

Abstract
MSK (mitogen- and stress-activated protein kinase) 1 and MSK2 are kinases activated downstream of either the ERK (extracellular-signal-regulated kinase) 1/2 or p38 MAPK (mitogen-activated protein kinase) pathways in vivo and are required for the phosphorylation of CREB (cAMP response element-binding protein) and histone H3. Here we show that the MSKs are involved in regulating the transcription of the immediate early gene Nur77. Stimulation of mouse embryonic fibroblasts with PMA, EGF (epidermal growth factor), TNF (tumour necrosis factor) or anisomycin resulted in induction of the Nur77 mRNA. The induction of Nur77 by TNF and anisomycin was abolished in MSK1/2 double-knockout cells, whereas induction was significantly reduced in response to PMA or EGF. The MSK responsive elements were mapped to two AP (activator protein)-1-like elements in the Nur77 promoter. The induction of Nur77 was also blocked by A-CREB, suggesting that MSKs control Nur77 transcription by phosphorylating CREB bound to the two AP-1-like elements. Consistent with the decrease in Nur77 mRNA levels in the MSK1/2-knockout cells, it was also found that MSKs were required for the induction of Nur77 protein by PMA and TNF. MSKs were also found to be required for the transcription of two genes related to Nur77, Nurr1 and Nor1, which were also transcribed in a CREB- or ATF1 (activating transcription factor-1)-dependent manner. Downstream of anisomycin signalling, a second ERK-dependent pathway, independent of MSK and CREB, was also required for the transcription of Nurr1 and Nor1.
Keywords: extracellular-signal-regulated kinase (ERK), mitogen- and stress-activated protein kinase (MSK), Nur77, Nurr1, Nor1, p38
Abbreviations: AP-1, activator protein-1; ATF1, activating transcription factor-1; CRE, cAMP response element; CREB, CRE-binding protein; DMEM, Dulbecco's modified Eagle's medium; EGF, epidermal growth factor; ERK, extracellular-signal-regulated kinase; MAPK, mitogen-activated protein kinase; MSK, mitogen- and stress-activated protein kinase; MEF, murine embryonic fibroblast; MEF2D, myocyte-enhancer-binding factor 2D; NGF, nerve growth factor; NGFI-B, NGF-induced B factor; NBRE, NGFI-B response element; PKA, protein kinase A; PKI, PKA inhibitor; TCR, T-cell receptor; TNF, tumour necrosis factor α
INTRODUCTION
MSK (mitogen- and stress-activated protein kinase)1 and MSK2 are dual kinase domain proteins that are activated by either the mitogenic stimulation of cells or by cellular stress. MSKs are direct substrates of the MAPK (mitogen-activated protein kinase) family members ERK (extracellular-signal-regulated kinase) 1/2 and p38 (stress-activated protein kinase 2, SAPK2), and the phosphorylation of MSKs by these kinases is required for MSK activity [1–4]. In vivo, both the ERK1/2 and p38 pathways are able to activate MSKs, although the relative contribution of each pathway depends on the stimulus and cell type in question. MSKs are constitutively localized to the nucleus suggesting that they may play a role in nuclear function [1]. Knockout mice for either MSK1 or MSK2, or both, are viable and fertile and show no apparent adverse phenotype [5]. Despite this, analysis of cells derived from these mice has shown that MSKs are required for the mitogen or stress induced phosphorylation of the transcription factors CREB (cAMP response element-binding protein) and ATF1 (activating transcription factor-1) [5,6], and the chromatin proteins histone H3 and high mobility group-14 in vivo [7–10]. In addition MSKs have also been suggested to phosphorylate other transcription factors including nuclear-factor-κB-binding protein and ER81 [11,12] although genetic evidence for this is still lacking. Studies on cells from knockout mice have further shown a role for MSKs in the regulation of mitogen- or stress-induced transcription of the immediate early genes c-fos and junB [5,8,13,14]. MSKs have also been suggested to be involved in the transcription of Cyr61 [15] and MUC5AC [16]. However, MSKs are not involved in the regulation of all immediate early genes as they do not regulate mitogen- or stress-induced transcription of early growth response factor-1 and c-jun [5,8]. The NR4A proteins, Nur77, Nor1 and Nurr1, form a subfamily of nuclear receptors [17–19]. Nur77 [also called N10, NGFI-B (nerve-growth-factor-induced B factor), NAK1 or TR3] was the first NR4A protein to identified, and was initially described as an immediate early gene induced by mitogens [20]. Since then transcription of Nur77 has been shown to occur in response to a variety of other signals. The two other NR4A genes, Nurr1 and Nor1, have also been shown to act as immediate early genes, and can be induced by similar stimuli [18,21,22]. NR4A nuclear receptors have been implicated in the regulation of cell survival and apoptosis. Both Nur77 and Nor1 have been shown to promote apoptosis and negative selection in T-cells [23–25] and more recently have also been shown to promote cell death in cancer cells [26–28] and macrophages [29]. In contrast Nur77 has also been shown to promote cell survival and growth in some cancer cell lines [30,31]. Nurr1 is required for the development and survival of dopaminergic neurons [32,33], and has been linked to Parkinson's disease [34,35].
Similar to other steroid receptors, Nur77, Nurr1 and Nor1 contain a DNA-binding domain, a transactivation domain, and a putative ligand-binding domain. NR4A proteins have been classed as orphan receptors as they have no known ligand, and there is now much evidence that they are true orphan receptors that do not require ligand binding for their physiological function. Nur77, as well as Nurr1 and Nor1, are able to bind as monomers to NBREs (NGFI-B response element) (AAAGGTCA) and as homodimers to NurRE (Nur response element) (TGATATTTX6AAATGCCA) in DNA [36]. Nur77 has been reported to bind to, and activate transcription of, NBRE and NuRE sequence reporter constructs, without the requirement for additional signalling inputs [36–38]. The function of the ligand-binding domain of Nur77 is unclear, but may be involved in dimerization or cofactor recuitment. The crystal structure of Nurr1 has shown that the ligand-binding domain exists in a closed form with an inaccessible ligand-binding site, supporting the idea that these proteins act independently of ligand binding [39]. As the NR4A nuclear receptors appear to function in a ligand-independent manner, other mechanisms such as control of NR4A transcription or post-translational modification are likely to exist, to allow the control of these nuclear orphan receptors in vivo. For instance phosphorylation by ERK1/2 has been shown to affect Nur77 localization [40], while phosphorylation of Ser350 in Nur77 has been suggested to inhibit the transcriptional activity of Nur77 [41].
Transcription of Nur77 has been shown to be upregulated by a variety of stimuli including TCR (T-cell receptor) activation in T-cells, serum in fibroblasts [42], NGF (nerve growth factor) and membrane depolarization in PC12 cells [43]. The kinetics and control of Nur77 transcription vary depending on the stimuli and cell type studied. In T-cells TCR activation causes a prolonged upregulation of Nur77 transcription, which involves the binding of the transcription factor MEF2D (myocyte-enhancer-binding factor 2D) to the Nur77 promoter. In unstimulated T-cells Nur77 transcription is repressed by the association of MEF2D with histone deacetylases and the repressor protein cabin1. Stimulation of the TCR inhibits the association of MEF2D with these repressor complexes allowing Nur77 transcription [29,44–48]. In PC12 cells induction of Nur77 transcription by both NGF and membrane depolarization has been shown to require two AP-1 (activator protein-1) (TGCGTCA)-like elements adjacent to the transcriptional start site in the Nur77 promoter [43]. These AP-1-like elements were reported to bind JunD, but not CREB, in PC12 cells, and expression of dominant-negative junD was found to block Nur77 transcription in response to both NGF and membrane depolarization [49]. Nur77 transcription induced by T-lymphotropic virus type I-tax-protein [50] and prostaglandin F2α [51] has also been reported to involve JunD binding to these AP-1-like elements. Induction of Nur77 by prostaglandin was further shown to require the phosphorylation of JunD by ERK1/2 [52]. Transcription of Nur77 has also been shown to be stimulated by treatments such as forskolin, which elevates the level of cyclic AMP and activates PKA (protein kinase A) [53,54], which has recently been linked to the phosphorylation and activation of CREB by PKA [22]. It has also been suggested that in some cell types both PKA and ERK1/2 are involved in the regulation of Nur77 transcription [55].
