

Abstract
Experimental infection with mouse cytomegalovirus (MCMV) has been used to elucidate the intricate host-pathogen mechanisms that determine innate resistance to infection. Linkage analyses in F2 progeny from MCMV-resistant MA/My (H2k) and MCMV-susceptible BALB/c (H2d) and BALB.K (H2k) mouse strains indicated that only the combination of alleles encoded by a gene in the Klra (also called Ly49) cluster on chromosome 6, and one in the major histocompatibility complex (H2) on chromosome 17, is associated with virus resistance. We found that natural killer cell–activating receptor Ly49P specifically recognized MCMV-infected cells, dependent on the presence of the H2k haplotype. This binding was blocked using antibodies to H-2Dk but not antibodies to H-2Kk. These results are suggestive of a new natural killer cell mechanism implicated in MCMV resistance, which depends on the functional interaction of the Ly49P receptor and the major histocompatibility complex class I molecule H-2Dk on MCMV-infected cells.
Worldwide, seven of ten individuals are infected with human cyto-megalovirus1, which causes severe and fatal disease in neonates and immunocompromised individuals2–4. The crucial role of natural killer (NK) cells in the control of this infection has been extensively documented5,6. NK cells are important in the innate response to a variety of viral infections through lysis of infected target cells and production of an array of cytokines6. MCMV infections share many pathophysiological aspects with the human disease, providing an excellent model for elucidating the role of NK cells in the host response to viral infection. In addition, different inbred strains of mice vary markedly in susceptibility to MCMV, enabling a genetic approach to the mapping of susceptibility traits and to the identification of key genes governing NK cell function.
H2 genes in the major histocompatibility complex (MHC) and non-MHC genes determine resistance or susceptibility to MCMV in inbred strains of mice7,8. The H2 complex influences susceptibility to the lethal effect of MCMV7. Strains such as C3H and CBA carry the protective H2k haplotype, whereas mice with the H2b (129) or H2d (BALB/c) haplotypes are more susceptible; susceptibility, in this case, is inherited as a dominant trait. At low-dose infection, in mouse strains of the C57BL/6 genetic background, H2-determined susceptibility is over-ridden by a dominant allele at the Cmv1 locus, which acts early after infection at the level of host NK cells9,10.
The Cmv1 locus has two alleles in inbred mice: a dominant resistance allele, Cmv1r, and a recessive susceptibility allele, Cmv1s. Mouse strains possessing Cmv1r are five times more resistant to lethal MCMV infection and their viral titers in the spleen are 1,000–10,000 times lower than those of mouse strains possessing Cmv1s (ref. 9). Cmv1 maps to distal mouse chromosome 6, in a region known as the NK cell gene complex (NKC)11,12 because it encompasses clusters of genes and gene families of C-lectin-like–type receptors related to NK cell functions13,14. Among these are Klrk1 (also called Nkg2d) and a group of more than 20 genes collectively known as Klra genes13–15. Recognition of classical MHC class I molecules by inhibitory Ly49 receptors is thought to provide a dominant inhibitory signal to NK cells against self in normal conditions16.
Cmv1r encodes the activating receptor Ly49H17–20, which recognizes MCMV-infected cells by a direct interaction with the m157 MCMV gene product17,18. Haplotype studies using genetic markers in the vicinity of Cmv1 established that although the Cmv1r haplotype is unique to C57BL/6 strains, there are two independent origins for MCMV susceptibility in mice. On the basis of haplotype relatedness, MCMV-susceptible strains of mice were clustered into two groups, with mice similar to strain 129P3 (hereafter called 129) forming one subset and mice similar to strain BALB/c forming a second, unrelated subset19.
The MCMV-resistant strain MA/My12 lacks Klra8 (also called Ly49h) mRNA and protein (M.P. Desrosiers, S. Girard Adam, A. Kiel-czewska and S.M. Vidal, unpublished data). In addition, depletion of MA/My NK cells with monoclonal antibody to NK1.1 abolished this resistance12, suggesting the presence of another type of Cmv1r allele in this mouse strain. Here we show by haplotype analysis that the resistant strain MA/My is genetically divergent from the resistant strain C57BL/6 in the region of Cmv1 and highly related to the MCMV-susceptible strains FVB/N (hereafter called FVB) and 129. Linkage analysis of the resistance phenotype indicated that MA/My mice possess a key determinant of resistance to MCMV, which maps to the NKC. Genetic and molecular analysis of this phenotype uncovered a new mechanism of host resistance against MCMV infection that involves interaction between the MA/My-encoded activating receptor Ly49P on NK cells and the MHC class I molecule H-2Dk on virus-infected cells.
RESULTS
The MA/My NKC haplotype
To understand the molecular basis of resistance in the absence of Ly49H, we characterized the MA/My Cmv1 region by haplotype analysis. We used 30 informative markers, either clustered in the Klra region or distributed in the NKC genomic domain and covering a physical distance of 5 Mb (Fig. 1). Haplotype analysis (Fig. 1b) showed that MA/My is genetically distinct from C57BL/6 in the Cmv1 region (16 of 30 markers in common). In particular, markers in the Klra region were polymorphic between these two strains, indicating the presence of a different Klra gene repertoire (7 of 10 different markers). Conversely, the MA/My haplotype was more similar to that of the MCMV-susceptible strains 129 and FVB, particularly in the Klra region (8 of 10 identical markers), indicating that this allele sharing might be the result of a common origin. Although the resistant MA/My strain and the susceptible FVB and 129 strains have a similar Cmv1 haplotype, the difference in resistance to MCMV infection between the three strains could be explained, at least in part, by their H2 haplotype, also implicated in MCMV infection. In fact, MA/My carries the protective H2k haplotype, whereas 129 and FVB have the susceptible H2b and H2q haplotypes, respectively.
Figure 1.
