

Abstract
Alzheimer’s disease (AD) remains a formidable challenge, necessitating the discovery of effective therapeutic agents targeting β-site amyloid precursor protein cleaving enzyme 1 (BACE1). This study investigates the inhibitory potential of phytochemicals derived from Bacopa monnieri, a plant renowned for its cognitive-enhancing properties, in comparison to established synthetic inhibitors such as Atabecestat, Lanabecestat, and Verubecestat. Utilizing molecular docking and advanced computational simulations, we demonstrate that Bacopaside I exhibits superior binding affinity and a unique interaction profile with BACE1, suggesting a more nuanced inhibitory mechanism. Our findings highlight the promising role of Bacopa monnieri phytochemicals as viable alternatives to synthetic drugs, emphasizing their potential to overcome limitations faced in clinical settings. Furthermore, the development of the SIMANA (https://simana.streamlit.app/) platform enhances the visualization and analysis of protein–ligand interactions, facilitating a deeper understanding of the dynamics involved. This research not only underscores the therapeutic promise of natural compounds in AD treatment but also advocates for a paradigm shift towards integrating traditional medicinal knowledge into contemporary drug discovery efforts.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-92644-y.
Keywords: Bacopa monnieri, Molecular docking, Molecular dynamic simulation, Alzheimers, BACE1
Subject terms: Biotechnology, Computational biology and bioinformatics, Drug discovery
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder that ranks among the most prevalent causes of dementia globally, primarily affecting the aging population1,2. AD is characterized by the progressive deterioration of cognitive functions, memory loss, and the gradual impairment of daily activities. At the molecular level, the disease’s hallmark features include the accumulation of extracellular amyloid plaques and intracellular neurofibrillary tangles in regions of the brain such as the hippocampus and cortical gray matter3–5. These plaques are primarily composed of amyloid-beta (Aβ) peptides, which arise from the sequential cleavage of the amyloid precursor protein (APP) by two key enzymes—β-secretase (BACE1) and γ-secretase. The β-secretase enzyme, in particular, plays a critical role in the initial and rate-limiting step of this proteolytic process, making it a prime therapeutic target for AD.
BACE1 (β-site of Amyloid Precursor Protein Cleaving Enzyme) is a membrane-bound aspartyl protease that plays a critical role in the production of amyloid-beta (Aβ) peptides, central to Alzheimer’s disease pathology6–9. Its active site contains two essential aspartate residues, Asp32 and Asp228 (Fig. 1), which coordinate a water molecule, facilitating the nucleophilic attack necessary for peptide bond hydrolysis. A unique feature of BACE1 is the flap region (residues 67–77), a hairpin loop that partially covers the active site cleft. This flap remains open in the enzyme’s inactive state8,9 but closes over the substrate or an inhibitor during catalysis, stabilizing the enzyme–substrate complex. Additionally, the 10 s loop (residues 9–14) (Fig. 1), undergoes conformational changes that regulate substrate access to the active site. These structural features, particularly the dynamic nature of the flap and 10 s loop, are key to understanding BACE1’s substrate specificity and catalytic mechanism, making them critical targets in the design of Alzheimer’s therapeutics.
Fig. 1.

Structure of BACE1 in surface view (left) and cartoon view (right). Important regions of BACE1 are highlighted in their respective colour and residue range.
Molecular dynamics simulations approaches have elucidated how structural variations in regions such as inserts A, D, and F, along with the 10 s loop, affect substrate binding and catalytic activity10. Various inhibitors have been developed to target BACE1, but their success has been limited by challenges such as off-target effects11,12. Atabecestat13, Lanabecestat14,15, and Verubecestat16 were among the most promising BACE1 inhibitors developed to target Alzheimer’s disease by reducing amyloid-beta (Aβ) production. These inhibitors progressed through clinical trials with hopes of curbing Aβ accumulation and slowing disease progression. However, despite their ability to effectively reduce Aβ levels, all three compounds failed in advanced clinical trials. Atabecestat was discontinued due to liver toxicity17, while Lanabecestat and Verubecestat were halted after showing no cognitive benefit in patients18,19. In particular, Verubecestat, which reached Phase III trials, revealed that although Aβ reduction occurred, the cognitive decline continued unabated, raising concerns about the complexity of Aβ’s role in Alzheimer’s pathology20. These failures highlight the challenges in translating BACE1 inhibition into an effective therapeutic strategy, as simply reducing Aβ levels may not be sufficient to alter the course of Alzheimer’s disease.
Bacopa monnieri, a traditional medicinal herb, has garnered significant attention for its neuroprotective properties, particularly in combating neurodegenerative disorders like Alzheimer’s disease21–24. Rich in bioactive compounds such as bacosides, flavonoids, and alkaloids, B. monnieri’s phytochemicals have demonstrated potent antioxidant, anti-inflammatory, and neuroprotective effects. It has been shown that Bacopa monnieri rejuvenates nerve cells and improves cognition and memory25. Studies have shown that Bacopa monnieri not only reduces Aβ deposition but also enhances cognitive function and neuronal health, making it a compelling natural candidate for Alzheimer’s therapy26,27. Moreover, we have shown that the phytochemicals of Bacopa monnieri has the potential to act as potent anti-neurodegenerative molecule by inhibiting the neurotrophins related neurological disorders28. In the current study, we have harnessed the therapeutic potential of B. monnieri’s phytochemicals to explore their efficacy in targeting Alzheimer’s-related neurodegeneration. By leveraging these compounds, we aim to investigate their role as promising alternatives or adjuncts to synthetic BACE1 inhibitors, potentially offering a safer and more holistic approach to mitigating cognitive decline and Aβ toxicity.
Results and discussion
Structural similarity and pharmacophoric analysis
Tanimoto similarity analysis and pharmacophoric feature comparisons were used to evaluate the structural link between the phytochemicals found in Bacopa monnieri and the well-known BACE1 inhibitors (Atabecestat, Lanabecestat, and Verubecestat). The Tanimoto similarity scores (see Supplementary Fig. S1) reveal low structural similarity between the phytochemicals and the known inhibitors, with an average similarity of 0.048 across all compounds. This suggests that the phytochemicals occupy a distinct chemical space compared to synthetic inhibitors. While low similarity may indicate divergence in chemical structure, it highlights the potential of Bacopa-derived compounds to serve as novel scaffolds for BACE1 inhibition, broadening the range of chemical diversity available for drug discovery.
A comprehensive comparison of molecular descriptors between control compounds and phytochemicals revealed several significant differences. The number of hydrogen bond acceptors (nHA) was higher in phytochemicals (13.27 ± 5.61) (see Table S1) compared to control compounds (6.33 ± 1.53), though this difference approached but did not reach statistical significance (p = 0.0610). Similarly, phytochemicals exhibited significantly more hydrogen bond donors (nHD: 6.82 ± 3.06 vs 2.67 ± 0.58; p = 0.0422, see Supplementary Fig. S2). Phytochemicals showed more rotatable bonds (nRot: 7.18 ± 3.66) than control compounds (3.33 ± 1.15), however this difference was not statistically significant (p = 0.1051) in terms of molecular flexibility. Both groups had similar levels of total molecular flexibility (Flex) (phytochemicals: 0.16 ± 0.08; controls: 0.15 ± 0.07; p = 0.8704). The structural properties showed the most notable differences. Ring systems (nRing: phytochemicals: 8.09 ± 1.14 vs controls: 3.67 ± 1.15; p = 0.0001) and aromatic rings (nAR: phytochemicals: 8.09 ± 1.14 vs controls: 3.67 ± 1.15; p = 0.0001) were substantially more prevalent in phytochemicals. Furthermore, the most statistically significant difference between the groups (p < 0.0001) was the fraction of sp3 carbons (Fsp3), which was significantly greater in phytochemicals (0.98 ± 0.03) than in control compounds (0.26 ± 0.16) (see Supplementary Fig. S2). These pharmacophoric analyses reveal that while the phytochemicals share minimal structural similarity with known BACE1 inhibitors, they possess enhanced molecular features—particularly increased hydrogen bonding capacity, ring systems, and three-dimensional character—that are crucial for protein–ligand interactions. Collectively, these findings suggest that Bacopa-derived compounds represent structurally distinct scaffolds that maintain essential physicochemical properties potentially offering new avenues for drug development against Alzheimer’s disease.