Nurr1 and Nor1 also act as immediate early genes and their transcription has been reported to be upregulated by similar stimuli to Nur77. In contrast to Nur77, however, both Nurr1 and Nor1 have one or more classical CRE (cAMP-response element) sites in their promoters and have been reported to be CREB dependent genes [28,56–58].
Here we show that in fibroblasts both mitogens and cellular stress are able to activate transcription of all three NR4A genes. We also demonstrate a role for MSKs and CREB/ATF1 in the regulation of Nur77, Nurr1 and Nor1 transcription in response to these stimuli.
MATERIALS AND METHODS
Antibodies
Antibodies which recognize total ERK1/ERK2, phospho-ERK1/2, total p38 and phospho-p38 were from Cell Signalling. The antibody against phospho-CREB was from Upstate. The anti-Nur77 antibody was generated against a specific peptide (VRDHLTGDPLALEFGK) from this protein (A. D. Wingate, unpublished work).
Plasmids and promoters
The Nur77 promoter was amplified by PCR from mouse genomic DNA using the primers gcctcgagTGCGCTCCGCTCCGCAGTC (bp+66) and gcgagctcCTAGGTGTCCAGGACAGACTGGG (bp −533). The fragment was cloned into the basic pGL3 R2.1 vector (Promega) as a SacI/XhoI fragment. Truncated fragments from this clone were used to generate fragments bp −274 to bp +66 and bp −100 to bp +66 of the promoter by PCR. To mutate the TGCGTCA AP-1-like elements in the promoter to TATATCA, mutagenesis was carried out using the Quickchange System (Stratagene). All constructs were confirmed by DNA sequencing using the Sequencing Service (School of Life Sciences, University of Dundee, Scotland, U.K.; www.dnaseq.co.uk) and Applied Biosystems’ BigDye v3.1 kit on an Applied Biosystems model 3730 automated capillary DNA sequencer. The dominant-negative junD and A-CREB constructs have been described previously [49,59]. The MSK1 expression vectors have also been described previously [1].
Cell culture
Primary murine embryonic fibroblast (MEF) cells from wild-type or MSK1/MSK2 double knockout (MSK1/MSK2−/−) mice were isolated and cultured as described previously [5]. Primary cells were used for most of the experimental work on MSK; however, for the luciferase reporter experiments, cells immortalized with simian-virus-40-large-tumour-antigen were used to allow for more efficient transfection. As knockout of p38α is embryonic lethal [60], the use of primary MEF cells was not feasible; instead, immortalized MEF cells from p38α knockout mice were used [61].
MEF cells were grown in DMEM (Dulbecco's modified Eagle's medium) containing 10% serum (Sigma), 2 mM L-glutamine, 50 units/ml penicillin G and 50 μg/ml streptomycin (Invitrogen). HeLa cells were grown in DMEM containing 10% serum (Sigma) and 1 mM sodium pyruvate (Invitrogen). Before stimulation, cells were serum-starved overnight. Where indicated, cells were pre-treated with inhibitors (2 μM PD 184352, 5 μM SB 203580, 5 μM Ro 318220, 10 μM H89 or 10 μM U0126), and then stimulated with either anisomycin (10 μg/ml), TNF (tumour necrosis factor) (10 ng/ml), EGF (epidermal growth factor) (100 ng/ml) or PMA (400 ng/ml) for the times indicated. When required, cells were transfected using FuGene 6 (Roche), in the presence of serum but in the absence of antibiotics. After 24 h, transfected cells were serum-starved for 16 h and then stimulated as described above.
Immunoblotting
Cells were lysed in 50 mM Tris/HCl, pH 7.5, 1 mM EGTA, 1 mM EDTA, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 1 mM sodium pyrophosphate, 0.27 M sucrose, 1% (v/v) Triton X-100, 0.1% (v/v) 2-mercaptoethanol, and then centrifuged at 13000 g for 5 min to remove insoluble material. Then 20–30 μg of soluble protein extract was run on 4–12% polyacrylamide gels (Novex, Invitrogen) and transferred to nitrocellulose membranes. Proteins were detected using primary antibodies in combination with horseradish peroxidase (Pierce) and chemiluminescent substrate (Amersham).
Kinase assays
For kinase assays, MSK1 was immunoprecipitated from 0.5 mg of pre-cleared cell lysate using 2 μg of anti-MSK1 antibody coupled to Protein-G–Sepharose. Precipitates were washed twice in 0.5 M NaCl, 50 mM Tris/HCl, pH 7.5, 0.1 mM EGTA and 0.1% (v/v) 2-mercaptoethanol and once in 50 mM Tris/HCl, pH 7.5, 0.1 mM EGTA and 0.1% (v/v) 2-mercaptoethanol. Precipitates were then resuspended in 35 μl of reaction buffer [containing Tris/HCl, pH 7.5, EGTA, PKI (protein kinase A inhibitor) and Crosstide peptide, GRPRTSSFAEG], and the reaction was started by the addition of 10 μl of 50 mM magnesium acetate, 0.5 mM [32P]ATP and incubated at 30 °C for 20 min. Final concentrations of reagents in the assay were 50 mM Tris/HCl, pH 7.5, 0.1 mM EGTA, 0.1% 2-mercaptoethanol, 2.5 μM PKI, 30 μM Crosstide peptide, 10 mM magnesium acetate and 0.1 mM [32P]ATP [5]. Reactions were stopped by transfer on to P81 paper and washing in 75 mM orthophosphoric acid. One unit was defined as the incorporation of 1 nmol of phosphate into the substrate peptide in 1 min.
Real-time PCR
Cells were treated as indicated, then lysed and RNA isolated using the NucleoSpin RNA purification method (Macherey-Nagel). RNA was reverse-transcribed (iScript, Bio-Rad) and real-time PCR was carried out using Sybr Green based detection. Levels of 18 S rRNA were used as normalization controls and relative mRNA levels calculated using the equation:
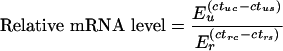 |
where E is the efficiency of the PCR, ct is the threshold cycle, u is the mRNA of interest, r is the reference gene (18 S RNA or β2-microglobulin), s is the sample and c is the unstimulated control sample [62]. Primer sequences for the amplification of mouse and human Nurr1, Nor1 and Nur77, mouse 18 S RNA and human-β2 microglobulin are shown in Table 1.
