

Abstract
Accurate diagnosis of schistosomiasis is crucial to achieve disease elimination as a public health problem. Rapid and highly sensitive diagnostic tools that can be used in decentralized environments and/or at the point-of-care are needed. This work optimises and simplifies an existing isothermal molecular diagnostic platform (recombinase polymerase amplification, RPA) for urogenital schistosomiasis, the ShDraI-RPA, with a focus on delivering a more accurate diagnosis in endemic settings. The standard ShDraI-RPA oligonucleotides were modified, incorporating a phosphorothioate backbone into the reverse primer and inverting the probe fluorophore and quencher, to prevent false positive results due to secondary structure formation. The sensitivity and specificity of the modified assay were evaluated on a donor urine spiked with one S. haematobium egg and an array of other schistosomes and human urinary tract pathogens. The stability of RPA reagents was assessed by storing them at ambient temperature (± 27 °C) in a dark environment for up to 90 days. Sample preparations were explored to develop a simple, rapid and low resource methodology that would complement the ShDraI-RPA platform when used in remote settings. The modified ShDraI-RPA assay was robust, sensitive and specific to S. haematobium group species, detecting down to 10 fg of gDNA and ten synthetic Dra I copies. DNA amplification was achieved at 42 °C within 20 min and results could easily be visualized using a portable fluorometer or under blue light. RPA reagents remained stable when stored in the absence of light at ± 27 °C for up to 30 days. A two-step DNA extraction method proved optimal for extracting DNA from single S. haematobium eggs in spiked urine. The optimized ShDraI-RPA platform shows improved specificity and sensitivity and has now reached several of the target product profile requirements set out by the WHO for the ideal diagnostic test for schistosomiasis.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-95887-x.
Keywords: Schistosoma haematobium, RPA, Optimisation, Schistosomiasis, Urogenital schistosomiasis, Isothermal molecular test
Subject terms: Diagnostic markers, Parasitic infection, Infectious diseases
Introduction
Schistosoma is a diverse genus of parasitic trematodes comprised of 25 species, with six of these being pathogenic to humans and causing schistosomiasis, a neglected tropical disease (NTD). At present, schistosomiasis affects more than 250 million people globally, mainly in Sub-Saharan Africa1,2. Schistosoma mansoni and Schistosoma haematobium are the most widespread species, causing intestinal and urogenital schistosomiasis, respectively. Urogenital schistosomiasis is often highly endemic in African countries with haematuria, urinary tract infections (UTIs), granuloma formation and chronic inflammation being the most common symptoms3. Furthermore, there is a clear correlation between urogenital schistosomiasis and an increased risk of bladder and kidney cancer, infertility and stigmatization, with the last two associated with female genital schistosomiasis (FGS)4,5.
Although highly prevalent, urogenital schistosomiasis is a preventable disease. In 2020, the world health organization (WHO) launched the “Road map for neglected tropical diseases”, in which it established the goal of eliminating schistosomiasis as a public health problem by 20306. Accurate diagnostics play a crucial role in achieving this goal, as it has been defined as “less than 1% prevalence of heavy-intensity infections” in a given area7. The WHO-defined target product profile (TPP) for ideal schistosomiasis diagnostics requires improvements in existing tests, with the ambitious goal of developing highly specific and sensitive point-of-care (POC) tests suitable for low infrastructure settings, which could then be used for monitoring, evaluation and surveillance purposes8.
Currently, urine filtration followed by the microscopic search for expelled S. haematobium eggs is the most routinely used method of diagnosing infections and estimating the disease prevalence in a given region. The estimated prevalence will then, in turn, determine the treatment approach that should be conducted8. However, diagnosis by microscopy lacks sensitivity, particularly when attempting to detect low-intensity and/or early stages of infection2. Moreover, microscopy is time-consuming and labour-intensive, requiring appropriately trained and skilled microscopists. Recently, an automated mobile phone associated POC tool, the SchistoScope, was developed for rapid microscopy screening of urogenital schistosomiasis9. Although the device has shown promising results, it has also shown some flaws with field of vision, issues with image and filter design, and manufacturing. Furthermore, biological aspects of the parasites’ lifecycle (e.g., daily fluctuation in egg release) limit its use in large-scale control programmes. Likewise, artificial intelligence-based image analysis has been developed to detect eggs on a robotised microscope10. However, this still does not overcome the inherent limitations of egg-based detection, which ultimately require the presence of schistosome eggs in the sample.