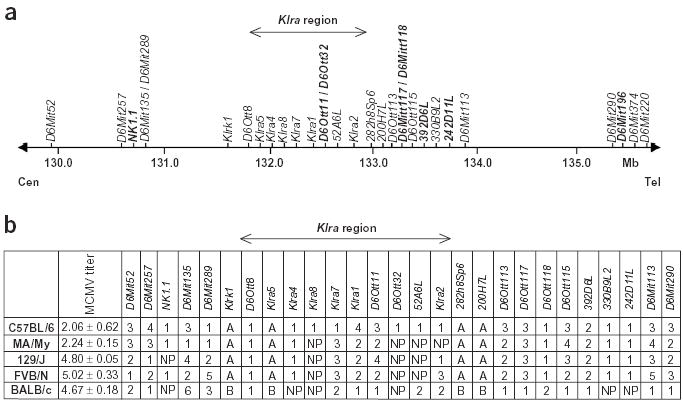
Haplotype mapping on chromosome 6 in the vicinity of Cmv1. (a) Physical distance of markers used for haplotype mapping and genetic analysis. Markers in bold were arbitrarily positioned between well-defined markers. (b) Haplotype map of 30 polymorphic markers. Numbers indicate the relative size of PCR products (1 being the shortest) for microsatellite markers; letters indicate PCR–restriction-fragment length polymorphism markers. NP, no product.
Genetic analysis of MCMV resistance in MA/My
We investigated the mode of inheritance of the MA/My resistance trait in F1 and F2 progeny from crosses between MCMV-resistant MA/My and MCMV-susceptible BALB.K mice. Both strains share the same H2k haplotype. The read-out phenotype was spleen viral load 3 d after infection measured in log10 plaque-forming units (PFUs) recovered. (MA/My × BALB.K) F1 mice had a mean viral titer (× 1 s.d.) of 3.35 ± 0.13 log10 PFUs, intermediate between the viral titers of MA/My (2.24 ± 0.62, P < 2.8 × 10–6) and BALB.K (4.43 ± 0.17, P < 2.3 × 10−6) mice (Fig. 2a), suggesting that resistance might be controlled by codominant alleles. In addition, the phenotypic distribution of the F2 progeny was discontinuous, with three means at 2.5, 3.3 and 4.0 log10 PFUs (Fig. 2b), consistent with the presence of a major codominant gene effect.
Figure 2.
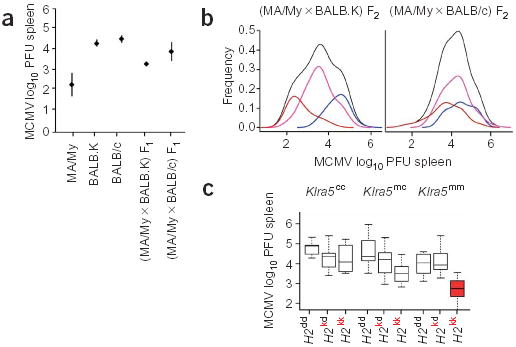
Genetic analysis of MCMV resistance in MA/My mice. (a) MCMV viral titers in the spleen of MA/My (n = 16), BALB.K (n = 25), BALB/c (n = 15), (MA/My × BALB.K) F1 (n = 6) and (MA/My × BALB/c) F1 (n = 10) mice. Bars show standard deviation. (b) Empirical density (black line) of phenotypes of 226 (MA/My × BALB.K) F2 mice (left) and 119 (MA/My × BALB/c) F2 mice (right). Colored lines indicate the empirical density by genotypes at Klra5. In blue, distribution of homozygous BALB.K (n = 55) or BALB/c (n = 30) genotypes; in pink, distribution of the heterozygous genotype (MA/My × BALB.K, n = 118, or MA/My × BALB/c, n = 58); in brown, distribution of the homozygous MA/My genotypes (left, n = 53; right, n = 31) (c) Box plots of log10 PFU counts of H2-Klra5 genotypes showing the combined effects of H2 (MA/My, H2k; BALB/c, H2d) and NKC (MA/My, Klra5m; BALB/c, Klra5c) loci on spleen viral titers of 119 (MA/My × BALB/c) F2 progeny. The median and interquartile range are shown. Whiskers extend to the most extreme value, which is less than 1.5 times the interquartile range. Solid dots denote outliers.
Although MA/My mice do not express Ly49H, we did not exclude the possibility that another member of the Klra gene family or another NK cell receptor gene could contribute to MCMV resistance in MA/My. Therefore, we genotyped Klra5 (also called Ly49e) and five additional markers spanning a total of 14 Mb in individual F2 mice. All these markers were linked to the NKC and were common to both parental strains of mice (Fig. 1a and Table 1). On the basis of the empirical frequencies of the genotypic classes for Klra5, mice homozygous with respect to BALB.K alleles clustered toward the susceptible end of the distribution, and mice homozygous with respect to the MA/My alleles (Klra5m) clustered toward the resistant end of the phenotypic spectrum (Fig. 2b). The logarithm of odds (lod) scores for the MCMV-resistance trait showed the strongest association with marker genes Klrk1 and Klra5 (Table 1), with a lod score of 22.7 (P < 1.0 × 10−6) under an additive mode of inheritance. The lod scores at the flanking markers D6MIT135 (proximal) and D6MIT291 (distal) were 20.8 and 18.7, respectively (Table 1). This linkage analysis located a new resistance locus that we named Cmv3. The one-lod support interval suggests that Cmv3 is located in a region of ~8 Mb, indicating that Klrk1 and Klra genes are good candidates for underlying the resistance trait. But analysis of MA/My and BALB/c Klrk1 cDNA sequences indicated that they were identical and equivalently expressed in these mouse strains, excluding Klrk1 as a potential resistance gene (A. Kielczewska and S.M. Vidal, unpublished observations).
Table 1.
Linkage analysis in (MA/My × BALB.K) F2 progeny
| QTL analysis
|
|||
|---|---|---|---|
| Locus | Mba | lodb | Variance (%)c |
| D6MIT300 | 123.6 | 18.6 | 32 |
| D6MIT52 | 128.4 | 20.9 | 35 |
| D6MIT135 | 129.5 | 20.8 | 35 |
| Klrk1 | 130.3 | 22.7 | 37 |
| Klra5 | 130.6 | 22.7 | 37 |
| D6MIT291 | 137.5 | 18.7 | 32 |
The analysis was done on 226 (MA/My × BALB.K) F2 mice.
The position of each marker obtained from Ensembl.
The lod ratio was used to measure the significance of each potential association of the trait with a locus.
The amount of total trait variance explained by a quantitative trait locus (QTL) is expressed as a percentage.
