

Abstract
The A1 subunits of verotoxin-1 (VT1) and VT2 genes were cloned into pGEX-4T-2 for the expression of glutathione S-transferase (GST) fusion proteins. The N-terminal and the transmembrane regions of the A1 subunits were excluded from the constructs in order to increase the product yields. Polyclonal anti-VT1A1 and anti-VT2A1 antibodies were produced by immunizing rabbits with GST-VT1A1 and GST-VT2A1 fusion proteins, respectively. The antibodies were tested for their ability to neutralize active toxins from 45 VT-producing Escherichia coli (VTEC) strains. The antibodies had significantly high neutralizing activities against their homologous toxins. The average percentages of neutralization of VT1 by anti-GST-VT1A1 and anti-GST-VT2A1 were 76.7% ± 7.9% and 3.6% ± 2.3%, respectively, and those of VT2 were 1.7% ± 2.3% and 82.5% ± 13.9%, respectively. VT2 variant toxin was neutralized by anti-GST-VT2A1, with cross neutralization being a possible consequence of sequence homology between VT2 and a VT2 variant. To our knowledge, this is the first report on the production of polyclonal antibodies from GST-VT fusion proteins. The antibodies were shown to exhibit specific toxin neutralizing activities and may be useful for immunological diagnosis of VTEC infections.
Verotoxin (VT) is the principal virulence factor of verotoxigenic Escherichia coli (VTEC), an emerging food-borne pathogen associated with diseases ranging from uncomplicated diarrhea to the hemolytic-uremic syndrome (1, 24). There are two types of VT, VT1 and VT2; the latter type has variants, including VT2c and VT2e. All VTs belong to the Shiga toxin family, in which the C terminus of the A subunit is encircled by a pentameric ring formed by five identical B subunits (7). This A-B bipartite molecule first binds to a eukaryotic glycolipid receptor via the pentamer of the VT B subunits. Then the catalytic VT A subunit dissociates from the VT B subunit pentamer and translocates into the cytoplasm through a retrograde secretory pathway. Subsequently, inhibition of the 28S rRNA in 60S ribosomal subunits induces programmed cell death (12, 14, 19). The N terminus of the VT A subunit is the A1 fragment, which is a catalytic domain essential for the cytotoxicity of VT (8). The C terminus A2 fragment facilitates the noncovalent interaction between subunits A and B (2). The A1 and A2 fragments are separated by a trypsin-sensitive region. Finally, there is a transmembrane region at the C terminus of fragment A1. This region is involved in toxin translocation across the endoplasmic reticulum membrane (23). The minimal sequence of VT1A required for activity includes the transmembrane region, and the deletion of this region retarded the functional activity of VT1A (11). Structural and biological properties of the VTs have been studied extensively (15); subunits of VTs have been prepared in large quantities as fusion proteins and used in seroepidemiology (10).
Antibodies against VTs have been produced for diagnostic or therapeutic purposes. These antibodies were produced by immunizing animals with VT or VT subunit toxoids (4, 16). The use of a VT fusion protein as an immunogen has not been reported. In this study, we describe the production of VT1A1 and VT2A1 subunits by the glutathione S-transferase (GST) fusion protein technique. The GST fusion constructs were prepared without the N-terminal signal peptide and C-terminal transmembrane region, which enabled hyperexpression of soluble protein products and a single-step purification. As the transmembrane region is required for cytotoxicity (11), it was unknown whether the fusion proteins lacking these regions elicited neutralizing antibodies. Hence, we also used the purified fusion proteins to raise polyclonal antibodies and evaluated the neutralization activities of the antibodies on standard VTEC strains as well as animal and human strains reported in our earlier study (13).
MATERIALS AND METHODS
Bacterial strains, culture conditions, and preparation of VT.
Forty-five VTEC strains (41 from animals, 4 from humans) isolated in Hong Kong (13) and 8 standard VTEC and non-VTEC strains were included in this study. Details of these strains are listed in Table 1. A single colony was inoculated into 2 ml of brain heart infusion broth (Oxoid, Basingstoke, United Kingdom) and was then incubated overnight at 37°C with agitation. One milliliter of culture was centrifuged at 20,000 × g for 15 min, and the supernatant was collected for a neutralization assay.