Diagnostic nucleic acids amplification tests (NAATs), also known as molecular tests, offer many advantages over traditional parasitological diagnosis due to their high sensitivity and specificity11. Polymerase chain reaction (PCR)-based tests have been widely used in the past decades and have made possible real progress in the diagnostics field. Real-time quantitative PCR (qPCR) is considered more sensitive than PCR, with the additional benefit of providing a quantitative result that can be correlated to the intensity of infection12. When using qPCR to detect S. haematobium DNA in urine samples, Keller et al.13 detected twice as many positive cases compared to urine-egg microscopy. Esiere et al.14 reported a 20-fold higher schistosomiasis prevalence using PCR compared to when egg microscopy was the test of choice. These results reinforce how powerful NAATs can be.
Despite the notable advantages of PCR-based tests, the financial costs and the need for adequate laboratory infrastructure to perform them limit their use in infection surveillance at the POC and in low resource areas13. In this context, isothermal DNA amplification methods stand out as promising alternatives for application at the POC, as they provide a rapid and sensitive diagnosis that can be performed with few resources and equipment, and without the need for temperature cycling to amplify DNA15. Several isothermal amplification methods have been described and two have already been adapted for schistosomiasis diagnosis: loop-mediated isothermal amplification (LAMP)16–19 and recombinase polymerase amplification (RPA).
The use of RPA for a molecular detection of S. haematobium was initially described by Rosser et al.20 who developed the lateral flow ShDraI-RPA which targets the genomic region Dra I (GenBank accession number: DQ157698.1). The Dra I region is a 121-base pair (bp) tandemly arranged repetitive sequence that corresponds to approximately 15% of S. haematobium genome21. It has similarities with DNA sequences from Schistosoma bovis, Schistosoma magrebowiei, Schistosoma mattheei, Schistosoma curassoni, and Schistosoma intercalatum, species that are part of the S. haematobium group21,22. Indeed Rosser et al.20 reported that the lateral flow test ShDraI-RPA cross-reacted with S. curassoni, S. bovis, but also S. mansoni. The cross-reactivity to the latter could be due to the concentrations used in the tests. Later, Roston et al.23 developed a real-time fluorescence-based ShDraI-RPA assay targeting the same tandem repeat in the S. haematobium genome, however the specificity of the assay was not reassessed by these authors. The ShDraI-RPA assay has been used to detect S. haematobium DNA in clinical samples, firstly reported by Archer et al.24, by testing urine samples which were egg-positive by microscopy achieving a high accuracy rate (93.7% sensitivity; 100% specificity). It was also reported an impressive sensitivity of the ShDraI-RPA with a limit of detection (LOD) of 10 synthetic copies of the target DNA region and one S. haematobium egg24. Frimpong et al.25 reported cross-reactivity with S. mansoni and Schistosoma japonicum, but no cross-reactivity was found with a range of other medically important protozoa or bacteria. Later, the ShDraI-RPA was used to detect FGS in cervicovaginal lavage (CVL) and vaginal self-swab samples26. The accuracy of the ShDraI-RPA, when compared to qPCR results, varied according to the sample examined and the DNA extraction method used. As a reaction enhancement strategy to improve ShDraI-RPA performance, Donnelly et al.27 assessed the effect of adding various amounts of betaine on the analytical sensitivity and specificity of the assay. It was found that by adding 2.5 M of betaine, the specificity of the assay was improved while the sensitivity remained the same as observed in studies previously mentioned.
The use of reaction enhancement strategies is a common approach with isothermal molecular methods, aiming to reduce secondary structure formation, improve reaction sensitivity and specificity, and decrease the incubation temperature of the reaction28. Assay optimizations should be considered for assays when moving from centralized laboratory to the POC, where resources are not abundant. Crucially, the pre-analytical stage (i.e., sample collection, storage, preparation, and DNA extraction) and the way fluorometric assay results are visualised should also be adapted to this context29.
With a view to use the ShDraI-RPA test at the POC, this paper focuses on modifications to the assay including simplifications to the initial two-step DNA extraction approach from urine samples, enhancements to the oligonucleotides used in the RPA assay, and improvements in alternative way of visualising results. Additionally, the robustness of the RPA reagents storage at different temperatures was tested to simulate lack of access to cold chain, and to evaluate their portability and stability in tropical environments. This work significantly advances the ShDraI-RPA platform towards portability and implementation in low resource settings, with results indicating that this newly described platform is a promising approach to support the diagnosis of urogenital schistosomiasis in resource-limited settings.
Results
Analytical specificity and sensitivity of the ShDraI-RPA with modified oligonucleotides
The ShDraI-RPA assay was carried out using the modified ShDraI reverse primer and fluorescent probe, and was incubated at different temperatures (39, 40 and 42 ºC), with reactions performed at 42 ºC found to be optimal (Supplementary Table S1). The ShDraI-RPA assay, using the modified oligonucleotides and incubated at 42 ºC, proved not to be specific to S. haematobium. However, the cross-reactivity was limited to members of the S. haematobium group: S. curassoni, S. bovis, Schistosoma guineensis, Schistosoma intercalatum, S. magrebowiei and S. mattheei. For S. mansoni, reactions demonstrated a weak fluorescence signal, but this did not cross the cut-off for positivity of 500 RFU above the baseline (Fig. 1A) and were therefore deemed negative. No cross-reactivity was observed in reactions containing gDNA from S. japonicum, S. rodhaini, other medically relevant helminths, or UTI bacteria (Fig. 1B–D).