H2 and Klra5 interaction is associated with MCMV resistance
To investigate a possible role of H2 in MA/My resistance, we studied the segregation of MCMV resistance in F1 and F2 crosses between MCMV-resistant MA/My (H2k) and MCMV-susceptible BALB/c (H2d) mice. Viral load in spleen 3 d after infection was used as the read-out phenotype. The mean value for the log10 of the PFUs recovered from the spleen of the (MA/My × BALB/c) F1 population was 4.0 ±0.5 (Fig. 2a). This value was closer to that of the susceptible parental strain BALB/c (4.67 ± 0.19) than to that of the resistant parental strain MA/My (2.24 ±0.62; Fig. 2a), suggesting that MCMV resistance segregated as a recessive trait in this cross. Frequencies of viral titers in the spleen of 119 (MA/My × BALB/c) F2 mice showed a distribution that was suggestive of a more complex genetic control than in the previous cross, as shown by the clustering of the modes of Klra5 genotype distributions (Fig. 2b).
To evaluate further the contribution of H2 and NKC genes to MCMV resistance, we used the markers IAA1 (located in H2) and Klra5 (located in the NKC) as candidate genes to carry out an analysis of variance on viral spleen loads of 119 (MA/My × BALB/c) F2 mice. The four additive-recessive combinations of H2-NKC models with and without an interaction term were fitted. All models that included H2, NKC and their interaction fitted consistently better. The most parsimonious model had a joint lod score of 9.3 and accounted for 30% of the phenotypic variation, consistent with the level of variation explained by Cmv3 (Table 2). The H2 recessive effect was almost twice as strong as the other two components combined. But less parsimonious models with other modes of inheritance were also highly significant. This might reflect a complex pattern of gene interactions with different effects on different H2-NKC haplotype combinations (Fig. 2c). The evidence for interaction was consistent across all the models (data not shown). The strength of the additive effect of Klra5 (or other genes closely linked to it) was strongest in the presence of the H2kk background (Fig. 2c). Only mice homozygous with respect to Klra5m and H2k alleles were fully resistant to MCMV. Mice showed average log10 PFUs of 2.71 ± 0.35, in the range of MA/My controls (Fig. 2a,c), indicating that homozygosity with respect to the two loci was required for MCMV resistance.
Table 2.
Analysis of variance of log10 PFUs in spleen in (MA/My × BALB/c) F2 progeny
| Sum of squares | F value | P value | Partial η2 | |
|---|---|---|---|---|
| Model | 20.80 | 16.53 | <1.0 × 10−6 | 0.301 |
| NKC additive | 1.66 | 3.95 | 4.9 × 10−2 | 0.033 |
| H2 recessive | 12.11 | 28.87 | <1.0 × 10−6 | 0.201 |
| Interaction | 4.60 | 10.97 | 1.2 × 10−3 | 0.087 |
The analysis was done on 119 (MA/My × BALB/c) F2 mice. P values are from 1,000,000 bootstrapped samples.
Activating receptor Ly49P recognizes MCMV-infected cells
In accordance with haplotype analysis, we found that Ly49 receptors cloned from the MA/My strain were highly homologous to those of MCMV-susceptible strain 129 (ref. 20), suggesting that the two strains share a Klra gene repertoire. The Klra9 (also called Ly49i) cDNA sequence from MA/My was identical to that of the 129 strain. The inhibitory receptor Ly49I129 binds to the m157 viral protein21, suggesting this as a possible susceptibility mechanism. Ten percent of MA/My NK cells stained with the m157-Ig fusion protein21 (Table 3), excluding a hypothetical lack of Ly49I-m157 interaction as a means of MCMV resistance. The activating receptors, including Ly49P, Ly49R and Ly49U, shared 98–100% identity at the nucleotide level with their 129 counterparts (data not shown). These receptors were expressed on MA/My NK cells (Supplementary Fig. 1 online) and are good candidates for MCMV resistance in this strain through direct recognition of infected cells.
Table 3.
Characteristics of mouse strains used in this study
| Mouse strain | H2 haplotype | MCMV | Ly49H | Ly49I isoform | Ly49P | m157-Ig–reactive NK cells* |
|---|---|---|---|---|---|---|
| C57BL/6 | H2b | Resistant | + | Ly49IC57BL/6 | − | 60% |
| BALB/c | H2d | Susceptible | − | Ly49IBALB/c | − | 0% |
| BALB.K | H2k | Susceptible | − | Ly49IBALB.K | − | 0% |
| FVB/N | H2q | Susceptible | − | Ly49I129 | + | 10% |
| 129P3 | H2b | Susceptible | − | Ly49I129 | + | 10% |
| MA/My | H2k | Resistant | − | Ly49I129 | + | 10% |
The Ly49I129 isoform binds to the m157-Ig fusion protein. Although the presence of Ly49H receptor is sufficient to provide MCMV resistance, Ly49P must be in combination with H2k haplotype.
To test this hypothesis, we used a mouse T-cell hybridoma carrying the DAP12 adaptor signaling protein and a reporter gene NFAT-GFP and transfected it with cDNAs encoding Ly49P, Ly49R or Ly49U amplified from MA/My (Fig. 3). Growing the reporter cells on culture plates coated with monoclonal antibodies specific to the transfected receptors induced expression of green fluorescent protein (GFP), confirming that the reporters were functional (Fig. 3c). When Ly49 reporter cells were cultured with MCMV-infected mouse embryo fibroblasts (MEFs) from MA/My, only Ly49P reporters turned green, indicating the presence of a Ly49P ligand (Fig. 3a). This was not observed in the presence of uninfected MEF cells (Fig. 3a). Stimulation of NFAT-GFP was also absent when Ly49P reporter cells were cultured with Ba/F3 mouse pro-B cells expressing the MCMV-encoded ligand of Ly49HC57BL/6 receptor, m157 (Fig. 3b), indicating that Ly49PMA/My recognized MCMV-infected cells by a mechanism that did not involve m157 detection.
Figure 3.
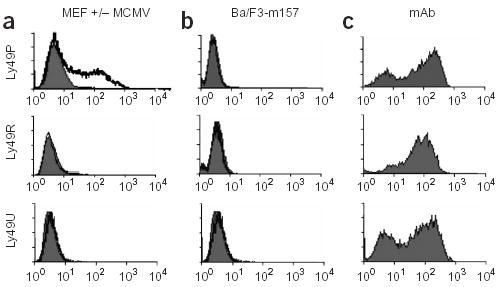
Activation of Ly49P reporter cells by MCMV-infected cells. NFAT-GFP expression by reporter cells carrying individual Ly49 receptors was measured and GFP expression was analyzed by flow cytometry following culture of reporter cells: (a) with uninfected (filled histograms) or MCMV-infected (hollow histograms) MA/My MEF cells; (b) with Ba/F3 cells (filled histograms) or Ba/F3 cells expressing the MCMV protein, m157 (hollow histograms); (c) on plastic plates coated with monoclonal antibodies (mAb) to Ly49H-Ly49U, Ly49P and Ly49R.