ADME-Tox (ADMET) studies
Comprehensive ADMET analysis of Bacopa monnieri phytochemicals revealed nuanced pharmacokinetic profiles compared to established BACE1 inhibitors (Atabecestat, Lanabecestat, and Verubecestat). Bacopasaponins (A-D, G, and X) demonstrated consistent MDCK permeability (score: 3) (Fig. S3), comparable to control compounds, indicating enhanced blood–brain barrier penetration potential. Notably, Bacopasaponins (A-D, G) exhibited optimal F20 and F30 bioavailability scores, suggesting improved oral absorption characteristics. Distribution analysis showed comparable Blood–Brain Barrier (BBB) penetration between the phytochemicals (score: 2–3) and control drugs (score: 3) (Fig. S4). The phytochemicals displayed optimized Volume Distribution (VDss) profiles, while maintaining reduced transporter interactions (OATP1B1/1B3, BCRP, MRP1, BSEP) comparable to control compounds (Fig. S4). Metabolic profiling revealed minimal CYP450 interactions for Bacopasaponins (A-D, G, X) and Bacopaside 1 (scores: 0–0.3), contrasting with moderate to strong interactions observed in control drugs, particularly for CYP2C8i and CYP2C9i (Fig. S5). Pharmacokinetic analysis demonstrated favourable plasma clearance rates (< 5 L/hr) for Bacopasaponins A-G and X, comparable to control drugs (3.5–4.5 L/hr) (Fig. S6a). Half-life profiles of Bacopa compounds (2–3 h) showed improvement over control drugs (< 1 h), with Bacoside A3 exhibiting an optimal 3.2-h half-life (Fig. S6b). Toxicological assessment revealed significantly reduced cardiac liability (hERG score: 3 vs. 1–2 in controls) and minimal hepatotoxicity for the phytochemicals (Fig. S7). Systemic toxicity evaluations demonstrated reduced cytotoxicity across multiple cell lines (HEK293, A549, RPMI-8226) for Bacopa compounds, particularly Bacopasaponins (A-D, G). These compounds also exhibited minimal organ-specific toxicity (neurotoxicity, nephrotoxicity, ototoxicity, hematotoxicity) and showed better alignment with FDA maximum daily dose guidelines compared to control drugs (Fig. S7).
Phytochemical and drug selection
We selected a suite of eleven biologically active phytochemicals, based on their reported efficacy in previous studies29. The molecular structures of these compounds are depicted in Fig. 2. To ensure accuracy, we cross-referenced the structural configurations and molecular masses of these phytochemicals against the PubChem database (https://pubchem.ncbi.nlm.nih.gov/)30. To benchmark the efficacy of the selected phytochemicals, we considered three known BACE1 inhibitors: Atabecestat, Lanabecestat and Verubecestat (Fig. 2).
Fig. 2.

Known inhibitor of BACE1 along with the selected phytochemicals of Bacopa monnieri.
Molecular docking
The docking protocol’s reliability was systematically validated through redocking experiments using N~3~-benzylpyridine-2,3-diamine, the co-crystallized inhibitor extracted from the BACE1 crystal structure (PDB ID: 2OHM). Root-mean-square deviation (RMSD) calculations between the predicted and crystallographic poses yielded 1.1 Å (Fig. S8), demonstrating high spatial agreement. This value falls well within the accepted threshold of ≤ 2.0 Å for successful pose reproduction31–33, indicating reliable performance of the docking protocol in recapitulating experimentally determined binding conformations. The molecular docking analysis revealed distinct binding patterns between control drugs and phytochemicals with the target enzyme. Among the control drugs, Lanabecestat emerged as the most potent binder with a binding energy of − 8.8 kcal/mol (Table 1). Its binding mechanism primarily involved a specific hydrogen bond between its N4 atom and the backbone carbonyl oxygen of Thr72, maintaining an optimal interaction distance of 2.96 Å (Fig. 3b). This primary interaction was reinforced by a network of hydrophobic interactions involving eight residues, creating a stable binding pocket. Atabecestat showed moderate binding affinity (− 7.5 kcal/mol) but demonstrated a more complex hydrogen bonding network, establishing three distinct interactions: the OG1 atom of Thr231 at 3.12 Å, the OD1 atom of Asp228 at 2.85 Å, and notably, an interaction with the backbone oxygen of Gly230 at 3.14 Å (Fig. 3a). Verubecestat, with a binding energy of − 7.4 kcal/mol, utilized its N4 atom to form two strategic hydrogen bonds—one with the OG1 atom of Thr231 (2.99 Å) and another with the OD2 atom of Asp228 (2.95 Å) (Fig. 3c). These interactions were complemented by nine hydrophobic contacts, contributing to the overall stability of the complex.
Table 1.
Molecular Docking details of residues of BACE1 interacting via Hydrogen Bonding and Hydrophobic Interactions with respective ligands.
| Ligand | Interacting residues (Hydrogen Bonding) | Distance (Å) | Binding energy (kcal/mol) |
|---|---|---|---|
| Atabecestat | Asp228 | 2.85 | − 7.5 |
| Gly230 | 3.14 | ||
| Thr231 | 3.12 | ||
| Lanabecestat | Thr72 | 2.96 | − 8.8 |
| Verubecestat | Asp228 | 2.95 | − 7.4 |
| Thr231 | 2.99 | ||
| Bacopasaponin A | Gly34 | 2.90 | − 8.6 |
| Asp228 | 2.88 | ||
| Thr231 | 2.96 | ||
| Leu263 | 2.88 | ||
| Gly264 | 3.15 | ||
| Bacopasaponin D | Asp32 | 2.76 | − 8.6 |
| Gly34 | 3.04 | ||
| Asp228 | 2.99 | ||
| Arg235 | 2.82 | ||
| Thr329 | 2.75 | ||
| Bacopaside 1 | Asp32 | 2.75 | − 8.6 |
| Gly34 | 3.13 | ||
| Thr72 | 2.30 | ||
| Phe109 | 3.31 | ||
| Asn111 | 2.97 | ||
| Asp228 | 2.85 | ||
| Gly230 | 2.78 | ||
| Thr231 | 2.97 | ||
| Arg235 | 2.80 | ||
| Thr329 | 3.00 | ||
| Bacopasaponin X | Asp32 | 3.12 | − 8.2 |
| Thr72 | 3.13 | ||
| Lys107 | 2.79 | ||
| Asp228 | 2.79 | ||
| Bacoside A3 | Gly11 | 3.15 | − 8.7 |
| Gln12 | 3.19 | ||
| Asp32 | 3.08 | ||
| Tyr71 | 3.13 | ||
| Lys107 | 3.16 | ||
| Asp228 | 2.78 | ||
| Gly230 | 3.26 | ||
| Thr231 | 2.78 | ||
| Thr232 | 3.24 | ||
| Jujubogenin | Lys107 | 2.70 | − 8.9 |
| Ligand | Interacting residues (Hydrophobic Interactions) | ||
|---|---|---|---|
| Atabecestat | Leu30, Asp32, Gly34, Ser35, Tyr71, Phe108, Tyr198, Lys224, Ile226, Thr329 | ||
| Lanabecestat | Asp32, Gly34, Tyr71, Lys107, Phe108, Ile118, Ile226, Asp228 | ||
| Verubecestat | Leu30, Asp32, Gly34, Tyr71, Ile110, Ile118, Trp132, Tyr198, Gly230 | ||
| Bacopasaponin A | Leu30, Tyr71, Phe108, Trp115, Ile118, Tyr198, Ile226, Asn233, Arg235, Glu265, Ser325 | ||
| Bacopasaponin D | Ser35, Tyr71, Lys107, Phe109, Ile110, Ile118, Asn111, Arg128, Tyr198, Ile226, Gly230, Thr231, Val332 | ||
| Bacopaside 1 | Ser35, Tyr71, Lys107, Ile110, Ile118, Tyr198 | ||
| Bacopasaponin X | Gly34, Tyr71, Gly74, Phe108, Ile118, Ser325, Gly230, Thr231, Arg235, Leu263, Gly264, Gln326 | ||
| Bacoside A3 | Gly13, Leu30, Gly34, Gly74, Phe108, Ile110, Trp115, Ile118, Arg235, Leu263, Gly264, Ser325, Gln326 | ||
| Jujubogenin | Gly34, Tyr71, Phe108, Tyr198, Ile226, Asp228, Thr329 | ||
Fig. 3.

Molecular Docking interaction of BACE1 with control drugs and phytochemicals. BACE1 is shown in molecular surface view (cyan) and the interactions are shown in the zoomed in plots. Hydrogen bonding is shown in green colour and hydrophobic interaction are shown in red colour.
The phytochemicals demonstrated binding characteristics, with several compounds showing binding energies comparable to the best control drug. Bacopasaponin A, Bacopasaponin D, and Bacopaside 1 all achieved binding energies of − 8.6 kcal/mol (Table 1), approaching Lanabecestat’s affinity. Particularly, Bacopaside 1, established the most extensive hydrogen bonding network. Its interactions included a precise bond between OG1 and Asp32 at 2.74 Å. Additional interactions included Gly34 (3.13 Å), and dual bonds with both Thr72 (2.85 Å, 2.97 Å) and Asp228 (2.85 Å, 3.00 Å). The compound’s NH1 atom formed strategic bonds with Thr231 (2.80 Å) and Arg235 (2.78 Å), while Asn111 participated in two distinct interactions (2.97 Å, 3.31 Å) (Fig. 3f). Bacopasaponin A exhibited a unique binding pattern where its O10 atom established dual bonds with Gly264 at distances of 3.15 Å and 3.23 Å. The compound’s OG1 atoms formed a triangulated interaction network with Asp228 and Thr231, maintaining distances of 2.84 Å, 2.86 Å, and 2.90 Å (Fig. 3d). Bacopasaponin D demonstrated binding with its OG1 atom forming a hydrogen bond with Asp32 (2.76 Å), while its NH1 and NH2 atoms engaged in parallel interactions with Arg235 (2.82 Å and 2.84 Å respectively). A critical interaction was also observed between its OG2 atom and Asp228 at 2.99 Å (Fig. 3e). Bacoside A3 contributed to the diverse binding patterns observed, forming five specific hydrogen bonds with catalytic residues (Fig. 3g), while Jujubogenin displayed a distinct interaction profile highlighted by a strong hydrogen bond with Tyr107 at 2.70 Å (Fig. 3h). A crucial feature across all phytochemicals was their consistent engagement with the catalytic aspartates (Asp32/Asp228) and their optimal positioning within hydrophobic cavities formed by Tyr71 and Phe108. This strategic placement, combined with their extensive hydrogen bonding networks and the preservation of key interactions with catalytic residues, strongly suggests that these phytochemicals could serve as effective alternatives to synthetic inhibitors, potentially offering new scaffolds for drug development.
