

Abstract
VP1 is the most suitable region for use in the identification of enterovirus. Although VP1 sequencing methods may vary, it is necessary to agree on a common strategy of sequence analysis. Identification of a strain type may be achieved by three different approaches: pairwise sequence alignment, multiple-sequence alignment, and phylogenetic inference. Other methods are also available, but they are not simple enough to be performed at a virology laboratory. The performances of these methods were evaluated with nucleotide and protein sequences obtained from 32 original samples, 8 enterovirus isolates, and 64 GenBank sequences. Pairwise sequence alignment methods had very different results. The DNASTAR package identified only 28.8% of enterovirus strains, while the Genetics Computer Group package identified 50.0 or 72.1% of enterovirus strains when nucleotide or amino acid sequences were analyzed, respectively. Multiple-sequence alignment methods identified 94.2% (Clustal W program) or 92.3% (Pileup program) of the enterovirus strains, while the phylogenetic method increased this rate to 99.0%. Comparative evaluation of these analysis methods showed that the Clustal W program (version 1.81), a freely available multiple-sequence alignment program, presented one of the best performances when used with the correct criteria. Other commercial and expensive programs did not achieve the same performances, making them less suitable for molecular typing of enteroviruses. Finally, although phylogenetic inference is the most demanding method in terms of knowledge of the user, it remained the best option analyzed.
Enteroviruses (EVs) are a large genus belonging to the Picornaviridae family, and 64 immunologically distinct serotypes are known to cause infections in humans. They are the etiological agents responsible for several diseases (poliomyelitis, acute myocarditis, and aseptic meningitis) and play an important role in common chronic diseases, including dilated cardiomyopathy and insulin-dependent diabetes mellitus. Identification of EVs is essential for epidemiological surveillance, identification of polioviruses, the study of correlations between EV subtypes and diseases, identification of new EV types, and adequate treatment of EV infections in neonates and immunodeficient patients (for a complete review, see reference 15). Although the serotype identity can be determined by neutralization of infectivity with serotype-specific antisera, individual typing of all 64 serotypes by neutralization is clearly impractical. To overcome this problem, intersecting pools of individual antisera which allow the identification of a serotype were developed. Despite that, the method is still time-consuming, labor-intensive, and costly; and the supply of antisera is limited. Moreover, there are frequent problems related to untypeable EVs that have been associated with mixtures of EVs, the existence of certain EV serotypes that cannot be identified with intersecting pools, the formation of aggregates (9), and the existence of antigenic variants of recognized EVs (13). Finally, some unrecognized serotypes are also untypeable by this standard method.
Because of these problems, several methods were developed for the molecular characterization of the genus (1, 5–7, 17). The coupling of reverse transcription and amplification of the enteroviral RNA by PCR, followed by direct sequencing of the amplified products, is the general approach. Some of these methods analyzed the 5′ noncoding, the VP2, or the 3D region of the EVs genome; but the sequence did not always correlate with the corresponding serotype (1, 10, 18). The VP1 region was the most suitable target due to the high correlation between serotypes and sequences and the availability of a large database of EVs sequences (17). Two methods were developed for the typing of EV strains by partial sequencing of VP1 from cell cultures (17) or clinical samples (5). These two approaches were based on the sequencing of amplified products and comparison of the sequences with the VP1 sequences in a database of EVs reference strains by pairwise local alignment (17). However, several problems were found for only a handful of field strains, suggesting that this method of analysis could be inconsistent (5).
Although methods for the sequencing of VP1 may vary, there must be widespread agreement as to the method of analysis so that it can be universally applied. Several methods of analysis are quick and standardized: pairwise sequence alignment, multiple-sequence alignment, and phylogenetic reconstruction. Although there are several choices, users generally apply the available methods instead of first performing an evaluation of them. Nevertheless, the election of the analysis method should be an essential part of the design process, since each method of analysis is based on different assumptions.
Previously proposed sequence analysis methods were based on the similarity index of the pairwise local alignment as a measure of identity of the strain sequence (17). The Gap program (Genetics Computer Group [GCG] software package) uses the algorithm of Needleman and Wunsch (16). The same algorithm is implemented by other commercial packages (e.g., the MegAlign program of the DNASTAR package [3]).
Multiple-sequence alignment is an extension of pairwise sequence alignment but is considered a more thorough method of analysis than pairwise sequence alignment. Two programs, a freeware version of the Clustal W program (version 1.81) (23) and the Pileup program, another part of the GCG package, were evaluated.
Phylogenetic reconstruction of gene trees from sequence data is considered the “gold standard” method of molecular analysis. The best results are obtained when the proper method is chosen and the quality control measures are followed. However, there are several available methods based on distinct assumptions.
In the present study, we compared the efficiencies of analysis methods over a range of values for the various parameters. The methods are simple enough to be performed in virology reference laboratories. A completely characterized database of EV strain sequences from different origins was used for comparison.
MATERIALS AND METHODS
Clinical specimens.
Clinical specimens (8 EVs isolates from 3 stool samples and 5 pharyngeal swabs or nasopharyngeal aspirates and 32 original samples [28 cerebrospinal fluid and 4 stool samples]) were collected at both the Diagnostic Microbiology Service (Centro Nacional de Microbiología, Instituto de Salud Carlos III, Madrid, Spain) and the Neurovirosis Division (Administracion Nacional de Laboratorio e Institutos de Salud, “Dr. Carlos G. Malbrán,” Buenos Aires, Argentina). Sequences from the original samples (see Table 1) were directly obtained from clinical specimens.
TABLE 1.