Fig. 1.

Graphs showing the fluorescent signal from the modified ShDraI-RPA assay when tested on different Schistosoma species and other human pathogens. The graphs show the analytical specificity of the ShDraI-RPA at 42 ºC using the modified oligonucleotides and 100 pg of: (A) gDNA of species from the S. haematobium group; (B) gDNA of other Schistosoma species; (C) gDNA from other helminths and, (D) gDNA from pathogenic bacteria. The assay was specific to the S. haematobium group species and did not cross-react with other medically important schistosome species, helminths, nor common urinary tract infection-causing bacteria. Samples were considered positive when the amplification curve crossed the threshold of 500 RFU, which is indicated by the dashed lines. Non uniform fluorescent curves can occur for gDNA samples extracted using the Qiagen DNeasy Blood and Tissue kit. The reason for this is currently unknown. RFU relative fluorescence units.
The incorporation of the modified oligonucleotides in the ShDraI-RPA improved analytical sensitivity, resulting in a lower limit of detection (LOD) of 10 fg of S. haematobium gDNA (Fig. 2A). This is a tenfold improvement on the previously reported 100 fg analytical sensitivity of the standard assay27, regardless of whether betaine was added to enhance reaction specificity. When synthetic copies of the Dra I target region were tested, assay sensitivity was 10 copies, which confirms the findings from previous studies using the standard ShDraI-RPA design (Fig. 2B)24,27. The ShDraI-RPA also robustly detected S. haematobium DNA isolated from a single egg (Fig. 2A) with 100% sensitivity.
Fig. 2.

Graphs showing the fluorescent signal from the modified ShDraI-RPA assay when tested on different amounts of S. haematobium DNA. The analytical sensitivity of the modified ShDraI-RPA tested with (A) a single S. haematobium egg and dilutions of S. haematobium gDNA; (B) synthetic copies of the Dra I target. Samples were considered positive when the amplification curve crossed the threshold of 500 RFU, indicated by the dashed lines. The ShDraI-RPA assay robustly detects a single S. haematobium egg, 10 fg of gDNA, and 1 × 101 copies of the Dra I sequence. RFU relative fluorescence units.
Performance of the modified ShDraI-RPA assay with DNA from egg- and gDNA-spiked urine samples
All egg- and gDNA-spiked urine samples had positive RPA results when the gDNA was extracted following the short protocol from the SwiftX DNA kit, regardless of the thermal conditions in which the lysis incubation was held (Table 1). Conversely, false negative qPCR result was observed when samples were incubated for 15 min which could have been due to the high DNA concentration. In urine samples that had not been spiked with eggs or gDNA, the only extracts in which all three replicates remained negative (i.e. which demonstrated no false positives) by both RPA and qPCR were those incubated at room temperature (regardless of the duration of incubation).
Table 1.
Table of ShDraI-RPA and qPCR results with gDNA from different extraction methods, and three different sample types. ShDraI-RPA and qPCR results, and nanodrop quantification of the spiked and non-spiked urine extracts obtained using the SwiftX DNA (Xpedite) and DNeasy blood & tissue (Qiagen) kits. Both assays were performed in triplicate for each sample used. Green cells represent three positive results among the three replicates performed; yellow cells represent two positive results among the three replicates performed; orange cells represent one positive result among the three replicates performed and, red cells represent three negative results among the three replicates performed. RT room temperature, 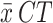 average of the threshold cycles of three qPCR replicates.
average of the threshold cycles of three qPCR replicates.

The spiked urine samples extracted using the long protocol demonstrated a reduced sensitivity and reproducibility in the ShDraI-RPA when compared to the short protocol, with false negative results obtained in urine samples spiked with one egg or with 1 ng of S. haematobium gDNA. One negative urine sample (which had not been spiked) gave a false positive result in one of the RPA repeats performed (Table 1).
The urine samples processed using the short protocol of the SwiftX DNA kit, with the lysis incubation step being performed at room temperature for both 5 and 15 min, presented 100% accuracy with no false positive nor false negative ShDraI-RPA results. However, a false negative qPCR result was observed when the short protocol was performed at room temperature incubation for 15 min, whilst a 5-minute incubation was 100% accurate with both ShDraI-RPA and qPCR, indicating that the latter may be the optimal method for the molecular detection of S. haematobium in urine samples (Table 1).