Table 1. Primer sequences used for real-time PCR.
| Primer | Sense | Antisense |
|---|---|---|
| Nur77 (mouse) | CCTGTTGCTAGAGTCTGCCTTC | CAATCCAATCACCAAAGCCACG |
| Nurr1 (mouse) | GAAGAGAGCGGACAAGGAGATC | AAGGCATGGCTTCAGCAGAG |
| Nor1 (mouse) | GCCATCTCCTCCGATCTGTATG | GAGGCCGTCAGAAGGTTGTAG |
| 18 S RNA (mouse) | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
| Nur77 (human) | ACAGGAGAGTTTGACACC | AACTTGAAGGAGGCAGAG |
| β2 Microglobulin (human) | TGCCGTGTGAACCATGTGAC | ACCTCCATGATGCTGCTTACA |
Luciferase assays
Cells were transfected with the appropriate Nur77 promoter vectors along with a Renilla luciferase vector as a transfection control. After 24 h transfected cells were serum-starved for 16 h. Cells were then left untreated or stimulated for 60 min with 400 ng/ml PMA, 100 ng/ml EGF or 10 ng/ml TNF. Following stimulation cells were lysed and the activities of firefly and Renilla luciferase measured using the Dual-Glo system (Promega). Luciferase activities were normalized to the Renilla control, and then fold stimulation was calculated relative to the bp −533 (Figure 2B) or bp −274 (Figures 2C and 2D) Nur77 promoter in unstimulated cells.
Figure 2. Reporter analysis of the murine Nur77 promoter.

(A) Fragments from bp −533, bp −274 and bp −100 to bp +66 of the murine Nur77 promoter were cloned into pGL3 to give Nur77 promoter luciferase reporters. Two proximal AP-1 sites were also mutated. Grey circles indicate AP-1-like elements while white circles indicate the mutated sites. (B) The 533 (cross-hatched bars), 274 (black bars) and 100 (grey bars) Nur77 reporter vectors were transfected into wild-type immortalized MEF cells. After 24 h transfected cells were serum-starved for a further 16 h and then stimulated with PMA (400 ng/ml, 60 min), EGF (100 ng/ml, 60 min) or TNF (10 ng/ml, 60 min). Cells were then lysed and luciferase activity measured as described in the Materials and methods section. (C) The 274 (black bars), AP1–1 (cross hatched bars), AP1–2 (horizontal hatched bars) and AP1–1/2 (grey bars) vectors were transfected into wild-type fibroblasts. After 24 h transfected cells were serum-starved for a further 16 h and then stimulated with PMA (400 ng/ml, 60 min) EGF (100 ng/ml, 60 min) or TNF (10 ng/ml, 60 min). Cells were then lysed and luciferase activity measured as described in the Materials and methods section. (D) The bp −274 Nur77 reporter vectors were co-transfected with either empty vector (black bars), A-CREB (white bars) or dominant-negative junD (cross-hatched bars) expression vectors into wild-type MEF cells. After 24 h transfected cells were serum-starved for a further 16 h and then stimulated with PMA (400 ng/ml, 60 min) EGF (100 ng/ml, 60 min) or TNF (10 ng/ml, 60 min). Cells were then lysed and luciferase activity measured as described in the Materials and methods section.
RESULTS
CREB is required for the transcription of Nur77, Nor1 and Nurr1
CREB, and the related transcription factor ATF1, are phosphorylated in response to a range of mitogenic stimuli and cellular stresses and have been implicated in the induction of some immediate early genes. Using GALA prediction software, analysis of the promoter regions of Nurr1 and Nor1 reveals the presence of two potential CRE sequences that are conserved in the human, mouse, rat and canine promoters (Figure 1A). Analysis of the Nur77 promoter did not identify a conserved CRE sequence but did reveal the presence of four conserved AP-1-like sequences. Three of these were found in the human, mouse, rat and canine promoters; however, the fourth was not conserved in the canine Nur77 promoter region. The role of CREB or ATF1 in the induction of these genes was tested in HeLa cells using A-CREB. This is a dominant-negative form of CREB that lacks the DNA binding domain, but that can dimerize to endogenous CREB or ATF1 and prevent its binding to DNA. PMA, EGF, TNF and anisomycin were all found to induce the mRNAs for Nur77, Nurr1 and Nor1 in HeLa cells. In response to these stimuli, A-CREB either blocked or significantly reduced the expression of Nur77, Nurr1 and Nor1 (Figure 1B). As junD has also been suggested to play a role in the induction of Nur77, we also tested the effect of dominant-negative junD expression (Figure 1C). In response to PMA, dominant-negative junD did not prevent the induction of Nur77, Nurr1 or Nor1. In response to EGF and TNF the dominant-negative junD was able to reduce the induction of Nur77, Nurr1 and Nor1; however, this decrease was less marked than with A-CREB. In response to anisomycin, the dominant-negative junD did reduce the induction of Nur77, but not of Nurr1 or Nor1. The dominant-negative junD did however completely block the induction of a known AP-1 target, IL-6 (the gene encoding interleukin-6), in response to PMA (results not shown).
Figure 1. NR4A gene transcription requires CREB.

(A) The promoter region bp −2000 to bp +1000 of the transcriptional start of the Nur77, Nor1 and Nurr1 transcription factors were analysed by GALA prediction software (http://gala.cse.psu.edu/). Potential CRE (closed circle) or AP-1-like (grey rectangle in human, mouse, rat and canine promoters, black rectangle indicating a non-conserved region in the canine promoter) as well as potential sites for MEF cells (black diamond), Elk (star), USF (upstream stimulatory factor; black triangle) and nuclear factor κB (grey triangle) were identified. Sequences predicted as CRE sites were TGACGTAG and TGACGTCT in Nor1, and TGACG.CA and TGACGTCA in Nurr1, against a concensus CRE site of TGACGT(A/C)A. (B) HeLa cells were transfected with either an empty vector (light grey bars) or A-CREB expression vector (dark grey bars). After 24 h transfected cells were serum-starved for 16 h and left unstimulated (control) or stimulated for 60 min with 400 ng/ml PMA, 100 ng/ml EGF, 10 ng/ml TNF or 10 μg/ml anisomycin. Cells were then lysed and RNA isolated and analysed by real-time PCR. Levels of Nur77 mRNA were determined relative to β2-microglobulin controls as described in the Materials and methods section. Error bars represent the S.E.M. of three individual stimulations. (C) As in (B) except a dominant-negative junD vector (black bars) was used in place of A-CREB.
To map the response element in the Nur77 promoter, various regions of the mouse Nur77 promoter region were cloned into a luciferase reporter vector (Figure 2A), and transfected into immortalized MEF cells. A vector containing bp −533 to bp +66 of the Nur77 promoter was able to drive expression of the luciferase reporter, and this could be further stimulated by PMA and EGF and weakly stimulated by TNF. Truncation of the promoter to bp −274 to bp +66 (a fragment still containing all four AP-1-like elements) did not affect this activity. Further truncation to a bp −100 to bp +66 fragment decreased the basal activity of the promoter and prevented the induction by PMA, EGF and TNF, suggesting that the response elements lie between bp −274 to bp −100 (Figure 2B). This region contains two AP-1-like elements; mutation of either or both of these AP-1-like sequences prevented the induction of the reporter by PMA, EGF or TNF (Figure 2C). To confirm that this region was the region targeted by CREB, the bp −274 to bp +66 vector was co-transfected with an expression vector for either A-CREB or dominant-negative junD (Figure 2C). A-CREB was found to significantly decrease the induction of the reporter by PMA, EGF and TNF. Consistent with the results obtained for the endogenous Nur77 gene in HeLa cells, the dominant-negative junD vector was less effective at blocking the induction of the reporter than A-CREB.