Description of EVs studieda
| Clinical sampleb | GenBank accession no. | Neutrali-zation sero-typesc | Phylogenetic genotype | Boot-strap value | Clustal homology test index (%)d | Genotype determined by clustal homology teste | Pileup homology test index(%)e | Genotype determined by pileup homol-ogy teste | Highest score NW-GCG similarity test index (%)f | Second highest score NW-GCG similarity test index (%)g | Genotype determined by NW-GCG similarity teste |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TX92-1647 | AF081634 | CAV14 | CAV14 | 100 | 87.7 | CAV14 | 87.5 | CAV14 | 87.2 | 63.5 | CAV14 |
| GA95-2095 | AF081613 | CAV16 | CAV16 | 86 | 78.9 | CAV16 | 78.9 | CAV16 | 78.1 | 70.1 | CAV16 |
| PA94-5753 | AF081628 | CAV16 | CAV16 | 86 | 78.9 | CAV16 | 78.9 | CAV16 | 78.1 | 69.3 | CAV16 |
| TX95-2147 | AF081635 | CAV16 | CAV16 | 86 | 77.7 | CAV16 | 77.7 | CAV16 | 76.8 | 71.2 | CAV16 |
| PA89-9262 | AF152288 | CAV16 | CAV16 | 86 | 78.9 | CAV16 | 78.9 | CAV16 | 78.1 | 70.2 | CAV16 |
| TAI84-5839 | AF152293 | CAV16 | CAV16 | 86 | 79.1 | CAV16 | 79.1 | CAV16 | 78.3 | 70.2 | CAV16 |
| 707F99 | AF290902 | CAV16 | CAV16 | 86 | 81 | CAV16 | 81.5 | CAV16 | 80.8 | 64.9 | CAV16 |
| TN88-8321 | AF152294 | CAV17 | CAV17 | 99 | 79.5 | CAV17 | 79.6 | CAV17 | 79.3 | 70.2 | CAV17 |
| MOR83-6282 | AF152273 | CAV20 | CAV20 | 100 | 81.1 | CAV20 | 81.1 | CAV20 | 80.9 | 73.1 | CAV20 |
| GUT88-8020 | AF152258 | CAV21 | CAV21 | 100 | 78.5 | CAV21 | 78.5 | CAV21 | 78.3 | 66.3 | CAV21 |
| GUT88-8438 | AF152259 | CAV21 | CAV21 | 100 | 78.5 | CAV21 | 78.5 | CAV21 | 78.3 | 65.6 | CAV21 |
| MD86-7277 | AF152265 | CAV21 | CAV21 | 100 | 93 | CAV21 | 93 | CAV21 | 92.9 | 67.2 | CAV21 |
| WA89-9165 | AF152300 | CAV21 | CAV21 | 100 | 93.3 | CAV21 | 93.3 | CAV21 | 93.2 | 67.2 | CAV21 |
| DOR93-1657 | AF081603 | CAV24 | CAV24 | 100 | 88 | CAV24 | 88.2 | CAV24 | 88.1 | 67.4 | CAV24 |
| 2106NE98 | AF252189 | CAV4 | CAV4 | 100 | 85.7 | CAV4 | 85.7 | CAV4 | 85.1 | 65 | CAV4 |
| 1734O99 | AF290899 | CAV4 | CAV4 | 100 | 86.6 | CAV4 | 86.7 | CAV4 | 86.1 | 66.2 | CAV4 |
| 727F99 | AF290903 | CAV6 | CAV6 | 100 | 84 | CAV6 | 84.3 | CAV6 | 83.6 | 67 | CAV6 |
| R15797 | AF252183 | CAV9 | CAV9 | 100 | 85.1 | CAV9 | 85.1 | CAV9 | 84.1 | 70.2 | CAV9 |
| 1712NE99 | AF290898 | CAV9 | CAV9 | 100 | 82.3 | CAV9 | 82.5 | CAV9 | 81.3 | 68.6 | CAV9 |
| R7596 | AF252169 | CBV1 | CBV1 | 94 | 79.9 | CBV1 | 80.1 | CBV1 | 79 | 71.7 | CBV1 |
| R6696 | AF252170 | CBV1 | CBV1 | 94 | 79.7 | CBV1 | 80.3 | CBV1 | 78.7 | 70.9 | CBV1 |
| HON84-6016 | AF152260 | CBV2 | CBV2 | 100 | 86.2 | CBV2 | 86.2 | CBV2 | 85.2 | 69.8 | CBV2 |
| MD84-5914 | AF152263 | CBV2 | CBV2 | 100 | 87.4 | CBV2 | 87.4 | CBV2 | 86.5 | 68 | CBV2 |
| NH97-2342 | AF081622 | CBV3 | CBV3 | 100 | 80.8 | CBV3 | 80.8 | CBV3 | 79.4 | 69.6 | CBV3 |
| BRA98-9169 | AF152249 | CBV3 | CBV3 | 100 | 84.8 | CBV3 | 85 | CBV3 | 84 | 70.1 | CBV3 |
| BRA98-9171 | AF152250 | CBV3 | CBV3 | 100 | 82.9 | CBV3 | 82.9 | CBV3 | 81.7 | 69 | CBV3 |
| BRA88-9172 | AF152251 | CBV3 | CBV3 | 100 | 84.8 | CBV3 | 85 | CBV3 | 84 | 70.1 | CBV3 |
| BRA88-9173 | AF152252 | CBV3 | CBV3 | 100 | 85.1 | CBV3 | 85.3 | CBV3 | 84.3 | 70.4 | CBV3 |
| PER89-9426 | AF152291 | CBV3 | CBV3 | 100 | 81.8 | CBV3 | 81.6 | CBV3 | 80.4 | 70.4 | CBV3 |
| R8797 | AF252182 | CBV4 | CBV4 | 100 | 86.2 | CBV4 | 86.2 | CBV4 | 85.2 | 68.3 | CBV4 |
| MEX88-8931 | AF152270 | CBV5 | CBV5 | 100 | 87.2 | CBV5 | 87.2 | CBV5 | 86.3 | 70.3 | CBV5 |
| PA88-8885 | AF152287 | CBV5 | CBV5 | 100 | 85.1 | CBV5 | 85 | CBV5 | 84 | 70.3 | CBV5 |
| R3597 | AF252177 | CBV5 | CBV5 | 100 | 93.1 | CBV5 | 93.1 | CBV5 | 92.6 | 70.9 | CBV5 |
| R8697 | AF252178 | CBV5 | CBV5 | 100 | 81.5 | CBV5 | 61.5 | CBV5 | 80.2 | 67.3 | CBV5 |
| R9497 | AF252179 | CBV5 | CBV5 | 100 | 93.1 | CBV5 | 79.3 | CBV5 | 77.9 | 67.8 | CBV5 |
| 1800NE99 | AF290900 | CBV5 | CBV5 | 100 | 92.4 | CBV5 | 92.4 | CBV5 | 91.9 | 70.4 | CBV5 |
| CB6IS2 | AF225470 | CBV6 | CBV6 | 96 | 80.2 | CBV6 | 80.2 | CBV6 | 78.6 | 72.6 | CBV6 |
| GA92-1616 | AF081606 | EV11 | EV11 | 99 | 81.6 | EV11 | 82.2 | EV11 | 81.1 | 72.3 | EV11 |
| WA92-1516 | AF081642 | EV11 | EV11 | 99 | 80.5 | EV11 | 81.1 | EV11 | 79.9 | 73.7 | EV11 |
| 834NE99 | AF290904 | EV11 | EV11 | 99 | 83.4 | EV11 | 83.6 | EV11 | 82.6 | 72.5 | EV11 |
| 2257NE99 | AF290908 | EV11 | EV11 | 99 | 85.7 | EV11 | 83.8 | EV11 | 84.9 | 72.1 | EV11 |
| ELS88-8236 | AF152256 | EV12 | EV12 | 100 | 82.2 | EV12 | 82.1 | EV12 | 81 | 71.2 | EV12 |
| GV34 | AF252184 | EV12 | EV12 | 100 | 80.7 | EV12 | 80.9 | EV12 | 79.5 | 71.8 | EV12 |
| M250 | AF252185 | EV12 | EV12 | 100 | 77.9 | EV12 | 77.9 | EV12 | 76.4 | 71.4 | EV12 |
| VA86-6776 | AF152299 | EV13 | ND (ENV69-EV13) | 96 | 74.2 | EV13 | 74.4 | EV13 | 72.7 | NT | EV13 |
| 47-98 | AF290901 | EV14 | EV14 | 100 | 81.1 | EV14 | 81 | EV14 | 79.8 | 69.6 | EV14 |
| CT92-1465 | AF081599 | EV16 | EV16 | 100 | 84.5 | EV16 | 84.5 | EV16 | 83.4 | 72.6 | EV16 |
| 875NE99 | AF290905 | EV17 | EV17 | 100 | 82.9 | EV17 | 83.1 | EV17 | 82 | 70.1 | EV17 |
| 1068NE99 | AF290907 | EV17 | EV17 | 100 | 84.1 | EV17 | 84.3 | EV17 | 83.2 | 71.3 | EV17 |
| CT96-2182 | AF081601 | EV18 | EV18 | 100 | 78 | EV18 | 78.2 | EV18 | 76.8 | 70.2 | EV18 |
| MD88-8208 | AF152269 | EV18 | EV18 | 100 | 79.1 | EV18 | 79.3 | EV18 | 78 | 70.2 | EV18 |