TABLE 1.
Characteristics of VTEC and standard strains used in neutralization studies
| Source or strain | vt genotype(s) | Serotype(s) (no. of strains) |
|---|---|---|
| Human | 2 | O157:H7 (3), OR:H45 (1) |
| Cattle | 1 | O84:H− (1), Ont:H− (3), O121:H19 (1), O110:H16 (1) |
| 2 | O15:H− (4), O157:H7 (5), O172:H− (10), Ont:H− (3), OR:H− (3) | |
| 1, 2 | O157:H7 (3), O91:H14 (1), Ont:H− (1) | |
| Variant | O2:H51 (1) | |
| Pig | 1, 2 | Ont:H14 (1) |
| 2, 2e | Ont:H21 (1) | |
| 2e | O20:H− (1), Ont:H27 (1) | |
| ATCC 43888 | NTa | O157:H7 |
| ATCC 25922 (non-VTEC) | NT | NAb |
| ATCC 43890 | 1 | O157:H7 |
| ATCC 43889 | 2 | O157:H7 |
| UK O157:H7 | 2 | O157:H7 |
| ATCC 43895 | 1, 2 | O157:H7 |
| WHO O157:H7 | 1, 2 | O157:H7 |
| S1191 | 2e | O139 |
NT, non-toxin producer.
NA, not available.
PCR amplification of vt1A1 and vt2A1 genes
The A1 subunits of the vt1 and vt2 genes were amplified from standard strains ATCC 43890 and ATCC 43889, respectively. Upon analysis with the computer program TMpred (18), two major hydrophobic regions were found at amino acid positions 3 to 23 and 242 to 263 (nucleotide positions 338 to 400 and 1055 to 1120), respectively, in subunit A of vt1 (GenBank accession number M16625 [6]) (Fig. 1). Similarly, two hydrophobic regions were found in amino acid positions 3 to 19 and 245 to 261 (nucleotide positions 203 to 253 and 929 to 979), respectively, in subunit A of vt2 (GenBank accession number M59432 [20]). Primers were then designed to exclude these hydrophobic regions. BamHI and XhoI restriction enzyme sites, stop codons, and CCC-GGG triplets were included to facilitate subsequent cloning procedures. The primers used for amplification of vt1A1 were KW3 (positions 398 to 415), 5′-CCCGGATCCAAGGAATTTACCTTAGAC-3′, and KW4 (positions 1052 to 1066), 5′-GGGGTCGAG(TCA)TCTTCCTACACGAAC-3′ (Fig. 1). The primers used for amplification of vt2A1 were KW7 (positions 263 to 280), 5′-CCCGGATCCCGGGAGTTTACGATAGAC-3′, and KW8 (positions 914 to 928), 5′-GGGCTCGAG(TCA)TCTCCCCACTCTGAC-3′. In each case, the amplified sequence was located between the two hydrophobic regions. BamHI and XhoI restriction sites are underlined, and stop codons are in parentheses. In order to amplify the entire vt1A1 and vt2A1 subunits, primers were also designed to include the hydrophobic regions of these sequences. The primers used for amplification of the entire vt1A1 gene were F380 (positions 380 to 394), 5′-CCCGGATCCTCAGTTAATGTGGTC-3′, and R1166 (positions 1163 to 1177), 5′-GGGGTCGAG(TCA)CATAGAAGGAAACTC-3′. The primers used for amplification of the entire vt2A1 gene were F212 (positions 212 to 226), 5′-CCCGGATCCTTTAAATGGGTACTG-3′, and R1039 (positions 1025 to 1039), 5′-GGGCTCGAG(TCA)TTCTGGTTGACTCTC-3′. PCR products were electrophoresed, stained, and visualized by UV transillumination, and products were then confirmed by DNA sequencing.
FIG. 1.

TMpred output for vt1 subunit A. The middle and upper bars represent the amino acid and nucleotide sequences of vt1A1, respectively. Two hydrophobic regions exist in amino acid positions 3 to 23 and 242 to 263. The relative positions of primers KW3 and KW4 are indicated.
Cloning of PCR products.
