

ABSTRACT
Myocardial ischemia–reperfusion injury (MIRI) significantly affects the prognosis of cardiac surgery patients. The anesthetic dexmedetomidine (Dex) has shown protective effects against ischemia–reperfusion injury in cardiomyocytes; however, its exact mechanism remains unclear. In this study, hypoxia/reoxygenation (H/R) and ischemia/reperfusion (I/R) models were used to investigate the effects of Dex on H9c2 cells and MIRI in mice. The roles of the Sirtuin 1/Forkhead box O3a (Sirt1/FoxO3a) pathway in the protective effects of Dex were explored using the Sirt1 inhibitor EX527 and FoxO3a gene silencing. Results showed that H/R significantly reduced H9c2 cell viability, increased Lactate Dehydrogenase (LDH) leakage, and elevated reactive oxygen species (ROS) production. Dex pretreatment reversed these effects. Additionally, Dex significantly reduced the expression of Bcl‐2‐associated X protein/B‐cell lymphoma 2 (Bax/Bcl‐2), cleaved caspase‐3, Beclin‐1, and microtubule‐associated protein 1A/1B‐light chain 3B (LC3B), inhibiting apoptosis and autophagy while increasing the expression of p62, Sirt1, and FoxO3a. The protective effects of Dex against H/R injury were abolished by EX527 or FoxO3a silencing. In the mouse MIRI model, Dex pretreatment decreased serum LDH and Creatine Kinase‐MB (CK‐MB) levels, reduced myocardial infarct size and cardiac injury, and significantly improved left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS). These protective effects were markedly reversed by EX527. These findings indicate that Dex alleviates MIRI by restoring Sirt1 expression and activating FoxO3a.
Keywords: apoptosis, autophagy, dexmedetomidine, FoxO3a, myocardial ischemia–reperfusion injury, Sirt1
The Mechanism of Dexmedetomidine in Treating Myocardial Ischemia–Reperfusion Injury via the Sirt1/Foxo3a Pathway.

Abbreviations
- AAR
the area at risk
- ANOVA
The one‐way analysis of variance
- ATCC
the American Type Culture Collection
- CCK‐8
cell counting kit‐8
- CST
Cell Signaling Technology
- DAPI
4,6‐diamino‐2‐phenyl indole
- Dex
Dexmedetomidine
- DHE
Dihydroethidium
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulfoxide
- ELISA
enzyme‐linked immunosorbent assay
- FBS
fetal bovine serum
- FoxO
the class O of forkhead box transcription factors
- FoxO3a
forkhead box O3a
- H/R
hypoxia/reoxygenation
- I/R
ischemia/reperfusion
- LC3B
Microtubule‐associated protein light chain 3B
- LDH
Lactate dehydrogenase
- LVEF
the left ventricular ejection fraction
- LVFS
the left ventricular fractional shortening
- MDA
Malondialdehyde
- MIRI
Myocardial ischemia/reperfusion injury
- OD
the optical density
- PBS
phosphate buffer saline
- PS
penicillin/streptomycin
- ROS
Reactive oxygen species
- SDS‐PAGE
sodium dodecyl sulfate‐polyacrylamide gel electrophoresis
- Sirt1
the silent information regulator 1
- TTC
2, 3, 5‐triphenyl tetrazolium chloride
- TUNEL
TdT‐mediated dUTP Nick‐End Labeling
1. Introduction
Acute myocardial infarction is characterized by myocardial necrosis and is caused by acute and prolonged ischemia and hypoxia in the coronary arteries. The increasing prevalence of this condition worldwide poses a substantial risk to human life and to overall wellbeing (Arora et al. 2019). Restoring coronary blood flow, whether through therapeutic approaches or primary percutaneous coronary intervention, is the most effective strategy to decrease the severity of myocardial infarction and thus improve outcomes. Although essential, the process of reperfusion may, however, paradoxically trigger additional injury (Davidson et al. 2019). This counterintuitive effect is termed myocardial ischemia/reperfusion injury (MIRI).
MIRI and its underlying mechanisms have been extensively studied, with a focus on apoptosis, autophagy, and oxidative stress (Deng and Zhou 2023). Reactive oxygen species (ROS) are small reactive molecules involved in the regulation of cellular functions and biological processes. ROS play a pivotal role in MIRI due to their ability to induce cell death (Wang et al. 2023b). Oxidative stress occurs when ROS production surpasses the capacity for its clearance. This causes an imbalance in the oxidation and antioxidant systems, ultimately resulting in cellular and tissue damage (He et al. 2022).
During cardiac surgery, intraoperative cardiac arrest may induce myocardial ischemia. The subsequent restoration of blood flow to the heart can trigger MIRI and severely impair heart function. Consequently, the identification of anesthetic drugs with organ‐protective properties is paramount. Dex is a highly specific α2‐adrenergic agonist known for its analgesic, antistress, and antiinflammatory effects. It is commonly used as a sedative and anesthetic agent in various clinical settings (Tang et al. 2022). Numerous studies have demonstrated the protective effects of Dex in diverse organ injuries mediated by I/R, notably in the heart (Sun et al. 2022), brain (Guo et al. 2023), lung (Zhang et al. 2019), and kidney (Si et al. 2013). Dex is particularly recommended as an adjunctive sedative for cardiac patients. Dex can reduce CK‐MB and Cardiac Troponin I (cTn‐I) concentrations and shorten the length of intensive care unit (ICU) stay for patients undergoing cardiac surgery with CPB and provide myocardial protection for MIRI (Chen et al. 2022; Poon et al. 2023).
Sirtuin proteins (Sirt1 to Sirt7) are a family of Nicotinamide adenine dinucleotide (NAD+)‐dependent deacetylases (Carrico et al. 2018). Sirt1 is primarily localized in the nucleus and regulates oxidative stress, cellular metabolism, autophagy, and apoptosis through deacetylation (Chen et al. 2020). The FoxO subfamily of forkhead transcription factors is a critical downstream target of Sirt1, with the acetylation/deacetylation process exerting a significant influence on their biological function (Zhao et al. 2019). FoxO3a is a crucial member of the FoxO subfamily and has significant involvement in cell apoptosis, proliferation, and DNA damage. Deacetylation of FoxO3a mediated by Sirt1 is known to protect against apoptosis. It was previously reported that Dex can mitigate MIRI through multiple pathways, including the Sirt1/mechanistic target of rapamycin (mTOR)/C/EBP homologous protein (CHOP) axis (Zhang et al. 2021). However, the precise mechanism implicating the Sirt1/FoxO3a interaction with MIRI during Dex treatment remains unknown, as well as its potential as a biomarker for MIRI therapy. Therefore, in the present study, we investigated the role of the Sirt1/FoxO3a signaling pathway in Dex‐mediated protection from I/R and H/R injuries in mice and H9c2 cells, respectively. Our findings provide novel insights into therapeutic targeting of the Sirt1/FoxO3a pathway as a potential treatment strategy for MIRI.