| OK89-9448 | AF152282 | EV18 | EV18 | 100 | 78.9 | EV18 | 79.1 | EV18 | 77.8 | 70.2 | EV18 |
| OR85-6323 | AF152284 | EV18 | EV18 | 100 | 80.3 | EV18 | 80.2 | EV18 | 78.9 | 70.2 | EV18 |
| SC87-7477 | AF152292 | EV18 | EV18 | 100 | 82.1 | EV18 | 82 | EV18 | 80.9 | 70.2 | EV18 |
| R100 | AF252187 | EV18 | EV18 | 100 | 76.4 | EV18 | 76.9 | EV18 | 75.4 | 68 | EV18 |
| 270N97 | AF252188 | EV18 | EV18 | 100 | 79.4 | EV18 | 79.6 | EV18 | 78.2 | 67.5 | EV18 |
| M256 | AF290906 | EV2 | EV2 | 100 | 77.9 | EV2 | 82.6 | EV2 | 81.5 | 71.4 | EV2 |
| RI94-1959 | AF081633 | EV21 | EV21 | 100 | 79.8 | EV21 | 79.8 | EV21 | 78.5 | 70.8 | EV21 |
| NC83-5515 | AF152275 | EV24 | EV24 | 100 | 78 | EV24 | 78.1 | EV24 | 76.7 | 69.9 | EV24 |
| NC84-5530 | AF152276 | EV24 | EV24 | 100 | 78.2 | EV24 | 78.3 | EV24 | 77 | 69.6 | EV24 |
| MD92-1649 | AF081615 | EV25 | EV25 | 100 | 78.5 | EV25 | 78.9 | EV25 | 77.5 | 69 | EV25 |
| MN94-1828 | AF081618 | EV25 | EV25 | 100 | 78.3 | EV25 | 78.7 | EV25 | 77.3 | 68.7 | EV25 |
| MO93-1808 | AF081619 | EV25 | EV25 | 100 | 78.9 | EV25 | 79.4 | EV25 | 78 | 68.3 | EV25 |
| OR93-1817 | AF081627 | EV25 | EV25 | 100 | 79.1 | EV25 | 79.6 | EV25 | 78.2 | 69 | EV25 |
| HON86-6843 | AF152261 | EV25 | EV25 | 100 | 80.9 | EV25 | 80.9 | EV25 | 79.7 | 69.8 | EV25 |
| NC84-5531 | AF152277 | EV25 | EV25 | 100 | 79.4 | EV25 | 79.6 | EV25 | 78.2 | 70.6 | EV25 |
| 1498N98 | AF252181 | EV25 | EV25 | 100 | 81.8 | EV25 | 81.8 | EV25 | 80.6 | 68.9 | EV25 |
| 1573NE99 | AF290909 | EV25 | EV25 | 100 | 81.8 | EV25 | 81.8 | EV25 | 80.3 | 69.1 | EV25 |
| T185 | AF290896 | EV29 | EV29 | 100 | 79.1 | EV29 | 70.4 | EV6 | 81.1 | 70.1 | EV29 |
| MD88-8157 | AF152266 | EV3 | EV3 | 100 | 77.1 | EV3 | 77.5 | EV3 | 76 | 69.1 | EV3 |
| MT87-7421 | AF152274 | EV3 | EV3 | 100 | 76 | EV3 | 78.3 | EV3 | 77 | 69.1 | EV3 |
| 1431N98 | AF252171 | EV30 | EV30 | 100 | 87.8 | EV30 | 88.5 | EV30 | 87.1 | 69.9 | EV30 |
| 1432N98 | AF252172 | EV30 | EV30 | 100 | 87.8 | EV30 | 88.5 | EV30 | 87.7 | 69.9 | EV30 |
| HC56 | AF252173 | EV30 | EV30 | 100 | 86.7 | EV30 | 87.3 | EV30 | 86 | 69.9 | EV30 |
| 127N88 | AF252174 | EV30 | EV30 | 100 | 88.7 | EV30 | 88.9 | EV30 | 88.2 | 70.1 | EV30 |
| PER98-2558 | AF081632 | EV33 | EV33 | 100 | 80.6 | EV33 | 80.6 | EV33 | 79.2 | 70.4 | EV33 |
| PA88-8412 | AF152286 | EV4 | EV4 | 100 | 83.3 | EV4 | 83.3 | EV4 | 82.1 | 75.6 | EV4 |
| T99 | AF252180 | EV4 | EV4 | 100 | 82.1 | EV4 | 82 | EV4 | 80.8 | 73 | EV4 |
| CT96-2181 | AF081602 | EV5 | EV5 | 100 | 87.4 | EV5 | 87.3 | EV5 | 86.5 | 72.5 | EV5 |
| NM95-2070 | AF081625 | EV6 | EV6 | 100 | 80.1 | EV6 | 80.5 | EV6 | 79.2 | 69.7 | EV6 |
| 1201C99 | AF290897 | EV6 | EV6 | 100 | 81 | EV6 | 80.4 | EV6 | 80.2 | 71.7 | EV6 |
| 1351N98 | AF252186 | EV7 | EV7 | 100 | 82.3 | EV7 | 82.2 | EV7 | 81.1 | 69.8 | EV7 |
| AR95-2139 | AF081596 | EV9 | EV9 | 100 | 83.3 | EV9 | 83.3 | EV9 | 82.2 | 69.2 | EV9 |
| NC92-1612 | AF081620 | EV9 | EV9 | 100 | 84.9 | EV9 | 84.8 | EV9 | 83.8 | 69.9 | EV9 |
| WI95-2151 | AF081645 | EV9 | EV9 | 100 | 84 | EV9 | 83.9 | EV9 | 78.2 | 69 | EV9 |
| SE74 | AF252166 | EV9 | EV9 | 100 | 83.7 | EV9 | 83.9 | EV9 | 82.3 | 68.5 | EV9 |
| MP211 | AF252167 | EV9 | EV9 | 100 | 83.3 | EV9 | 83.5 | EV9 | 82.4 | 68.8 | EV9 |
| T94 | AF252168 | EV9 | EV9 | 100 | 82.6 | EV9 | 82.8 | EV9 | 81.7 | 68.5 | EV9 |
| T22 | AF252175 | EV9 | EV9 | 100 | 86.4 | EV9 | 86.4 | EV9 | 85.5 | 69.4 | EV9 |
| R1192 | AF252176 | EV9 | EV9 | 100 | 84.2 | EV9 | 84.2 | EV9 | 83.1 | 69.7 | EV9 |
| M8/72 | D17604 | ENV70 | ENV70 | 100 | 98.7 | ENV70 | 96.7 | ENV70 | 98.6 | 68.9 | ENV70 |
| V1250/81 | D17611 | ENV70 | ENV70 | 100 | 93.2 | ENV70 | 93.4 | ENV70 | 93.1 | 69.5 | ENV70 |
| AL88-8149 | AF152248 | ENV71 | ENV71 | 100 | 99.1 | ENV71 | 99 | ENV71 | 99 | 70.7 | ENV71 |
| MD87-9256 | AF152267 | ENV71 | ENV71 | 100 | 98.6 | ENV71 | 98.6 | ENV71 | 98.5 | 70.2 | ENV71 |
| NM90-9873 | AF152278 | ENV71 | ENV71 | 100 | 83.6 | ENV71 | 83.6 | ENV71 | 83 | 66 | ENV71 |
| OK89-9243 | AF152279 | ENV71 | ENV71 | 100 | 84.4 | ENV71 | 84.2 | ENV71 | 83.7 | 66.2 | ENV71 |
| OK89-9452 | AF152283 | ENV71 | ENV71 | 100 | 83.4 | ENV71 | 83.3 | ENV71 | 82.7 | 65.9 | ENV71 |
| TX89-9166 | AF152297 | ENV71 | ENV71 | 100 | 83.6 | ENV71 | 83.6 | ENV71 | 83 | 65.8 | ENV71 |
| T100 | AF252190 | ENV71 | ENV71 | 100 | 84 | ENV71 | 84 | ENV71 | 83.4 | 66.7 | ENV71 |
| OK85-6388 | AF152280 | NT | UNT-GenB | 100 | 72.3 | CBV6 | 70.8 | EV1 | 67.5 | 67.5 | EV4 |
| VA86-6765 | AF152298 | NT | UNT-GenB | 100 | 73.5 | ENV69 | 72.1 | EV1 | 70.3 | 68.8 | EV1 |
| CT87-7122 | AF152254 | NT | UNT-GenB | 100 | 74.1 | CBV6 | 69.7 | EV8 | 67.7 | 67.7 | EV3 |
| CT87-7123 | AF152255 | NT | UNT-GenB | 100 | 74.1 | CBV6 | 69.7 | EV8 | 67.5 | 67.3 | EV4 |
| 1827N98 | AF252165 | PV1 | PV1 | 100 | 100 | PV1 | 100 | PV1 | 100 | 71.2 | PV1 |
Abbreviations: CAV, coxsackie A virus; CBV, coxsackie B virus; PV, poliovirus; NA, isolate not available for neutralization test. EV was directly detected and sequenced from the clinical sample; ND, not identified (bootstrap value, <65%); UNT, no reference strain was grouped into same cluster; Gen B, strain located in Cluster B of EV whole genome classification. NT, untypeable enterovirus (19).