PCR products were purified with the QIAquick kit (Qiagen, Santa Clarita, Calif.) before and after restriction digestion with BamHI and XhoI (Promega, Madison, Wis.). Each of the digested products was cloned into BamHI- and XhoI-digested pGEX-4T-2 (Amersham Pharmacia Biotech, Uppsala, Sweden), and each construct encoded a fusion protein consisting of GST at the N terminus and VT1A1 or VT2A1 at the C terminus. The recombinant constructs were electroporated into E. coli XL1-Blue MRF′ (Stratagene, La Jolla, Calif.). The presence of inserts was confirmed by PCR. The forward primer 5PGEX, 5′-GGGCTGGCAAGCCACGTTTGGTG-3′, was derived from the 3′ end of the GST gene. The reverse primers were KW4 and R1166 for truncated and entire vt1A1, respectively, and KW8 and R1039 for truncated and entire vt2A1, respectively.
Expression and purification of GST-VT fusion proteins.
The recombinant proteins were expressed and purified as described by the GST Gene Fusion System (Amersham Pharmacia Biotech). Briefly, 300 ml of E. coli XL1-Blue MRF′ transformed with the recombinant plasmid or plasmid without insert was grown at 37°C in Luria-Bertani medium to an optical density of 0.5 at 600 nm. Isopropyl-β-d-thiogalactopyranoside (IPTG) was then added to the culture at a final concentration of 1 mM. The cells were grown for an additional 2 h at 30°C. The cells were harvested by centrifugation at 7,700 × g for 10 min at 4°C followed by suspension in 12 ml of cold phosphate-buffered saline (PBS) with 1 mM phenylmethanesulfonyl fluoride and lysis by sonication on ice. Cell debris was removed by centrifugation at 12,000 × g for 10 min at 4°C. Fusion protein present in the supernatant was purified by affinity chromatography on glutathione-Sepharose 4B (Amersham Pharmacia Biotech) according to the manufacturer's protocol. The purity of protein products was assessed by sodium dodecyl sulfate-12.5% polyacrylamide gel electrophoresis (SDS-12.5% PAGE), and protein concentrations were estimated by absorbance at 280 nm with bovine serum albumin as the standard.
Immunization of rabbits.
New Zealand White rabbits were injected subcutaneously with 150 μg (0.5 ml) of purified GST-VT1A1, GST-VT2A1, or GST mixed with an equal volume of complete Freund's adjuvant. The immunization was repeated at 4 and 6 weeks after the first injection. The animals were bled 1 week after the third injection and then once monthly. Antibody titers were determined by a neutralization assay, and the animals were sacrificed when there was no further increase in antibody titers.
Purification of polyclonal antibodies.
Before performing immunoblot analysis, polyclonal antibodies obtained from the immunized rabbits were purified to remove anti-GST present in the sera. This was done by incubating the sera with GST immobilized on glutathione-Sepharose beads. The immunoadsorbent beads were prepared according to the manufacturer's recommendations. Briefly, 200 μl of glutathione-Sepharose beads was coupled with 210 mg of GST. The GST was expressed from a 300-ml culture of E. coli XL1-Blue MRF′ transformed with the plasmid pGEX-4T-2. After coupling of GST, the beads were washed and pelleted, 1 ml of 1/100-diluted polyclonal antibody was added to the beads, and the mixture was incubated at 4°C overnight with gentle agitation. The purified polyclonal antibody was separated from the beads by centrifugation at 500 × g for 5 min.
Immunoblot analysis of polyclonal antibodies.
Fusion protein samples were run on SDS-12.5% polyacrylamide gels under reducing conditions and subsequently electroblotted onto nitrocellulose membranes (Bio-Rad Laboratories, Hercules, Calif.). The membrane was blocked at 4°C overnight in blocking buffer (10% nonfat milk-0.03% Tween 20-PBS) and then washed three times in 0.03% Tween 20-PBS. The washed membrane was incubated with 1:2,000-diluted anti-GST-VT at room temperature for 45 min, and the membrane was washed again and incubated for 30 min at room temperature in 1:2,000-diluted anti-rabbit immunoglobulin conjugated with alkaline phosphatase. The membrane was washed again and incubated with the chromogenic substrates 5-bromo-4-chloro-3-indolylphosphate (BCIP) and nitroblue tetrazolium (Boehringer Mannheim, Mannheim, Germany) for 1 to 5 min.