The DNeasy blood & tissue kit (Qiagen) was used according to the standard protocol from the manufacturer and gave accurate results in all replicates with no false positive or false negative results recorded.
Six microliters of urine sample spiked with a single S. haematobium egg were also directly added to the RPA reaction tube without any prior DNA extraction step. All the replicates tested provided a positive result, indicating that the RPA assay tolerates the presence of typical PCR inhibitors present in urine samples, such as urea (Fig. 3).
Fig. 3.

Graph showing the fluorescent signal from the modified ShDraI-RPA assay when tested on S. haematobium eggs without a preceding DNA extraction step. Six µL of urine spiked with a single S. haematobium egg was directly added to the RPA reaction, without any prior sample preparation. Samples were considered positive when the amplification curve crossed the threshold of 500 RFU, which is indicated by the dashed line. RFU relative fluorescence units; S. haematobium = gDNA control; DA direct addition replicates 1,2 and 3.
Blue-light exposure
The fluorescence from all the RPA assays performed in this study was visualised in real-time via the T16-ISO Software, and at the endpoint, by exposing the reaction tubes to blue-light. The results observed by both approaches gave concordant results and representative images are displayed in (Fig. 4).
Fig. 4.

(A) Graph showing the fluorescent signal from the modified ShDraI-RPA assay when tested on gDNA extracted from urine samples containing a single S. haematobium egg and with 1 ng of S. haematobium gDNA following the short protocol from SwiftX DNA (Xpedite diagnostics- i.e. protocol one; incubation step at room temperature and 95 °C for 5 min). Samples were considered positive when the amplification curve crossed the threshold of 500 RFU. Dashed lines indicate the 500 RFU cut-off. RFU relative fluorescence units, RT room temperature; 95 = 95 °C. (B) Reaction tubes exposed to a source of blue light after ShDraI-RPA reactions. Exposing to blue light proved to be a robust and simple alternative method of visualising the results of ShDraI-RPA reactions. +ve- Schistosoma haematobium gDNA control, -ve- negative control.
The reaction tubes containing samples in which gDNA amplification occurred glowed when under the blue-light exposure, whilst negative samples remained dark. The tubes containing samples that presented a slight fluorescence that did not cross the 500 RFU threshold or those that had a background fluorescence observed at the fluorometer, appeared turbid under the blue-light exposure. However, this could clearly be differentiated from the glow observed in positive samples (Fig. 4A,B).
Stability and longevity of the ShDraI-RPA
The ShDraI-RPA performed well and consistently when all the reagents were stored in the absence of light at ± 27 °C for up to 30 days. One replicate on both days one and two provided a false positive result in the negative control (i.e. nuclease-free water) which is likely to be a result of contamination during the assay set-up. At the 60-day time point, two out of three replicates gave a negative result, which may relate to the degradation of the reagents (Table 2).
Table 2.
Data showing the stability of the ShDra1-RPA assay reagents stored ± 27 °C for up to 30 days. False positive results were observed in one replicate on days 1 and 2 after the start of the test. False negative results were observed after 60 days after the start of the test. PCT positive control (100 pg of S. haematobium gDNA); NCT negative control (nuclease-free water); green cells- positive result; red cells- negative result.

Discussion
The present study aimed to advance the performance of the real-time fluorescence-based ShDraI-RPA platform by modifying essential components of the reaction such as the reverse primer and the fluorophore. The reverse primer was modified by incorporating a phosphorothioate backbone to its sequence. Several benefits and applications of phosphorothioate-modified (PM) oligonucleotides have been reported since its first use30. PM primers have been used in PCR30, LAMP31, CRISPR-Cas12a diagnostics (Clustered Regularly Interspaced Short Palindromic Repeats/ CRISPR- associated protein 12a)32, NPSA (Nested Phosphorothioated Hybrid Primer-mediated Isothermal Amplification)33, and RPA34,35 assays. They have been shown to provide better results in terms of specificity, stability against degradation by nucleases, reduction in the incubation temperature and/or reaction time, and prevention of primer-dimer formation, as well as providing more accurate discrimination between matched and mismatched regions of the target28,36. To further enhance the efficiency of the assay, the positions of the fluorophore (6FAM) and the quencher (BHQ1) were inverted within the fluorescent probe sequence, as previously proposed by Frimpong et al.25. This modification aims to keep the 6FAM away from the nearest guanine, since this base is known to act as a quencher for FAM due to its electron-donating properties37. With this alteration, the assay sensitivity increased, detecting down to 10 fg of S. haematobium gDNA.