Ly49P-MCMV specific activation depends on H-2Dk
To address the role of H2 in Ly49P recognition of infected cells, we monitored stimulation of reporter cells transfected with Ly49P or with Ly49H (as a positive control) after culture with MCMV-infected MEF cells originating from mouse strains with different H2 haplotypes (Fig. 4). Ly49P reporter cells did not respond when the infected target cells originated from BALB/c (H2d), FVB (H2q) or C57BL/6 (H2b) mice. In contrast, Ly49P receptors were activated in the presence of infected cells from MA/My (H2k) or BALB.K (H2k) mice (Fig. 4), indicative of a functional interaction between the H2 molecule and Ly49P receptor in the presence of MCMV infection.
Figure 4.
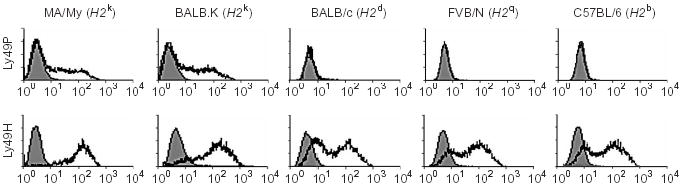
Ly49P recognition of MCMV-infected cells is target MHC-dependent. NFAT-GFP stimulation by Ly49P and Ly49H reporter cells was determined after culture with MEF cells from MA/My (H2k), BALB.K (H2k), BALB/c (H2d) and FVB (H2q) inbred mouse strains and TpnT fibroblasts (H2b) in the presence (hollow histograms) and absence (filled histograms) of MCMV infection. Ly49H reporter cells, which directly recognize the m157 MCMV protein even in the total absence of H2 molecules on the MCMV-infected cells17, were equally activated when cultured with infected target cells from all of the above strains.
Because H2 molecules are well-documented ligands of Ly49 receptors, we had to verify that Ly49P activation in reporter cells was MCMV-specific and not a result of an over-expression of MHC class I during infection. Whereas MEF cells that were cultured in the presence of recombinant interferon-β (IFN-β) had increased H2-Dk expression, as verified by surface staining (data not shown), they did not activate Ly49P reporters (Fig. 5a). Similarly, MEF cells infected with mouse herpesvirus 68 (MHV68) also did not activate Ly49P reporters, confirming the specificity of Ly49P for MCMV (Fig. 5a).
Figure 5.
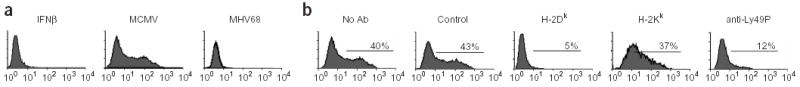
Characterization of Ly49P interaction with an infected cell. (a) Ly49P activation is MCMV-specific. Ly49P reporter cell activation after culture with target MEF cells from BALB.K mice. Target BALB.K MEFs were either treated with IFNβ or infected with MCMV or mouse herpesvirus 68 (MHV68). (b) Ly49P recognition of MCMV-infected cells is blocked by antibodies to H-2Dk and Ly49P (YE1/48) but not by antibodies to H-2Kk. The antibody to Ly49P was used to block recognition of the infected cells by the Ly49P receptor to ensure its proper expression. NFAT-GFP stimulation of Ly49P reporter cells was measured after culture with MCMV-infected BALB.K MEF cells in the absence of blocking antibody or in the presence of isotype control antibody or antibodies to H-2Dk, H-2Kk or Ly49P.
The previous results suggested that there is an H2k product (or products) recognized by Ly49P receptors. We determined that a monoclonal antibody to H-2Dk (15-5-5S) efficiently blocked the activation of Ly49P reporter cells by BALB.K MCMV-infected cells (H2k; Fig. 5b). In contrast, neither a monoclonal antibody recognizing H-2Kk (36-7-5) nor an isotype-matched control antibody prevented Ly49P reporter activation. Furthermore, the presence of antibody against Ly49P, YE1/48, resulted in blocking of GFP production, showing that the reporter cell activation was Ly49P-specific. These results, together with the observation that Ly49R or Ly49U reporters were not activated (Fig. 3), support the role of Ly49P in mediating recognition of H-2Dk on MCMV-infected cells.
DISCUSSION
In C57BL/6 mice, innate resistance to MCMV is mediated by the activating receptor Ly49H. We report here that MCMV resistance in MA/My is associated with Cmv3, a new NKC resistance locus linked to the Klra gene cluster. A fundamental difference between C57BL/6 and MA/My resistance alleles resides in their sensitivity to the H2 background. Whereas the Cmv1r-Ly49H phenotype is independent of H2 environment, results from crosses between MA/My and BALB/c show that only a combination of homozygous Cmv3m and H2k alleles is associated with MCMV resistance, indicative of different mechanisms of action. BALB/c has the H2d haplotype, and many of the inhibitory Ly49 receptors recognize H2d gene products as ligands. Therefore, it is possible that H2d contributes to MCMV susceptibility of BALB/c mice because many of their NK cells possess inhibitory Ly49 receptors, which bind to the products of these H2 alleles. Heterozygous H2kd mice are also susceptible, suggesting that the H2d haplotype encodes additional ligands for inhibitory receptors in MA/My, resulting in increased NK cell inhibition22. These potential susceptibility mechanisms would, however, be overridden by the effect of Cmv3m-H2k epistatic interaction.
In the investigation of the candidate genes for MCMV resistance, we provide evidence that Klra16MA/My (also called Ly49pMA/My) is a resistance allele of Cmv3. Klra16 maps to the confidence interval of Cmv3 in the two crosses that we analyzed, totaling more than 690 meioses. Ly49P is associated with MCMV resistance in the H2k strain MA/My but not in the MCMV-susceptible FVB (H2q) and 129 (H2b) mice, even though the Klra16 cDNAs are identical in these mouse strains (A. Makrigiannis, A. Kielczewska, S. Girard Adam, M.-P. Desrosiers and S.M. Vidal, unpublished observations). Moreover, Ly49P seems to be expressed on MA/My NK cells as far as we could establish using the YE1/48 monoclonal antibodies, which were previously used in the characterization of this receptor in 129 and NOD strains23,24. The exact number of Ly49P-positive NK cells cannot be determined owing to cross-reactivity of these antibodies with other Ly49 molecules in these mouse strains.