Evaluation of anti-VT1A1 and anti-VT2A1 by sandwich enzyme-linked immunosorbent assay (ELISA).
A 96-well microtiter plate precoated with VT-specific monoclonal antibodies was used. The plate was obtained from the Premier EHEC Assay kit (Meridian Diagnostics, Cincinnati, Ohio). A 50-μl aliquot of culture supernatant obtained from a VT- or non-VT-producing strain was diluted in 200 μl of diluent supplied with the assay kit. The diluted supernatant was added to the well and incubated at 37°C for 30 min. The microtiter plate was washed three times with PBS with 0.5% Tween 20, blocked with 2% bovine serum albumin at 37°C for 30 min, and washed as described above. A 100-μl volume of 1/400-diluted anti-VT1A1 or 1/25-diluted anti-VT2A1 was added to the well, incubated at 37°C for 30 min, and washed. A 100-μl aliquot of horseradish peroxidase-conjugated goat anti-rabbit serum (diluted 1/1,200) was added, incubated, and washed as before. After washing, 100 μl of para-nitrophenyl phosphate substrate (Sigma, St. Louis, Mo.) was added and then incubated at 37°C for 10 min. The absorbance was read at 405 nm with a Spectrafluor Plus microplate reader (TECAN GmbH, Salzburg, Austria).
Neutralization assay.
The neutralization assay was performed on Vero cell monolayers in Eagle minimum essential medium (Gibco-BRL, Gaithersburg, Md.) with 10% fetal calf serum (Gibco-BRL) grown on 96-well plates. For determination of antibody titers, 50 μl of serially diluted antibody was mixed with an equal volume of culture supernatant prepared from ATCC 43890 or ATCC 43889 containing 40 50% cytotoxic doses (CD50) (20).
For testing of VTEC isolates, 50 μl of 1/400-diluted anti-GST-VT1A1 or 1/25-diluted anti-GST-VT2A1 (dilutions twofold higher than the 50% neutralization dose) was preincubated with an equal volume of diluted culture supernatant from standard strains or animal or human VTEC isolates containing 40 CD50. For toxin control, 50 μl of PBS was added instead of antibody. The mixture was incubated at 37°C for 1 h in a moist chamber, added to the Vero cell monolayer, and incubated for 2 days. The Vero cell monolayer was fixed and stained with crystal violet as described previously (9). The intensity of the color of the stained cells was measured at 620 nm, and the absorbance (A) was proportional to the amount of viable cells. Percentage neutralization was calculated by using the following formula (20):
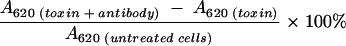 |
The Wilcoxon signed-rank test was used to assess the statistical significance of differences in neutralizing activities between the two antibodies when reacting against VT1 or VT2.
RESULTS
PCR amplification and cloning of vt1A1 and vt2A1 genes.
The vt1A1 and vt2A1 genes were amplified to the expected sizes, and DNA sequencing confirmed that the selected regions were amplified. Without taking the restriction enzyme sites and the stop codons into account, the amplicon of truncated vt1A1 was a 669-bp fragment between nucleotide positions 398 and 1066 of the vt1 gene. The amplicon of truncated vt2A1 was a 666-bp fragment between nucleotide positions 263 and 928 of the vt2 gene. Amplicons of the entire vt1A1 and vt2A1 subunits had sizes of 798 bp (nucleotide positions 380 to 1180) and 828 bp (nucleotide positions 212 to 1039), respectively. BamHI- and XhoI-digested amplicons were cloned into pGEX-4T-2. The resultant plasmids carrying individual truncated A1 subunits were designated pGEX-vt1A1 and pGEX-vt2A1; plasmids carrying the entire A1 subunit were designated pGEX-vt1A1′ and pGEX-vt2A1′. PCR amplification of these recombinant plasmids confirmed the presence of inserts in these constructs.
Expression and purification of fusion proteins.