MSKs are required downstream of MAPK signalling for the transcription of Nur77, Nurr1 and Nor1
PMA, EGF, TNF and anisomycin are all known to stimulate CREB phosphorylation downstream of either the ERK1/2 or p38 MAPK cascades. In MEF cells we found that TNF stimulation resulted in a transient activation of ERK1/2 and a more prolonged activation of p38. TNF also activated MSK1 and MSK2, resulting in the phosphorylation of CREB (results are shown in Supplementary Figure 1S at http://www.BiochemJ.org/bj/390/bj3900749add.htm). We have previously shown that in primary MEF cells, PMA stimulation resulted in a strong and sustained activation of the ERK1/2 cascade but did not activate p38α. EGF both strongly activates ERK1/2 and weakly activates p38α, although the activation of ERK1/2 was more transient than with PMA. In contrast anisomycin strongly activates p38 but only weakly activates ERK1/2 ([5]; results are shown in Supplementary Figure 2S at http://www.BiochemJ.org/bj/390/bj3900749add.htm). PMA and EGF were also found to activate MSK1 and MSK2, and phosphorylate CREB downstream of the ERK1/2 cascade in MEF cells, while anisomycin activated MSKs and phosphorylated CREB downstream of p38 [5]. We therefore examined the transcription of NR4A genes in these cells using real-time PCR. Nur77, Nurr1 and Nor1 mRNA levels were found to be induced by PMA, EGF, TNF and anisomycin (Figure 3). To determine the role of MAPK signalling in this process, the p38α/β inhibitor SB 203580 [63] and the MKK (MAPK kinase) 1/2 inhibitor PD 184352 (which blocks the activation of ERK1/2 in vivo [63]) were used. In response to PMA and EGF, Nur77, Nurr1 and Nor1 induction were completely blocked by PD 184352, but unaffected by SB 203580, demonstrating a role for ERK1/2 but not p38α/β in the transcription of these genes (Figures 3A and 3B). The induction of Nur77, Nurr1 and Nor1 mRNAs by TNF was partially inhibited by either PD 184352 or SB 203580 but completely blocked by a combination of both inhibitors (Figure 3C). The induction of Nur77, Nurr1 and Nor1 by anisomycin was almost completely blocked by SB 203580 demonstrating a major role for p38 in this process (Figure 3D). As SB 203580 inhibits both p38α and p38β it does not distinguish between the roles of these two isoforms. In vitro both p38α and p38β can phosphorylate and activate MSK; however, it is not known if both isoforms contribute to MSK activation and CREB phosphorylation in vivo [1]. We therefore used immortalized MEF cells from p38α knockout and wild-type mice to determine the role of p38α in MSK activation and NR4A transcription. As expected Nur77, Nurr1 and Nor1 transcription was induced in wild-type MEF cells and the induction was inhibited by SB 203580. No induction of these genes was seen in p38α knockout MEF cells in response to anisomycin (Figure 4A). Knockout of p38α was also found to prevent the activation of the CREB kinase MSK1 in response to anisomycin (Figure 4B). Consistent with this, CREB was phosphorylated in response to anisomycin in wild-type MEF cells, but not in p38α knockout MEF cells (Figure 4C).
Figure 3. NR4A transcription requires MAPK signalling.

Wild-type primary MEF cells were serum-starved for 16 h and then treated for 1 h with 2 μM PD 184352 (PD), 5 μM SB 203580 (SB), or no inhibitor as indicated. Cells were then left unstimulated (control) or stimulated (st) for 60 min with 400 ng/ml PMA (A), 100 ng/ml EGF (B), 10 ng/ml TNF (C) or 10 μg/ml anisomycin (D). Cells were lysed and the RNA isolated. Nur77 (grey bars), Nurr1 (white bars) and Nor1 (black bars) mRNA levels were determined by real-time PCR as described in the Materials and methods section. Error bars represent the S.E.M. of three individual stimulations.
Figure 4. ERK1/2 and p38α are required for NR4A induction by anisomycin.

Immortalized p38α knockout (white bars) and wild-type (black bars) MEF cells (A–C) were serum-starved for 16 h, incubated with 5 μM SB 203580 (SB) for 1 h where indicated and then left unstimulated (control) or stimulated with 10 μg/ml anisomycin (Anis) for 1 h. After stimulation, total RNA was isolated and the expression of Nur77, Nurr1 and Nor1 determined by real-time PCR (A). Levels of MSK1 activity were determined by immunoprecipitation kinase assay as under the Materials and methods section (B). To determine if the ERK1/2 and p38 pathways were activated, lysates were run on SDS/polyacrylamide gels. The levels of phospho-CREB, phospho-p38, (p-p38), phospho-ERK1/2, total p38α and total ERK1/2 were determined by immunoblotting with specific antibodies (C). (D) Primary wild-type MEF cells were serum-starved for 16 h, incubated for 1 h with 5 μM SB 203580 (SB) and/or 2 μM PD 184352 (PD) where indicated and then stimulated with 10 μg/ml anisomycin (Anis) for 1 h. Cells were then lysed and the levels of phospho-CREB, phospho-p38, phospho-ERK1/2, total p38α and total ERK1/2 were determined by immunoblotting with specific antibodies. (E) Primary wild-type MEF cells were serum-starved for 16 h, incubated for 1 h with 10 μM U0126 (grey bars) or DMSO control (black bars) and then stimulated with 10 μg/ml anisomycin for 1 h. The induction of Nur77, Nurr1 and Nor1 mRNA was then determined by real-time PCR as described in the Materials and methods section. Error bars represent the S.E.M. of three individual stimulations.
Interestingly PD 184352 partially inhibited Nur77 induction and almost completely blocked Nurr1 and Nor1 mRNA induction in response to anisomycin, suggesting that ERK1/2 was involved in this process. This was unexpected as anisomycin is only a weak activator of ERK1/2 in MEF cells [5]. Inhibition of ERK1/2 by PD 184352 did not prevent p38α activation, as judged by immunoblotting with an antibody against the phosphorylated TGY motif of p38. PD 184352 also did not affect the phosphorylation of CREB downstream of anisomycin in MEF cells (Figure 4D). Together this suggests that, for the transcription of Nurr1 and Nor1 in response to anisomycin, a second pathway, independent of CREB and p38, may be required. To confirm that the effect of PD 184352 was due to inhibition of the ERK1/2 cascade and not a non-specific effect, another MEK1/2 inhibitor, U0126, was tested. U0126 was able to reduce the Nur77 transcription induced by ansiomycin by half and the Nurr1 and Nor1 transcription by approx. 70% (Figure 4E).
We have shown previously that MSK1 and MSK2 phosphorylate CREB and ATF1 downstream of PMA, EGF and anisomycin in MEF cells. MSK1 and 2 are also required for the TNF-induced phosphorylation of CREB and ATF1 in these cells (results are shown in Supplementary Figure 2S at http://www.BiochemJ.org/bj/390/bj3900749add.htm). This suggests that MSKs may be required for NR4A transcription downstream of MAPK signalling. We therefore examined the induction of Nur77, Nor1 and Nurr1 in MSK1/2 knockout primary MEF cell lines. In wild-type cells PMA strongly induced Nur77, Nurr1 and Nor1 mRNA levels up to a maximal time point of 60 min, and the induction of all three genes was significantly reduced in MSK1/MSK2−/− cells (Figure 5A). EGF stimulated the transcription of all three genes in wild-type cells; however, Nur77 induction was maximal by 30 min whereas induction of Nurr1 and Nor1 was maximal by 60 min. The induction of Nur77 was decreased in MSK1/MSK2−/− cells, while the induction of both Nurr1 and Nor1 was blocked in MSK1/MSK2−/− cells (Figure 5B). TNF stimulation also induced all three genes in wild-type cells, reaching maximal levels after 60 min of stimulation, and this was significantly reduced in the MSK1/MSK2−/− cells (Figure 5C). Anisomycin produced a more prolonged increase in Nur77, Nor1 and Nurr1 mRNA levels in wild-type cells, and this was abolished in MSK1/MSK2−/− cells (Figure 5D). Consistent with the decrease in the mRNA levels for Nur77 seen in the MSK1/MSK2−/− cells, the increase in Nur77 protein levels seen in MSK1/MSK2−/− cells in response to PMA and TNF was much lower than in wild-type cells (Figure 5E).