Finally, testing the candidate genes in a reporter cell assay provides evidence of Ly49P-specific recognition of MCMV-infected cells. From a panel of reporter cells expressing MA/My activating receptors, only the Ly49P reporters expressed GFP when cultured with MCMV-infected cells, indicating the presence of a ligand binding to Ly49P but not the other receptors. In addition, Ly49P recognition of the infected cells depended strictly on the H2 background of the target cell because only H2k haplotype targets stimulated Ly49P, in concordance with the observed genetic interaction between the NKC and H2 loci. Furthermore, we conclude from antibody blocking experiments that H-2Dk in the presence of MCMV infection is a potential ligand for the Ly49P receptor. Together, these results support the idea that Ly49P receptor has a role in MCMV resistance through the NK cell-specific destruction of MCMV-infected cells.
Ly49P receptor activity was previously characterized in two other mouse strains. Using the rat NK cell line RNK-16 transfected with Ly49P from the NOD strain, it was shown that Ly49P induced lysis of susceptible targets23, whereas immunoprecipitation assays showed that Ly49P from 129 precipitated with the adaptor protein DAP12 (ref. 24), confirming the role of Ly49P as an activating receptor. But the killing assay indicated that NOD Ly49P activity was dependent on H-2Dd–bearing targets23, whereas the studies in the 129 strain showed that Ly49P interacted very weakly with H-2Dk and H-2Dd soluble tetramers. None of the previous studies tested the role of these MHC class I molecules in the context of MCMV infection. We present evidence that Ly49P target recognition is strictly dependent on the availability of H-2Dk molecules on MCMV-infected cells. Depletion of MA/My NK cells with the YE1/48 monoclonal antibody before MCMV injection rendered mice susceptible to infection, underscoring the biological relevance of Ly49P in this model.
Activating and inhibitory isoforms of Ly49 MHC class I receptors are highly homologous in their extracellular domains. This probably results from the generation of the activating and inhibitory receptors by gene duplication and gene conversion. In cases where related activating and inhibitory receptors have been shown to bind to the same MHC class I ligand, the activating receptors generally seem to bind with much lower affinity to H2 than do the inhibitory receptors25,26. Our results suggest that Ly49P recognition of H-2Dk in the infected cell is a new mechanism of activating NK cell receptor function, involving specific recognition of infected cells through classical MHC class I ligands. Although the exact nature of the interaction between Ly49P and H-2Dk in MCMV-infected cells remains to be defined, it is plausible that ligation of the H-2Dk molecule depends on the presence of a MCMV-specific peptide. Therefore, although the precise molecular machinery of target recognition is not yet known, our results indicate the possibility of peptide-specific recognition of MCMV by the Ly49P receptor. The requirement for peptide selectivity, previously well described for target-specific recognition by inhibitory MHC class I receptors27,28, and H2 allelic specificity renders the Ly49P–MCMV-infected cell encounter a highly specific mechanism of innate immunity mediated by NK cells, previously thought to be confined to cytotoxic T cells.
Peptide preference in MHC class I recognition by NK cell receptors is well documented for both Ly49 receptors and their functional homologs, human killer cell immunoglobulin receptors (KIR). In the case of the interaction between receptor KIR2DL2 and HLA-Cw3, direct contact between the KIR and the presented peptide occurs. There is less receptor contact to the side chain of the peptide than in T-cell receptor binding; therefore, the interaction is less sensitive to peptide content29. On the other hand, in the case of the Ly49C peptide-selective receptor, crystallographic studies show no contact between the receptor and the peptide. In fact, Ly49C interacts with the MHC class I molecule over a broad region beneath the peptide-binding platform. Dam and colleagues30 proposed that, with Ly49C, certain peptides might exert interactions through the floor of the H-2Kb binding grove, transmitted to the NK cell receptor by β2M. This is a plausible mechanism for the Ly49P recognition of infected cells, resulting in the protection of the host against pathogens. Analogous to the mechanism of ligand recognition by the promiscuous receptor Ly49C, Ly49P may also be able to recognize allosteric effects caused by a virus-encoded peptide. Alternatively, Ly49P may recognize an H2 molecule presenting a host-encoded peptide resulting from MCMV infection. Future studies will be necessary to determine whether recognition is specific for a single peptide or whether it results from a particular allosteric change in the H-2Dk molecule. Furthermore, if the peptide is of viral origin, the MCMV gene product implicated also awaits identification.
Although Ly49 and human KIR receptors are structurally unrelated, the two gene families share certain features, including polymorphism and variation in gene content and a similar pattern of clonal expression on different NK cells31, as recently confirmed by transgenic models of KIR in mice32. Most notably, members of both receptor families recognize MHC class I antigens and use similar mechanisms for signal transduction. In humans, genetic epidemiological studies have identified several combinations of KIR and MHC loci associated with disease outcome, including those reported for human KIR2DL3 and HLA-C protection against hepatitis C33, and human KIR3DS1 and HLA-Bw4 activity delaying the progression of AIDS34. The evidence presented in this study emphasizes the importance of activating MHC class I NK cell receptors in recognition of infection, warranting further investigation of their role in host resistance to pathogens.
METHODS
Mice.
We purchased inbred mouse strains C57BL/6, MA/My, 129P3, FVB/N, BALB/c and BALB.K from the Jackson Laboratory. We produced F1 and F2 progeny from MA/My × BALB.K and MA/My × BALB/c crosses. Mice were kept at the animal facility of the University of Ottawa, in agreement with guidelines and regulations of the Canadian Council of Animal Care.
Virus stocks, mouse infections and virus titration.
We obtained the Smith strain of MCMV from the American Type Culture Collection. We produced virus stocks by salivary gland propagation in 3-week-old BALB/c mice as previously described35. To study susceptibility of mice to MCMV, we infected 6- to 8-week-old mice intraperitoneally with 5 × 103 PFUs MCMV. We determined viral titers on MA/My (n = 16), BALB.K (n = 25), BALB/c (n = 15), C57BL/6 (n = 22), 129 (n = 18), FVB (n = 20), (MA/My × BALB.K) F1 (n = 6), (MA/My × BALB.K) F1 (n = 10), (MA/My × BALB.K) F2 (n = 226) and (MA/My × BALB/c) F2 (n = 119) mice by plaque assay as described35. We collected spleens of mice 3 d after infection, homogenized them and centrifuged them at 500g for 20 min in a refrigerated centrifuge. We plated the supernatants for 1 h on monolayers of BALB/c MEF cells. After 3 d of incubation, we washed, fixed and stained the infected cells and counted MCMV plaques. Viral titers in the spleen are expressed as MCMV log10 PFUs. For antibody-depletion experiments, we injected mice intraperitoneally with 100–400 μg of antibody 2 d before infection.