Molecular dynamic simulation
The BACE1 protein–ligand complexes were subjected to a 150 ns MD simulation. To account for potential sampling bias, we ran the MD Simulations three times. The details of the triplicate simulation are provided in the supplementary file (see Supplementary Fig. S9-S17).
Root mean square deviation (RMSD)
The Root Mean Square Deviation (RMSD) for BACE1 in complex with three known inhibitors and phytochemicals from Bacopa monnieri was analysed over a 150 ns molecular dynamics (MD) simulation to assess stability. RMSD values for all complexes were generally stable, ranging between 0.1 and 0.3 nm, with an initial adjustment phase observed during the first 20 ns. After equilibration, the RMSD profiles stabilized, indicating that the complexes reached a steady conformation. Among the inhibitors, Verubecestat exhibited the highest RMSD values, slightly exceeding 0.3 nm, suggesting greater conformational flexibility (Fig. 4a, blue). Lanabecestat (Fig. 4a, green) and Atabecestat (Fig. 4a, red) displayed more conservative profiles, maintaining RMSD values within 0.2 to 0.3 nm, indicating similar stabilization of BACE1. The Bacopa monnieri phytochemicals demonstrated promising stability. Bacopaside I closely matched Lanabecestat, with RMSD values between 0.2 and 0.25 nm (Fig. 4a, maroon), suggesting it induces similar stability in BACE1. Bacopasaponin A (Fig. 4a, magenta) and Bacopasaponin D (Fig. 4a, cyan) also showed stable profiles, with RMSD values ranging from 0.2 to 0.3 nm. Bacopasaponin D exhibited slightly higher flexibility, akin to Verubecestat, potentially indicating dynamic binding behaviour. Bacoside A3 (Fig. 4a, orange) and Jujubogenin (Fig. 4a, violet) maintained consistent RMSD values around 0.2–0.25 nm. RMSD distribution analysis revealed overlapping conformational populations, with Jujubogenin showing the narrowest distribution centered at ~ 0.22 nm, suggesting a well-defined binding mode. Overall, the phytochemicals exhibited RMSD profiles comparable to the known inhibitors, suggesting they stabilize BACE1 similarly without introducing excessive conformational strain.
Fig. 4.

150 ns Molecular Dynamic Simulation analysis of BACE1 showing (a) RMSD (b) RMSF (c) Radius of Gyration and (d) SASA analysis in apo form (black) and in association with Atabecestat (red), Lanabecestat (green), Verubecestat (blue), Bacopasaponin A (magenta), Bacopasaponin D (cyan) and Bacopaside 1 (maroon). Each analysis has their corresponding probability distribution curve with the same colour code.
Root mean square fluctuation (RMSF)
The Root Mean Square Fluctuation (RMSF) analysis was performed to assess the flexibility of specific BACE1 residues bound to different ligands, providing insights into their binding dynamics and stability. Overall, BACE1 exhibited low RMSF values, indicating structural stability across ligand-bound states. Key active site residues, Asp32 and Asp228, remained highly stable (RMSF < 0.2 nm) in all cases, confirming that the ligands effectively stabilize the catalytic core, preserving BACE1’s enzymatic function. The flap region, crucial for substrate binding, displayed moderate flexibility (RMSF ~ 0.2–0.3 nm) with ligand-specific variations. Bacopasaponin A induced the highest flap flexibility (RMSF ~ 0.4 nm), suggesting a unique inhibitory mechanism through flap modulation (Fig. 4b, magenta). Atabecestat (Fig. 4b, red) and Verubecestat (Fig. 4b, blue) showed moderate effects, while Lanabecestat (Fig. 4b, green), Bacopaside I (Fig. 4b, maroon), and Bacopasaponin D (Fig. 4b, cyan) resulted in lower flap movement, indicating tighter binding. The N-terminal 10S loop exhibited high flexibility (RMSF > 0.3 nm) in all ligand-bound states, largely independent of ligand binding, potentially playing a role in substrate recognition or product release. Bacopasaponin D and Lanabecestat induced slightly higher fluctuations in this region. Similarly, the 113S loop displayed moderate flexibility (RMSF ~ 0.2–0.3 nm) with ligand-specific effects. Bacopasaponin A and Bacopaside I caused higher flexibility (~ 0.4 nm), which might impact substrate access to the active site, while the known inhibitors (Atabecestat, Lanabecestat, Verubecestat) showed moderate effects. A hyper-flexible region near Insert A (residues 175–185) exhibited the highest flexibility among all regions (RMSF > 0.8 nm), with notable ligand-dependent variations. Bacopasaponin A induced the most significant fluctuations (RMSF > 1.0 nm), indicating a unique interaction mode that might alter BACE1’s overall conformation. Lanabecestat and Verubecestat had moderate effects (~ 0.8 nm), while Atabecestat, Bacopasaponin D, and Bacopaside I induced lower fluctuations. Insert D (residues 270–274), a short loop region, showed moderate flexibility (~ 0.2–0.3 nm) with distinct ligand effects. Bacopasaponin D and Bacopasaponin A induced the highest flexibility (~ 0.4 nm), suggesting potential interactions that may affect BACE1’s C-terminal domain. Atabecestat and Verubecestat caused moderate effects, while Lanabecestat and Bacopaside I led to minimal movement, possibly stabilizing this region of the protein. Finally, Insert F (residues 311–317) exhibited similar flexibility to Insert D, with RMSF values ranging from 0.05 to 0.1 nm across ligands. Bacopasaponin D caused the most significant fluctuations (~ 0.1 nm), indicating a unique interaction mode that may influence the conformation of BACE1’s C-terminal domain. Lanabecestat, Verubecestat, and Bacopasaponin A showed moderate effects (~ 0.15 nm), while Atabecestat and Bacopaside I induced lower fluctuations (~ 0.1 nm). Bacoside A3 induced moderate flexibility (0.25–0.3 nm) (Fig. 4b, orange), positioning between the higher fluctuations of Bacopasaponin A and the more restrained movement of Atabecestat. At Insert A, Bacoside A3 caused significant but controlled fluctuations (~ 0.8 nm), matching Verubecestat’s profile. The compound uniquely stabilized the 113S loop (RMSF ~ 0.3 nm) while maintaining moderate flexibility in Insert D and F regions (0.2–0.3 nm). Jujubogenin effectively stabilized the catalytic core (RMSF < 0.2 nm) (Fig. 4b, violet) and demonstrated remarkable control over flap dynamics (0.2–0.25 nm), closely aligning with Lanabecestat’s profile. At Insert A, Jujubogenin induced moderate fluctuations (~ 0.75 nm), lower than other phytocompounds. The N-terminal 10S loop showed reduced flexibility under Jujubogenin binding compared to other ligands. Most notably, Jujubogenin maintained consistent, moderate flexibility across Insert D and F regions (0.2–0.25 nm), suggesting a well-balanced interaction profile that neither over-rigidifies nor excessively mobilizes these regulatory regions. RMSF analysis indicates that Bacopa monnieri phytochemicals, show stability and flexibility profiles comparable to known inhibitors, particularly Atabecestat and Lanabecestat. The slightly higher flexibility observed with Bacopasaponin A and Bacopasaponin D may reflect a more adaptable binding mode, which could be advantageous for targeting different conformations of BACE1 but might also introduce some instability. RMSF profile of Bacopaside I, Bacoside A3 and Jujubogenin aligns closely with those of the synthetic inhibitors, suggesting it may stabilize key protein regions while potentially avoiding the issues that led to the clinical failure of Lanabecestat and Atabecestat.