2. Materials and Methods
2.1. Materials
Dimethyl sulfoxide (DMSO, 472301) was purchased from Sigma Aldrich. Dex (HY‐12719) was purchased from MedChemExpress. Reagents for SDS‐PAGE (P0012A) and ECL Western blotting (P0018S), as well as 4% paraformaldehyde (P0099‐3 L) were purchased from Beyotime (China). Cell culture and transfection reagents (11668500) were purchased from Thermo‐Invitrogen (Massachusetts, USA). Small interfering RNA (siRNA) fragments targeting FoxO3a, as well as control scrambled siRNA (siRNA NC: siN0000001‐1‐5), were procured from Ribibio (Guangzhou). The cell counting kit‐8 (CCK‐8, 40203ES60) was purchased from Yeasen (China), the lactate dehydrogenase (LDH) assay kit (MAK464) and creatine kinase (CK‐MB) activity assay kit (MAK116) from Sigma Aldrich. The Lipid Peroxidation Malondialdehyde (MDA) Assay Kit (S0131S), Dihydroethidium (DHE) assay kit (S0064S), ROS assay kit (S0033S), Annexin V‐PE apoptosis assay kit (C1065S), Hematoxylin and Eosin (HE) Staining Kit (C0105S), Evans blue staining kit (ST3273), and 2,3,5‐triphenyltetrazolium chloride (TTC) staining kit (C0651) were purchased from Beyotime (China). The Evo M‐MLV RT Kit (AG11706) and the SYBR Green Pro Taq HS qPCR Master Mix (AG11759) were purchased from Accurate Biology (China). Primary antibodies against Sirt1 (9475 T), FoxO3a (2497 T), Bcl‐2 (28,150 T), LC3B (2775S), β‐actin (4967S), Glyceraldehyde‐3‐Phosphate Dehydrogenase (GAPDH) (2118 T), Bax (2772 T), Cleaved Caspase‐3 (9661 T), p62 (5114 T), and Beclin‐1 (3495 T), as well as secondary antibodies (7074P2), were purchased from Cell Signaling Technology (CST). The H9c2 cardiac cell strain derived from the left ventricle of an S‐D rat was purchased from the Shanghai Institute for Biological Sciences, Chinese Academy of Science (Shanghai, China). The cells were authenticated by the Genomics Unit at CIMA using Short Tandem Repeat profiling. Mycoplasma testing was performed every second week using the MycoAlert Mycoplasma Detection Kit (LT07‐118, LONZA). Male C57BL/6J mice (weight 20 ± 5 g, age 6–8 weeks) were obtained from the Guangdong Medical Laboratory Animal Center. The experimental use of animals in this study was authorized by the Committee for the Use of Live Animals in Teaching and Research, Guangdong Medical University (Approval no. GDY2002072).
2.2. Establishment of an In Vitro H/R Injury Model
H9c2 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were grown in a humidified incubator consisting of 95% air and 5% CO2 at 37°C. After treatment with 10 μM Dex dissolved in DMSO for 2 h, cells were placed in a hypoxic incubator (37°C, 95% N2 and 5% CO2) with culture medium lacking glucose and serum. After 6 h of hypoxia, cells were reoxygenated for 12 h in regular medium (Tian et al. 2019).
2.3. FoxO3a‐Specific siRNA Transfection
Oxidative stress‐induced cellular damage was evaluated using the reactive oxygen species (ROS) detection kit as recommended by the manufacturer and using the ROS fluorescent probe dihydroethidium (DHE). Cells were exposed to 50 μmol/L DHE at a temperature of 37°C for 30 min in a dark environment. To assist with cell counting, the nuclei were stained with 4,6‐diamino‐2‐phenyl indole (DAPI) and the cells were subsequently washed twice with cold phosphate‐buffered saline (PBS). The fluorescence intensity of ROS within the cells was recorded using a light microscope (Olympus IX51). In these images, DHE staining indicative of ROS appeared as red color, while the nuclei stained with DAPI were blue color. ImageJ software (The National Institutes of Health) was employed for quantitative analysis of the ROS level.
2.4. Measurement of Reactive Oxygen Species
The small interfering RNA (siRNA) fragments targeting FoxO3a and the control scrambled siRNA (siRNA NC: siN0000001‐1‐5) were procured from Guangzhou Ribibio company. The specific siRNA sequences for FoxO3a included si‐FoxO3a‐001 (5′‐GCTCTTGGTGGATCATCAA‐3′; 5’‐UUGATGAUCCACCAAGAGC‐3′), si‐FoxO3a‐003 (5′‐GGAACGTGATGCTTCGCAA‐3′; 5’‐UUGCAAGCAUCACGUUCC‐3′), and si‐FoxO3a‐004 (5′‐GATTCGGCCAGGCTGCTGT‐3′; 5’‐ACAGCAGCCUGGCCGAAUC‐3′). Before the transfection procedure, H9c2 cells were plated at a density of 5 × 10^4 cells per well in 6‐well plates and incubated under suitable conditions. Subsequently, the cells were transfected with siRNAs using Lipofectamine 2000 (ThermoFisher Scientific, MA, USA) for a duration of 48 h. Following this transfection period, the H9c2 cells were prepared for subsequent experimental procedures.
2.5. Measurement of MDA Release
The extent of myocardial cellular injury, as indicated by the release of MDA, was assessed using the lipid peroxidation MDA assay kit. This procedure involved the collection of supernatant from the cells of all experimental groups. The supernatant and relevant reagents were then methodically added to the enzyme‐linked immunosorbent assay (ELISA) plates as described in the kit protocol. MDA release was quantified by measuring the absorbance at a wavelength of 532 nm using a microplate reader. This allowed the precise estimation of myocardial cellular injury in different experimental conditions.
2.6. Cell Proliferation Assay
The CCK‐8 cytotoxicity assay was used to evaluate cell proliferation, as described by the manufacturer. This is a reliable and sensitive method for measuring cell viability and proliferation.
2.7. Cell Apoptosis Assay
H9c2 cells were seeded into 12‐well culture plates and subjected to the previously mentioned manipulations. Apoptotic cells were identified with the one‐step TdT‐mediated dUTP nick‐end labeling (TUNEL) apoptosis assay kit following the guidelines provided by the manufacturer. Apoptotic cells were identified as TUNEL‐positive cells staining red, while nuclei counterstained with DAPI were blue. Quantification of TUNEL‐positive cells was carried out using ImageJ software. The percentage of TUNEL‐positive cells was determined by normalizing to the number of DAPI‐positive cells.
2.8. Measurement of Lactate Dehydrogenase (LDH) in the Supernatant of Cultured Cells
The LDH release assay kit was used to assess cytotoxicity. This also served as an indicator of cell membrane integrity. Following the establishment of the H/R model, H9c2 cells were seeded into 96‐well culture plates at a density of 8000 cells/well. The assay was performed strictly as recommended by the manufacturer. Subsequently, the optical density (OD) at a wavelength of 492 nm was measured using a microplate reader. The OD value served as an indirect measure of cell viability and the extent of cell damage.
2.9. Establishment of an In Vivo Model of I/R‐Induced Injury
Mice were randomly divided into four groups consisting of 8 mice each: sham, I/R, I/R + Dex, and I/R + Dex + EX527. Those in the I/R + Dex group received an intravenous injection of Dex dissolved in DMSO (20 μg/kg) (Du et al. 2019) 1 day prior to the experiment. The I/R + Dex + EX527 group additionally received an intraperitoneal injection of EX527 dissolved in DMSO (10 mg/kg/day) (Luo et al. 2019) for 1 week before the induction of MIRI. Prior to the surgical procedure, mice were anesthetized with an intraperitoneal injection of 1.5% pentobarbital sodium (80 mg/kg). Tracheal intubation was then performed and the mice ventilated using an animal ventilator (ALC‐ANE6, Shanghai, China). All mice in the MIRI groups underwent a 30 min occlusion of the left anterior descending coronary artery, followed by 120 min of reperfusion (Ge et al. 2023). Ligation was considered successful if the myocardium was cyanotic on the ligature line and white below the ligature line. The criterion for reperfusion was that the color of the blood supplying the myocardium in the lower part of the ligated vessel changed from cyanotic to light or dark red.
2.10. Analysis of Heart Tissue by Western Blot (WB) and Assessment of Myocardial Infarction Size
During reperfusion, four mice in each group were kept in deep anesthesia with 1.5% pentobarbital sodium. Serum was then collected for LDH and CK‐MB measurement, and heart tissue for Western blot analysis.
The remaining four mice in each group were anesthetized with isoflurane for cardiac ultrasound to assess their hearts. Subsequently, the mice were euthanized under isoflurane anesthesia, and the hearts were collected for double staining with Evans blue and 2,3,5‐triphenyltetrazolium chloride (TTC) to determine the area at risk and the infarct area. The resulting images were captured and quantified using ImageJ.