The field clinical samples added in this work are in boldface.
The neutralization serotypes of the 64 sequences obtained from GenBank are as in the corresponding publications (17, 19).
Highest value obtained after the application of equation 1 for the chosen alignment.
Results that differing from the neutralization serotype are in boldface.
Highest value obtained by the NW-GCG similarity test with GOP equal to 50 and GEP equal to 3. Values <75% are shaded in boldface since they do not meet the criterion for identification.
Values higher than 70% are in boldface since they do not achieve the criterion for identification. Values higher than 75% are shaded in boldface and italic.
Virus isolation and neutralization.
Isolation of EV in cell cultures was attempted for every clinical specimen. The specimens were inoculated in human embryonic fibroblast, buffalo green monkey kidney, human rhabdomyosarcoma, and human lung carcinoma (A549) cell lines. The isolates were typed by neutralization with a panel of antiserum pools (Lim-Benyesh-Melnick) and by evaluation of virus growth inhibition.
Extraction, amplification, and sequencing.
Nucleic acids from clinical samples and isolates were extracted as described previously (4). The dried pellet was resuspended in 15 μl of RNase-free sterile water (Sigma Chemical Co., St. Louis, Mo.) and used immediately. A reverse transcription-nested PCR was used to amplify the VP1 genome region, and a 656-bp product (5) which was located between nucleotide 2874 and nucleotide 3529 of echovirus type 9 strain Barty (GenBank accession no. X92886) was obtained.
Cycle sequencing reactions were performed with the Big Dye terminator kit (Perkin-Elmer Applied Biosystems) by using the same primers used for the nested PCR. Raw sequence data were first analyzed with CHROMAS software (version 1.3; C. McCarthy, 1996, Griffith University, Brisbane, Queensland, Australia), and the forward and reverse sequence data for each sample were aligned by using the MegAlign program (DNASTAR Inc. Software, Madison, Wis.) to obtain the final consensus sequence.
EV sequences used.
The sequences of 40 original EV strains were used. Another 64 EV sequences were obtained from the GenBank database, and their accession numbers are included in Table 1. VP1 reference strains from a previously described database (19) were included in every analysis method. The 2A region and the primer sequences were excised from the sequences before analysis.
Pairwise alignment analysis. (i) Martinez-Needleman and Wunsch algorithm.
The Martinez (11)-Needleman and Wunsch (16) algorithm (M-NW similarity test) is implemented by the MegAlign program of the DNASTAR commercial package. A similarity score between each pair of sequences was obtained manually after sequential pairwise alignment (M-NW similarity test) was performed. The quality of the alignment could not be measured, and the gap opening penalty (GOP) and the gap extension penalty (GEP) values were used as default values.
(ii) Needleman and Wunsch algorithm.
The Needleman and Wunsch (16) algorithm (NW similarity test) is implemented by the Gap program of the GCG package (NW-GCG similarity test), and it was performed with both nucleotide and amino acid sequences.
Given that strains of the same serotype rarely have gaps in their alignments, increasing the GOP and GEP parameters will not modify the similarity score for very related sequences but will decrease it for more diverging sequences. Therefore, to allow the separation of the peak of the similarity score for strains of the same serotype from the peak of the similarity score for strains of related serotypes, GOP options were set as default, double, and quadruple (50, 100, and 200, respectively) with GEP options of 3, 10, and 20, respectively. The randomization option was set on 100 to process the quality control of the alignment.
The quality of the alignment was recorded as the difference between the quality of the best alignment (V) and the mean of the Monte Carlo randomized alignments (m) divided by the standard deviation (SD) of the Monte Carlo simulation ([V − m]/SD), as established previously (2). Significance scores above 15 SDs indicated a nearly ideal alignment, while scores above 5 SDs suggested a good alignment and scores below 5 SDs were regarded with caution.
The conditions used to identify the sequences in both nucleotide schemes were the same as those described previously (19). Even though these conditions had been determined for the GCG program, the same criteria were used for the DNASTAR program since both programs are based on the same algorithm.
A sequence identity of ≥75% for any EV prototype strain indicated a homologous serotype, provided that the second-highest score was <70% (next closest serotype). A highest score between 70 and 75% or a second-highest score greater than 70% indicated a tentative identification, while a highest score lower than 70% indicated a nonmatching sequence.
Amino acid analysis conditions were set on the basis of a highest score of 90% and a second-highest score of 85%. These cutoff scores were inferred from a frequency distribution graphic of the pairwise sequence analysis scores when the intraserotype peak differentiated (25). The same kind of analysis had been used before for potyvirus classification (25).
Multiple-sequence alignment analysis.
Two widely used multiple-sequence alignment programs, the Clustal W program (version 1.81) and the Pileup program, were compared (23). The Pileup program (which is part of the GCG package) is a progressive pairwise alignment program which implements a simplified version of the Feng and Doolittle algorithm (8a). Sequence similarity is used to cluster sequences, and then a dendrogram is constructed by the unweighted pair group method with arithmetic means, which orders the subsequent pairwise alignments based on the method of Needleman and Wunsch (16).
The Clustal W program (the slow, accurate option) is also based on the Feng and Doolitle algorithm but is improved through dynamic assignment of penalties. The Clustal W program varies gap penalties in relation to sites along the sequence and the relative evolutionary distance among sequences (and subsets of sequences), and the dendrogram is constructed by the neighbor joining method. GOP and GEP costs can be specified separately for the pairwise sequence and multiple-sequence alignments, while the delay divergent sequences option delays the inclusion of sequences with less than the specified sequence identity until the most similar sequences are aligned.
The key parameters for both programs are the cost assigned to the opening of the gap (gap cost), the cost for extension of a gap (gap-length cost) (total gap cost = gap cost + [gap−length cost × gap length]), and the delay divergent sequence option, which is available only in the Clustal W program. These parameters were modified to evaluate the performances of the programs. The alignments of the deduced amino acid sequences of all strains were first compared. The methods were then tested by varying the GOP value between 5 and 100 and by using different GEP values (1, 5, and 10 for the Pileup program and 6.66, 20, 100, and 200 for the Clustal W program). A similar analysis was performed for the nucleotide sequences with the Pileup program and a GEP value of 0.3. The Clustal W program delay divergent option was also evaluated. This value was set at 80 since the limit value of the identity score for two strains of the same serotype was estimated to be 75%.