Radius of gyration (Rg)
The Radius of Gyration (Rg) measures the compactness of a protein, providing insights into its stability and folding during a molecular dynamics (MD) simulation. The unbound BACE1 protein (Fig. 4c, black) fluctuates around 2.1 nm, indicating a stable, compact structure. This serves as a baseline for evaluating ligand effects. Atabecestat (Fig. 4c, red) and Lanabecestat (Fig. 4c, green) closely mirror the unbound state, with Rg values around 2.1 nm, indicating that these ligands stabilize the protein without significant structural changes. Verubecestat (Fig. 4c, blue) shows slightly higher fluctuations, occasionally reaching 2.15 nm, suggesting minor conformational changes that slightly reduce compactness but still maintain overall stability. Bacopasaponin A (Fig. 4c, magenta) and Bacopasaponin D (Fig. 4c, cyan) also show similar Rg profiles to the known inhibitors. However, Bacopasaponin D exhibits a slight increase in Rg after 70 ns, reaching up to 2.25 nm, indicating potential expansion and a more flexible interaction. Bacopaside I (Fig. 4c, maroon) maintains a stable Rg around 2.1 nm, suggesting that it stabilizes the protein similarly to Atabecestat and Lanabecestat. Bacoside A3 (Fig. 4c, orange) exhibited dynamic behaviour, with Rg fluctuating between 2.1 and 2.15 nm. The distribution analysis shows a broader peak centered at ~ 2.13 nm, indicating moderate conformational flexibility while maintaining overall protein stability. Jujubogenin (Fig. 4c, violet) demonstrated unique characteristics, consistently maintaining lower Rg values (~ 2.08 nm) compared to other ligands. Its narrow distribution peak suggests a compact, well-defined conformational state that may enhance protein stability. This tighter packing could indicate favourable binding interactions that stabilize BACE1’s structure. Overall, Rg values for BACE1-ligand complexes range from 2.05 to 2.25 nm. The slight increases for Verubecestat and Bacopasaponin D suggest some flexibility in binding, while Atabecestat, Lanabecestat, and Bacopaside I, Bacoside A3 and Jujubogenin stabilize the protein in a compact conformation. This stability is likely beneficial for inhibiting BACE1 activity, as it may prevent excessive conformational changes that could affect the enzyme’s function.
Solvent accessible surface area (SASA)
The Solvent Accessible Surface Area (SASA) simulation over 150 ns provides key insights into the dynamic interactions between BACE1 and its ligands, including both known inhibitors and phytochemicals. The SASA plot reveals time-dependent fluctuations for all compounds, highlighting dynamic protein–ligand interactions. Notably, the phytochemicals display SASA profiles like the clinically tested inhibitors, suggesting that they may bind to BACE1 in a comparable manner, potentially offering similar inhibitory effects. Among the phytochemicals, Bacopasaponin D (Fig. 4d, cyan) exhibits the highest average SASA values (around 190–200 nm2) and the most significant fluctuations, paralleling Atabecestat’s behaviour. These elevated values suggest extensive interactions with BACE1’s surface, possibly due to Bacopasaponin D’s structural complexity. This dynamic interaction may contribute to a stable binding mode, enhancing its potential as an effective inhibitor. Bacopasaponin A (Fig. 4d, magenta) and Bacopaside I (Fig. 4d, maroon) display SASA profiles like Verubecestat (Fig. 4d, blue) and Lanabecestat (Fig. 4d, green) (approximately 180–190 nm2), indicating that these phytochemicals may occupy a similar volume within the BACE1 binding site. Their consistent SASA values throughout the simulation suggest stable interactions with BACE1, a trait often linked with effective inhibitors. Bacoside A3 (Fig. 4d, orange) displayed dynamic SASA behaviour, fluctuating between 185 and 190 nm2. Its distribution profile centers around 188 nm2, indicating expanded protein surface exposure compared to the apo state. These moderate fluctuations suggest a balance between stable binding and conformational flexibility. Jujubogenin (Fig. 4d, violet) exhibited the lowest SASA values (175–180 nm2) among all compounds, with a narrow distribution peak centered at ~ 178 nm2. This reduced surface exposure suggests a compact binding mode that minimizes protein-solvent interactions, potentially indicating deep pocket engagement. The periodic fluctuations in SASA values for all compounds, including both inhibitors and phytochemicals, reflect conformational changes in the protein–ligand complexes. This suggests that the phytochemicals induce structural adaptations in BACE1 akin to those triggered by the known inhibitors, contributing to their inhibitory potential. Bacopasaponin D and Atabecestat show synchronized increases in SASA values and fluctuations after 90 ns, indicating that both compounds may induce large-scale conformational changes in BACE1 over time. This sustained effect could signify prolonged inhibitory action, an advantageous feature for therapeutic agents. Bacopasaponin A, Bacopaside I, Bacoside A3 and Jujubogenin maintain stable SASA ranges, mirroring Verubecestat and Lanabecestat’s behaviour, indicating robust interactions with BACE1. Slightly lower SASA values for Bacopaside I and Jujubogenin suggest a more compact binding mode, possibly leading to higher binding affinity.
Molecular mechanics Poisson-Boltzmann surface area (MMPBSA)
The MMPBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) analysis reveals binding free energies (ΔG) of various ligands interacting with BACE1, offering key insights into their binding affinities. Lower (more negative) ΔG values represent stronger binding. Among the clinically tested inhibitors, Atabecestat has the most favourable ΔG (− 29.21 kcal/mol) (Fig. 5), while Lanabecestat and Verubecestat showed a moderately favourable binding energy of − 11.94 kcal/mol and − 9.17 kcal/mol respectively. The Bacopa monnieri phytochemicals demonstrate highly favourable binding energies, surpassing or matching those of the clinically tested inhibitors. Bacopaside 1 shows the most negative ΔG (− 36.71 kcal/mol) (Fig. 5), suggesting the strongest binding affinity among all tested compounds. Bacopasaponin D also exhibits a strong ΔG (− 26.16 kcal/mol) (Fig. 5), comparable to Atabecestat. Bacopasaponin A, with a ΔG of − 10.48 kcal/mol (Fig. 5), shows a binding affinity similar to Lanabecestat. Bacoside A3 exhibited moderate binding affinity (− 20.60 kcal/mol), positioning it between Atabecestat and Lanabecestat. The relatively small error bars suggest consistent binding mode throughout the simulation. Jujubogenin showed comparable binding strength (− 11.72 kcal/mol) to Lanabecestat, with stable interactions indicated by narrow error margins. The detailed energetics have been given in Table 2. These findings suggest that Bacopa monnieri phytochemicals, especially Bacopaside 1, Bacopasaponin D and Bacoside A3, may overcome some limitations faced by synthetic inhibitors in clinical trials. The strong binding affinities of these phytochemicals indicate that they could form stable interactions with BACE1, potentially leading to effective enzyme inhibition. Overall, the MMPBSA results align with SASA findings, further supporting the potential of Bacopa monnieri phytochemicals as BACE1 inhibitors, with Bacopaside 1 being particularly promising for future drug development.
Fig. 5.

MM-PBSA analysis of BACE1 with respective ligands. The error bar denotes the standard deviation associated with each ligand’s energy.
Table 2.
MMPBSA analysis of BACE1 in complex with different ligands.
| Ligand |
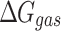 (kcal/mol) (kcal/mol) |
 (kcal/mol) (kcal/mol) |
 (kcal/mol) (kcal/mol) |
|---|---|---|---|
| Atabecestat | − 88.12 ± 9.33 | 58.91 ± 7.34 | − 29.21 ± 5.40 |
| Lanabecestat | − 78.41 ± 2.36 | 66.47 ± 2.16 | − 11.94 ± 5.17 |
| Verubecestat | − 82.56 ± 5.44 | 73.39 ± 4.89 | − 9.17 ± 5.75 |
| Bacopasaponin A | − 43.72 ± 11.10 | 33.25 ± 9.12 | − 10.48 ± 4.0 |
| Bacopasaponin D | − 107.65 ± 7.26 | 81.49 ± 3.89 | − 26.16 ± 4.58 |
| Bacopaside 1 | − 145.05 ± 7.40 | 108.34 ± 7.55 | − 36.71 ± 2.83 |
| Bacoside A3 | − 93.06 ± 10.98 | 72.46 ± 6.39 | − 20.60 ± 5.39 |
| Jujubogenin | − 56.79 ± 3.92 | 45.07 ± 2.86 | − 11.72 ± 4.32 |
Dynamic cross correlation matrix (DCCM) analysis
The Dynamic Cross-Correlation Matrix (DCCM) analysis provides crucial insights into the correlated motions of residues within the BACE1 active site when bound to different ligands. This comprehensive examination reveals intriguing patterns of residue interactions that may explain their potential effectiveness as BACE1 inhibitors. The DCCM heatmaps showcase a range of correlations from strong positive (red, + 1) to strong negative (blue, − 1) between residues 225–235 and 25–35, which encompass the critical catalytic dyad (residues 32 and 228) of BACE1. This region is pivotal for the enzyme’s function, and alterations in its dynamic behaviour can significantly impact BACE1’s catalytic activity. The apo protein (Fig. 6a) exhibits baseline anticorrelations, particularly between residues 27–31 and 229–233, indicating intrinsic conformational dynamics essential for catalytic function. Upon binding of clinically tested inhibitors, modulation of these intrinsic dynamics with compound-specific signatures was observed. Atabecestat (Fig. 6b) induces enhanced anticorrelations across the matrix, suggesting a stabilizing effect on the binding pocket through coordinated opposing motions. This pattern is notably intensified around the catalytic dyad, potentially explaining its mechanism of inhibition. Lanabecestat (Fig. 6c) demonstrates a similar but more focused pattern of anticorrelations, with distinct localization around residues 32–34, indicating targeted perturbation of catalytic residues. Verubecestat (Fig. 6d) exhibits a markedly different correlation landscape, characterized by regions of both positive and negative correlations. The mixed correlation pattern, particularly evident in residues 32–34 and their interaction with 229–233, suggests a unique mode of binding that may explain its distinct pharmacological profile. Among the phytochemicals, Bacopasaponin A (Fig. 6e) demonstrates remarkable anticorrelations between residues 26–31 and 230–234, exceeding the intensity observed with synthetic inhibitors. This enhanced coordination of opposing motions suggests a potentially more robust mechanism of conformational control. Bacopasaponin D (Fig. 6f) exhibits the most extensive network of anticorrelated motions among all compounds studied, with pronounced effects spanning residues 26–32 and 228–234. This comprehensive perturbation of active site dynamics indicates a potentially novel mechanism of inhibition that extends beyond the catalytic dyad. Bacopaside 1 (Fig. 6g) shows moderate but widespread anticorrelations, suggesting a more distributed effect on enzyme dynamics. Bacoside A3 (Fig. 6h) and Jujubogenin (Fig. 6i) demonstrate intermediate patterns of anticorrelation, with distinct signatures in the catalytic dyad region. Notably, the phytochemicals, particularly Bacopasaponin A and D, induce more pronounced perturbations in BACE1 dynamics compared to clinically tested inhibitors. This enhanced modulation of protein motion, especially around the catalytic residues, suggests a potentially more effective mechanism of enzyme inhibition.