Cell cultures.
We constructed 2B4 NFAT-GFP reporter cells and m157-transfected mouse Ba/F3 cells as described17 and maintained them in complete RPMI-1640 medium (supplemented with 10% fetal bovine serum, penicillin, streptomycin and glutamine). We prepared MEF cells from strains MA/My, BALB/c, BALB.K, 129 and FVB as previously described35, with modifications. We obtained embryos at 14–16 d of pregnancy, dissected them out of the embryonic sacs and removed livers and heads. We then finely minced the embryonic tissue with two forceps, washed it with phosphate-buffered saline and incubated it at 4 °C with trypsin overnight. The next, we dissociated the cells into a single-cell suspension and plated them on T175 flasks in complete DMEM medium (10% fetal bovine serum, 25 mM HEPES, penicillin, streptomycin and glutamine). TpnT fibroblasts (an SV40-immortalized MEF cell line from C57BL/6 mice) were a gift from A. Hill (Oregon Health Sciences University, Portland, Oregon).
Genotyping.
We extracted genomic DNA from individual mouse tail tips using the alkaline method36. We carried out the haplotype analysis using 30 markers clustered in the Klra region or distributed in the NKC genomic domain. The DNA markers were microsatellites (D6Mit52, D6Mit257, D6Mit135, D6Mit289, D6Ott8, Ly49d, Klra8, Ly49g, Ly49a, D6Ott11, D6Ott32, 52A6L, D6Ott113, D6Ott117, D6Ott118, D6Ott115, 392D6L, 330B9L2, 242D11L, D6Mit13, D6Mit290, D6Mit196, D6Mit374 and D6Mit220) or restriction-fragment length polymorphisms (Klrk1, Klra5, 282h8Sp6 and 200H7L) or were detected by single-strand conformational polymorphism (Ly49b). The oligonucleotide primers and genotyping conditions were as described11,19,37,38. We analyzed NK1.1 expression by staining the NK cells with the PK136 monoclonal antibody (BD Pharmingen) using previously described conditions12. Together, the markers span a physical distance of 5 Mb (D6MIT52–D6MIT220 interval). For the linkage analysis, we genotyped 226 (MA/My × BALB.K) F2 mice and 119 (MA/My × BALB/c) F2 using the Klra5 marker representing the NKC region and IAA1 markers representing the H2 region. To discriminate between the Klra5c (BALB/c, BALB.K) and Klra5m (MA/My) alleles, we carried out amplification with the Klra5 marker followed by digestion with HincII (New England Biolabs) as described13. To distinguish the H2d (BALB/c) and H2k (MA/My, BALB.K) haplotypes, we carried out amplification with IAA1 primers followed by digestion with PstI (New England Biolabs). PCR and enzyme digestion conditions and oligonucleotide sequences for the IAA1 primers were previously reported39. For fine-mapping on chromosome 6, we genotyped (MA/My × BALB.K) F2 mice using the Klrk1 restriction-fragment length polymorphism marker and the microsatellite markers D6MIT300, D6MIT52, D6MIT135 and D6MIT291 (Mouse MapPairs; Research Genetics), spanning 14 Mb. Genotyping with Klrk1 and microsatellite markers was done as previously described11,37.
MA/My Klra cDNA repertoire.
We isolated total RNA from MA/My mice with TRIzol reagent (Life Technologies) and enriched it for poly (A)+ using an Oligotex mRNA Midi kit (Qiagen). We used oligo(dT) primer and SuperScript II polymerase (Life Technologies) for reverse transcription. We amplified Klra transcripts with Advantage cDNA polymerase (Clonetech) by using Klra universal primers40. We purified the PCR product with a QIAEX II Gel Extraction Kit (Qiagen) and directly ligated it into pGEM T-Easy Vector System, in accordance with the manufacturer’s instructions (Promega). We transformed ligation products using competent DH5α bacterial cells (InVitro-Gen) and grew them overnight at 37 °C on nylon membranes on Luria-Bertani broth agar plates. We duplicated the nylon membranes and treated them with 10% SDS buffer for 3 min to obtain optimal lysis, followed by a 5-min denaturing, neutralizing and washing step. We fixed DNA on membranes by ultraviolet cross-linking (70,000 μJ cm−2). The duplicated set of membranes was independently hybridized with ITIM (inhibitory)- and non-ITIM (activating)-labeled (γ32-P-dATP) degenerated primers to discriminate inhibitory and activating Klra transcripts40. We exposed filters on Kodak X-ray films for 16 h and identified positive clones on the transformed plates. We dissolved single positive colonies in water and used them as DNA templates for PCR using M13 universal primers to amplify the cDNA inserts. We purified plasmids from 20 positive clones with a Qiagen mini-kit and sequenced them by using a Licor automated system with SequiTherm EXCEL Kits (LC technology, Epicentre). We carried out DNA analysis of these clones using standard parameters of nucleotide-nucleotide BLAST (blastn) and protein-protein BLAST (blastp) programs. We used the ClustalW program for multiple sequence alignments.
Statistical analysis.
For the 226 (MA/My × BALB.K) F2 mice, the genotypic data of markers on chromosome 6 were associated with their respective MCMV log10 PFU counts in the spleen using the MapManager QTb program41. This program, based on single-marker regression, allows identification of the loci that affect quantitative traits. For the 119 (MA/My × BALB/c) F2 mice, the contribution of Klra5 and H2 alleles to the segregation of the phenotype was estimated under the linear model phenotype = m + NKC + H2 + NKC:H2 + e, where NKC and H2 represent factors that depend on the mode of inheritance proposed, m is the common mean value, NKC:H2 is an interaction term and e is the usual independent normally distributed random deviations. For the additive mode of inheritance, NKC and H2 represent the number of MA/My alleles at each locus. For the recessive mode of inheritance, NKC and H2 are the indicator variables of the homozygous BALB/c and MA/My background, respectively. The four possible additive-recessive combinations of H2-NKC models with and without an interaction term were fitted. We assessed the magnitude of the contribution for each term in the model by its P value, obtained by 1,000,000 bootstrapped samples, and partial η2. Partial η2 is computed as η2 = SSfactor/(SSfactor + SSerror), where SSfactor is the type 3 associated sum of squares with the factor in the analysis of variance table, and SSerror is the sum of squares corresponding to the residual variation42. We carried out statistical and graphical analyses using R software.