The quality of the alignments should also be assessed in order to evaluate and compare alignment programs. Two different scores were calculated to estimate the quality of the alignment. The sum-of-pairs score (SPS) increases with the number of sequences correctly aligned and is used to determine the abilities of the programs to align some or all sequences within an alignment. The column score (CS) is a binary score which tests the abilities of the programs to align all the sequences correctly. Both scores were previously developed and tested to evaluate multiple-sequence alignment programs (24), but no reference alignment is considered to normalize the corresponding results in the present study.
Finally, the most consistent alignment was chosen to calculate the homology index to identify the serotype. For multiple-sequence alignments, this score for each pair of sequences was calculated as follows:
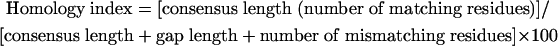 |
A homology index higher than 75% assigned a strain to a serotype.
Phylogenetic approach.
The most consistent alignment was used as the input for the phylogenetic methods. Phylogenetic relationships were inferred with the DNAPARS program (as a character state method) and the DNADIST or PROTDIST program (as a distance method) and then with the NEIGHBOUR and KITSCH programs (PHYLIP, version 3.57) (8). The PROTPARS, DNAML, and FITCH programs were not evaluated due to the excessive analysis time needed for large numbers of sequences with a common personal computer system. The statistical significance of phylogenies was estimated by using the SEQBOOT program by bootstrap analysis with 100 pseudoreplicate data sets. Only groupings with bootstrap values higher than 65 were considered significant. The consensus tree obtained with CONSENSE was plotted by TREEVIEW (version 1.2; R. Page, 1995, Glasgow, United Kingdom).
Nucleotide sequence accession numbers.
The original sequences detected in the present study were submitted to the GenBank sequence database under accession numbers AF252165 to AF252190 and AF290896 to AF290909.
RESULTS
Table 1 describes the viruses analyzed and the values obtained for their sequences by each analysis method.
Pairwise sequence alignment methods. (i) Quality analysis.
The quality analysis showed that each program obtained different results. Since the DNASTAR package did not provide an option to perform the quality analysis, the results were compared with these obtained with the GCG package on a pairwise basis to test its quality. One of the most divergent results was observed when isolate R100, identified by seroneutralization as EV type 18 (EV18), was aligned with the reference strain, EV18 Metcalf. The overall length of the Gap alignment was 419 bp, while the MegAlign alignment had an overall length of 531 bp. No gaps were inserted by the Gap program, and the similarity score was 75.3%. On the other hand, the MegAlign program inserted 224 gaps, resulting in a similarity score of 46.3%. The same discordant insertion of gaps was found frequently in other pairs of alignments (data not shown).
The randomization option of the Gap program was used to evaluate the quality of the result by performing a Monte Carlo simulation. The more similar or related the sequences were, the higher the quality of the alignment was (data not shown).
The parameters selected for the analysis of nucleotide sequences were GOP equal to 50 and GEP equal to 3, considering the fact that the lowest-quality scores were 4.99, 2.74, and 1.09 with GOP equal to 50, 100, and 200, respectively (Table 2). By using the selected options, only one alignment resulted in a score lower than 5 (4.99 [1 of 8,216 comparisons; 0.0001%]), while 170 (2.1%) and 946 (11.5%) of the pairwise alignments had scores lower than 5 by using GOP values of 100 and 200, respectively.
TABLE 2.
Summary of resultsa
| Method and parameter values | Quality control
|
Identification
|
Method performance
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Quality (boot-strapping)
|
No. of samples with quality <5 (boot-strapping <65) | No. of strains with:
|
No. of strains affected by multiple-reference-strain factor (n = 28) | Failure to identify:
|
No. of strains correctly identifiedb | % Correctly identified | ||||||
| Maximum | Minimum | Highest scores <75% nt or <90% aa | Second-highest score >70% nt or >85% aa | Highest scores not correspond-ing to its serotype | Second-highest scores >75% | EV13 strain | New sero-type | |||||
| Pairwise sequence alignment | ||||||||||||
| DNASTAR, nucleotides (default GOP and GEP values) | NA | NA | NA | 24 | 50 | 4 | 4 | 13 | Yes | Yes | 30 | 28.85 |
| GCG | ||||||||||||
| Nucleotides | ||||||||||||
| GOP = 50, GEP = 3 | 77.97 | 4.99 | 1 | 5 | 46 | 0 | 1 | 1 | Yes | Yes | 52 | 50.00 |
| GOP = 100, GEP = 10 | 77.24 | 2.75 | 170 | 5 | 45 | 0 | 1 | 1 | Yes | Yes | 51 | 49.03 |
| GOP = 200, GEP = 20 | 70.03 | 1.09 | 946 | 5 | 45 | 0 | 1 | 1 | Yes | Yes | 51 | 49.03 |
| Proteins (GOP = 8, GEP = 2) | 145 | 24.67 | 0 | 4 | 25 | 0 | 0 | 0 | No | Yes | 75 | 72.10 |
| Multiple-sequence alignment | ||||||||||||
| Pileup, nucleotides (GOP = 5, GEP = 1) | NA | NA | NA | 6 | NA | 0 | 2 | 0 | Yes | Yes | 96 | 92.30 |
| Clustal W, nucleotides (GOP = 100, GEP = 100) | NA | NA | NA | 5 | NA | 0 | 1 | 0 | Yes | Yes | 98 | 94.20 |
| Phylogenetic approach (PHYLIP package with nucleotides) | 100 | 86 | 0 | NA | NA | NA | NA | 0 | Yes | No | 103 | 99.04 |
Abbreviations: NA, not applicable; nt, nucleotides; aa, amino acids.
Number of strains among 104 strains tested that fulfilled all established criteria.
Finally, selection of parameters for amino acid alignments was not decisive since all similarity scores had high-quality values (lowest score, 24.67).
(ii) Similarity test as method for EV identification.
The values obtained by the NW-GCG similarity test of nucleotide sequences from clinical samples are shown in Table 1. Only the EV13 strain and four other field strains (untypeable strains OK85-6388, VA86-6765, CT87-7122, and CT87-7123) resulted in similarities scores <75%, as reported previously (19).
However, the second-highest scores were higher than 70% in more than 60 cases under any condition, resulting in more than 45 different untypeable EV serotypes (Table 2). This was also true for isolate PA88-8412, whose second-highest score was also higher than 75% (the score for strain EV8 Bryson was 75.6%, which was independent of the parameter chosen).
On the other hand, deduced amino acid sequence analysis grouped all tested strains except the untypeable strains with their respective reference strains (including the EV13 field strain). Again, 25 different strains had in second-highest scores higher than 85% (data not shown).
The similarity values obtained by the M-NW similarity test were lower than the values by the NW-GCG similarity test (mean score by the NW-GCG similarity test of 81.5% versus mean score by the M-NW similarity test of 78.8%). A total of 19.2% of the strains, including the untypeable strains, could not be typed because of low similarity scores or because the score attributed the strain to a serotype different from the serotype established by the other methods (Table 3). In conclusion, the M-NW similarity test appears to be less suitable for EV identification.
TABLE 3.