A broader evaluation of the assay specificity was performed in this study compared to previous ones. The ShDraI-RPA using the modified oligonucleotides and incubated at 42 °C was specific to the S. haematobium species group with no cross-reactivity against S. mansoni, S. japonicum, S. rodhaini, nor to other helminths and bacteria that were tested. The S. haematobium species group is composed of S. haematobium, S. intercalatum, S. guineensis, S. margrebowiei, Schistosoma leiperi, S. mattheei, S. bovis and S. curassoni. Three of these (S. haematobium, S. intercalatum and S. guineensis) are of medical importance as causative agents of human schistosomiasis22, whereas the remaining species have veterinary importance. The occurrence of hybrids between human and veterinary schistosomes have also been reported38. Since the ShDraI-RPA is not specific to S. haematobium, this approach should not be used for studies that require species-specific identification of the parasite. Nevertheless, in a human diagnostic context, detecting infections caused by medically relevant schistosome species with high sensitivity is an asset. Currently, praziquantel is the drug of choice recommended by the WHO to treat schistosomiasis, regardless of the of the causative species39. New molecular targets should be explored in the future to enable the development of a species-specific assay, that would be used in cases where detecting the exact species is important. Alternatively, a genus-specific assay that can detect all schistosome species, including the epidemiologically important S. mansoni and S. japonicum, would be extremely useful for worldwide public health and disease control measures.
In this study, naive urine samples were spiked with single S. haematobium eggs to evaluate the efficacy of the ShDraI-RPA assay using newly modified oligonucleotides on clinical samples. Naive urine samples were also spiked with 1 ng of S. haematobium DNA to simulate the detection of cell-free DNA (cfDNA). Spiked samples had their DNA isolated using two types of DNA extraction methods, a pure column-based method (Qiagen DNeasy Blood & Tissue) and a crude bead-based one (Xpedite SwiftX DNA). Archer et al.26 reported that the sensitivity and specificity of the ShDraI-RPA differed depending on the DNA extraction approach used in CVL samples, with the highest sensitivity and specificity values observed in gDNA samples that were extracted with the discontinued SpeedXtract Nucleic Acid Kit (Qiagen, Germany), now superseded by the SwiftX DNA kit which uses similar technology. Donnelly et al.27 evaluated the performance of the ShDraI-RPA on DNA extracts obtained using the SwiftX DNA and SpeedXtract Nucleic Acid Kits, and no difference was observed in terms of reproducibility among the replicates or compatibility with the RPA components. Likewise, in the present work, the SwiftX DNA kit showed promising RPA results with all variations of the protocol evaluated (i.e. short and long protocols with different time and temperature for lysis incubation). However, false-negative results were observed in urine samples spiked with 1 ng of gDNA when the long protocol was followed, indicating that this protocol may not be appropriate for the identification of cfDNA that may be present in urine samples of infected individuals40. False-positive results have occasionally also been observed and reported by others26,27. Here, all the samples were also analysed by qPCR (used as reference test), with false-negative results being observed in only one of the egg-spiked samples and the gDNA-spiked samples. Additionally, two false-positive results were observed when non-spiked urine samples were analysed by qPCR, with one of these also being RPA-positive. The short protocol, modified for the lysis incubation at room temperature for five minutes, was the only one that provided samples that had 100% ShDraI-RPA and qPCR specificity, indicating that this may be the best SwiftX DNA protocol to follow for this assay. This is a promising result considering potential POC use, as the short protocol with the five-minute incubation at room temperature is faster and simpler. Additionally, this is a rapid protocol that takes approximately 15 min to process eight samples, simple to perform as it is a two-step approach, with an estimated cost of 3.20 USD27. However, to confirm these findings, further studies should be conducted using a larger clinical sample set, ideally from endemic areas. The data also highlights the importance of robust methods to mitigate against cross-sample contamination.
Interestingly, despite the SwiftX DNA kit being designed to isolate the gDNA through cell-capture followed by a cell-lysis step, it was possible to recover and detect S. haematobium DNA from gDNA spiked urine samples, designed to mimic cfDNA. The potential detection of S. haematobium cfDNA in urine samples should be further investigated using clinical samples from endemic areas.
Naive urine samples spiked with S. haematobium single eggs (without any prior extraction or other sample processing) were directly added to an ShDraI-RPA reaction, with a positive result observed in all three replicates. The direct addition of schistosomes eggs has been previously proved to be possible27,35 but this is the first time that it was tested with eggs resuspended in urine, suggesting that RPA may tolerate common inhibitors in clinical samples that must be removed prior to using PCR41. Again, although very promising, this finding should be further evaluated and confirmed by repeating the test in a larger number of clinical samples from endemic areas.