Aliquots of the sonicated bacterial cell lysate and pre- and postpurified supernatant were analyzed by SDS-PAGE after IPTG induction. A 46-kDa band was observed only in bacterial cells incubated with IPTG (data not shown), and the presence of the induced proteins in the supernatant suggested that these fusion proteins are soluble. The impurities were completely removed after purification by affinity chromatography (Fig. 2). Expression of the GST fusion protein was only observed in bacterial cells transformed with pGEX-vt1A1 but not with pGEX-vt1A1′ (Fig. 3). Similarly, the fusion protein was produced from pGEX-vt2A1 only. The yields of GST-VT1A1 and GST-VT2A1 were estimated to be 1.03 and 0.5 g per liter of culture, respectively.
FIG. 2.

SDS-PAGE of total bacterial cell lysates and supernatants of E. coli transformed with pGEX-vt1A1 (lanes 1 to 4) and pGEX-vt2A1 (lanes 6 to 9) after IPTG induction. Lanes: 1 and 6, bacterial cell lysates; 2 and 7, cell debris; 3 and 8, supernatants; 4 and 9, supernatants after purification by affinity chromatography; 5, molecular mass marker.
FIG. 3.

SDS-PAGE of total bacterial cell lysates and supernatants of E. coli transformed with pGEX-vt1A1′ (lanes 1 to 4) and pGEX-vt1A1 (lanes 6 to 9) after IPTG induction. Lanes: 1 and 6, bacterial cell lysates after 1.5-h induction; 2 and 7, bacterial cell lysates after 3-h induction; 3 and 8, cell debris; 4 and 9, supernatants of the cell lysates; 5, molecular mass marker.
Generation of polyclonal antibodies.
Purified anti-GST-VT1A1 and anti-GST-VT2A1 were tested for reactivity to the fusion proteins by immunoblot analysis. Both antibodies reacted with their homologous antigens. There was no reaction with the GST protein after removal of anti-GST by affinity chromatography (Fig. 4). The preimmune rabbit serum showed no reactivity with any of the fusion proteins (data not shown).
FIG. 4.

Immunoblot analysis of reactivities of anti-GST-VT1A1 and anti-GST-VT2A1. Fusion proteins were subjected to SDS-PAGE, immobilized onto nylon membranes, and then blotted with prepurified (A) and postpurified (B) anti-GST-VT1A1. Similar procedures were done on prepurified (C) and postpurified (D) anti-GST-VT2A1. Lanes: 1, GST-VT1A1; 2, GST-VT2A1; 3, GST. Molecular sizes (in kilodaltons) are indicated to the right.
Evaluation of the polyclonal antibodies by sandwich ELISA.
The specificities of anti-VT1A1 and anti-VT2A1 were assessed by using three VT1-, five VT2-, and two non-VT-producing strains. When anti-VT1A1 was tested against the culture supernatant from VT1-producing strains, the mean absorbance ± standard deviation was 0.79 ± 0.15; when tested against VT2-producing and non-VT-producing strains, the mean absorbance values were 0.03 ± 0.006 and 0.03 ± 0.01, respectively. When anti-VT2A1 was tested against culture supernatant from VT1-, VT2-, and non-VT-producing strains, the mean absorbance values were 0.52 ± 0.10, 0.57 ± 0.19, and 0.38 ± 0.10, respectively.
In vitro cytotoxicity neutralization of the rabbit antisera.
The highest dilutions of anti-GST-VT1A1 and anti-GST-VT2A1 able to achieve 50% neutralization of their homologous antigens were 1/1,600 and 1/100, respectively. When tested with standard VTEC strains, both antibodies at a dilution twofold higher than the 50% neutralization dose achieved 80 to 100% neutralization of toxins (40 CD50) produced from homologous strains and there was 3 to 5% neutralization activity with toxins from heterologous strains. Culture supernatants from the nontoxigenic strains ATCC 43888 and ATCC 25922 had no cytotoxic effect on the Vero cells. Anti-GST-VT2A1 neutralized VT2e produced from strain S1191. Antiserum raised from rabbits immunized with the GST protein had no neutralizing activities against the toxins.