2.11. Assessment of Myocardial Injury Extent
Heart tissues from mice were collected and fixed in a 4% paraformaldehyde solution. Subsequently, the tissues were embedded in paraffin and sectioned. HE staining was performed to assess myocardial injury. The scoring standard was as follows: 0 indicated no damage, 1 indicated damage affecting less than 25%, 2 indicated damage affecting 25%–50%, 3 indicated damage affecting 50%–75%, and 4 indicated damage exceeding 75% (Xu et al. 2021).
2.12. Echocardiography Assessment of Cardiac Function
Cardiac ultrasonography was performed using a Visual Sonics Vevo 2100 high‐resolution ultrasound system equipped with a 40 MHz probe. To induce anesthesia, mice were initially placed in an airtight chamber containing a 3% isoflurane–oxygen gas mixture. Once anesthetized, as indicated by a reduced ability to move freely, the mice were transferred and secured on the ultrasound table. The isoflurane concentration was adjusted to 1.0% to maintain light anesthesia throughout the procedure. Prior to imaging, the chest area was shaved and coated with an ultrasound coupling agent to facilitate image acquisition. Two‐dimensional echocardiographic images were captured at both the long‐axis and short‐axis levels of the left ventricle. M‐mode images were also obtained and preserved for further analysis. Cardiac function parameters, including the left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS), were assessed using analysis software integrated into the ultrasound imaging system (Luo et al. 2020).
2.13. Western Blotting
Western blot analysis was performed as described previously (Han et al. 2021), using primary antibodies against Sirt1, FoxO3a, Bax, Bcl‐2, Cleaved caspase‐3, LC3B, p62, Beclin‐1, β‐Actin, and GAPDH. Incubation with primary antibodies was for 12–16 h at 4°C, followed by incubation with secondary antibodies (1:5000 or 1:10,000) for 1 h. After washing the membrane, protein bands were revealed using BeyoECL Plus, and the band intensity evaluated using Image J software (National Institutes of Health, USA). Data are presented as the percent change relative to β‐actin or GAPDH references.
2.14. Real‐Time Quantitative PCR (RT‐qPCR)
For the analysis of gene expression related to apoptosis, autophagy, and oxidative stress, first‐strand cDNA was synthesized using the Evo M‐MLV RT Kit. This cDNA then served as a template for RT‐qPCR using the SYBR Green Pro Taq HS qPCR Master Mix. Gene expression was quantified utilizing the CFX Connect Real‐Time PCR Detection System (Bio‐Rad, USA). Primers specific to the genes of interest are detailed in Table S1. The relative expression level of these genes was calculated using the 2−ΔΔCt method, with normalization against the expression level of the housekeeping gene β‐actin. To ensure reproducibility and accuracy, assays were performed in duplicate for each sample.
2.15. Statistical Analysis
The data are presented as the mean ± standard error of the mean (SEM). Statistical analyses were performed using GraphPad Prism version 9.0 (GraphPad Software, La Jolla, CA). To ensure objectivity, all analyses were conducted in a blinded manner. The statistical significance of differences between the various treatment groups was determined using one‐way analysis of variance (ANOVA) or multifactorial ANOVA and Tukey's post hoc tests. Statistical significance was considered at p < 0.05.
3. Results
3.1. Evaluation of an In Vitro H/R Injury Model After Dex Treatment
To evaluate the toxic effects of Dex on H9c2 cells, the cells were treated with three different concentrations of Dex (5, 10, and 20 μM) for a duration of 2 h. As shown in Figure 1A, no significant differences in cell viability were observed between cells treated with 5 or 10 μM Dex and the control group (untreated cells), suggesting that these concentrations of Dex did not exert notable cytotoxic effects on H9c2 cells. However, treatment with 20 μM Dex significantly decreased cell activity, indicating that it was toxic to the cells. Pretreatment with 10 μM Dex prior to H/R significantly increased cell viability compared to 5 μM Dex (Figure 1B). Moreover, pretreatment with 10 μM Dex before H/R mitigated the release of LDH, in contrast to the effect observed with 5 μM Dex (Figure 1C). Collectively, these findings indicate that treatment with 10 μM Dex was optimal for the in vitro model of H/R injury.
FIGURE 1.

Construction of the H/R injury model in H9c2 cells under Dex treatment. (A) Effect of Dex concentrations on the cell viability. (B) Effect of Dex concentrations on the cell viability following H/R injury. (C) Effect of Dex concentrations on the LDH release following H/R injury. The cells were subjected to hypoxia treatment for 6 h and reoxygenation for 12 h. Then, LDH release was measured. CN: Control, H/R: Hypoxia/reoxygenation, Dex: Dexmedetomidine. ns p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. One‐way ANOVA was applied, followed by Tukey's post hoc test, a multiple comparison test.
3.2. Treatment of H9c2 Cells With 10 μM Dex Attenuates H/R‐Induced Apoptosis, Autophagy, and Oxidative Stress, While Upregulating Sirt1 and FoxO3a Expression
H/R‐induced apoptosis and autophagy were observed in H9c2 cells (Figure 2B,C). This was accompanied by increased ratios of Bax/Bcl‐2 and LC3B II/I, elevated expression of the pro‐apoptosis protein Cleaved caspase‐3 and the pro‐autophagy protein Beclin‐1, and decreased expression of the autophagy inhibitory protein p62. However, Dex treatment reversed the expression of these proteins, leading to the inhibition of cell apoptosis and autophagy. The results of TUNEL staining confirmed these findings and provided additional validation (Figure 2F).
FIGURE 2.
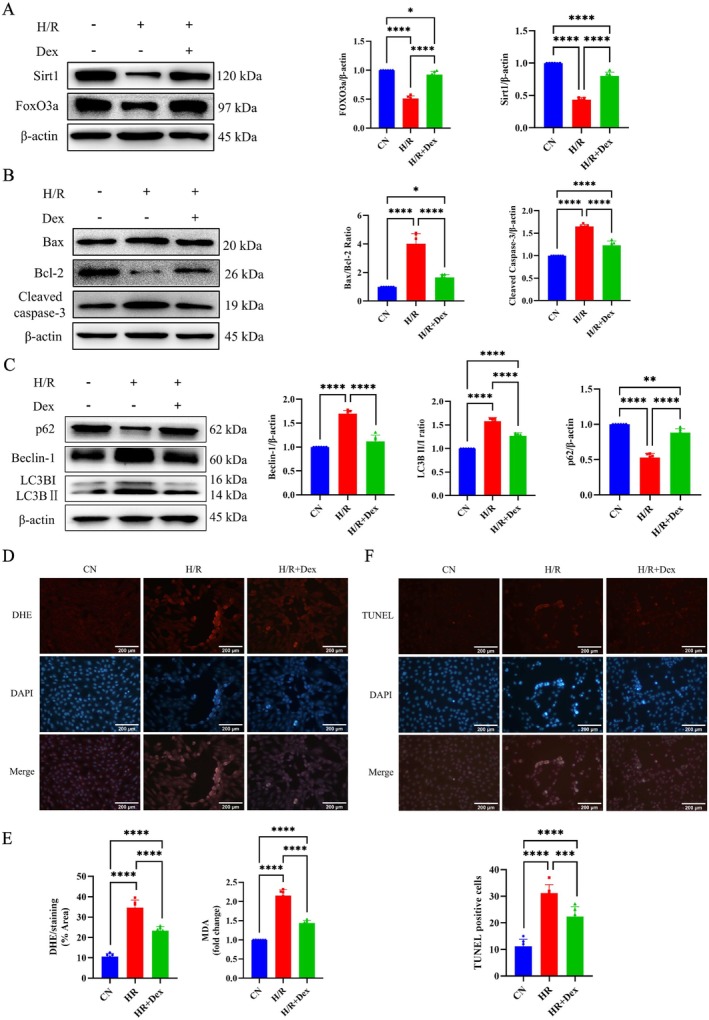
Effect of treatment with Dex at 10 μM on apoptosis, autophagy, and oxidative stress of H/R‐induced H9c2 cells. (A) Western blot analysis of Sirt1, FoxO3a, and densitometry analysis of proteins. (B) Western blot analysis of Bax, Bcl‐2, Cleaved caspase‐3, and densitometry analysis of proteins. (C) Western blot analysis of p62, Beclin‐1, LC3B, and densitometry analysis of proteins. (D) ROS degrees measured by DHE‐DAPI staining (20×) and statistical graph of DHE. (E) The content of MDA. (F) Representative TUNEL‐staining images (20×) and statistical graph of TUNEL‐positive cells. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. One‐way ANOVA was applied, followed by Tukey's post hoc test, a multiple comparison test.