A significant consideration when developing and validating new diagnostic tests is their accessibility and affordability to those most in need of them42. Schistosomiasis is a neglected tropical disease, and endemic areas usually lack significant financial and technical resources. In this context, we assessed an alternative method to visualise the RPA result, independent of the fluorometer, which consists of exposing the reaction tubes to a source of blue light after a 20-minute incubation at 42 °C, so that positive samples would glow. Blue-light results obtained in this work corresponded to results from the Axxin fluorometer, with the additional benefit of simpler result interpretation. The stability and longevity evaluation of the ShDraI-RPA using the modified oligonucleotides indicated that the assay components may be stable for up to one month at 27 °C, facilitating their transportation and storage in the absence of a cold-chain, which is of significant benefit when considering outreach from a regional centre to more remote disease-endemic settings. The tests were conducted in triplicate and false-positive results on negative controls were observed at the first two days of the test. Since this did not occur on the following days and in the other replicates, we consider this to be unlikely to be due to degradation of the RPA components and instead may represent contamination during assay set-up.
In conclusion, the optimized ShDraI-RPA assay looks to be a promising and efficient alternative for a fast and portable schistosomiasis diagnostic, able to support the detection of schistosomiasis infection, especially in low-resource settings.
Methods
ShDraI-RPA modifications and optimisation
Modified oligonucleotides
RPA primers and probe targeting the S. haematobium Dra I tandem repeat region (GenBank accession number: DQ157698.1) described by Rostron et al.23 were modified with the aim of increasing assay specificity, whilst maintaining high levels of sensitivity. Modifications included the addition of a phosphorothioate backbone (denoted as “s” in the primer sequence) before the last 3’ base of the reverse primer, and inversion of the position of the fluorophore 6-carboxyfluorescein (6FAM) and the black-hole quencher 1 (BHQ1) within the fluorescent probe (Table 3).
Table 3.
Sequences of the primers and the fluorescent probe for the modified ShDra1-RPA assay. Modifications were made to the original ShDra1-RPA assay reverse primer and probe, however, the forward primer remained unchanged.
| Oligonucleotide | Modified | Sequence (5’-3’) | |
|---|---|---|---|
| Yes | No | ||
| Forward primer | x | ATC TCA CCT ATC AGA CGA AAC AAA GAA AAT | |
| Reverse primer | x | AAT ATG AAA CAA TTT TCA CAA CGA TAC GAsC | |
| Fluorescent probe | x | AAT TGT TGG TGG AAG TGC CTG TTT CGC AA (BHQ1) (THF) (6FAM) CTC CGG AAT GGT TG (C3) | |
s phosphorothioate backbone, BHQ1 black hole quencher 1, THF tetrahydrofuran, 6FAM 6-carboxyfluorescein, C3 spacer C3.
RPA reaction and visualisation of the results
ShDraI-RPA reactions were performed using the TwistAmp Exo Kit (TwistDx, Cambridge, UK) following the manufacturer instructions and as described by Rostron et al.23. All reactions were performed in triplicate, and positive (100 pg of S. haematobium DNA) and negative (nuclease-free water) controls were included with each RPA run of 16 reactions. RPA reaction tubes were incubated at 42 °C for 20 min in the portable fluorometer Axxin T16-ISO (Axxin Diagnostics, Australia) where the fluorescence was measured, and results were observed in real-time via T16-ISO Software (V2.1.0.3- Axxin Diagnostics, Australia). Samples were considered positive for S. haematobium when an increase of ≥ 500 relative units of fluorescence (RFU) above the baseline was observed27. Additionally, to verify whether the assay could provide a qualitative binary diagnosis without the need for a fluorometer, the end-point fluorescence was observed by exposing the reaction tubes to a source of blue light after the amplification. The blue light has a wavelength of approximately 450 to 495 nm and the absorbance of 6FAM is 493 nm. This means that in positive samples (i.e. which contain amplified DNA), where the fluorophore, 6FAM, has been released from the quencher, is able to absorb the blue light and produce a fluorescence that is visible to the naked eye.
Source of biological samples
Genomic DNA (gDNA) isolated from S. haematobium and other Schistosoma species (S. mansoni, S. curassoni, S. bovis, S. rodhaini, S. mattheei, S. japonicum, S. margrebowiei and S. guineensis) were obtained from the Schistosomiasis Collection at The Natural History Museum- UK (SCAN)43 repository. The Medical Malacology Collection at René Rachou Institute, Fiocruz Minas- Brazil (Fiocruz-CMM) provided samples from trematodes belonging to the families Clinostomidae, Echinostomatidae, Fasciolidae, Notocotylidae, Spirorchiidae and Strigeidae, and SCAN provided trematodes belonging to the genus Tricobilharzia. Genomic DNA from helminths of medical importance (i.e. Ascaris lumbricoides, Enterobius vermicularis, Trichuris trichiura, and hookworms) were provided by the Helminthology and Medical Malacology Laboratory from the René Rachou Institute, Fiocruz Minas- Brazil. To test for Gram-negative bacteria which commonly cause UTIs, gDNA previously obtained by Rosser et al.20from Escherichia coli and Klebsiella pneumoniae were used. In addition, Charing Cross Hospital (London- UK) supplied a reference colony of Proteus mirabilis (ATCC strain 35659; https://www.atcc.org/products/35659) from their microbiology laboratory. Single S. haematobium ova were provided frozen by the Snail Schistosome Resource (SSR, Natural History Museum, UK) via the NIAID Schistosomiasis Resource Centre (SRC, Biomedical Research Institute, USA). All DNA samples were stored at -20 °C before use.