Fig. 6.

Dynamic Cross Correlation of BACE1 active site 1 (residue 25–35, including Asp32) and active site 2 (residues 225–235, including Asp228) with (a) Protein (Apo) (b) Protein with Atabecestat (c) Protein with Lanabecestat (d) Protein with Verubecestat (e) Protein with Bacopasaponin A (f) Protein with Bacopasaponin D (g) Protein with Bacopaside 1 (h) Protein with Bacoside A3 (i) Protein with Jujubogenin.
Moreover, the apo structure (Fig. 7a) exhibits moderate positive correlations, particularly between residues 71–72 (flap region) and 29–34 (active site 1), suggesting inherent coordinated movements that may facilitate substrate access. A distinct anticorrelation pattern is observed between residues 74–75 and the catalytic region (30–32), indicating the flap’s natural flexibility essential for substrate binding. Atabecestat binding (Fig. 7b) induces a shift toward anticorrelated motions between residues 70–73 and 26–29, while maintaining positive correlations near the catalytic residues (32–35). This mixed pattern suggests a mechanism where the inhibitor modulates flap dynamics while stabilizing specific catalytic interactions. Lanabecestat (Fig. 7c) demonstrates a striking enhancement of positive correlations across the matrix, particularly pronounced between residues 71–74 and 31–35, indicating a distinct mode of flap stabilization that may contribute to its inhibitory mechanism. Verubecestat (Fig. 7d) exhibits the strongest positive correlations among all compounds, especially between residues 69–72 and 32–35, suggesting a unique mechanism of flap stabilization that may lock the enzyme in an inactive conformation. The phytochemicals display diverse effects on flap-active site coupling. Bacopasaponin A (Fig. 7e) shows strong positive correlations between residues 70–74 and 31–35, similar to Verubecestat, but with distinct patterns in the lower flap region (67–69). Bacopasaponin D (Fig. 7f) presents a balanced distribution of correlations, with anticorrelated motions in the upper flap region (73–75) and positive correlations near the catalytic residues. This pattern suggests a sophisticated mechanism of conformational control. Bacopaside 1 (Fig. 7g) predominantly shows anticorrelated motions, particularly between residues 71–75 and 26–31, indicating a different mode of flap regulation that may enhance inhibitor effectiveness. Bacoside A3 (Fig. 7h) and Jujubogenin (Fig. 7i) demonstrate intermediate correlation patterns with gradual transitions from anticorrelated to positively correlated motions across the matrix. This suggests these compounds may induce more subtle modifications to the flap dynamics.
Fig. 7.

Dynamic Cross Correlation of BACE1 active site 1 (residue 25–35, including Asp32) and Flap region (residues 67–75) with (a) Protein (Apo) (b) Protein with Atabecestat (c) Protein with Lanabecestat (d) Protein with Verubecestat (e) Protein with Bacopasaponin A (f) Protein with Bacopasaponin D (g) Protein with Bacopaside 1 (h) Protein with Bacoside A3 (i) Protein with Jujubogenin.
The distinct patterns of flap-active site coupling observed with different ligands provide fundamental insights into their mechanisms of action. The phytochemicals demonstrate correlation patterns that suggest effective modulation of both local and allosteric dynamics. These findings highlight the importance of considering both active site and flap dynamics in the rational design of BACE1 inhibitors and suggest that natural compounds may offer unique advantages through their sophisticated regulation of enzyme dynamics.
In the apo state, strong positive correlations (≥ 0.75) indicate a highly coordinated motion between Asp228 and the flap (Fig. 8a), reinforcing its closed conformation and stabilizing the active site architecture. Atabecestat binding induces a slight reduction in correlation (Fig. 8b), increasing flap flexibility, which may facilitate conformational transitions. Conversely, Lanabecestat enhances correlated motion (Fig. 8c), stabilizing the active site in a manner akin to the apo state, potentially contributing to its high inhibitory efficacy. Verubecestat largely preserves the apo-like correlation pattern, suggesting minimal perturbation of the native dynamic behaviour (Fig. 8d). Among the phytochemicals, Bacopasaponin A reduces correlation moderately, indicating partial decoupling of Asp228 from the flap (Fig. 8e). Bacopasaponin D (Fig. 8f) and Bacopaside I (Fig. 8g) induce localized anti-correlated motions, suggesting a potential destabilization of the active site, which could impair substrate recognition and enzymatic function. In contrast, Bacoside A3 (Fig. 8h) and Jujubogenin (Fig. 8i) maintain correlation patterns like the apo protein, suggesting negligible impact on flap dynamics. These observations underscore ligand-specific effects on BACE1 conformational plasticity, with high-affinity inhibitors maintaining or reinforcing native flap-active site coupling, while select phytochemicals introduce destabilizing motions. A detailed DCCM analysis of other relevant regions is provided in the Supplementary Information (Fig. S19-S28).
Fig. 8.

Dynamic Cross Correlation of BACE1 active site 2 (residue 225–235, including Asp228) and Flap region (residues 67–75) with (a) Protein (Apo) (b) Protein with Atabecestat (c) Protein with Lanabecestat (d) Protein with Verubecestat (e) Protein with Bacopasaponin A (f) Protein with Bacopasaponin D (g) Protein with Bacopaside 1 (h) Protein with Bacoside A3 (i) Protein with Jujubogenin.
UMAP based free energy landscape
Using dimensionality reduction through UMAP analysis, we investigated the conformational landscape of BACE1 in its apo form and in complex with the ligands. The apo BACE1 exhibited a relatively diffuse energy landscape with multiple local minima (Fig. 9a), suggesting significant conformational flexibility in the absence of ligands. Upon Atabecestat binding (Fig. 9b), the energy landscape showed a notable shift in the conformational ensemble, with a more focused distribution of states compared to the apo form. Structural comparison between the lowest energy conformers revealed that Atabecestat binding induces subtle but significant changes in the insert A region (residues 158–167), which adopts a more closed configuration suggesting a ligand-induced stabilisation of this regulatory element. The overlaid structures demonstrate that there are localized conformational adjustments in the substrate-binding cleft. The Lanabecestat complex (Fig. 9c) displayed a distinct energy landscape with a prominent low-energy basin, indicating a stabilized conformational state. The structural alignment with the apo form reveals that Lanabecestat binding promotes a unique reorganization of the insert A region, with moderate changes in the positioning of the 10 s loop (residues 9–14) that forms part of the active site. These modifications likely contribute to the compound’s reported high binding affinity and selectivity profile. Verubecestat binding (Fig. 9d) resulted in the most altered energy landscape, characterized by a sharp low-energy minimum and distinct high-energy regions. The structural comparison shows a proximity to apo form which likely induces a more rigid and closed conformation of the enzyme’s active site. Moreover, Verubecestat showed the closest structural similarity to the apo form with an RMSD of 1.97 Å, suggesting more subtle conformational changes despite its potent inhibitory effects. Atabecestat induced moderate structural changes (RMSD = 2.52 Å), while Lanabecestat showed more substantial conformational differences (RMSD = 2.91 Å).
Fig. 9.

Uniform Manifold Approximation and projection (UMAP) of (a) BACE1 (apo form) and in presence of (b) Atabecestat (red) (c) Lanabecestat (green) (d) Verubecestat (blue). The overlayed structure is the energetically minimised structure of BACE1 compared with the apo form (violet).