Next, we evaluated ROS and MDA levels under H/R conditions in order to assess the impact of Dex on oxidative stress in cardiomyocytes. H/R was found to significantly increase intracellular ROS production and MDA levels (Figure 2D,E). However, pretreatment with Dex caused a significant reversal and led to a marked reduction in ROS and MDA levels.
In addition, Western blot experiments revealed that Sirt1 and FoxO3a protein expression was significantly downregulated in the H/R group compared with the control group, but significantly upregulated by pretreatment with 10 μM Dex (Figure 2A).
Based on the above findings, we postulated that Dex could alleviate myocardial apoptosis, autophagy, and oxidative stress by upregulating the expression of Sirt1 and FoxO3a.
3.3. Effects of Sirt1 Targeting on the Apoptosis, Autophagy, and Oxidative Stress of H9c2 Cells Subjected to H/R In Vitro
We next investigated the regulatory role of Sirt1 in H9c2 cells subjected to H/R stress, with or without Dex treatment, by using EX527 to inhibit Sirt1 expression. The suppression of Sirt1 led to a marked decrease in cell viability (Figure 3A) and an increase in LDH leakage (Figure 3B). The expression of both Sirt1 and FoxO3a decreased after Sirt1 inhibition (Figure 3C). Moreover, in the H/R + Dex + EX527 group, we observed increased ratios of Bax/Bcl‐2 and LC3B II/I, increased expression of Cleaved caspase‐3 and Beclin‐1, and a concomitant decrease in p62 expression (Figure 3D,E). Inhibition of Sirt1 led to a significant increase in cellular oxidative stress markers, including ROS and MDA levels, compared to the H/R + Dex group (Figure 3F,G). TUNEL staining also revealed that inhibition of Sirt1 caused an increase in the proportion of apoptotic cells (Figure 3H). These findings highlight the pivotal role of Sirt1 in modulating apoptosis, oxidative stress, and autophagy in H9c2 cells exposed to H/R conditions in an in vitro setting.
FIGURE 3.

Effect of Dex treatment on apoptosis, autophagy, and oxidative stress of H/R‐induced H9c2 cells through the inhibition of Sirt1 expression. (A) Cell viability. (B) LDH leakage. (C) Western blot analysis of Sirt1, FoxO3a, and densitometry analysis of proteins. (D) Western blot analysis of Bax, Bcl‐2, Cleaved caspase‐3, and densitometry analysis of proteins. (E) Western blot analysis of p62, Beclin‐1, LC3B, and densitometry analysis of proteins. (F) ROS degrees measured by DHE‐DAPI staining (20×) and statistical graph of DHE. (G) The content of MDA. (H) Representative TUNEL‐staining images 20×) and statistical graph of TUNEL‐positive cells. (I) qPCR shows the mRNA expression levels of genes related to apoptosis, oxidative stress, and autophagy. ns p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. One‐way ANOVA or two‐way ANOVA was applied, followed by Tukey's post hoc test, a multiple comparison test.
The expression levels of apoptosis‐related genes (Fas Ligand (FasL), tumor necrosis factor superfamily member 10 (Tnfsf10), Bcl‐2, and Bcl‐6), autophagy‐related genes (BCL2/adenovirus E1B 19 kDa interacting protein 3 (Bnip3), autophagy‐related 12 (Atg12), and Gabarap), and oxidative stress‐related genes (Catalase, Superoxide Dismutase 2 (Sod2), growth arrest and DNA damage‐inducible alpha (Gadd45a), and ataxia telangiectasia mutated (Atm)) in H9c2 cells were evaluated under conditions of H/R, and with or without Dex pretreatment. As shown in Figure 4I, H/R significantly upregulated the mRNA expression of FasL, Tnfsf10, Bnip3, Atg12, Gabarap, Catalase, Sod2, Gadd45a, and Atm, while downregulating the mRNA expression of Bcl‐2 and Bcl‐6. The administration of Dex reversed these changes, but treatment with EX527 abolished the effects of Dex. These results suggested that Dex could attenuate H/R‐induced damage to H9c2 cells by regulating Sirt1 at the mRNA level.
FIGURE 4.

Effect of Dex treatment on apoptosis, autophagy, and oxidative stress of H/R‐induced H9c2 cells through the silencing of FoxO3a expression. (A) Western blot analysis of FoxO3a silence vs. CN or CN + NC siRNA. NC: Negative control of FoxO3a. (B) Cell viability. (C) LDH Leakage. (D) Western blot analysis of Sirt1, FoxO3a, and densitometry analysis of proteins. (E) Western blot analysis of Bax, Bcl‐2, Cleaved caspase‐3, and densitometry analysis of proteins. (F) Western blot analysis of p62, Beclin‐1, LC3B, and densitometry analysis of proteins. (G) ROS degrees measured by DHE‐DAPI staining (20×) and statistical graph of DHE. (H) The content of MDA. (I) Representative TUNEL‐staining images (20×) and statistical graph of TUNEL‐positive cells.*p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. One‐way ANOVA was applied, followed by Tukey's post hoc test, a multiple comparison test.
3.4. Effect of FoxO3a Targeting on the Apoptosis, Autophagy, and Oxidative Stress of H9c2 Cells Subjected to H/R In Vitro
A siRNA interference assay was used to investigate the regulatory role of FoxO3a in H/R‐induced H9c2 cells, with or without Dex treatment. As shown in Figure 4A, the siRNA4 fragment showed the strongest interference against FoxO3a expression and was therefore selected for further experiments. Silencing of FoxO3a significantly reduced cell viability and increased LDH leakage compared with the H/R + Dex group (Figure 4B,C). Western blot analysis showed that FoxO3a silencing did not significantly alter Sirt1 expression (p > 0.05, H/R + Dex + siFoxO3a) (Figure 4D). However, FoxO3a silencing in H9c2 cells counteracted the Dex‐mediated reductions in Bax/Bcl‐2 ratio, LC3B II/I ratio, protein levels of Cleaved caspase‐3 and Beclin‐1, formation of apoptotic bodies, and the increased expression of p62 (Figure 4E,F,I). Furthermore, the levels of cellular oxidative stress markers such as ROS and MDA were significantly increased in the H/R + Dex + siFoxO3a group compared to the H/R + Dex group (Figure 4G,H). These results suggested that FoxO3a also played an essential role in regulating apoptosis, autophagy, and oxidative stress in H9c2 cells exposed to H/R conditions in an in vitro setting.
3.5. Dex Alleviates H/R‐Induced Injury via the Sirt1/FoxO3a Signaling Pathway
In light of their known roles in the H9c2 cellular model, we next explored the interaction between Sirt1 and FoxO3a in H/R‐induced H9c2 cells. The experimental design included six distinct groups: control (CN), H/R, H/R + siFoxO3a, H/R + Dex, H/R + Dex + siFoxO3a, and H/R + Dex + siFoxO3a + EX527. These groups were comprehensively investigated in order to discern the interaction dynamics.
Dex administration was found to significantly enhance cell viability (Figure 5A), reduce LDH leakage (Figure 5B), decrease the MDA level (Figure 5C), and inhibit apoptosis (Figure 5D) compared to the H/R group. However, inhibition of Sirt1 expression or FoxO3a silencing reversed these protective effects of Dex (Figure 5A–D), indicating that both Sirt1 and FoxO3a were integral to the Dex‐mediated protective effects observed in H/R‐induced H9c2 cells.