Analysis of discordant M-NW similarity test valuesa
| Clinical sample | Neutralization serotype | Highest M-NW similarity index (%) with respec-tive reference strainb | Highest M-NW similarity index (%)c | Genotype determined by M-NW similarity indexd |
|---|---|---|---|---|
| GV34 | EV12 | 71.8 | 71.8 | EV12 |
| M250 | EV12 | 73.7 | 73.7 | EV12 |
| 270N97 | EV18 | 57.4 | 66.5 | EV6 Burgess |
| R100 | EV18 | 46.2 | 67.4 | EV15CH96 |
| MD92-1649 | EV25 | 74.9 | 74.9 | EV25 |
| MN94-1828 | EV25 | 74.1 | 74.1 | EV25 |
| PA94-5753 | CAV16 | 52.3 | 65.9 | ENV71 BrCr |
| GA95-2095 | CAV16 | 51.8 | 66.7 | ENV71 BrCr |
| TX95-2147 | CAV16 | 74.5 | 74.5 | CAV16 |
| NC83-5515 | EV24 | 75 | 75 | EV24 |
| TAI84-5839 | CAV16 | 73.8 | 73.8 | CAV16 |
| OK85-6388 | UNT | NA | 66.4 | EV7 |
| VA86-6765 | UNT | NA | 67.6 | EV24 |
| VA86-6776 | EV13 | 71.5 | 71.5 | EV13 |
| CT87-7122 | UNT | NA | 68.2 | EV15 CH96 |
| CT87-7123 | UNT | NA | 68.2 | EV15 CH96 |
| MT87-7421 | EV3 | 74.1 | 74.1 | EV3 |
| GUT88-8020 | CAV21 | 74 | 74 | CAV21 |
| MD88-8157 | EV3 | 73 | 73 | EV3 |
| GUT88-8436 | CAV21 | 74.3 | 74.3 | CAV21 |
| PA89-9262 | CAV16 | 74.8 | 74.8 | CAV16 |
| OK89-9448 | EV18 | 73.9 | 73.9 | EV18 |
| RI94-1959 | EV21 | 74.4 | 74.4 | EV21 |
| CB6IS2 | CBV6 | 73.6 | 73.6 | CBV6 |
Abbreviations: UNT, “untypeable” EV. See footnote a of Table 1 for definitions of the other abbreviations.
Values lower than 75% are in boldface.
Results higher than those obtained for the corresponding reference strain are in boldface.
Serotype determined by the highest score obtained elsewhere (17). Results that differed from the neutralization serotype are in boldface.
(iii) Analysis of test results over multiple reference strain groups.
Samples corresponding to EV serotypes represented by several reference strains were compared by both similarity tests. The M-NW similarity test could not correctly correlate the field strain to another reference strain of the same serotype in 13 (46.4%) cases. For example, the sequences of EV30 strains 127N88, 1431N98, 1432N98, and HC56 resulted in scores of 72.3, 58.3, 58.3, and 74.1%, respectively, with EV30 reference strain PR17. Strains 127N88 and HC56 also had similarity scores of 74.1 and 74.3%, respectively, with reference strain EV30 Giles. On the other hand, only one field strain (strain 127N88) had a value lower than 75% (74.9%) when its sequence was compared with sequence of EV30 PR17 by the NW-GCG nucleotide similarity test with any combination of parameters. Moreover, the NW-GCG amino acid similarity test was not affected by the multiple reference strain factor since all the strains in this group correlated with their corresponding reference strains.
Multiple-sequence alignment methods. (i) Quality analysis. (a) Deduced amino acid sequences.
The Pileup program generated multiple-sequence alignments with very different lengths and qualities. The Clustal W program generated alignments with more conserved lengths and qualities for different parameters instead. The performances measured as the overall similarity (SPS/length) and the CSs for the different methods of analysis with different GEP and GOP parameters are shown in Fig. 1a and b.
FIG. 1.

Results from each test comparing the performances of the programs. The program tested the combinations of GEP and GOP parameters shown for each indicator with each alignment test. The program and the GEP parameters used are shown in the rows. Columns show the GOP parameter for each alignment. The box to the right of each comparison describes the symbols used for the scores. (a) Column score result for deduced amino acid alignments. (b) Percent overall similarity result for each amino acid alignment. (c) Column score result for nucleotide alignments. (d) Percent overall similarity result for each nucleotide alignment. (e) Variation in overall similarity of the alignment considering different delay divergent values with the Clustal W program. The GOP and GEP scores were set equal to 100. The default delay divergent value was increased by 5 units up to a value of 95.
The Clustal W program generated the most conserved and consistent alignment when a GOP value of 75 and GEP value of 6.66 were used. The resulting alignment had an overall similarity of 43.52% and a length of 151 amino acids.
(b) Nucleotide sequences.
For the Clustal W program, the alignment of the divergent sequences was delayed with the delay divergent option (which was varied between 30 and 95) until all the remaining sequences were aligned. The effect of this optional parameter in the alignment quality is presented in Fig. 1e, showing that the best alignments were obtained with values higher than 85. Thus, the conditions mentioned above were tested by varying only the values of GOP and GEP. Again, the Clustal W program generated multiple-sequence alignments with more conserved lengths and qualities than the Pileup program for the different parameters tested (Fig. 1c and d).
The Clustal W program generated the most conserved and consistent alignment with GOP equal to 100 and GEP equal to 100 and a delay divergent option of 90%. The alignment had an overall similarity of 54.78% and was 453 nucleotides long. Instead, GOP equal to 5 and GEP equal to 0.3 or 1 generated the best alignment with the Pileup program, with an overall similarity of 54.61% and a length of 452 nucleotides. Other combinations of parameter values generated very different results.
(ii) Homology index as method of EV identification.
The results obtained by use of the homology index are shown in Table 1. The homology test with the Clustal W program identified every serotype but an EV13 strain (homology indices with prototype strains EV13 Del Carmen and ENV69 Tolucal, 74.2 and 73.5%, respectively) and the untypeable strains with values higher than 75%. The PA88-8412 sequence presented the same problem encountered with the pairwise sequence analysis, showing a second-highest score higher than 75%.
The homology test with the Pileup program also identified every serotype but an EV13 strain, the untypeable strains, and field strain T185. Strain T185, typed as EV29, and was correctly identified by the other methods studied (Table 1), while the score obtained with the Pileup program with its respective reference strain was 30.6%. Again, the PA88-8412 sequence resulted in a second-highest score higher than 75% (the score for EV8 Bryson was 77.5%).
(iii) Analysis of test results over multiple reference strain groups.
Neither the homology index obtained with the Clustal W program nor the homology index obtained with the Pileup program was affected by the multiple reference strain group factor since all the field strains tested correlated with their corresponding reference strains.
Phylogenetic methods.
All the phylogenetic methods generated the same tree topology with different bootstrap values, and the one with higher bootstrap values was obtained with the KITSCH program (Fig. 2). Bootstrap values for each serotype cluster are shown in Table 1. Four separate groups supported by high bootstrap values clustered similarly to other previously published studies (genogroups A, B, C, and D) (15, 20–22). The method identified all serotypes except an EV13 isolate (VA86-6776). This isolate was correctly grouped with its reference strain with a not significant bootstrap value (42%), while it was grouped with ENV69 and EV13 reference strains with higher values. An untypeable EV serotype that could not be identified by other methods of analysis (19) was correctly grouped into genogroup B of the EV genus with a high bootstrap value.
FIG. 2.