The BACE1-bacopasaponin A complex (Fig. 10a) exhibited a free energy landscape characterized by a prominent medium-energy basin in the central region, with several shallow minima distributed across the conformational space. Structural comparison with the apo form reveals that Bacopasaponin A binding induces moderate conformational changes in the flexible loops surrounding the active site, particularly in the flap region and insert A region, while maintaining the core structural integrity of the enzyme. Bacopasaponin D binding (Fig. 10b) resulted in a distinctly different energy landscape, featuring a well-defined low-energy basin in the lower region of the UMAP projection. The structural overlay demonstrates that this phytochemical promotes a unique reorganization of the substrate-binding pocket, with notable adjustments in the flap, 10S loop and insert A regions. The overlapped complex structure suggests a more open conformation of the active site compared to the apo form, potentially indicating a different mechanism of interaction compared to traditional BACE1 inhibitors. The BACE1-bacopaside 1 complex (Fig. 10c) showed a broader distribution of conformational states, with multiple interconnected energy basins. The structural alignment reveals moderate changes, suggesting that Bacopaside 1 may influence BACE1 dynamics through allosteric effects. Particularly notable are the conformational shifts in the insert A region and the flap region, which are critical for substrate recognition and catalysis. Bacoside A3 binding (Fig. 10d) generated a unique energy landscape with a distinctive medium-energy basin centered in the UMAP projection. The structure indicates substantial reorganization of the active site architecture, with the most pronounced changes observed in the insert A region aligning closely with apo form. These structural modifications suggest that bacoside A3 may modulate BACE1 activity through a combination of direct active site interactions and conformational selection. The Jujubogenin complex (Fig. 10e) displayed a more focused energy landscape with a well-defined low-energy basin, indicating a stabilized conformational state. The structural comparison with the apo form reveals subtle but significant changes in the enzyme’s dynamic regions, particularly in the 10S loop region and insert A region.
Fig. 10.

Uniform Manifold Approximation and projection (UMAP) of BACE1 (apo form) and in presence of (a) Bacopasaponin A (magenta) (b) Bacopasaponin D (cyan) (c) Bacopaside 1 (maroon) (d) Bacoside A3 (orange) (e) Jujubogenin (pink). The overlayed structure is the energetically minimised structure of BACE1 compared with the apo form (violet).
Notably, all five phytochemicals induced distinct conformational signatures in BACE1, different from both the apo form and from each other. The phytocompounds generally induced larger conformational changes compared to the synthetic inhibitors, suggesting potentially distinct mechanisms of interaction. Bacopasaponin A showed the most pronounced structural deviation with an RMSD of 3.48 Å from the apo structure, followed closely by Bacopasaponin D (RMSD = 3.31 Å) and Bacopaside 1 (RMSD = 3.10 Å). These larger RMSD values indicate significant reorganization of BACE1’s structure upon binding these saponins, possibly due to their larger molecular size and complex structural features. In contrast, Bacoside A3 and Jujubogenin induced more moderate structural changes (RMSDs of 2.43 Å and 2.36 Å respectively), comparable to those observed with synthetic inhibitors. This suggests that these compounds might interact with BACE1 through mechanisms more similar to traditional inhibitors, despite their natural origin. These unique structural modifications suggest that Bacopa monnieri compounds may modulate BACE1 activity through diverse mechanisms. The observed conformational changes predominantly affect the flexible regions and substrate-binding sites while preserving the overall fold of the enzyme, indicating that these phytochemicals might offer novel approaches to BACE1 inhibition distinct from traditional synthetic inhibitors.
Conclusion
Alzheimer’s disease (AD) remains an unmet medical challenge, necessitating novel therapeutic strategies that can effectively target its underlying molecular mechanisms. The inhibition of BACE1, a critical enzyme in the amyloidogenic pathway, has long been pursued as a promising strategy for reducing amyloid-beta (Aβ) accumulation. However, the failure of synthetic BACE1 inhibitors in clinical trials due to off target effects11,12, toxicity17, and lack of cognitive benefits18,19 underscores the necessity of alternative therapeutic approaches. In this study, we present evidence that phytochemicals derived from Bacopa monnieri might exhibit superior BACE1 inhibition through a mechanistically distinct and potentially safer approach compared to synthetic inhibitors.
Molecular docking and dynamic simulations reveal that phytochemicals engage in a highly stable and extensive network of interactions with BACE1’s catalytic dyad (Asp32 and Asp228), flap region, and adjacent hydrophobic pockets. Unlike synthetic inhibitors, which rigidly fix BACE1 in an inactive conformation, phytochemicals induce controlled dynamic flexibility within the active site, allowing for partial inhibition rather than complete enzymatic blockade. This unique mechanism holds significant therapeutic potential, as complete inhibition of BACE1 has been linked to adverse neurophysiological consequences34,35. The ability of phytochemicals to modulate rather than fully disrupt BACE1 function suggests a balanced approach to reducing toxic Aβ production while maintaining physiological roles in axonal guidance and synaptic function. Comparative binding energetics, as assessed via Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA) calculations, further reinforce the efficacy of Bacopa phytochemicals. Bacopaside I exhibit a remarkably low binding free energy (− 36.71 kcal/mol), surpassing the best-performing synthetic inhibitor, Atabecestat (− 29.21 kcal/mol), while Bacopasaponin D (− 26.16 kcal/mol) and Bacoside A3 (− 20.60 kcal/mol) demonstrate a comparable binding free energy. These findings indicate that Bacopa-derived compounds form thermodynamically stable complexes with BACE1, suggesting higher inhibitory potency and prolonged residence time. Notably, the hydrogen bonding network of phytochemicals involves Asp32, Gly34, Thr72, Phe109, Asn111, and Arg235—residues crucial for BACE1 catalysis. Such extensive engagement enhances binding affinity and provides structural stabilization to the protein–ligand complex.
Dynamic Cross-Correlation Matrix (DCCM) analysis further supports the mechanistic advantage of Bacopa phytochemicals. Unlike synthetic inhibitors, which induce rigid stabilization, Bacopaside I and Bacopasaponin D selectively disrupt the correlated motion of catalytic dyad residues while preserving overall structural integrity. This finding highlights the potential of these phytochemicals to act as dynamic inhibitors, disrupting BACE1’s catalytic efficiency without inducing complete enzymatic deactivation. The role of sugar moieties in Bacopa saponins is particularly noteworthy, as they contribute to hydrogen bonding with polar residues while anchoring hydrophobic interactions through aglycone cores. This dual-mode engagement likely explains the superior stability and specificity observed for Bacopa-derived compounds.
The Uniform Manifold Approximation and Projection (UMAP) free energy landscape analysis provides further structural insights, revealing that Bacopa phytochemicals induce distinct conformational shifts in BACE1 that are absent in synthetic inhibitor-bound states. These conformational adaptations involve key regulatory regions, including the flap and insert A, which govern substrate access and catalytic turnover. This mechanistic divergence highlights the potential for Bacopa phytochemicals to overcome the pitfalls of conventional BACE1 inhibitors and achieve more precise enzymatic modulation.
The clinical failures of synthetic inhibitors underscore the limitations of a simplistic BACE1 blockade strategy. While Atabecestat, Lanabecestat, and Verubecestat effectively reduced Aβ levels, they failed to translate this reduction into cognitive improvement, with some exhibiting severe off-target effects. Our findings suggest that the nuanced inhibitory mechanism of Bacopa phytochemicals may overcome these challenges by providing a fine-tuned modulation of BACE1 activity, rather than a complete shutdown. Furthermore, ADMET analysis suggests that Bacopa phytochemicals exhibit favourable pharmacokinetics, with improved blood–brain barrier permeability, minimal hepatotoxicity, and reduced cardiac liability compared to synthetic inhibitors. These properties enhance their viability as clinically relevant drug candidates. Future studies should focus on validating these findings through in vivo efficacy assessments and clinical trials to fully harness the therapeutic potential of Bacopa-derived BACE1 inhibitors in Alzheimer’s disease treatment.
Methodology
Structural similarity and pharmacophoric analysis
To assess the structural relationship between the Bacopa monnieri phytochemicals and the known BACE1 inhibitors (Atabecestat, Lanabecestat, Verubecestat), we employed a two-step approach comprising of Tanimoto similarity analysis and Pharmacophoric feature comparison using SIMANA server (https://simana.streamlit.app/). The Tanimoto coefficient was used to measure the molecular similarity between the phytochemicals found in Bacopa monnieri and the control inhibitors. This measure was calculated using the Morgan algorithm, which is based on chemical fingerprints. Chemical variety is highlighted by structural divergence, which is indicated by a low Tanimoto coefficient. The Bacopa monneiri phytochemicals and the inhibitors were then examined using important molecular characteristics such as fraction of sp3 centres (Fsp3), rotatable bonds (nRot), hydrogen bond donors (nHD), hydrogen bond acceptors (nHA), aromatic ring systems, and molecular flexibility. Using RDKit36 package, these pharmacophoric characteristics were retrieved, allowing for a thorough assessment of their ability to interact with the BACE1 active site.
ADME-Tox studies
Absorption, distribution, metabolism, excretion (ADME) properties and toxicity profiles were evaluated using ADMETlab 3.037. Molecular structures of both synthetic BACE1 inhibitors and Bacopa monnieri phytochemicals were submitted in SMILES format for comprehensive physicochemical characterization. The analysis encompassed evaluation of compliance with established medicinal chemistry guidelines along with a detailed ADME-Tox profiling.