Cell reporter assay.
We transduced NFAT-GFP 2B4 T-cell hybridoma reporter cells with cDNAs encoding MA/My Ly49P, Ly49R or Ly49U cloned into BamHI and EcoRI sites of the pMx-puro vector as previously described17. We prepared primary MEF cultures from MA/My, BALB/c, FVB or BALB.K mice, as described above, infected with MCMV (Smith strain) at a multiplicity of infection of 1.0 for 22 h and used these as stimulator cells for the reporter assay. We cultured reporter cells overnight with the stimulator cells in 48-well plates in a 1:1 ratio. Activation of the NFAT-GFP reporter gene was detected by flow cytometric analysis and analyzed on a FACSCalibur flow cytometer with Cell Quest software (Becton Dickinson) and WinMDI (J. Trotter, Scripps Research Institute). In antibody blocking assays, reporter or stimulator cells were preincubated with a saturating concentration (>50 μg ml−1) of an isotype-matched control immunoglobulin, 15-5-5S (anti-H-2Dk) or 16-3-22S (anti-H-2Kk) antibodies. We detected surface expression of H-2Kk and H-2Dk by using fluorescein isothiocyanate–conjugated 16-3-22S and 15-5-5S (anti-H-2Dk) antibodies (BD PharMingen) and measured fluorescence by flow cytometry.
URLs.
BLAST programs are available at http://www.ncbi.nlm.nih.gov/BLAST/. ClustalW is available at http://www.ebi.ac.uk/clustalw/. R software is available at http://www.R-project.org/.
GenBank accession numbers.
Klra18MA/My mRNA, AY971805; Klra21MA/My mRNA, AY971806; Klra16MA/My mRNA, AY971807.
Acknowledgments
We thank S.-H. Lee for the initial characterization of the MA/My strain; F. Takei for providing the YE1/48 antibody for depletion experiments; D. Albert and M.-H. Lacombe for technical support; M. Reuben for editorial assistance; and P. Gros, E. Schurr and M. Fujiwara for critical reading of the manuscript. This work was supported by grants from the Canadian Institutes of Health Research and the Canadian Genetic Diseases Network (Network of Centres of Excellence program). A.K., S.G.A. and J.-C.L.-O. were Canadian Institutes of Health Research Training Fellows in Infectious Diseases and Autoimmunity. A.K. was also supported by a McGill Majors Fellowship. M.B.L., T.P. and L.L.L. are supported by a grant from the US National Institutes of Health. L.L.L. is an American Cancer Society Research Professor. S.M.V. is a Canada Research Chair.
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
COMPETING INTERESTS STATEMENT The authors declare that they have no competing financial interests.
References
- 1.Britt, W.J. & Alford, C.A. Cytomegalovirus. in Fields Virology (eds. Fields, B.N., Knipe, D.M. & Howley, P.M.) 2493–2524 (Lippincott-Raven, Philadelphia, 1996).
- 2.Reusser P. Management of viral infections in immunocompromised cancer patients. Swiss Med Wkly. 2002;132:374–378. doi: 10.4414/smw.2002.09875. [DOI] [PubMed] [Google Scholar]
- 3.Soderberg-Naucler C, Emery VC. Viral infections and their impact on chronic renal allograft dysfunction. Transplantation. 2001;71:SS24–SS30. [PubMed] [Google Scholar]
- 4.Trincado DE, Rawlinson WD. Congenital and perinatal infections with cytomega-lovirus. J Paediatr Child Health. 2001;37:187–192. doi: 10.1046/j.1440-1754.2001.00645.x. [DOI] [PubMed] [Google Scholar]
- 5.Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. 1989;320:1731–1735. doi: 10.1056/NEJM198906293202605. [DOI] [PubMed] [Google Scholar]
- 6.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 7.Chalmer JE, Mackenzie JS, Stanley NF. Resistance to murine cytomegalovirus linked to the major histocompatibility complex of the mouse. J Gen Virol. 1977;37:107–114. doi: 10.1099/0022-1317-37-1-107. [DOI] [PubMed] [Google Scholar]
- 8.Grundy JE, Mackenzie JS, Stanley NF. Influence of H-2 and non-H-2 genes on resistance to murine cytomegalovirus infection. Infect Immun. 1981;32:277–286. doi: 10.1128/iai.32.1.277-286.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scalzo AA, Fitzgerald NA, Simmons A, La Vista AB, Shellam GR. Cmv-1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J Exp Med. 1990;171:1469–1483. doi: 10.1084/jem.171.5.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scalzo AA, et al. The effect of the Cmv-1 resistance gene, which is linked to the natural killer cell gene complex, is mediated by natural killer cells. J Immunol. 1992;149:581–589. [PubMed] [Google Scholar]
- 11.Depatie C, Muise E, Lepage P, Gros P, Vidal SM. High-resolution linkage map in the proximity of the host resistance locus Cmv1. Genomics. 1997;39:154–163. doi: 10.1006/geno.1996.4498. [DOI] [PubMed] [Google Scholar]
- 12.Scalzo AA, et al. Genetic mapping of Cmv1 in the region of mouse chromosome 6 encoding the NK gene complex-associated loci Ly49 and musNKR-P1. Genomics. 1995;27:435–441. doi: 10.1006/geno.1995.1074. [DOI] [PubMed] [Google Scholar]
- 13.Brown MG, et al. A 2-Mb YAC contig and physical map of the natural killer gene complex on mouse chromosome 6. Genomics. 1997;42:16–25. doi: 10.1006/geno.1997.4721. [DOI] [PubMed] [Google Scholar]
- 14.Brown MG, Scalzo AA, Matsumoto K, Yokoyama WM. The natural killer gene complex: a genetic basis for understanding natural killer cell function and innate immunity. Immunol Rev. 1997;155:53–65. doi: 10.1111/j.1600-065x.1997.tb00939.x. [DOI] [PubMed] [Google Scholar]
- 15.Yokoyama WM, Plougastel BF. Immune functions encoded by the natural killer gene complex. Nat Rev Immunol. 2003;3:304–316. doi: 10.1038/nri1055. [DOI] [PubMed] [Google Scholar]
