholar]
- Sengupta, R. and Tompa, M. 2002. Quality control in manufacturing oligo arrays: A combinatorial design approach. J. Comput. Biol. 9: 1-22. [DOI] [PubMed] [Google Scholar]
- Serino, G. and Deng, X.W. 2003. The COP9 signalosome: Regulating plant development through the control of proteolysis. Annu. Rev. Plant Biol. 54: 165-182. [DOI] [PubMed] [Google Scholar]
- Spellman, P.T. and Rubin, G.M. 2002. Evidence for large domains of similarly expressed genes in the Drosophila genome. J. Biol. 1: 1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, J.A., Shirasu, K., and Deng, X.W. 2003. The diverse roles of ubiquitin and the 26S proteasome in the life of plants. Nat. Rev. Genet. 4: 948-958. [DOI] [PubMed] [Google Scholar]
- Vierstra, R.D. 2003. The ubiquitin/26S proteasome pathway, the complex last chapter in the life of many plant proteins. Trends Plant Sci. 8: 135-142. [DOI] [PubMed] [Google Scholar]
- Wu, J., Maehara, T., Shimokawa, T., Yamamoto, S., Harada, C., Takazaki, Y., Ono, N., Mukai, Y., Koike, K., Yazaki, J., et al. 2002. A comprehensive rice transcript map containing 6591 expressed sequence tag sites. Plant Cell 14: 525-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, K., Lim, J., Dale, J.M., Chen, H., Shinn, P., Palm, C.J., Southwick, A.M., Wu, H.C., Kim, C., Nguyen, M., et al. 2003. Empirical analysis of transcriptional activity in the Arabidopsis genome. Science 302: 842-846. [DOI] [PubMed] [Google Scholar]
- Yu, J., Hu, S., Wang, J., Wong, G.K.S, Li, S., Liu, B., Deng, Y., Dai, L., Zhou, Y., Zhang, X., et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. indica) Science 296: 79-92. [DOI] [PubMed] [Google Scholar]
- Yu, J., Wang, J., Lin, W., Li, S., Li, H., Zhou, J., Ni, P., Dong, W., Hu, S., Zeng, C, et al. 2005. The genome sequence of indica and japonica rice. PLoS Biol. 3: e38.15685292 [Google Scholar]
- Zhao, W., Wang, J., He, X., Huang, X., Jiao, Y., Dai, M., Wei, S., Fu, J., Chen, Y., Ren, X., et al. 2004. BGI-RIS: An integrated information resource and comparative analysis workbench for rice genomic. Nucleic Acids Res. 32: D377-D382. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://info.med.yale.edu/wmkeck/dna_arrays.htm; Yale University DNA laboratory.
- http://rise.genomics.org.cn; Phase II rice genome assembly, October 2002.
- http://www.geneontology.org; Gene Ontology.
- http://rgp.dna.affrc.go.jp/index.html; Japan Rice Genome Program.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}