Ligand and phytochemicals selection
To investigate potential BACE1 inhibitors, we employed a comparative approach using both synthetic and natural compounds. Three clinically-studied BACE1 inhibitors—Atabecestat, Lanabecestat, and Verubecestat—were selected as reference ligands due to their established efficacy and progression to clinical trials for Alzheimer’s disease treatment. For exploring natural alternatives, we focused on eight specific bacosides from Bacopa monnieri, a plant traditionally used for cognitive enhancement: Bacopasaponin A, Bacopasaponin B, Bacopasaponin C, Bacopasaponin D, Bacopasaponin G, Bacopaside I, Bacopaside II, Sarsasapogenin, Bacopasaponin X, Bacoside A3 and Jujubogenin. These triterpenoid saponins, unique to B. monnieri, were chosen based on their prevalence in the plant and previous studies suggesting neuroprotective properties29.
Molecular docking
The BACE1 active site, comprising the catalytic dyad (Asp32 and Asp228) and surrounding residues (e.g., Thr72, Ser35, Gly34, Arg235, and Tyr71), was analysed for ligand interactions. The structures of the inhibitors and selected phytochemicals were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/)30 in .sdf format and converted to .pdb format using OpenBabel (https://openbabel.org/)38. The ligand structures were then subjected to energy minimization using the MM2 algorithm39, as implemented in the Chem3D software40. Molecular docking studies employed three platforms: AutoDock41, DockThor42, and CBDock43, enhancing result robustness (Table S2). For receptor preparation, the structure of BACE1 (PDB ID: 2OHM) was obtained from the RCSB Protein Data Bank44. Preparation involved the removal of unwanted residues (small ions) and water molecules, followed by energy minimization using Chimera45. The minimization was conducted using the Steepest Descent Algorithm with 1000 steps and a step size of 0.1 Å, followed by 100 conjugate gradient steps with a step size of 0.1 Å to ensure optimal energetic conformation. The default AMBER ff14SB force field46 was applied to standard residues, and the AMI-BCC force field was used for other residues. Ligand-receptor interactions were visualized using LigPlot47. The docking result for each docking procedure are provided in the Supplementary File (see Supplementary Table S1). Redocking was performed by docking N~3~-benzylpyridine-2,3-diamine (inhibitor present in 2OHM PDB structure) with the BACE1 crystal structure to validate the docking protocol. Validation was confirmed by calculating the RMSD between the docked and crystal-bound N~3~-benzylpyridine-2,3-diamine molecules.
Molecular dynamic simulation
Molecular dynamics (MD) simulations were performed to explore the dynamic interactions of known inhibitors and the top-ranked phytochemicals with BACE1. All simulations were executed in triplicate using GROMACS version 2018.148–50 for a duration of 150 ns. The protein–ligand complexes were prepared with the CHARMM36 force field, and ligand topologies were generated using CGenFF51. The simulation system was solvated in a 5 nm3 cubic box with the TIP3P water model, and charge neutrality was achieved by adding Na+ and Cl- ions. Energy minimization was carried out using the steepest descent algorithm for a maximum of 5000 steps. The system was then equilibrated through two consecutive 1 ns step: first under an NVT ensemble at 300 K using a V-rescale thermostat, followed by an NPT ensemble at 1 bar pressure using a Berendsen barostat. The production MD phase ran for 150 ns using the leap-frog algorithm with a 2-fs time step. Van der Waals interactions were treated with a 1.2 nm cutoff, while long-range electrostatic interactions were computed using Particle Mesh Ewald (PME). The Nosé-Hoover thermostat and the Parrinello-Rahman barostat were applied to maintain temperature and pressure stability throughout the simulation. Coordinates were saved every 20 ps for subsequent analysis. Post simulation analyses were carried out to assess the stability, flexibility, and conformational behaviour of the protein–ligand complexes.
Molecular mechanics Possoin-Boltzmann surface area (MM-PBSA)
The calculation of binding free energy ( ) between a protein and its ligand is crucial for understanding the molecular interactions within protein–ligand complexes. In this study, we employed the Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) approach, a widely accepted method for estimating binding free energy. In MM/PBSA, the binding free energy (
) between a protein and its ligand is crucial for understanding the molecular interactions within protein–ligand complexes. In this study, we employed the Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) approach, a widely accepted method for estimating binding free energy. In MM/PBSA, the binding free energy ( ) is derived as the difference between the free energy of the protein–ligand complex (
) is derived as the difference between the free energy of the protein–ligand complex ( ) and the sum of the free energies of the isolated protein (
) and the sum of the free energies of the isolated protein (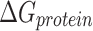 ) and ligand (
) and ligand (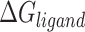 ). This relationship is mathematically expressed as:
). This relationship is mathematically expressed as:
 |
Here, 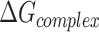 represents the free energy of the protein–ligand complex, while
represents the free energy of the protein–ligand complex, while 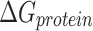 and
and  correspond to the free energies of the isolated protein and ligand, respectively. This method provides a detailed energetic perspective on the stability and binding affinity of the complexes.
correspond to the free energies of the isolated protein and ligand, respectively. This method provides a detailed energetic perspective on the stability and binding affinity of the complexes.
Dynamic cross correlation matrix (DCCM) analysis
Dynamic cross-correlation analysis (DCCM) was conducted to explore the associations between protein residues. A structural ensemble was generated from the MD simulation trajectories of the protein in complex with the top-ranked ligands. A matrix representing these correlations was generated, with elements visualized as a Dynamic Cross-Correlation Matrix (DCCM). To enhance the robustness of the analysis, we utilized three independent MD simulation trajectories (triplicates), averaged the results, and calculated the final DCCM.
Free energy landscape using uniform manifold approximation and projection (UMAP)
Uniform Manifold Approximation and Projection (UMAP) analysis was employed to construct the free energy landscape of the protein–ligand complex, providing insights into the conformational dynamics and energy states of the system. By reducing the high-dimensional conformational space to a two-dimensional representation while preserving both local and global topological features, UMAP enabled visualization of energetically favourable states and transition pathways. The analysis began with the construction of a high-dimensional dataset comprising atomic coordinates from molecular dynamics trajectories. UMAP then generated a lower-dimensional embedding that maintained the essential relationships between different conformational states, with the probability density of states being transformed into free energy values using the Boltzmann relationship:  , where
, where  is the free energy,
is the free energy,  is the Boltzmann constant,
is the Boltzmann constant,  is the temperature and
is the temperature and  is the probability density estimated from kernel density estimation. The topographical features of the landscape, including energy barriers and transition pathways between stable states, were clearly visualized through UMAP’s non-linear dimensionality reduction approach. This method proved particularly effective in capturing both local conformational fluctuations and global structural transitions, providing a more complete picture of the system’s energy landscape compared to traditional linear reduction techniques.
is the probability density estimated from kernel density estimation. The topographical features of the landscape, including energy barriers and transition pathways between stable states, were clearly visualized through UMAP’s non-linear dimensionality reduction approach. This method proved particularly effective in capturing both local conformational fluctuations and global structural transitions, providing a more complete picture of the system’s energy landscape compared to traditional linear reduction techniques.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Author contributions
S.S: Conceptualization, Data Curation, Formal Analysis, Investigation, Methodology, Visualisation, Writing—original draft, Writing—Review and editing. A.K: Methodology, Visualisation, Writing—original draft, Writing—Review and Editing All authors read and approved the final manuscript.
Funding
No funding was provided for the current study.