FIGURE 5.

Dex treatment attenuates H/R‐induced injury via Sirt1/FoxO3a signal pathway. (A) Cell viability. (B) LDH leakage. (C) The content of MDA. (D) Representative TUNEL‐staining images (20×) and statistical graph of TUNEL‐positive cells. (E) Western blot analysis of Sirt1, FoxO3a, and densitometry analysis of proteins. ns p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. One‐way ANOVA was applied, followed by Tukey's post hoc test, a multiple comparison test.
Silencing of FoxO3a significantly reduced FoxO3a protein expression without affecting the expression of Sirt1 protein (p > 0.05, H/R + siFoxO3a vs. H/R, H/R + Dex + siFoxO3a vs. H/R + Dex) (Figure 5E). Intriguingly, inhibition of Sirt1 expression markedly decreased the expression of both Sirt1 and FoxO3a. These findings suggest that Dex mitigates H/R‐induced cellular injury by inducing Sirt1 to activate FoxO3a.
3.6. Dex Attenuates MIRI In Vivo by Regulating the Sirt1/FoxO3a Signaling Pathway
The results of the above in vitro studies confirmed that pretreatment with Dex protected against H/R‐induced injury by regulating the expression of Sirt1 and FoxO3a. We next utilized a mouse model to examine whether a similar mechanism also occurs in vivo. The I/R group exhibited significant myocardial infarction and myocardial injury. The postischemic myocardial infarct size and myocardial injury in the I/R group were significantly larger than in the sham group (Figure 6A,B). Dex treatment significantly reduced infarct size and alleviated myocardial injury. The I/R group was accompanied by increased LDH and CK‐MB levels as well as marked reductions in LVEF and LVFS (Figure 6C,D). However, Dex administration significantly mitigated these adverse changes (Figure 6A–D). Furthermore, the protective effects of Dex were significantly reversed by inhibiting Sirt1 expression (p < 0.05, I/R + Dex + EX527 vs. I/R + Dex). Western blot analysis revealed significantly lower levels of Sirt1 and FoxO3a expression in mouse heart tissue from the I/R group compared to the sham group. This was reversed by Dex treatment. Moreover, inhibition of Sirt1 expression significantly decreased the expression of both Sirt1 and FoxO3a compared to the I/R + Dex group. This suggested that inhibition of Sirt1 expression could counteract the increase in Sirt1 and FoxO3a expression caused by Dex (Figure 6E).
FIGURE 6.
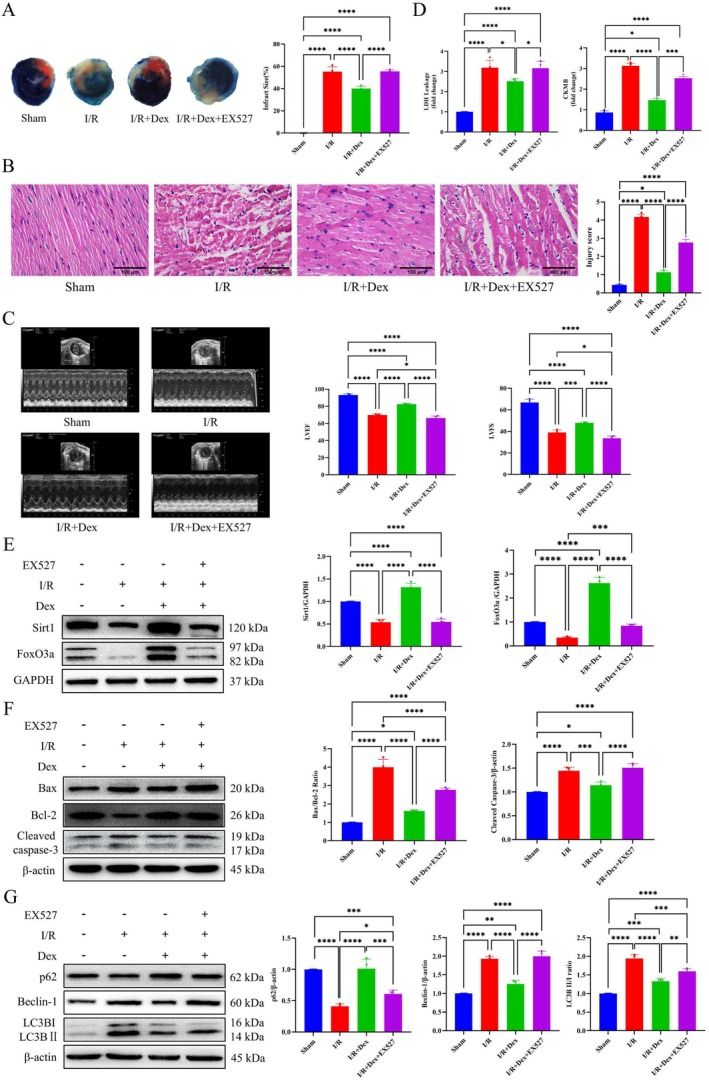
Dex treatment alleviates myocardial ischemia–reperfusion injury in vivo. (A) Evans blue and TTC double staining were used to evaluate the myocardial infarction area. The Evans blue‐staining area represented in blue, the TTC‐staining area in red (indicating the area at risk), and the TTC‐negative staining area appearing pale (indicating infarcted myocardium). (B) HE staining was used to evaluate myocardial injury (40×). (C) Echocardiography of mouse and statistical graph of LVEF and LVFS. (D) Serum LDH leakage and Serum CK‐MB levels were measured. (E) Western blot analysis of Sirt1, FoxO3a, and densitometry analysis of proteins. (F) Western blot analysis of Bax, Bcl‐2, Cleaved caspase‐3, and densitometry analysis of proteins. (G) Western blot analysis of p62, Beclin‐1, LC3B, and densitometry analysis of proteins. I/R: Ischemia/reperfusion. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. One‐way ANOVA was applied, followed by Tukey's post hoc test, a multiple comparison test.
We also analyzed the expression of proteins associated with apoptosis and autophagy in the different experimental groups. Significant increases in the Bax/Bcl‐2 ratio, LC3B II/I ratio, and protein levels of Beclin‐1 and Cleaved caspase‐3 were found in the I/R group, whereas the expression of p62 decreased. Dex treatment significantly modulated the expression levels of these proteins in the I/R + Dex group. Notably, inhibition of Sirt1 expression reversed the effects of Dex treatment (Figure 6F,G). Hence, these results suggested that Dex treatment in mice could reduce the size of myocardial infarction and enhance postischemic cardiac function by upregulating the expression of Sirt1 and FoxO3a.
4. Discussion
The results of this study indicated that Dex conferred myocardial protection through the induction of specific gene products, consistent with previous reports (Wang et al. 2022c) (Chen et al. 2023). Our investigation highlighted the pivotal role of Dex in regulating cellular apoptosis, autophagy, and oxidative stress through the modulation of Sirt1/FoxO3a expression. Dex may have exerted a certain protective effect against MIRI and H/R injury.
Dex is commonly used as a sedative and anesthetic in cardiac surgery. It has been shown to have a protective effect on the heart, thereby contributing to improved 5‐year patient survival rates (Peng et al. 2021). The efficacy of Dex in protecting against MIRI in cellular (Zhu et al. 2020) and animal models (Wang et al. 2023a) involves various intracellular signaling pathways. In the current study, Dex treatment reduced apoptosis, autophagy, and oxidative stress in H9c2 cells following H/R injury. This concurs with findings by Zhu et al. on the ability of Dex to attenuate cellular injury and apoptosis in H9c2 cardiomyocytes (Zhu et al. 2020). Yu et al. reported that Dex inhibits septic myocardial dysfunction in rats by activating α7nAChR and PI3K/Akt‐mediated autophagy (Yu et al. 2019). The present study also demonstrated the effectiveness of Dex in reducing myocardial infarct size and improving left ventricular function in mice subjected to I/R injury, thus corroborating earlier studies (He et al. 2019) (Xiong et al. 2021).