VTs from 41 animal and 4 human VTEC strains were similarly tested in the neutralization assay (Fig. 5). Culture supernatants were reacted with individual antibodies or mixtures of antibodies and then added to Vero cell monolayers to test for residual cytotoxicity of the culture supernatant. Cell supernatants from all VTEC strains were cytotoxic to Vero cells. For the six VTEC strains harboring only the vt1 gene, the average neutralization percentages ± standard deviations were 76.7% ± 7.9% and 3.6% ± 2.3% when reacted with anti-GST-VT1A1 and anti-GST-VT2A1, respectively. For the 29 VTEC strains (25 from animals and 4 from humans) harboring only the vt2 gene, the neutralization percentages were 1.7% ± 2.3% and 82.5% ± 13.9% when reacted with anti-GST-VT1A1 and anti-GST-VT2A1, respectively. In both cases, the differences were statistically significant (P = 0.03 for VT1 and P < 0.0001 for VT2). The antibodies had high neutralizing activities against their homologous toxins. For the six strains producing both VT1 and VT2, the levels of neutralization were low when reacted with each antibody individually. When a mixture of both antibodies was used, strains producing either or both toxins were neutralized ≥75% (Fig. 5). Culture supernatants from the three porcine VTEC strains harboring vt2e alone or with vt2 were neutralized by anti-GST-VT2A1 and antibody mixture but not by anti-GST-VT1A1. Similar results were observed in the cattle VTEC strains carrying the vt variant gene. In both cases, neutralization activities were lower than those against strains carrying vt2.
FIG. 5.

Neutralization activities of the antisera. VTs produced from human and animal VTEC isolates were reacted with antisera, and the neutralization of cytotoxicity was detected.
DISCUSSION
In this study, we report the use of GST-VT fusion proteins as immunogens for the production of polyclonal antibodies. The manipulations involved were straightforward: a selected region of VT was fused with GST and the fusion protein was purified from the bacterial lysate (5). Specific polyclonal antibodies were then produced from the fusion proteins by animal immunization. The A1 subunit was selected for the construction of the fusion protein, as this is the catalytic domain of the toxin molecule (11). Several investigators have encountered difficulties in expressing a sufficient amount of protein product due to the presence of the signal peptide at the N terminus, which causes rapid transportation of the product out of the bacterial cell (21, 22). Removal of the signal peptide leads to cytoplasmic retention of the toxin, resulting in higher yields of expressed products. In order to obtain a sufficient amount of fusion protein in soluble form, we have excluded the signal peptide at the N terminus and the hydrophobic transmembrane region at the C terminus during the preparation of the constructs. We showed that upon IPTG induction, bacterial cells transformed with these constructs, but not with constructs carrying entire A1 subunits, expressed large amounts of soluble fusion proteins.
As VT1A1 and VT2A1 were without the transmembrane region, which is required for cytotoxicity (11, 23), the ability of the fusion proteins to produce neutralizing antibodies needed confirmation. Our results demonstrated that the antibodies produced had high levels of neutralization against their homologous native VTs. The titer of the anti-GST-VT1A1 antiserum was higher than that of anti-GST-VT2A1. This may be due to differences in immunogenicity for the rabbits used or degradation of the fusion protein after injection into the animal. Cross neutralization between VT1 and VT2 was not evident. However, anti-GST-VT2A1 was able to neutralize VT2 variants. The cross neutralization between VT2 and VT2 variants is probably due to the high homologies of subunit A between these variants (15). This can be considered an advantage as coverage of the antibody activity is extended to the VT2 variants. When animal and human VTEC strains were tested, neutralization results were consistent with those of the vt1 and vt2 genotypes. Attempts have been made to cleave GST from the fusion proteins before immunization; however, the yields of end products after thrombin cleavage were very low. The entire fusion proteins were subsequently used for animal injections, and anti-GST was also present in the antisera. Our results showed that antiserum raised from rabbits immunized with GST did not neutralize toxins produced from any of the test or standard strains, indicating that anti-GST did not interfere with the neutralization assay. We have tried evaluating the polyclonal antibodies by using ELISA and found that the polyclonal anti-VT1A1 detected VT1 specifically. However, nonspecific reactivity occurred in the polyclonal anti-VT2A1.