Figure 5. MSKs are required for the transcription of Nur77.

(A–D) Wild-type (solid bars), MSK1/MSK2−/− (hatched bars) and primary MEF cells were serum-starved for 16 h. Cells were then stimulated with (A) PMA (400 ng/ml), (B) EGF (100 ng/ml), (C) TNF (10 ng/ml) or (D) anisomycin (10 μg/ml) for 0, 30, 60 or 120 min. Cells were lysed and the RNA isolated. Nur77, Nurr1 and Nor1 levels were determined by real-time PCR as described in the Materials and methods section. Error bars represent the S.E.M. of three individual stimulations. (E) Wild-type (WT) or MSK1/MSK2−/− cells were serum-starved (KO) for 16 h, and then stimulated with 400 ng/ml PMA or 10 ng/ml TNF for the times indicated. Cells were then lysed and Nur77 immunoprecipitated from 3 mg of cell lysate. Immunoprecipitates were then blotted for Nur77. Cell lysates were also blotted for total ERK.
To ensure that the loss of NR4A gene induction was due to the lack of MSKs, and not clonal differences between the different MEF cell preparations, we examined the effect of compounds that inhibit MSKs in cells, and also re-expressed MSK1 in immortalized MSK1/MSK2−/− MEF cells. H89 and Ro 318220 are two compounds that have been shown to inhibit MSKs both in vitro and in cells [63,64]. Both of these compounds were able to reduce or block the induction of Nur77, Nurr1 and Nor1 mRNAs in immortalized wild-type MEF cells stimulated with either EGF or anisomycin (Figures 6A and 6B). Similar effects were also seen using PMA or TNF stimulation (results not shown). Transient expression of MSK1 in immortalized MSK1/MSK2−/− MEF cells was tested for the ability to restore the induction of endogenous Nur77, Nor1 and Nurr1 transcription in knockout cells (Figure 6C). Consistent with observations in primary MSK1/MSK2−/− cells, EGF stimulation caused modest induction of Nur77, but not of Nor1 or Nurr1, whereas anisomycin stimulation resulted in a modest induction (between 2–10-fold) of all three genes. In all cases expression of a wild-type MSK1 protein increased the induction of these genes. This change was dependent on the kinase activity of MSK1, as expression of a kinase-dead mutant of MSK1 had no effect on Nur77, Nurr1 or Nor1 induction. The fold induction was less than was seen for wild-type cells, however, this probably reflects the low transfection efficiency of these cells (15–25%). Finally the bp −274 to bp +66 Nur77 promoter-reporter-vector was transiently transfected into MSK1/MSK2−/− MEF cells with either empty vector or MSK1-expression vectors encoding either wild-type or kinase-dead protein. The luciferase reporter was expressed in these cells at similar basal levels to wild-type cells. Unlike in wildtype cells, however, where the reporter was further stimulated by PMA, the reporter was not significantly stimulated by PMA in MSK1/MSK2−/− cells (Figure 6D). Co-transfection of the reporter with an expression construct for wild-type MSK1, however, restored the ability of the reporter to be induced by PMA in MSK1/MSK2−/− cells. However, co-transfection with a kinase-dead version of MSK1 did not restore induction of the reporter vector.
Figure 6. Expression of MSK1 in MSK1/MSK2−/− cells rescues NR4A expression.

(A) Wild-type immortalized MEF cells were serum-starved for 16 h and then incubated with 5 μM Ro 318220 (RO) or 10 μM H89 were indicated. Cells were then left unstimualed, or stimulated (St) with EGF (100 ng/ml) for 1 h. The induction of Nur77, Nurr1 and Nor1 mRNA was measured by real-time PCR as described in the Materials and methods section. Error bars represent the S.E.M. of three individual stimulations. (B) As in (A) except that cells were stimulated with anisomycin (10 μg/ml) for 1 h. (C) Immortalized MSK1/MSK2−/− MEF cells were transfected with either empty vector (white bars) or wild-type (light grey bars) or kinase-dead (black bars) MSK1 expression vectors. Cells were then serum-starved for 16 h and left unstimulated (control), or stimulated with either EGF (100 ng/ml) or anisomycin (10 μg/ml), for 1 h. The induction of Nur77, Nurr1 and Nor1 mRNA was measured by real-time PCR as described in the Materials and methods section. Error bars represent the S.E.M. three individual stimulations. (D) Immortalized wild-type (wt) or MSK1/MSK2−/− (ko) MEF cells were transfected with a bp −274 to bp +66 Nur77 promoter luciferase reporter vector, together with either an empty vector, wild-type or kinase-dead (KD) MSK1 expression vector, as indicated. Cells were then serum-starved for 16 h and left unstimulated (white bars), or stimulated with PMA (400 ng/ml) for 1 h. Luciferase activity was measured as described in the Materials and methods section. Error bars represent the S.E.M. of three individual stimulations.
DISCUSSION
Nur77, Nurr1 and Nor1 are a group of immediate early genes whose transcription has previously been shown to be induced by growth factors and mitogens, and this process is dependent on the activation of ERK1/2. Here we confirm these results and also demonstrate that MSK1 and MSK2 are involved in this process downstream of ERK1/2. We further demonstrate that in fibroblasts, transcription of Nur77, Nurr1 and Nor1 is also induced by the cellular stress-inducing anisomycin, and that this induction requires the activation of the p38 pathway and MSK1 and MSK2. The transcription of these genes was also induced by TNF in fibroblasts, and this required MSKs which acted downstream of both the ERK1/2 and p38α cascades. The induction of the NR4A genes by anisomycin or TNF was not seen in fibroblasts from MSK1/MSK2−/− mice, suggesting that these kinases may act downstream of p38α and ERK1/2 in controlling Nur77 transcription. MSKs are essential for the phosphorylation of CREB and ATF1 in response to anisomycin or TNF stimulation of fibroblasts ([5], results are shown in Supplementary Figure 1S at http://www.BiochemJ.org/bj/390/bj3900749add.htm). MSKs have previously been shown to activate transcription of CRE-dependent genes, suggesting a possible mechanism by which they could control Nur77 transcription [14]. The Nur77 promoter in mice does not contain a consensus CRE sequence; however, the induction of Nur77 by PMA, EGF, anisomycin and TNF was found to require the presence of two AP-1-like elements in the promoter. AP-1-like elements have a similar sequence to CRE elements and have been reported to be able to bind members of the CREB family of transcription factors [22]. Consistent with this, expression of a dominant-negative form of CREB, A-CREB, was able to completely block the expression of Nur77 in response to cellular stress. Transcription of Nur77 in response to PMA or EGF was also completely blocked by expression of A-CREB. In contrast, transcription of Nur77 in MSK1/MSK2−/− cells in response to PMA or EGF was decreased but not abolished. This may be because PMA is still able to induce residual phosphorylation of CREB and ATF1 in response to PMA in the MSK1/MSK2−/− cells [5], and this is enough to promote transcription of these genes. Consistent with our findings implicating CREB or ATF1 in the transcription of Nur77, it has recently been reported that forskolin-induced transcription of Nur77 involves CREB [22]. In addition it has also been shown that CREB is capable of binding to the endogenous Nur77 promoter in vivo [65]. These results are, however, in contrast with earlier reports suggesting a role for junD in regulating Nur77 transcription via the AP-1-like elements in response to NGF or KCl in PC12 cells or prostaglandin F2α in ovarian cells [49,52]. In these systems, junD was observed to bind to an oligonucleotide based on the Nur77 AP-1-like element in electrophoretic mobility shift assays and expression of a dominant-negative junD inhibited Nur77 transcription. The dominant-negative junD acts by heterodimerizing with endogenous AP-1 proteins including JunD and c-Fos and preventing them from binding to DNA. Using the same dominant-negative junD construct in MEF cells, we did observe a modest inhibition of Nur77 transcription; however, this decrease was less marked than with A-CREB. It is unlikely that MSKs could regulate the activity of junD directly by phosphorylation, as junD does not contain any consensus MSK phosphorylation sites. It is possible that MSKs could regulate the Nur77 promoter by controlling the transcription of AP-1 type transcription factors such as c-fos or junB. However, this also seems unlikely, as MSK is required for anisomycin-stimulated Nur77 transcription. As the concentration of anisomycin used inhibits protein synthesis [66], the induction of Nur77 must be an immediate early response that does not require the synthesis of more AP-1 proteins. It is possible, however, that both junD and CREB or ATF1 act co-operatively at the level of the AP-1 sites in the Nur77 promoter. The differences between effects of dominant-negative junD in the MEF cells reported here, and the previous findings in PC12 and ovarian cells, could therefore be due to different occupancy of CREB or ATF1 and junD on the AP-1-like elements in the Nur77 promoter in different cell types.