Sirt1 and FoxO3a play essential roles in the FoxO pathway. Sirt1 deacetylates FoxO3 and/or FoxO4, thereby reducing FoxO‐induced apoptosis and promoting FoxO‐induced cell‐cycle arrest (Giannakou and Partridge 2004; Lee et al. 2022). Dex has been shown to increase the expression of Sirt1. Conversely, a specific inhibitor of Sirt1, EX527, was found to reduce the expression of Sirt1 protein, similar to the effect observed with Sirt1‐specific siRNA (Li et al. 2023a). This inhibition notably attenuated the cardioprotective effects imparted by Dex (Chen et al. 2019). FoxO3a regulates cell differentiation, proliferation, and oxidative stress through various pathways, including Akt‐FoxO3a (Tan et al. 2023), Sirt1‐FoxO3a (Li et al. 2023c), AMPK‐FoxO3a‐MnSOD (Kim et al. 2021). Activation of the Sirt1/FoxO3a pathway can enhance antioxidant capacity (Li et al. 2023b) and regulate mitochondrial autophagy after ischemia in rats (Xie et al. 2022). Moreover, activation of the Akt‐FoxO3A‐mTOR signaling pathway can also reduce apoptosis and autophagy (Liu et al. 2023). Our animal study revealed a significant reduction in Sirt1 and FoxO3a protein levels after 30 min of ischemia followed by 2 h of reperfusion. This agrees with findings using a similar experimental model (Tian et al. 2019) and indicates that MIRI disrupts the mitochondrial energy balance in cardiac myocytes and impacts Sirt1‐mediated pathways. Sirt1 expression was significantly reduced after I/R, leading to the acetylation and subsequent reduction of FoxO3a. Interestingly, we observed a more than 2‐fold increase in FoxO3a expression compared to the control group following intraperitoneal injection of Dex in mice. Moreover, Sirt1 protein expression in the I/R + Dex group was also higher than in the control group. This finding suggests that the abnormal increase of Sirt1 and FoxO3a after intraperitoneal injection of Dex may be a compensatory mechanism adopted by the body to protect ischemia–reperfusion cardiomyocytes. The underlying in vivo mechanisms are likely to be quite complex, and the details remain to be clarified. In our in vitro study, the silencing of FoxO3a reduced FoxO3a expression but not Sirt1 expression. This suggests that in the current working model of cardiomyocyte H/R, Sirt1 acts upstream of FoxO3a.
Oxidative stress is pivotal in ischemic injury and may intensify during reperfusion (Zhang et al. 2024). Dex has shown protective effects against MIRI by inhibiting oxidative stress (Wang et al. 2022a). The present study found that inhibition of Sirt1 expression, or silencing of FoxO3a, reversed the protective effects of Dex. Previous research has highlighted the significance of Bax and Bcl‐2 in the intrinsic apoptosis pathway during ischemic myocardium (Li et al. 2018; Lv et al. 2020). Dex attenuates myocardial I/R‐induced intrinsic apoptosis by modulating the expression of antiapoptotic proteins (Deng et al. 2022; Wang et al. 2022b). Apoptosis and autophagy are both adaptive responses that are crucial for cell growth, survival, and homeostasis. Autophagy is responsible for eliminating and recycling misfolded proteins and damaged organelles. It plays a critical role in regulating cell survival and apoptotic pathways (Wang et al. 2017). However, excessive autophagy during the reperfusion phase can worsen cardiac injury. Li et al. showed that Dex can attenuate myocardial I/R injury in rats and reduce I/R‐induced autophagy by activating the PI3K/Akt pathway (Li et al. 2021). Similarly, our results showed that I/R and H/R injury promoted oxidative stress, cardiomyocyte apoptosis, and autophagy. Moreover, Dex preconditioning significantly inhibited oxidative stress, cardiomyocyte apoptosis, and autophagy by activating Sirt1/FoxO3a signaling pathways. Interestingly, Xiao et al. also found that the protective effect of Dex against I/R damage in pluripotent stem cells was associated with upregulation of autophagy (Xiao et al. 2021). The different results may in part be attributed to the use of different cell lines. Xiao et al. employed human‐induced pluripotent stem cells, whereas our study utilized rat cardiomyocytes (H9c2). The posthypoxia/reoxygenation effects of Dex on autophagy may vary depending on the specific cell type and the extent of the autophagy response. Our findings agree with those of Li et al. (2022) and further support the notion that Dex attenuates H/R‐ and I/R‐induced cardiac injury by inhibiting excessive postischemic autophagy. Li et al. previously suggested that inhibition of excessive autophagy by the PI3K/Akt–mTOR pathway was a possible mechanism by which pharmacological treatment mitigates MIRI in rats. This was in line with their predictions derived from network pharmacology.
While this study provides valuable insights into the cardioprotective effects of Dex via the Sirt1/FoxO3a pathway, several limitations exist. The use of H9c2 cells and mouse models may limit the direct applicability to humans, and the involvement of other signaling pathways in Dex's protective effects remains unexplored. Additionally, the long‐term effects of Dex treatment and its impact on chronic myocardial health were not evaluated, requiring further investigation. Future studies should address these aspects to better understand the full therapeutic potential of Dex.
5. Conclusions
This study highlights the crucial role of Sirt1/FoxO3a in cardiomyocytes subjected to H/R or I/R injury. Furthermore, our study found that Dex administration effectively mitigated apoptosis, autophagy, and oxidative stress, thereby exerting a protective effect in the context of MIRI. This protective mechanism was mediated through the Sirt1/FoxO3a signaling pathway and the modulation of key genes involved in apoptosis, autophagy, and oxidative stress, as illustrated in Figure 7. The novel insights provided by this research have enhanced our understanding of the mechanism underlying the therapeutic benefits of Dex in the treatment of MIRI. Such insights could potentially guide future clinical approaches for cardiac protection.
FIGURE 7.

Schematic diagram displays the role of Dex treatment alleviate myocardial ischemia–reperfusion injury. The upward red arrow represents upregulation of gene expression, and the downward blue arrow represents downregulation of gene expression.
Ethics Statement
The experimental use of animals in this study was authorized by the Committee for the Use of Live Animals in Teaching and Research, Guangdong Medical University (Approval no. GDY2002072).
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Table S1. Primer list for qRT‐PCR.
Acknowledgements
The authors have nothing to report.
Funding: This work was supported by The National Science Foundation of China (82172160).