Analytical specificity testing
Genomic DNA was isolated from all organisms listed above using the DNeasy Blood & Tissue (Qiagen GmbH, Germany) kit, according to the manufacturer’s instructions. All gDNA isolates were quantified using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and normalised to a working concentration of 100 pg/µL using nuclease-free water. All samples were analysed using the modified ShDraI-RPA oligonucleotides using various incubation temperature (39, 40 and 42 °C) to determine the optimal temperature for the assay specificity.
Analytical sensitivity testing
Genomic DNA isolated from adult worms of S. haematobium was used to obtain tenfold serial dilutions from 1 ng/µL to 1 fg/µL. Synthetic copies of the Dra I target region were obtained as described by Donnelly et al.27 and dilutions ranging from 1 × 105 to 1 × 100 copies/µL were prepared. Genomic DNA from single eggs was isolated using the SwiftX DNA extraction kit (Xpedite Diagnostics GmbH, Germany) following the manufacturer’s instruction (protocol 1 from the handbook). All DNA (genomic and synthetic) samples were analysed using the modified ShDraI-RPA, as described above, to determine assay sensitivity. The direct addition of single S. haematobium eggs (i.e. without their gDNA extracted) to the RPA reactions was also tested.
Spiked urine samples
Aliquots (100 µL) of urine from a donor (from a non-endemic country) were spiked with: (i) a single egg of S. haematobium and, (ii) 1 ng of S. haematobium gDNA. Also, non-spiked aliquots were processed as negative controls.
All of the spiked and non-spiked aliquots were processed and used as follows: (i) direct addition to the RPA reaction; (ii) DNA extraction using DNeasy Blood & Tissue kit (Qiagen), following the manufacturer’s protocol; (iii) DNA extraction using a rapid, low resource method for POC use, SwiftX DNA kit (Xpedite), according to the manufacturer’s protocols one and two, testing variations on the time and temperature of lysis incubation.
Briefly, the SwiftX DNA kit protocol one (hereon, the short protocol) consists of the addition of 100 µL of the lysis buffer DL and 30 µL of Beads A to the sample (100 µL). The mixture was homogenised for 10 s by vortexing and incubated at 95 °C (or alternatively, at room temperature- RT) for five or 15 min. The tubes were placed in a magnetic rack for one minute and the supernatant (containing the gDNA) was transferred to a fresh 1.5 mL microcentrifuge tube and stored at −20 °C until use. Thus, four variations of the short protocol were tested: (i) lysis incubation at 95 °C for 5 min; (ii) lysis incubation at 95 °C for 15 min; (iii) lysis incubation at RT for five min; (iv) lysis incubation at RT for 15 min.
The SwiftX DNA kit protocol two (hereon, the long protocol) consists of the addition of 100 µL of the buffer EN and 30 µL of Beads A to the sample (100 µL). The mixture was homogenised for 10 s by vortexing and incubated at RT for 3 min. The tubes were then placed in a magnetic stand for 1 min and the supernatant was discarded. 100 µL of the lysis buffer DN was added to each tube that was subsequently homogenised by vortexing. The mixture was incubated at 95 °C (or alternatively, at RT) for five or 15 min and then placed into a magnetic stand for 1 min. The supernatant (containing the gDNA) was transferred to a fresh 1.5 mL microcentrifuge tube and stored at -20 °C until use. Again, four variations of the long protocol were tested: (i) lysis incubation at 95 °C for 5 min; (ii) lysis incubation at 95 °C for 15 min; (iii) lysis incubation at RT for 5 min; (iv) lysis incubation at RT for 15 min.