Data availability
The data of the current study are provided in the main manuscript and supplementary material.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kandalepas, P. C. et al. The Alzheimer’s beta-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol.126, 329–352. 10.1007/s00401-013-1152-3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rajapaksha, T. W., Eimer,W. A., Bozza, T. C., & Vassar, R. The Alzheimer’s beta-secretase enzyme BACE1 is required for accurate axon guidance of olfactory sensory neurons and normal glomerulus formation in the olfactory bulb. Mol. Neurodegener.6. 10.1186/1750-1326-6-88 (2011). [DOI] [PMC free article] [PubMed]
- 3.Cole, S. L. & Vassar, R. The basic biology of BACE1: A key therapeutic target for Alzheimer’s disease. Curr. Genom.8, 509–530. 10.2174/138920207783769512 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hampel, H. & Shen, Y. Beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) as a biological candidate marker of Alzheimer’s disease. Scand. J. Clin. Lab. Invest.69, 8–12. 10.1080/00365510701864610 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Barman, A., Schürer, S. & Prabhakar, R. Computational modeling of substrate specificity and catalysis of the β-secretase (BACE1) enzyme. Biochemistry50, 4337–4349. 10.1021/bi200081h (2011). [DOI] [PubMed] [Google Scholar]
- 6.Chakraborty, S., Kumar, S. & Basu, S. Conformational transition in the substrate binding domain of beta-secretase exploited by NMA and its implication in inhibitor recognition: BACE1-myricetin a case study. Neurochem. Int.58, 914–923. 10.1016/j.neuint.2011.02.021 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Gorfe, A. A. & Caflisch, A. Functional plasticity in the substrate binding site of β-secretase. Structure13, 1487–1498. 10.1016/j.str.2005.06.015 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Shimizu, H. et al. Crystal structure of an active form of BACE1, an enzyme responsible for amyloid beta protein production. Mol. Cell. Biol.28, 3663–3671. 10.1128/MCB.02185-07 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu, Y. et al. Flexibility of the flap in the active site of BACE1 as revealed by crystal structures and molecular dynamics simulations. Acta Crystallogr. D Biol. Crystallogr.68, 13–25. 10.1107/S0907444911047251 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Kumalo, H. M., Bhakat, S. & Soliman, M. E. Investigation of flap flexibility of beta-secretase using molecular dynamic simulations. J. Biomol. Struct. Dyn.34, 1008–1019. 10.1080/07391102.2015.1064831 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Zuhl, A. M. et al. Chemoproteomic profiling reveals that cathepsin D off-target activity drives ocular toxicity of betasecretase inhibitors. Nat. Commun.7, 13042. 10.1038/ncomms13042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai, J. et al. β-Secretase (BACE1) inhibition causes retinal pathology by vascular dysregulation and accumulation of age pigment. EMBO Mol. Med.4, 980–991. 10.1002/emmm.201101084 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Timmers, M. et al. Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer’s disease: Randomized, double-blind, placebo-controlled study. Alzheimers Res. Ther.10, 85. 10.1186/s13195-018-0415-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eketjall, S. et al. AZD3293: A novel, orally active BACE1 inhibitor with high potency and permeability and markedly slow off-rate kinetics. J. Alzheimers Dis.50, 1109–1123. 10.3233/JAD-150834 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sims, J. R. et al. Development review of the BACE1 inhibitor lanabecestat (AZD3293/LY3314814). J. Prev. Alzheimers Dis.4, 247–254. 10.14283/jpad.2017.38 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Scott, J. D. et al. Discovery of the 3-imino-1,2,4-thiadiazinane 1,1-dioxide derivative verubecestat (MK-8931)–a β-site amyloid precursor protein cleaving enzyme 1 inhibitor for the treatment of Alzheimer’s disease. J. Med. Chem.59(23), 10435–10450 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Lahiri, D. K. et al. Lessons from a BACE1 inhibitor trial: Off-site but not off base. Alzheimers Dement.59, 10435–10450. 10.1021/acs.jmedchem.6b00307 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egan, M. F. et al. Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med.378, 1691–1703. 10.1056/NEJMoa1706441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wessels, A. M. et al. Efficacy and safety of lanabecestat for treatment of early and mild Alzheimer disease: The AMARANTH and DAYBREAK-ALZ randomized clinical trials. JAMA Neurol.77, 199–209. 10.1001/jamaneurol.2019.3988 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sur, C. et al. BACE inhibition causes rapid, regional, and non-progressive volume reduction in Alzheimer’s disease brain. Brain143, 3816–3826. 10.1093/brain/awaa332 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saraf, M. K., Prabhakar, S. & Anand, A. Neuroprotective effect of Bacopa monniera on ischemia induced brain injury. Pharmacol. Biochem. Behav97, 192–197. 10.1016/j.pbb.2010.07.017 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Stough, C. et al. Examining the cognitive effects of a special extract of Bacopa monniera (CDRI08: Keenmnd): A review of ten years of research at Swinburne University. J. Pharm. Pharm. Sci.16, 254–258. 10.18433/J35G6M (2013). [DOI] [PubMed] [Google Scholar]
- 23.Bhattacharya, S. K. & Ghosal, S. Anxiolytic activity of a standardized extract of Bacopa monniera: An experimental study. Phytomedicine5, 77–82. 10.1016/S0944-7113(98)80001-9 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Shinomol, G. K., Mythri, R. B., Srinivas Bharath, M. M. & Muralidhara,. Bacopa monnieri extract offsets rotenone-induced cytotoxicity in dopaminergic cells and oxidative impairments in mice brain. Cell. Mol. Neurobiol.32, 455–465. 10.1007/s10571-011-9776-0 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sukumaran, N. P., Amalraj, A. & Gopi, S. Neuropharmacological and cognitive effects of Bacopa monnieri (L.) Wettst-A review on its mechanistic aspects. Complement. Ther. Med.44, 68–82. 10.1016/j.ctim.2019.03.016 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Russo, A. & Borrelli, F. Bacopa monniera, a reputed nootropic plant: An overview. Phytomedicine12, 305–317. 10.1016/j.phymed.2003.12.008 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Kumar, S. S., Saraswathi, P. & Vijayaraghavan, R. Effect of Bacopa monniera on cold stress induced neurodegeneration in hippocampus of wistar rats: A histomorphometric study. J. Clin. Diagn. Res9, AF05–AF07. 10.7860/JCDR/2015/10199.5423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sangeet, S. et al. Computational analysis of Bacopa monnieri (L.) Wettst. Compounds for drug development against neurodegenerative disorders. Curr. Comput. Aided Drug Des.19, 24–36. 10.2174/1573409918666221010103652 (2023). [DOI] [PubMed] [Google Scholar]
- 29.Lal, U. R. & Lal, S. Bioactive molecules from Indian medicinal plants as possible candidates for the management of neurodegenerative disorders (ed. Sharma, K., Mishra, K., Senapati, K. K. & Danciu, C). 10.5772/intechopen.92043 (2019).
- 30.Kim, S. P. et al. update: Improved access to chemical data. Nucleic Acids Res.47, 1102–1109. 10.1093/nar/gky1033(2019) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramirez, D. & Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data?. Molecules23(5), 1038. 10.3390/molecules23051038 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mena-Ulecia, K., Tiznado, W. & Caballero, J. Study of the differential activity of thrombin inhibitors using docking, QSAR, molecular dynamics, and MM-GBSA. PLoS One10(11), e0142774. 10.1371/journal.pone.0142774 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gohlke, H., Hendlich, M. & Klebe, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol.295(2), 337–356. 10.1006/jmbi.1999.3371 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Blume, T. et al. BACE1 inhibitor MK-8931 alters formation but not stability of dendritic spines. Front. Aging Neurosci. 10, 229. 10.3389/fnagi.2018.00229 [DOI] [PMC free article] [PubMed]
- 35.Ulku, I. et al. Inhibition of BACE1 affected both its Aβ producing and degrading activites and increased Aβ42 and Aβ40 levels at high-level BACE1 expression. J. Biol. Chem.300, 107510. 10.1016/j.jbc.2024.107510 [DOI] [PMC free article] [PubMed]
- 36.Landrum, G. et al. RDKit: Open-source cheminformatics. https://www.rdkit.org. 10.5281/zenodo.591637 (2024).
- 37.Fu, L. et al. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res.52, W422–W431. 10.1093/nar/gkae236 [DOI] [PMC free article] [PubMed]
- 38.O’Boyle, N. M. et al. Open babel: An open chemical toolbox. J. Cheminform.3(1), 33. 10.1186/1758-2946-3-33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lii, J. H. et al. Molecular mechanics (MM2) calculations on peptides and on the protein Crambin using the CYBER 205. J. Comput. Chem.10, 503–513. 10.1002/jcc.540100408 (2004). [Google Scholar]
- 40.ChemOffice, 7.0.1. Cambridge Soft Corporation (Cambridge, 2002).
- 41.Morris, G. M. et al. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem.30, 2785–2791. 10.1002/jcc.21256 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guedes, I. A. et al. Drug Design and repurposing with DockThor-VS web server focusing on SARS-CoV-2 therapeutic targets and their non-synonym variants. Sci. Rep.11, 5543. 10.1038/s41598-021-84700-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu, Y. et al. CB-Dock: A web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol. Sin.41, 138–144. 10.1038/s41401-019-0228-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berman, H. M. et al. The protein data bank. Nucleic Acids Res.28, 235–242. 10.1093/nar/28.1.235 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pettersen, E. F. et al. USCF Chimera—A visualisation system for exploratory research and analysis. J. Comput. Chem.25, 1605–1612. 10.1002/jcc.20084 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Maier, J. A. et al. Ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theor. Comput.11, 3696–3713. 10.1021/acs.jctc.5b00255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laskowski, R. A. & Swindells, M. B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model51, 2778–2786. 10.1021/ci200227u (2011). [DOI] [PubMed] [Google Scholar]
- 48.Abraham, M. J. et al. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX1, 19–25. 10.1016/j.softx.2015.06.001 (2015). [Google Scholar]
- 49.Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced and scalable molecular simulation. J. Chem. Theory Comput.4, 435–447. 10.1021/CT700301Q (2008). [DOI] [PubMed] [Google Scholar]
- 50.Van Der Spoel, D. et al. GROMACS: Fast, flexible, and free. J. Comput. Chem.26, 1701–1718. 10.1002/JCC.20291 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Vanommeslaeghe, K. et al. CHARMM general force field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem.31, 671–690. 10.1002/jcc.21367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data of the current study are provided in the main manuscript and supplementary material.