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- Arora, S. , Stouffer G. A., Kucharska‐Newton A. M., et al. 2019. “Twenty Year Trends and Sex Differences in Young Adults Hospitalized With Acute Myocardial Infarction.” Circulation 139: 1047–1056. 10.1161/circulationaha.118.037137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrico, C. , Meyer J. G., He W., Gibson B. W., and Verdin E.. 2018. “The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications.” Cell Metabolism 27: 497–512. 10.1016/j.cmet.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Zhou M., Ge Y., and Wang X.. 2020. “SIRT1 and Aging Related Signaling Pathways.” Mechanisms of Ageing and Development 187: 111215. 10.1016/j.mad.2020.111215. [DOI] [PubMed] [Google Scholar]
- Chen, L. , Cao J., Cao D., et al. 2019. “Protective Effect of Dexmedetomidine Against Diabetic Hyperglycemia‐Exacerbated Cerebral Ischemia/Reperfusion Injury: An In Vivo and In Vitro Study.” Life Sciences 235: 116553. 10.1016/j.lfs.2019.116553. [DOI] [PubMed] [Google Scholar]
- Chen, M. , Li X., and Mu G.. 2022. “Myocardial Protective and Anti‐Inflammatory Effects of Dexmedetomidine in Patients Undergoing Cardiovascular Surgery With Cardiopulmonary Bypass: A Systematic Review and Meta‐Analysis.” Journal of Anesthesia 36: 5–16. 10.1007/s00540-021-02982-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. R. , Hong Y., Wen S. H., Zhan Y. Q., and Huang W. Q.. 2023. “Dexmedetomidine Pretreatment Protects Against Myocardial Ischemia/Reperfusion Injury by Activating STAT3 Signaling.” Anesthesia and Analgesia 137: 426–439. 10.1213/ane.0000000000006487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson, S. M. , Ferdinandy P., Andreadou I., et al. 2019. “Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week.” Journal of the American College of Cardiology 73: 89–99. 10.1016/j.jacc.2018.09.086. [DOI] [PubMed] [Google Scholar]
- Deng, R. M. , and Zhou J.. 2023. “The Role of PI3K/AKT Signaling Pathway in Myocardial Ischemia‐Reperfusion Injury.” International Immunopharmacology 123: 110714. 10.1016/j.intimp.2023.110714. [DOI] [PubMed] [Google Scholar]
- Deng, X. , Ye F., Zeng L., et al. 2022. “Dexmedetomidine Mitigates Myocardial Ischemia/Reperfusion‐Induced Mitochondrial Apoptosis Through Targeting lncRNA HCP5.” American Journal of Chinese Medicine 50: 1529–1551. 10.1142/s0192415x22500641. [DOI] [PubMed] [Google Scholar]
- Du, J. , Xu Z., Zhen J., et al. 2019. “Dexmedetomidine Attenuates Myocardial Ischemia/Reperfusion Injury Through Regulating Lactate Signaling Cascade in Mice.” European Review for Medical and Pharmacological Sciences 23: 3527–3532. 10.26355/eurrev_201904_17721. [DOI] [PubMed] [Google Scholar]
- Ge, C. , Peng Y., Li J., et al. 2023. “Hydroxysafflor Yellow A Alleviates Acute Myocardial Ischemia/Reperfusion Injury in Mice by Inhibiting Ferroptosis via the Activation of the HIF‐1α/SLC7A11/GPX4 Signaling Pathway.” Nutrients 15: 53411. 10.3390/nu15153411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakou, M. E. , and Partridge L.. 2004. “The Interaction Between FOXO and SIRT1: Tipping the Balance Towards Survival.” Trends in Cell Biology 14: 408–412. 10.1016/j.tcb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Guo, Q. , Ma M., Yu H., Han Y., and Zhang D.. 2023. “Dexmedetomidine Enables Copper Homeostasis in Cerebral Ischemia/Reperfusion via Ferredoxin 1.” Annals of Medicine 55: 2209735. 10.1080/07853890.2023.2209735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, R. H. , Huang H. M., Han H., et al. 2021. “Propofol Postconditioning Ameliorates Hypoxia/Reoxygenation Induced H9c2 Cell Apoptosis and Autophagy via Upregulating Forkhead Transcription Factors Under Hyperglycemia.” Military Medical Research 8: 58. 10.1186/s40779-021-00353-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, J. , Liu D., Zhao L., et al. 2022. “Myocardial Ischemia/Reperfusion Injury: Mechanisms of Injury and Implications for Management (Review).” Experimental and Therapeutic Medicine 23: 430. 10.3892/etm.2022.11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, L. , Hao S., Wang Y., et al. 2019. “Dexmedetomidine Preconditioning Attenuates Ischemia/Reperfusion Injury in Isolated Rat Hearts With Endothelial Dysfunction.” Biomedicine & Pharmacotherapy 114: 108837. 10.1016/j.biopha.2019.108837. [DOI] [PubMed] [Google Scholar]
- Kim, W. S. , Kim C. H., Lee J. M., et al. 2021. “Purple Corn Extract (PCE) Alleviates Cigarette Smoke (CS)‐Induced DNA Damage in Rodent Blood Cells by Activation of AMPK/Foxo3a/MnSOD Pathway.” Animal Cells Systems (Seoul) 25: 65–73. 10.1080/19768354.2021.1883734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. , Kim J., Lee J. H., et al. 2022. “SIRT1 Promotes Host Protective Immunity Against Toxoplasma Gondii by Controlling the FoxO‐Autophagy Axis via the AMPK and PI3K/AKT Signalling Pathways.” International Journal of Molecular Sciences 23: 13578. 10.3390/ijms232113578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. R. , Liu Q., Zhu C. L., et al. 2023a. “β‐Nicotinamide Mononucleotide Activates NAD+/SIRT1 Pathway and Attenuates Inflammatory and Oxidative Responses in the Hippocampus Regions of Septic Mice.” Redox Biology 63: 102745. 10.1016/j.redox.2023.102745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Hu H. P., Li Y., Shao W., Zhang J. Z., and Wang L. M.. 2018. “Influences of Remifentanil on Myocardial Ischemia‐Reperfusion Injury and the Expressions of Bax and Bcl‐2 in Rats.” European Review for Medical and Pharmacological Sciences 22: 8951–8960. 10.26355/eurrev_201812_16665. [DOI] [PubMed] [Google Scholar]
- Li, M. , Wang Y., Qi Z., et al. 2022. “QishenYiqi Dripping Pill Protects Against Myocardial Ischemia/Reperfusion Injury via Suppressing Excessive Autophagy and NLRP3 Inflammasome Based on Network Pharmacology and Experimental Pharmacology.” Frontiers in Pharmacology 13: 981206. 10.3389/fphar.2022.981206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Lin Z., Xiao H., et al. 2023b. “Fyn Deficiency Inhibits Oxidative Stress by Decreasing c‐Cbl‐Mediated Ubiquitination of Sirt1 to Attenuate Diabetic Renal Fibrosis.” Metabolism 139: 155378. 10.1016/j.metabol.2022.155378. [DOI] [PubMed] [Google Scholar]
- Li, S. , Xiao H., Sun X., et al. 2023c. “Connexin32 Promotes the Activation of Foxo3a to Ameliorate Diabetic Nephropathy via Inhibiting the Polyubiquitination and Degradation of Sirt1.” Antioxidants & Redox Signaling 39: 241–261. 10.1089/ars.2022.0108. [DOI] [PubMed] [Google Scholar]
- Li, Y. , Qu M., Xing F., et al. 2021. “The Protective Mechanism of Dexmedetomidine in Regulating Atg14L‐Beclin1‐Vps34 Complex Against Myocardial Ischemia‐Reperfusion Injury.” Journal of Cardiovascular Translational Research 14: 1063–1074. 10.1007/s12265-021-10125-9. [DOI] [PubMed] [Google Scholar]