VTs have an important role in the pathogenesis of hemolytic-uremic syndrome (17). Molecular methods for the detection of vt genes have been established. However, certain strains carrying vt genes may be nontoxigenic or produce low levels of toxin (25) and the detection of VT production in bacterial isolates by specific antibodies is of ultimate importance. ELISA-based kits such as Premier EHEC (Meridian Diagnostics) and Ridascreen Verotoxin (R-Biopharm GmbH) have been developed for VT detection by using monoclonal antibodies, but false-positive results have been documented in these systems (3). The polyclonal antibodies produced from the GST-VT fusion proteins are specific and useful alternatives for the detection and differentiation of VT1 and VT2. In conclusion, we showed that avoidance of the signal peptide and the hydrophobic transmembrane regions in vt1A1 and vt2A1 sequences resulted in hyperexpression of GST-VT fusion proteins. We also found that these fusion proteins elicited specific neutralizing polyclonal antibodies in rabbits. The availability of these antibodies and the purified VT antigens allows for the development of reliable systems for immunological diagnosis of VTEC infections. The strategy presented here can be a paradigm applied to other systems for the production of fusion proteins and antibodies for diagnostic and therapeutic purposes.
Acknowledgments
We thank Wong Ka-wing for excellent technical assistance in the preparation of the fusion proteins and N. A. Strockbine from the Centers for Disease Control and Prevention, Atlanta, Ga., for providing the standard VTEC strains. The cooperation of the government abattoir in the isolation of VTEC strains is also gratefully acknowledged.
This work was supported by a grant from the Hong Kong Research Grants Council (HKU 7314/97 M) and a SPACE research grant award (21386308.03982.70300.420.01).
REFERENCES
- 1.Agbodaze, D. 1999. Verocytotoxins (Shiga-like toxins) produced by Escherichia coli: a minireview of their classification, clinical presentations and management of a heterogeneous family of cytotoxins. Comp. Immunol. Microbiol. Infect. Dis. 22:221-230. [DOI] [PubMed] [Google Scholar]
- 2.Austin, P. R., P. E. Jablonski, G. A. Bohach, A. K. Dunker, and C. J. Hovde. 1994. Evidence that the A2 fragment of Shiga-like toxin type I is required for holotoxin integrity. Infect. Immun. 62:1768-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beutin, L., S. Zimmermann, and K. Gleier. 1996. Pseudomonas aeruginosa can cause false-positive identification of verotoxin (Shiga-like toxin) production by a commercial enzyme immune assay system for the detection of Shiga-like toxins (SLTs). Infection 24:267-268. [DOI] [PubMed] [Google Scholar]
- 4.Bielaszewska, M., I. Clarke, M. A. Karmali, and M. Petric. 1997. Localization of intravenously administered verocytotoxins (Shiga-like toxins) 1 and 2 in rabbits immunized with homologous and heterologous toxoids and toxin subunits. Infect. Immun. 65:2509-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calderwood, S. B., D. W. Acheson, M. B. Goldberg, S. A. Boyko, and A. Donohue-Rolfe. 1990. A system for production and rapid purification of large amounts of the Shiga toxin/Shiga-like toxin I B subunit. Infect. Immun. 58:2977-2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calderwood, S. B., F. Auclair, A. Donohue-Rolfe, G. T. Keusch, and J. J. Mekalanos. 1987. Nucleotide sequence of the Shiga-like toxin genes of Escherichia coli. Proc. Natl. Acad. Sci. USA 84:4364-4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fraser, M. E., M. M. Chernaia, Y. V. Kozlov, and M. N. James. 1994. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 A resolution. Nat. Struct. Biol. 1:59-64. [DOI] [PubMed] [Google Scholar]
- 8.Garred, O., B. van Deurs, and K. Sandvig. 1995. Furin-induced cleavage and activation of Shiga toxin. J. Biol. Chem. 270:10817-10821. [DOI] [PubMed] [Google Scholar]