Consensus phylogenetic tree constructed with the PHYLIP package for 104 EV sequences. Alignments were obtained with the Clustal W program. The statistical significance of the phylogenies constructed was estimated with the SEQBOOT program. The 100 pseudoreplicate data sets obtained were analyzed with the DNADIST program with the parameters of the Kimura 2 model of nucleotide substitution. The observed nucleotide distance matrix was then processed with the KITSCH program, and the tree generated was treated with the CONSENSE program. The tree was displayed with the TREEVIEW program. The numbers at the nodes represent the percentage of 100 bootstrap pseudoreplicates that contained the cluster distal to the node.
Finally, the remaining serotypes, including PA88-8412, clustered with very high bootstrap values.
DISCUSSION
Analysis of sequence information is becoming the major approach to the study and identification of human EVs. Three different levels of analysis of sequence information are discussed in this report: a pairwise sequence alignment of homologues, multiple-sequence alignment of homologues, and phylogenetic reconstruction of trees. The results showed that each analysis method has different EV identification results and that the accuracies of the results depend on the parameters chosen (Table 2).
The complexity of this genus makes the correct identification of EVs difficult, and problems associated with the seroneutralization method or analysis of untypeable EV strains appear frequently. Nevertheless, although identification of EVs through analysis of the VP1 genome region has been recognized as the best choice, it is also necessary to achieve consensus on a common system of sequence analysis.
The alignment of two sequences is critical for all sequence analysis methods. Differences in alignments and accuracies of methods have previously been reported with large sets of protein sequences with several programs (12). However, the biological validities of nucleotide sequence alignments have been studied less. This is because DNA sequences (which have 4 character states) are more difficult to align than amino acid sequences (which have 20 character states). It was also demonstrated for rRNA data and protein-encoding sequences that alignment accuracy depends on the alignment program and the parameters chosen (14). As was expected, the more that insertion or deletion events are incorporated, the greater the differences among sequences alignments are.
Like other steps in phylogenetic reconstruction, automated sequence alignment requires selection of the most appropriate parameters. Therefore, published studies should describe the parameter values used to create the alignments if they are to be repeatable. In addition, each method of analysis should provide an assessment of quality.
Pairwise nucleotide sequence alignment methods were very different. Although both programs use the same algorithm, the results obtained with the DNASTAR program were very poor.
Pairwise sequence alignment methods provide a simple way to assess the alignment quality by the Monte Carlo approach. The software developer indicates that the algorithm used in the MegAlign program is a slight modification (11) of the algorithm originally designed by Needleman and Wunsch (16) for proteins. This allows the user to align two nucleotide sequences, first by an approach described by Martinez (11), which identifies regions with perfect matches, and then by the method of Needleman and Wunsch (16), which optimizes the fit between perfect matches. The MegAlign program does not provide a quality assessment option, and the results obtained were different from those obtained with the Gap program. An example of this is for isolate R100, which was correctly identified by the Gap program, while the MegAlign program showed a very low similarity index (46.2%) in tests with the same reference strain. In addition, the M-NW similarity test score resulted in the worst performance, and only 32.7% of the strains fulfilled the criteria for identification. Therefore, the MegAlign program is useless for the identification of EVs by use of this genome region.
Instead, the GCG package provides an option that assesses the quality of the alignment obtained with the Gap program. The Monte Carlo simulation determined that the best pair of parameter values was GOP equal to 50 and GEP equal to 3. Other conditions resulted in scores for quality controls lower than the proposed 5 SDs for several pairs of alignments.
By use of these parameters, the highest scores were higher than 75% for 99 of 104 (94.6%) clinical strains. However, 46 clinical isolates could not be identified since they did not fulfill the second-highest-score criterion. Added to that isolate PA88-8412 had a score higher than 75% for two different serotypes, including the correct one (EV4). Scores higher than 70% slightly decreased as the GOP increased (data not shown), showing that these pairwise sequence alignments were relating divergent pairs of sequences, as suggested previously (17). Although the parameters had to be varied to increase the penalty to the similarity score for divergent sequences, the distances between similarity scores were not enough to increase the number of identifiable isolates.
In summary, this pairwise sequence alignment method did not allow the identification of 52 of 104 (50%) strains mainly due to problems related to the proposed criteria. It seems that the criterion of a second-highest score lower than 70% established for identification (19) is very stringent, since all the strains that did not fulfill this criterion had highest scores greater than 75% and the correct serotype.
An even more disappointing aspect was that the pattern of gaps in multiple-sequence alignments was often inconsistent with that in pairwise sequence alignments; thus, the optimal alignment between the two closest sequences was often altered in the presence of a third or a fourth sequence.
Analysis of deduced protein sequences with the Gap program improved the results. Unfortunately, the four untypeable isolates also could not be identified, but the EV13 field strain was correctly identified. The multiple-reference-strain effect was not observed, and the quality analysis had higher values. In this case, the second-highest-score criterion should be also discarded since 25 field strains would have been correctly identified if this criterion had not been considered. The analysis of deduced amino acid sequences could constitute a better approach than the analysis of nucleotide sequences.
The greater precision of the Clustal W program and the more consistent performance of the Clustal W program over that of the Pileup program is due to more sophisticated features such as neighbor joining that guide the alignments. Based on the same reasons considered for pairwise sequence alignment, two different methods were used to evaluate the accuracies of the alignments obtained.
Since length is one of the parameters that allows the user to test the quality of the alignment at first sight, the deduced amino acid sequences were evaluated by considering that if the overall length of a protein alignment is known, the nucleotide sequence alignment length could be calculated (overall amino acid length times 3). The results were different for the two methods. The alignments obtained with the Clustal W program were more consistent and conserved, and better methods to improve quality with the delay divergent sequence option were provided. Moreover, both methods of quality control, SPS and CS, were more consistent for the Clustal W program than for the Pileup program. The alignments of the nucleotide sequences obtained with the Clustal W program were also more reliable than those obtained with the Pileup program. Although the Clustal W program generated the exact 453-bp alignment, the Pileup program was not able to generate an alignment with the predicted overall length. Despite that, the most important features of the Clustal W program are its reproducibility and reliability, given that the resulting alignments were similar under all conditions selected.
The Clustal W program homology index identified all except six clinical strains; while the Pileup program homology index did not identify seven strains. The second-highest-score criterion should not be considered for these tests since their alignments are clearly more reliable than pairwise sequence alignments. In summary, the Clustal W program homology index was able to identify 98 of 104 (94.2%) field strains. Added to that, the Clustal W program is freely available on the Internet and can be performed with a wide variety of computer platforms or with a common personal computer in less than a half hour, while the GCG program is an expensive commercial package that must be run on a mainframe computer system and some Unix programming knowledge is needed to perform the process automatically. This should be considered when proposing a method easily transferable to all reference laboratories.
In any phylogenetic study, alignment of a less conserved region and regions with variable lengths is often problematic, even when sequence divergence is moderate. The alignment process is critical in phylogenetic analyses since inclusion of ambiguously aligned regions could result in erroneous patterns of branching, while removal of such regions could reduce the resolution. To avoid this reduction in the resolution, the terminal regions of the VP1 sequences were included, because it is known that the VP1 region is very divergent and generates a variable-length alignment.
Phylogenetic reconstruction allowed not only the correct clustering of all serotypes with a high bootstrap value (except for the EV13 field strain) but also the prediction and clustering of the new serotypes. This is shown for the cluster of untypeable isolates from clinical samples. Despite the location of the untypeable serotype near the EV20 JV1 reference strain, the cluster is clearly composed of strains of a new serotype since the observed nucleotide sequence distance among strains of the untypeable serotype is lower than 0.20 and the distance between strains of the untypeable serotype and EV20 strains is higher than 0.45 (data not shown).