- Liu, Q. , Yuan W., Yan Y., et al. 2023. “Identification of a Novel Small‐Molecule Inhibitor of miR‐29b Attenuates Muscle Atrophy.” Molecular Therapy ‐ Nucleic Acids 31: 527–540. 10.1016/j.omtn.2023.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, G. , Jian Z., Zhu Y., et al. 2019. “Sirt1 Promotes Autophagy and Inhibits Apoptosis to Protect Cardiomyocytes From Hypoxic Stress.” International Journal of Molecular Medicine 43: 2033–2043. 10.3892/ijmm.2019.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, J. , Yan D., Li S., et al. 2020. “Allopurinol Reduces Oxidative Stress and Activates Nrf2/p62 to Attenuate Diabetic Cardiomyopathy in Rats.” Journal of Cellular and Molecular Medicine 24: 1760–1773. 10.1111/jcmm.14870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv, X. , Lu P., Hu Y., and Xu T.. 2020. “miR‐346 Inhibited Apoptosis Against Myocardial Ischemia‐Reperfusion Injury via Targeting Bax in Rats.” Drug Design, Development and Therapy 14: 895–905. 10.2147/dddt.S245193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, K. , Shen Y. P., Ying Y. Y., et al. 2021. “Perioperative Dexmedetomidine and 5‐Year Survival in Patients Undergoing Cardiac Surgery.” British Journal of Anaesthesia 127: 215–223. 10.1016/j.bja.2021.03.040. [DOI] [PubMed] [Google Scholar]
- Poon, W. H. , Ling R. R., Yang I. X., et al. 2023. “Dexmedetomidine for Adult Cardiac Surgery: A Systematic Review, Meta‐Analysis and Trial Sequential Analysis.” Anaesthesia 78: 371–380. 10.1111/anae.15947. [DOI] [PubMed] [Google Scholar]
- Si, Y. , Bao H., Han L., et al. 2013. “Dexmedetomidine Protects Against Renal Ischemia and Reperfusion Injury by Inhibiting the JAK/STAT Signaling Activation.” Journal of Translational Medicine 11: 141. 10.1186/1479-5876-11-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, M. , Wang R., Xia R., Xia Z., Wu Z., and Wang T.. 2022. “Amelioration of Myocardial Ischemia/Reperfusion Injury in Diabetes: A Narrative Review of the Mechanisms and Clinical Applications of Dexmedetomidine.” Frontiers in Pharmacology 13: 949754. 10.3389/fphar.2022.949754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, J. , Xiang Y., Xiong Y., Zhang Y., Qiao B., and Zhang H.. 2023. “Crebanine Induces ROS‐Dependent Apoptosis in Human Hepatocellular Carcinoma Cells via the AKT/FoxO3a Signaling Pathway.” Frontiers in Pharmacology 14: 1069093. 10.3389/fphar.2023.1069093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, R. , Liu Y. Q., Zhong H. L., et al. 2022. “Evidence Basis for Using Dexmedetomidine to Enhance the Quality of Paravertebral Block: A Systematic Review and Meta‐Analysis of Randomized Controlled Trials.” Frontiers in Pharmacology 13: 952441. 10.3389/fphar.2022.952441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, L. , Cao W., Yue R., et al. 2019. “Pretreatment With Tilianin Improves Mitochondrial Energy Metabolism and Oxidative Stress in Rats With Myocardial Ischemia/Reperfusion Injury via AMPK/SIRT1/PGC‐1 Alpha Signaling Pathway.” Journal of Pharmacological Sciences 139: 352–360. 10.1016/j.jphs.2019.02.008. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Liu J., Wang Z., et al. 2023a. “Dexmedetomidine Abates Myocardial Ischemia Reperfusion Injury Through Inhibition of Pyroptosis via Regulation of miR‐665/MEF2D/Nrf2 Axis.” Biomedicine & Pharmacotherapy 165: 115255. 10.1016/j.biopha.2023.115255. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Liu X., Zhou X., et al. 2022a. “Dexmedetomidine Inhibits Parthanatos in Cardiomyocytes and in Aortic Banded Mice by the ROS‐Mediated NLRP3 Inflammasome Activation.” Journal of Cardiovascular Translational Research 16: 624–635. 10.1007/s12265-022-10340-y. [DOI] [PubMed] [Google Scholar]
- Wang, S. , Wang C., Yan F., et al. 2017. “N‐Acetylcysteine Attenuates Diabetic Myocardial Ischemia Reperfusion Injury Through Inhibiting Excessive Autophagy.” Mediators of Inflammation 2017: 9257291. 10.1155/2017/9257291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T. , Li Z., Xia S., Xu Z., Chen X., and Sun H.. 2022b. “Dexmedetomidine Promotes Cell Proliferation and Inhibits Cell Apoptosis by Regulating LINC00982 and Activating the Phosphoinositide‐3‐Kinase (PI3K)/protein Kinase B (AKT) Signaling in Hypoxia/Reoxygenation‐Induced H9c2 Cells.” Bioengineered 13: 10159–10167. 10.1080/21655979.2022.2060900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Yang N., Hou Y., et al. 2023b. “L‐Arginine‐Loaded Gold Nanocages Ameliorate Myocardial Ischemia/Reperfusion Injury by Promoting Nitric Oxide Production and Maintaining Mitochondrial Function.” Advanced Science (Weinh) 10: e2302123. 10.1002/advs.202302123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Yao M., Jiang L., et al. 2022c. “Dexmedetomidine Attenuates Myocardial Ischemia/Reperfusion‐Induced Ferroptosis via AMPK/GSK‐3β/Nrf2 Axis.” Biomedicine & Pharmacotherapy 154: 113572. 10.1016/j.biopha.2022.113572. [DOI] [PubMed] [Google Scholar]
- Xiao, Y. , Li J., Qiu L., et al. 2021. “Dexmedetomidine Protects Human Cardiomyocytes Against Ischemia‐Reperfusion Injury Through α2‐Adrenergic Receptor/AMPK‐Dependent Autophagy.” Frontiers in Pharmacology 12: 615424. 10.3389/fphar.2021.615424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, W. , Zhu T., Zhang S., and Sun X.. 2022. “Protective Effects of Gypenoside XVII Against Cerebral Ischemia/Reperfusion Injury via SIRT1‐FOXO3A‐ and Hif1a‐BNIP3‐Mediated Mitochondrial Autophagy.” Journal of Translational Medicine 20: 622. 10.1186/s12967-022-03830-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, W. , Zhou R., Qu Y., et al. 2021. “Dexmedetomidine Preconditioning Mitigates Myocardial Ischemia/Reperfusion Injury via Inhibition of Mast Cell Degranulation.” Biomedicine & Pharmacotherapy 141: 111853. 10.1016/j.biopha.2021.111853. [DOI] [PubMed] [Google Scholar]
- Xu, S. , Wu B., Zhong B., et al. 2021. “Naringenin Alleviates Myocardial Ischemia/Reperfusion Injury by Regulating the Nuclear Factor‐Erythroid Factor 2‐Related Factor 2 (Nrf2) /System xc−/ Glutathione Peroxidase 4 (GPX4) Axis to Inhibit Ferroptosis.” Bioengineered 12: 10924–10934. 10.1080/21655979.2021.1995994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, T. , Liu D., Gao M., et al. 2019. “Dexmedetomidine Prevents Septic Myocardial Dysfunction in Rats via Activation of α7nAChR and PI3K/Akt‐ Mediated Autophagy.” Biomedicine & Pharmacotherapy 120: 109231. 10.1016/j.biopha.2019.109231. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Sha J., Feng X., et al. 2019. “Dexmedetomidine Ameliorates LPS Induced Acute Lung Injury via GSK‐3β/STAT3‐NF‐κB Signaling Pathway in Rats.” International Immunopharmacology 74: 105717. 10.1016/j.intimp.2019.105717. [DOI] [PubMed] [Google Scholar]
- Zhang, M. , Liu Q., Meng H., et al. 2024. “Ischemia‐Reperfusion Injury: Molecular Mechanisms and Therapeutic Targets.” Signal Transduction and Targeted Therapy 9: 12. 10.1038/s41392-023-01688-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Zhao Q., Li X., and Ji F.. 2021. “Dexmedetomidine Reversed Hypoxia/Reoxygenation Injury‐Induced Oxidative Stress and Endoplasmic Reticulum Stress‐Dependent Apoptosis of Cardiomyocytes via SIRT1/CHOP Signaling Pathway.” Molecular and Cellular Biochemistry 476: 2803–2812. 10.1007/s11010-021-04102-8. [DOI] [PubMed] [Google Scholar]
- Zhao, X. , Liu Y., Zhu G., et al. 2019. “SIRT1 Downregulation Mediated Manganese‐Induced Neuronal Apoptosis Through Activation of FOXO3a‐Bim/PUMA Axis.” Science of the Total Environment 646: 1047–1055. 10.1016/j.scitotenv.2018.07.363. [DOI] [PubMed] [Google Scholar]
- Zhu, Z. , Ling X., Zhou H., Zhang C., and Yan W.. 2020. “Dexmedetomidine Attenuates Cellular Injury and Apoptosis in H9c2 Cardiomyocytes by Regulating p‐38MAPK and Endoplasmic Reticulum Stress.” Drug Design, Development and Therapy 14: 4231–4243. 10.2147/dddt.S265970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer list for qRT‐PCR.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.