As MSKs were required for the induction of Nur77, the involvement of MSKs in the transcription of the Nurr1 and Nor1 was also examined. Both Nurr1 and Nor1 have been reported to be CREB-dependent genes [28,57,58], and we found their expression to be inhibited by A-CREB. The transcription of both of these genes was severely compromised in MSK-deficient cells and it is likely that MSK regulates these genes through its ability to phosphorylate CREB or ATF1. For Nurr1, an alternative distal promoter and first exon has also been described upstream of the proximal promoter studied here. The distal promoter is thought not to require CREB for its activity [67]; however, we could find no evidence using real-time PCR that this distal promoter was active in fibroblasts (results not shown).
Interestingly the transcription of Nur77, Nor1 and Nurr1 in response to anisomycin was inhibited by the MSK1/2 inhibitors PD 184352 and U0126, which block the activation of ERK1/2, as well as by the p38α/β inhibitor SB 203580. In contrast, both MSK activation and CREB phosphorylation in response to anisomycin are blocked by SB 203580 but not PD 184352. This suggests that an additional input is required for Nur77, Nurr1 and Nor1 transcription, and that MSK activation and CREB phosphorylation are required but are not sufficient for transcription of these genes in response to anisomycin. As PMA, EGF and TNF phosphorylate CREB downstream of ERK1/2, it is not possible to tell if this second PD 184352-sensitive pathway also operates in NR4A induction by these stimuli. Further work will, however, be required in order to determine how ERK regulates these genes in response to anisomycin.
NR4A proteins have been described as playing roles in both dopaminergic neurons and T-cells [17,32,68]. It will therefore be interesting to examine if knocking out MSK affects the expression of NR4A proteins in these systems in mice, and if, as a consequence, the MSK knockout mice show defects in brain functioning and neuronal systems.
Online data
Acknowledgments
This research was supported by the UK Medical Research Council, Astra-Zeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck and Co, Merck KGaA and Pfizer. We thank Julia Carr for the isolation of MEF cells, and the protein production and antibody purification teams (Division of Signal Transduction Therapy, University of Dundee) co-ordinated by Hilary McLauchlan and James Hastie for affinity purification of antibodies. We would also like to thank Dr Angel Nebreda for provision of p38−/− MEF cells, Professor Lester F. Lau for the gift of the dominant-negative JunD construct and Dr Soren Impey for providing the A-CREB construct.
References
- 1.Deak M., Clifton A. D., Lucocq L. M., Alessi D. R. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierrat B., Correia J. S., Mary J.-L., Tomas-Zuber M., Lesslauer W. RSK-B, a novel ribosomal S6 kinase family member, is a CREB kinase under dominant control of p38alpha mitogen-activated protein kinase (p38alpha MAPK) J. Biol. Chem. 1998;273:29661–29671. doi: 10.1074/jbc.273.45.29661. [DOI] [PubMed] [Google Scholar]
- 3.New L., Zhao M., Li Y., Bassett W. W., Feng Y., Ludwig S., Padova F. D., Gram H., Han J. Cloning and characterization of RLPK, a novel RSK-related protein kinase. J. Biol. Chem. 1999;274:1026–1032. doi: 10.1074/jbc.274.2.1026. [DOI] [PubMed] [Google Scholar]
- 4.McCoy C. E., Campbell D. G., Deak M., Bloomberg G. B., Arthur J. S. MSK1 activity is controlled by multiple phosphorylation sites. Biochem. J. 2005;387:507–517. doi: 10.1042/BJ20041501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiggin G. R., Soloaga A., Foster J. M., Murray-Tait V., Cohen P., Arthur J. S. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol. Cell. Biol. 2002;22:2871–2881. doi: 10.1128/MCB.22.8.2871-2881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arthur J. S., Cohen P. MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS. Lett. 2000;482:44–48. doi: 10.1016/s0014-5793(00)02031-7. [DOI] [PubMed] [Google Scholar]
- 7.Thomson S., Clayton A. L., Hazzalin C. A., Rose S., Barratt M. J., Mahadevan L. C. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 1999;18:4779–4793. doi: 10.1093/emboj/18.17.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soloaga A., Thomson S., Wiggin G. R., Rampersaud N., Dyson M. H., Hazzalin C. A., Mahadevan L. C., Arthur J. S. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788–2797. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strelkov I. S., Davie J. R. Ser-10 phosphorylation of histone H3 and immediate early gene expression in oncogene-transformed mouse fibroblasts. Cancer Res. 2002;62:75–78. [PubMed] [Google Scholar]
- 10.Davie J. R. MSK1 and MSK2 mediate mitogen- and stress-induced phosphorylation of histone H3: a controversy resolved. Sci. STKE. 2003;2003:PE33. doi: 10.1126/stke.2003.195.pe33. [DOI] [PubMed] [Google Scholar]
- 11.Vermeulen L., De Wilde G., Van Damme P., Vanden Berghe W., Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1) EMBO J. 2003;22:1313–1324. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janknecht R. Regulation of the ER81 transcription factor and its coactivators by mitogen- and stress-activated protein kinase 1 (MSK1) Oncogene. 2003;22:746–755. doi: 10.1038/sj.onc.1206185. [DOI] [PubMed] [Google Scholar]
- 13.Schuck S., Soloaga A., Schratt G., Arthur J. S., Nordheim A. The kinase MSK1 is required for induction of c-fos by lysophosphatidic acid in mouse embryonic stem cells. BMC Mol. Biol. 2003;4:6. doi: 10.1186/1471-2199-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arthur J. S. C., Fong A. L., Dwyer J. M., Davare M., Reese E., Obrietan K., Impey S. Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J. Neurosci. 2004;24:4324–4332. doi: 10.1523/JNEUROSCI.5227-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han J. S., Macarak E., Rosenbloom J., Chung K. C., Chaqour B. Regulation of Cyr61/CCN1 gene expression through RhoA GTPase and p38MAPK signaling pathways. Eur. J. Biochem. 2003;270:3408–3421. doi: 10.1046/j.1432-1033.2003.03723.x. [DOI] [PubMed] [Google Scholar]
- 16.Song K. S., Lee W. J., Chung K. C., Koo J. S., Yang E. J., Choi J. Y., Yoon J. H. Interleukin-1 beta and tumor necrosis factor-alpha induce MUC5AC overexpression through a mechanism involving ERK/p38 mitogen-activated protein kinases-MSK1-CREB activation in human airway epithelial cells. J. Biol. Chem. 2003;278:23243–23250. doi: 10.1074/jbc.M300096200. [DOI] [PubMed] [Google Scholar]