Identification of the EV13 field strain is a special case since it was clustered with both EV13 and enterovirus type 69 (ENV69) reference strains with high bootstrap values. It was also the only strain that could not be correctly identified by all the methods tested. This could mean that the three strains are of the same serotype, but this hypothesis should be further evaluated with more sequence data from field strains of the EV13 or ENV69 serotype.
In addition, 16 serotypes of EV were not represented in the data. Field isolates of these serotypes are not available in GenBank or in the set of samples sequenced so far. The results should be evaluated again when fields samples of these serotypes are available.
The results of the present study indicate that phylogenetic inference is the best method. Multiple-sequence alignment programs or pairwise sequence alignment of deduced amino acid sequences appears to be the second choice, although adoption of universal criteria for selection of parameters and assessment of the quality of the alignment is needed before multiple-sequence alignment programs can be used. The parameters selected should be also reported in order to repeat and compare the experiments.
Finally, the present study shows that accurate identification of EVs can easily be achieved directly from clinical samples with freely available software.
Acknowledgments
We thank Jorge Gomez for continuous support during this work and for helpful discussions and critical reading of the manuscript.
The work was supported by institute funds from INEI-ANLIS “Dr. Carlos G. Malbrán” and by Fondo de Investigaciones Sanitarias grant FIS98/0229 from the Spanish Ministry of Health. I.C. is a postdoctoral fellow funded by the Instituto de Salud Carlos III, Becas de Perfeccionamiento.
REFERENCES
- 1.Arola, A., J. Santti, O. Ruuskanen, P. Halonen, and T. Hyypia. 1996. Identification of enteroviruses in clinical specimens by competitive PCR followed by genetic typing using sequence analysis. J. Clin. Microbiol. 34:313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barton, G. 1990. Protein multiple sequence alignment and flexible pattern matching. Methods Enzymol. 183:403–428. [DOI] [PubMed] [Google Scholar]
- 3.Burland, T. G. 2000. DNASTAR’s Lasergene sequence analysis software. Methods Mol. Biol. 132:71–91. [DOI] [PubMed] [Google Scholar]
- 4.Casas, I., P. E. Klapper, G. M. Cleator, J. E. Echevarría, A. Tenorio, and J. M. Echevarría. 1995. Two different PCR assays to detect enteroviral RNA in CSF samples from patients with acute aseptic meningitis. J. Med. Virol. 47:378–385. [DOI] [PubMed] [Google Scholar]
- 5.Casas, I. P., G. Trallero, D. Cisterna, M. Freire, and A. Tenorio. 2001. Molecular characterization of human enteroviruses in clinical samples: comparison between VP2, VP1, and RNA polymerase regions using RT nested PCR assays and direct sequencing of products. J. Med. Virol. 65:138–148. [PubMed] [Google Scholar]
- 6.Drebot, M. A., J. J. Campbell, and S. H. Lee. 1999. A genotypic characterization of enteroviral antigenic variants isolated in eastern Canada. Virus Res. 59:131–140. [DOI] [PubMed] [Google Scholar]
- 7.Drebot, M. A., C. Y. Nguan, J. J. Campbell, S. H. Lee, and K. R. Forward. 1994. Molecular epidemiology of enterovirus outbreaks in Canada during 1991–1992: identification of echovirus 30 and coxsackievirus B1 strains by amplicon sequencing. J. Med. Virol. 44:340–7. [DOI] [PubMed] [Google Scholar]
- 8.Felsenstein, J. 1993. PHYLIP: phylogeny inference package, version 3.57. University of Washington, Seattle.
- 8a.Feng, D. F., and R. F. Doolittle. 1996. Progressive alignment of amino acid sequences and construction of phylogenetic trees from them. Methods Enzymol. 266:368–382. [DOI] [PubMed] [Google Scholar]
- 9.Kapsenberg, J. G., A. Ras, and J. Korte. 1980. Improvement of enterovirus neutralization by treatment with sodium deoxycholate or chloroform. Intervirology 12:329–334. [DOI] [PubMed] [Google Scholar]
- 10.Kopecka, H., B. Brown, and M. Pallansch. 1995. Genotypic variation in coxsackievirus B5 isolates from three different outbreaks in the United States. Virus Res. 38:125–136. [DOI] [PubMed] [Google Scholar]
- 11.Martinez, H. 1983. An efficient method for finding repeats in molecular sequences. Nucleic Acids Res. 11:4629–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McClure, M. A., T. K. Vasi, and W. M. Fitch. 1994. Comparative analysis of multiple protein-sequence alignment methods. Mol. Biol. Evol. 11:571–592. (Erratum, 11:811.) [DOI] [PubMed] [Google Scholar]
- 13.Melnick, J. L. 1996. Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses, 3rd ed. Lippincott-Raven, Philadelphia, Pa.
- 14.Morrison, D. A., and J. T. Ellis. 1997. Effects of nucleotide sequence alignment on phylogeny estimation: a case study of 18S rDNAs of apicomplexa. Mol. Biol. Evol. 14:428–441. [DOI] [PubMed] [Google Scholar]
- 15.Muir, P., U. Kammerer, K. Korn, M. N. Mulders, T. Poyry, B. Weissbrich, R. Kandolf, G. M. Cleator, and A. M. van Loon for The European Union Concerted Action on Virus Meningitis and Encephalitis. 1998. Molecular typing of enteroviruses: current status and future requirements. Clin. Microbiol. Rev. 11:202–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Needleman, S. B., and C. D. Wunsch. 1970. A general method applicable to the search for similarities in the amino acid sequences of two proteins. J. Mol. Biol. 48:444–453. [DOI] [PubMed] [Google Scholar]
- 17.Oberste, M. S., K. Maher, D. R. Kilpatrick, M. R. Flemister, B. A. Brown, and M. A. Pallansch. 1999. Typing of human enteroviruses by partial sequencing of VP1. J. Clin. Microbiol. 37:1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oberste, M. S., K. Maher, and M. A. Pallansch. 1998. Molecular phylogeny of all human enterovirus serotypes based on comparison of sequences at the 5′ end of the region encoding VP2. Virus Res. 58:35–43. [DOI] [PubMed] [Google Scholar]
- 19.Oberste, M. S., K. Maher, M. R. Flemister, G. Marchetti, D. R. Kilpatrick, and M. A. Pallansch. 2000. Comparison of classic and molecular approaches for the identification of untypeable enterovirus. J. Clin. Microbiol. 38:1170–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poyry, T., T. Hyypia, C. Horsnell, L. Kinnunen, T. Hovi, and G. Stanway. 1994. Molecular analysis of coxsackievirus A16 reveals a new genetic group of enteroviruses. Virology 202:982–987. [DOI] [PubMed] [Google Scholar]
- 21.Pöyry, T., L. Kinnunen, T. Hyypiä, B. Brown, C. Horsnell, T. Hovi, and G. Stanway. 1996. Genetic and phylogenetic clustering of enteroviruses. J. Gen. Virol. 77:(Pt 8):1699–1717. [DOI] [PubMed] [Google Scholar]
- 22.Pulli, T., P. Koskimies, and T. Hyypiä. 1995. Molecular comparison of coxsackie A virus serotypes. Virology 212:30–38. [DOI] [PubMed] [Google Scholar]
- 23.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson, J. D., F. Plewniak, and O. Poch. 1999. A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res. 27:2682–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ward, C. W., N. M. McKern, M. J. Frenkel, and D. D. Shuckla. 1992. Sequence data as the major criterion for potyvirus classification. Arch Virol. 5(Suppl.):283–297. [DOI] [PubMed] [Google Scholar]