- 16.Long EO. Regulation of immune responses through inhibitory receptors. Annu Rev Immunol. 1999;17:875–904. doi: 10.1146/annurev.immunol.17.1.875. [DOI] [PubMed] [Google Scholar]
- 17.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 18.Smith HR, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci USA. 2002;99:8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SH, et al. Haplotype mapping indicates two independent origins for the Cmv1s susceptibility allele to cytomegalovirus infection and refines its localization within the Ly49 cluster. Immunogenetics. 2001;53:501–505. doi: 10.1007/s002510100359. [DOI] [PubMed] [Google Scholar]
- 20.Makrigiannis AP, et al. A BAC contig map of the Ly49 gene cluster in 129 mice reveals extensive differences in gene content relative to C57BL/6 mice. Genomics. 2002;79:437–444. doi: 10.1006/geno.2002.6724. [DOI] [PubMed] [Google Scholar]
- 21.Arase H, Lanier LL. Specific recognition of virus-infected cells by paired NK receptors. Rev Med Virol. 2004;14:83–93. doi: 10.1002/rmv.422. [DOI] [PubMed] [Google Scholar]
- 22.Makrigiannis AP, et al. Class I MHC-binding characteristics of the 129/J Ly49 repertoire. J Immunol. 2001;166:5034–5043. doi: 10.4049/jimmunol.166.8.5034. [DOI] [PubMed] [Google Scholar]
- 23.Silver ET, et al. Ly-49P activates NK-mediated lysis by recognizing H-2Dd. J Immunol. 2000;165:1771–1781. doi: 10.4049/jimmunol.165.4.1771. [DOI] [PubMed] [Google Scholar]
- 24.Makrigiannis AP, et al. Cloning and characterization of a novel activating Ly49 closely related to Ly49A. J Immunol. 1999;163:4931–4938. [PubMed] [Google Scholar]
- 25.Nakamura MC, Hayashi S, Niemi EC, Ryan JC, Seaman WE. Activating Ly-49D and inhibitory Ly-49A natural killer cell receptors demonstrate distinct requirements for interaction with H2-D(d) J Exp Med. 2000;192:447–454. doi: 10.1084/jem.192.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura MC, Seaman WE. Ligand interactions by activating and inhibitory Ly-49 receptors. Immunol Rev. 2001;181:138–148. doi: 10.1034/j.1600-065x.2001.1810111.x. [DOI] [PubMed] [Google Scholar]
- 27.Peruzzi M, Wagtmann N, Long EO. A p70 killer cell inhibitory receptor specific for several HLA-B allotypes discriminates among peptides bound to HLA-B*2705. J. Exp. Med. 1996;184:1585–1590. doi: 10.1084/jem.184.4.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franksson L, et al. Peptide dependency and selectivity of the NK cell inhibitory receptor Ly-49C. Eur J Immunol. 1999;29:2748–2758. doi: 10.1002/(SICI)1521-4141(199909)29:09<2748::AID-IMMU2748>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 29.Radaev S, Sun PD. Structure and function of natural killer cell surface receptors. Annu. Rev. Biophys. Biomol. Struct. 2003;32:93–114. doi: 10.1146/annurev.biophys.32.110601.142347. [DOI] [PubMed] [Google Scholar]
- 30.Dam J, et al. Variable MHC class I engagement by Ly49 natural killer cell receptors demonstrated by the crystal structure of Ly49C bound to H-2K(b) Nat Immunol. 2003;4:1213–1222. doi: 10.1038/ni1006. [DOI] [PubMed] [Google Scholar]
- 31.Trowsdale J, et al. The genomic context of natural killer receptor extended gene families. Immunol Rev. 2001;181:20–38. doi: 10.1034/j.1600-065x.2001.1810102.x. [DOI] [PubMed] [Google Scholar]
- 32.Belkin D, et al. Killer cell Ig-like receptor and leukocyte Ig-like receptor transgenic mice exhibit tissue- and cell-specific transgene expression. J Immunol. 2003;171:3056–3063. doi: 10.4049/jimmunol.171.6.3056. [DOI] [PubMed] [Google Scholar]
- 33.Khakoo SI, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science. 2004;305:872–874. doi: 10.1126/science.1097670. [DOI] [PubMed] [Google Scholar]
- 34.Martin MP, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002;31:429–434. doi: 10.1038/ng934. [DOI] [PubMed] [Google Scholar]
- 35.Scalzo, A.A., Farrell, H. & Karupiah, G. Techniques for studying murine natural killer cells in defense against viral infection. in Natural Killer Cell Protocols, Cellular and Molecular Methods (eds. Campbell, K.S. & Colonna, M.) 163–177 (Humana, Totowa, New Jersey, 2000). [DOI] [PubMed]
- 36.Truett GE, et al. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) Biotechniques 29. 2000;52:54. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- 37.Depatie C, et al. Assessment of Cmv1 candidates by genetic mapping and in vivo antibody depletion of NK cell subsets. Int Immunol. 1999;11:1541–1551. doi: 10.1093/intimm/11.9.1541. [DOI] [PubMed] [Google Scholar]
- 38.Depatie C, et al. Sequence-ready BAC contig, physical, and transcriptional map of a 2-Mb region overlapping the mouse chromosome 6 host-resistance locus Cmv1. Genomics. 2000;66:161–174. doi: 10.1006/geno.2000.6186. [DOI] [PubMed] [Google Scholar]
- 39.Peng SL, Craft J. PCR-RFLP genotyping of murine MHC haplotypes. Biotechniques. 1996;21 :362–368. doi: 10.2144/96213bm03. [DOI] [PubMed] [Google Scholar]
- 40.Silver ET, Gong D, Hazes B, Kane KP. Ly-49W, an activating receptor of nonobese diabetic mice with close homology to the inhibitory receptor Ly-49G, recognizes H-2D(k) and H-2D(d) J Immunol. 2001;166:2333–2341. doi: 10.4049/jimmunol.166.4.2333. [DOI] [PubMed] [Google Scholar]
- 41.Manly KF, Olson JM. Overview of QTL mapping software and introduction to map manager QT. Mamm Genome. 1999;10:327–334. doi: 10.1007/s003359900997. [DOI] [PubMed] [Google Scholar]
- 42.Pierce CA, Block RA, Aquinis H. Cautionary note on reporting eta-squared from multifactor ANOVA designs. Educ Psychol Meas. 2004;64:916–924. [Google Scholar]