- 17.Winoto A., Littman D. R. Nuclear hormone receptors in T lymphocytes. Cell. 2002;109:S57–S66. doi: 10.1016/s0092-8674(02)00710-9. [DOI] [PubMed] [Google Scholar]
- 18.Maruyama K., Tsukada T., Ohkura N., Bandoh S., Hosono T., Yamaguchi K. The NGFI-B subfamily of the nuclear receptor superfamily. Int. J. Oncol. 1998;12:1237–1243. doi: 10.3892/ijo.12.6.1237. [DOI] [PubMed] [Google Scholar]
- 19.Sladek R., Giguere V. Orphan nuclear receptors: an emerging family of metabolic regulators. Adv. Pharmacol. 2000;47:23–87. doi: 10.1016/s1054-3589(08)60109-x. [DOI] [PubMed] [Google Scholar]
- 20.Ryseck R. P., Macdonald-Bravo H., Mattei M. G., Ruppert S., Bravo R. Structure, mapping and expression of a growth factor inducible gene encoding a putative nuclear hormonal binding receptor. EMBO J. 1989;8:3327–3335. doi: 10.1002/j.1460-2075.1989.tb08494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park J. I., Park H. J., Lee Y. I., Seo Y. M., Chun S. Y. Regulation of NGFI-B expression during the ovulatory process. Mol. Cell. Endocrinol. 2003;202:25–29. doi: 10.1016/s0303-7207(03)00057-1. [DOI] [PubMed] [Google Scholar]
- 22.Fass D. M., Butler J. E., Goodman R. H. Deacetylase activity is required for cAMP activation of a subset of CREB target genes. J. Biol. Chem. 2003;278:43014–43019. doi: 10.1074/jbc.M305905200. [DOI] [PubMed] [Google Scholar]
- 23.Zhou T., Cheng J., Yang P., Wang Z., Liu C., Su X., Bluethmann H., Mountz J. D. Inhibition of Nur77/Nurr1 leads to inefficient clonal deletion of self-reactive T cells. J. Exp. Med. 1996;183:1879–1892. doi: 10.1084/jem.183.4.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng L. E., Chan F. K., Cado D., Winoto A. Functional redundancy of the Nur77 and Nor-1 orphan steroid receptors in T-cell apoptosis. EMBO J. 1997;16:1865–1875. doi: 10.1093/emboj/16.8.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calnan B. J., Szychowski S., Chan F. K., Cado D., Winoto A. A role for the orphan steroid receptor Nur77 in apoptosis accompanying antigen-induced negative selection. Immunity. 1995;3:273–282. doi: 10.1016/1074-7613(95)90113-2. [DOI] [PubMed] [Google Scholar]
- 26.Mu X., Chang C. TR3 orphan nuclear receptor mediates apoptosis through up-regulating E2F1 in human prostate cancer LNCaP cells. J. Biol. Chem. 2003;278:42840–42845. doi: 10.1074/jbc.M305594200. [DOI] [PubMed] [Google Scholar]
- 27.Wu Q., Liu S., Ye X. F., Huang Z. W., Su W. J. Dual roles of Nur77 in selective regulation of apoptosis and cell cycle by TPA and ATRA in gastric cancer cells. Carcinogenesis. 2002;23:1583–1592. doi: 10.1093/carcin/23.10.1583. [DOI] [PubMed] [Google Scholar]
- 28.Ohkubo T., Ohkura N., Maruyama K., Sasaki K., Nagasaki K., Hanzawa H., Tsukada T., Yamaguchi K. Early induction of the orphan nuclear receptor NOR-1 during cell death of the human breast cancer cell line MCF-7. Mol. Cell. Endocrinol. 2000;162:151–156. doi: 10.1016/s0303-7207(00)00222-7. [DOI] [PubMed] [Google Scholar]
- 29.Kim S. O., Ono K., Tobias P. S., Han J. Orphan nuclear receptor Nur77 is involved in caspase-independent macrophage cell death. J. Exp. Med. 2003;197:1441–1452. doi: 10.1084/jem.20021842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki S., Suzuki N., Mirtsos C., Horacek T., Lye E., Noh S. K., Ho A., Bouchard D., Mak T. W., Yeh W. C. Nur77 as a survival factor in tumor necrosis factor signaling. Proc. Natl. Acad. Sci. U.S.A. 2003;100:8276–8280. doi: 10.1073/pnas.0932598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolluri S. K., Bruey-Sedano N., Cao X., Lin B., Lin F., Han Y. H., Dawson M. I., Zhang X. K. Mitogenic effect of orphan receptor TR3 and its regulation by MEKK1 in lung cancer cells. Mol. Cell. Biol. 2003;23:8651–8667. doi: 10.1128/MCB.23.23.8651-8667.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zetterstrom R. H., Solomin L., Jansson L., Hoffer B. J., Olson L., Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science (Washington D.C.) 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- 33.Wagner J., Akerud P., Castro D. S., Holm P. C., Canals J. M., Snyder E. Y., Perlmann T., Arenas E. Induction of a midbrain dopaminergic phenotype in Nurr1-overexpressing neural stem cells by type 1 astrocytes. Nat. Biotechnol. 1999;17:653–659. doi: 10.1038/10862. [DOI] [PubMed] [Google Scholar]
- 34.Eells J. B. The control of dopamine neuron development, function and survival: insights from transgenic mice and the relevance to human disease. Curr. Med. Chem. 2003;10:857–870. doi: 10.2174/0929867033457700. [DOI] [PubMed] [Google Scholar]
- 35.Le W. D., Xu P., Jankovic J., Jiang H., Appel S. H., Smith R. G., Vassilatis D. K. Mutations in NR4A2 associated with familial Parkinson disease. Nat. Genet. 2003;33:85–89. doi: 10.1038/ng1066. [DOI] [PubMed] [Google Scholar]
- 36.Philips A., Lesage S., Gingras R., Maira M. H., Gauthier Y., Hugo P., Drouin J. Novel dimeric Nur77 signaling mechanism in endocrine and lymphoid cells. Mol. Cell. Biol. 1997;17:5946–5951. doi: 10.1128/mcb.17.10.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis I. J., Hazel T. G., Lau L. F. Transcriptional activation by Nur77, a growth factor-inducible member of the steroid hormone receptor superfamily. Mol. Endocrinol. 1991;5:854–859. doi: 10.1210/mend-5-6-854. [DOI] [PubMed] [Google Scholar]
- 38.Wilson T. E., Fahrner T. J., Johnston M., Milbrandt J. Identification of the DNA binding site for NGFI-B by genetic selection in yeast. Science (Washington D.C.) 1991;252:1296–1300. doi: 10.1126/science.1925541. [DOI] [PubMed] [Google Scholar]
- 39.Wang Z., Benoit G., Liu J., Prasad S., Aarnisalo P., Liu X., Xu H., Walker N. P., Perlmann T. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature (London) 2003;423:555–560. doi: 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]
- 40.Katagiri Y., Takeda K., Yu Z. X., Ferrans V. J., Ozato K., Guroff G. Modulation of retinoid signalling through NGF-induced nuclear export of NGFI-B. Nat. Cell Biol. 2000;2:435–440. doi: 10.1038/35017072. [DOI] [PubMed] [Google Scholar]
- 41.Katagiri Y., Hirata Y., Milbrandt J., Guroff G. Differential regulation of the transcriptional activity of the orphan nuclear receptor NGFI-B by membrane depolarization and nerve growth factor. J. Biol. Chem. 1997;272:31278–31284. doi: 10.1074/jbc.272.50.31278. [DOI] [PubMed] [Google Scholar]