- 9.Gentry, M. K., and J. M. Dalrymple. 1980. Quantitative microtiter cytotoxicity assay for Shigella toxin. J. Clin. Microbiol. 12:361-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunzer, F., and H. Karch. 1993. Expression of A and B subunits of Shiga-like toxin II as fusions with glutathione S-transferase and their potential for use in seroepidemiology. J. Clin. Microbiol. 31:2604-2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haddad, J. E., A. Y. al-Jaufy, and M. P. Jackson. 1993. Minimum domain of the Shiga toxin A subunit required for enzymatic activity. J. Bacteriol. 175:4970-4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inward, C. D., J. Williams, I. Chant, J. Crocker, D. V. Milford, P. E. Rose, and C. M. Taylor. 1995. Verocytotoxin-1 induces apoptosis in Vero cells. J. Infect. 30:213-218. [DOI] [PubMed] [Google Scholar]
- 13.Leung, P. H. M., W. C. Yam, W. W. S. Ng, and J. M. S. Peiris. 2001. The prevalence and characterization of verotoxin-producing Escherichia coli isolated from cattle and pigs in an abattoir in Hong Kong. Epidemiol. Infect. 126:173-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lord, J. M., and L. M. Roberts. 1998. Toxin entry: retrograde transport through the secretory pathway. J. Cell Biol. 140:733-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melton-Celsa, A. R., and A. D. O'Brien. 1998. Structure, biology, and relative toxicity of Shiga toxin family members for cells and animals, p. 121-128. In J. B. Kaper and A. D. O'Brien (ed.), Escherichia coli O157:H7 and other Shiga toxin-producing E. coli strains. American Society for Microbiology, Washington, D.C.
- 16.Nakao, H., N. Kiyokawa, J. Fujimoto, S. Yamasaki, and T. Takeda. 1999. Monoclonal antibody to Shiga toxin 2 which blocks receptor binding and neutralizes cytotoxicity. Infect. Immun. 67:5717-5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paton, J. C., and A. W. Paton. 1998. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev. 11:450-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Persson, B., and P. Argos. 1994. Prediction of transmembrane segments in proteins utilising multiple sequence alignments. J. Mol. Biol. 237:182-192. [DOI] [PubMed] [Google Scholar]
- 19.Saxena, S. K., A. D. O'Brien, and E. J. Ackerman. 1989. Shiga toxin, Shiga-like toxin II variant, and ricin are all single-site RNA N-glycosidases of 28S RNA when microinjected into Xenopus oocytes. J. Biol. Chem. 264:596-601. [PubMed] [Google Scholar]
- 20.Schmitt, C. K., M. L. McKee, and A. D. O'Brien. 1991. Two copies of Shiga-like toxin II-related genes common in enterohemorrhagic Escherichia coli strains are responsible for the antigenic heterogeneity of the O157:H− strain E32511. Infect. Immun. 59:1065-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skinner, L. M., and M. P. Jackson. 1998. Inhibition of prokaryotic translation by the Shiga toxin enzymatic subunit. Microb. Pathog. 24:117-122. [DOI] [PubMed] [Google Scholar]
- 22.Suh, J. K., C. J. Hovde, and J. D. Robertus. 1998. Shiga toxin attacks bacterial ribosomes as effectively as eucaryotic ribosomes. Biochemistry 37:9394-9398. [DOI] [PubMed] [Google Scholar]
- 23.Suhan, M. L., and C. J. Hovde. 1998. Disruption of an internal membrane-spanning region in Shiga toxin 1 reduces cytotoxicity. Infect. Immun. 66:5252-5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong, S. S. Y., W. C. Yam, P. H. M. Leung, P. C. Y. Woo, and K. Y. Yuen. 1998. Verocytotoxin-producing Escherichia coli infection: the Hong Kong experience. J. Gastroenterol. Hepatol. 13(Suppl.):S289-S293. [DOI] [PubMed] [Google Scholar]
- 25.Yam, W. C., D. N. Tsang, T. L. Que, M. Peiris, W. H. Seto, and K. Y. Yuen. 1998. A unique strain of Escherichia coli O157:H7 that produces low verocytotoxin levels not detected by use of a commercial enzyme immunoassay kit. Clin. Infect. Dis. 27:905-906. [DOI] [PubMed] [Google Scholar]