- 42.Williams G. T., Lau L. F. Activation of the inducible orphan receptor gene Nur77 by serum-starved growth factors: dissociation of immediate-early and delayed-early responses. Mol. Cell. Biol. 1993;13:6124–6136. doi: 10.1128/mcb.13.10.6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoon J. K., Lau L. F. Transcriptional activation of the inducible nuclear receptor gene Nur77 by nerve growth factor and membrane depolarization in PC12 cells. J. Biol. Chem. 1993;268:9148–9155. [PubMed] [Google Scholar]
- 44.Kasler H. G., Victoria J., Duramad O., Winoto A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol. 2000;20:8382–8389. doi: 10.1128/mcb.20.22.8382-8389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dequiedt F., Kasler H., Fischle W., Kiermer V., Weinstein M., Herndier B. G., Verdin E. HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity. 2003;18:687–698. doi: 10.1016/s1074-7613(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 46.Liu W., Youn H. D., Liu J. O. Thapsigargin-induced apoptosis involves Cabin1-MEF2-mediated induction of Nur77. Eur. J. Immunol. 2001;31:1757–1764. doi: 10.1002/1521-4141(200106)31:6<1757::aid-immu1757>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 47.Esau C., Boes M., Youn H. D., Tatterson L., Liu J. O., Chen J. Deletion of calcineurin and myocyte enhancer factor 2 (MEF2) binding domain of Cabin1 results in enhanced cytokine gene expression in T cells. J. Exp. Med. 2001;194:1449–1459. doi: 10.1084/jem.194.10.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blaeser F., Ho N., Prywes R., Chatila T. A. Ca(2+)-dependent gene expression mediated by MEF2 transcription factors. J. Biol. Chem. 2000;275:197–209. doi: 10.1074/jbc.275.1.197. [DOI] [PubMed] [Google Scholar]
- 49.Yoon J. K., Lau L. F. Involvement of JunD in transcriptional activation of the orphan receptor gene Nur77 by nerve growth factor and membrane depolarization in PC12 cells. Mol. Cell. Biol. 1994;14:7731–7743. doi: 10.1128/mcb.14.12.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X., Chen X., Zachar V., Chang C., Ebbesen P. Transcriptional activation of human TR3/Nur77 gene expression by human T-lymphotropic virus type I Tax protein through two AP-1-like elements. J. Gen. Virol. 1999;80:3073–3081. doi: 10.1099/0022-1317-80-12-3073. [DOI] [PubMed] [Google Scholar]
- 51.Stocco C. O., Zhong L., Sugimoto Y., Ichikawa A., Lau L. F., Gibori G. Prostaglandin F2α-induced expression of 20alpha-hydroxysteroid dehydrogenase involves the transcription factor Nur77. J. Biol. Chem. 2000;275:37202–37211. doi: 10.1074/jbc.M006016200. [DOI] [PubMed] [Google Scholar]
- 52.Stocco C. O., Lau L. F., Gibori G. A calcium/calmodulin-dependent activation of ERK1/2 mediates JunD phosphorylation and induction of Nur77 and 20alpha-hsd genes by prostaglandin F2alpha in ovarian cells. J. Biol. Chem. 2002;277:3293–3302. doi: 10.1074/jbc.M110936200. [DOI] [PubMed] [Google Scholar]
- 53.Ciani E., Paulsen R. E. Activation of a reporter gene responsive to NGFI-B in cultured neurons and astrocytes. J. Mol. Neurosci. 1995;6:131–139. doi: 10.1007/BF02736772. [DOI] [PubMed] [Google Scholar]
- 54.Maira M., Martens C., Batsche E., Gauthier Y., Drouin J. Dimer-specific potentiation of NGFI-B (Nur77) transcriptional activity by the protein kinase A pathway and AF-1-dependent coactivator recruitment. Mol. Cell. Biol. 2003;23:763–776. doi: 10.1128/MCB.23.3.763-776.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kovalovsky D., Refojo D., Liberman A. C., Hochbaum D., Pereda M. P., Coso O. A., Stalla G. K., Holsboer F., Arzt E. Activation and induction of Nur77/Nurr1 in corticotrophs by CRH/cAMP: involvement of calcium, protein kinase A, and MAPK pathways. Mol. Endocrinol. 2002;16:1638–1651. doi: 10.1210/mend.16.7.0863. [DOI] [PubMed] [Google Scholar]
- 56.Martinez-Gonzalez J., Rius J., Castello A., Cases-Langhoff C., Badimon L. Neuron-derived orphan receptor-1 (NOR-1) modulates vascular smooth muscle cell proliferation. Circ. Res. 2003;92:96–103. doi: 10.1161/01.es.0000050921.53008.47. [DOI] [PubMed] [Google Scholar]
- 57.McEvoy A. N., Murphy E. A., Ponnio T., Conneely O. M., Bresnihan B., FitzGerald O., Murphy E. P. Activation of nuclear orphan receptor Nurr1 transcription by NF-kappa B and cyclic adenosine 5′-monophosphate response element-binding protein in rheumatoid arthritis synovial tissue. J. Immunol. 2002;168:2979–2987. doi: 10.4049/jimmunol.168.6.2979. [DOI] [PubMed] [Google Scholar]
- 58.Saucedo-Cardenas O., Kardon R., Ediger T. R., Lydon J. P., Conneely O. M. Cloning and structural organization of the gene encoding the murine nuclear receptor transcription factor, Nurr1. Gene. 1997;187:135–139. doi: 10.1016/s0378-1119(96)00736-6. [DOI] [PubMed] [Google Scholar]
- 59.Ahn S., Olive M., Aggarwal S., Krylov D., Ginty D. D., Vinson C. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol. Cell. Biol. 1998;18:967–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adams R. H., Porras A., Alonso G., Jones M., Vintersten K., Panelli S., Valladares A., Perez L., Klein R., Nebreda A. R. Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell. 2000;6:109–116. [PubMed] [Google Scholar]
- 61.Porras A., Zuluaga S., Black E., Valladares A., Alvarez A. M., Ambrosino C., Benito M., Nebreda A. R. P38 alpha mitogen-activated protein kinase sensitizes cells to apoptosis induced by different stimuli. Mol. Biol. Cell. 2004;15:922–933. doi: 10.1091/mbc.E03-08-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pfaffl M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davies S. P., Reddy H., Caivano M., Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caivano M., Cohen P. Role of mitogen-activated protein kinase cascades in mediating lipopolysaccharide-stimulated induction of cyclooxygenase-2 and IL-1 beta in RAW264 macrophages. J. Immunol. 2000;164:3018–3025. doi: 10.4049/jimmunol.164.6.3018. [DOI] [PubMed] [Google Scholar]
- 65.Impey S., McCorkle S. R., Cha-Molstad H., Dwyer J. M., Yochum G. S., Boss J. M., McWeeney S., Dunn J. J., Mandel G., Goodman R. H. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 66.Grollman A. P. Inhibitors of protein biosynthesis. II. Mode of action of anisomycin. J. Biol. Chem. 1967;242:3226–3233. [PubMed] [Google Scholar]
- 67.Ohkura N., Hosono T., Maruyama K., Tsukada T., Yamaguchi K. An isoform of Nurr1 functions as a negative inhibitor of the NGFI-B family signaling. Biochim. Biophys. Acta. 1999;1444:69–79. doi: 10.1016/s0167-4781(98)00247-4. [DOI] [PubMed] [Google Scholar]
- 68.Woronicz J. D., Calnan B., Ngo V., Winoto A. Requirement for the orphan steroid receptor Nur77 in apoptosis of T-cell hybridomas. Nature (London) 1994;367:277–281. doi: 10.1038/367277a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.