In total, eight protocols were tested for the SwiftX DNA kit. The concentration of gDNA in all the extracts was measured using a Nanodrop™ spectrophotometer.
qPCR analysis
DNA isolated from all egg- and gDNA-spiked urine samples extracted using the different methods were analysed by qPCR. This was performed as a confirmatory test for the isolation of S. haematobium DNA from each sample type by the different methods tested27,44,45. The quantitative nature of this assay also allowed comparison of the DNA yield of the different sample preparation methodologies described above. Schistosoma genus-specific oligonucleotides targeting the internal transcribed spacer region 2 (ITS 2) were used in a 20 µl reaction containing 0.3 µM of each primer (Forward- Ssp48F: 5’-GGTCTAGATGACTTGATYGAGATGCT- 3’; Reverse- Ssp124R: 5’-TCCCGAGCGYGTATAATGTCATTA-3’), 0.1 µM of the probe (Ssp78T: 5’- FAM-TGGGTTGTG/ZEN/CTCGAGTCGTGGC-IowaBlack-FQ -3’), 1× Luna Universal Probe qPCR MasterMix (New England Biolabs), and 1 µl of template DNA. The StepOne Plus thermocycler (Applied Biosystems) was used with the following cycling conditions: 2 min initial denaturation at 95 °C, and 50 cycles of 95 °C denaturation for 15 s followed by combined 60 °C annealing/extension for 60 s. Samples were considered positive when the cycle threshold (Ct) value was < 38. This step was particularly important for assessing the different sample preparation methodologies.
Assay stability and longevity
To determine the stability of the ShDraI-RPA reagents (i.e. primers, probe, and components from the TwistAmp exo kit- RPA pellet, RPA rehydration buffer and magnesium acetate MgAc) at ambient temperature in schistosomiasis-endemic settings, all reagents were stored at ± 27 °C in a dark environment without the addition of any stabilizers. The reagents were then used to perform ShDraI-RPA reactions on days: 0, 1, 2, 3, 7, 14, 21, 30, 60 and 90. The ShDraI-RPA reactions were set up as described above and positive (100 pg of S. haematobium DNA) and negative (nuclease-free water) controls were included in all tests. Results were observed in real time via the Axxin T16- ISO portable fluorometer and software, and post-amplification exposure of the tubes to a blue light.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank the Wellcome Trust funded Schistosome and Snail Resource (SSR) Wellcome Trust Biomedical Resource Grant 221368/Z/20/Z (2021–2026), the Collection of Medical Malacology (Fiocruz-CMM- Brazil), the Helminthology and Medical Malacology Laboratory (Fiocruz, Brazil), Schistosomiasis Collection at the Natural History Museum (SCAN- UK) and the Charing Cross Hospital (London, UK) for the provision the samples used in this study; Andy Wende and Ludovic Ebert from Xpedite Diagnostics for advice and support during the study. This work received financial support from the Coalition for Operational Research on Neglected Tropical Diseases (COR-NTD), which is funded at The Task Force for Global Health primarily by the Bill & Melinda Gates Foundation (OPP1190754), by UK aid from the British government, and by the United States Agency for International Development through its Neglected Tropical Diseases Program. Under the grant conditions of the Foundation, a Creative Commons Attribution 4.0 Generic License has already been assigned to the Author Accepted Manuscript version that might arise from this submission. Support was also provided via the Departmental Investment Fund of the Natural History Museum.
Author contributions
SGM, OD and BLW conceived the experiment(s); SGM, OD, EBL conducted the experiment(s); SGM, OD, JA, EBL and BLW analysed the results. All authors reviewed the manuscript.
Data availability
All data generated and analysed during this study are included in this publication (and its Supplementary Information file).
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Silvia G. Mesquita and Owain Donnelly contributed equally to this work.
References
- 1.WHO & Schistosomiasis. https://www.who.int/news-room/fact-sheets/detail/schistosomiasis (2023).
- 2.McManus, D. P. et al. Schistosomiasis. Nat. Rev. Dis. Primers4, (2018). [DOI] [PubMed]
- 3.Colley, D. G. & Secor, W. E. Immunology of human schistosomiasis. Parasite Immunol.36, 347–357 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ally, O. et al. Schistosomiasis diagnosis: challenges and opportunities for elimination. PLoS Negl. Trop. Dis.1810.1371/journal.pntd.0012282 (2024). [DOI] [PMC free article] [PubMed]
- 5.Bustinduy, A. L. et al. An update on female and male genital schistosomiasis and a call to integrate efforts to escalate diagnosis, treatment and awareness in endemic and non-endemic settings: the time is now. 1–44. 10.1016/bs.apar.2021.12.003 (2022). [DOI] [PubMed]
- 6.WHO World Health Organization. Ending the neglected to attain the sustainable development goals—A road map for neglected tropical diseases 2021–2030 (55). (2020).
- 7.Wiegand, R. E. et al. Defining elimination as a public health problem for schistosomiasis control programmes: beyond prevalence of heavy-intensity infections. Lancet Global Health10, e1355–e1359. 10.1016/S2214-109X(22)00287-X (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.WHO World Health Organization. Diagnostic target product profiles for monitoring, evaluation and surveillance of schistosomiasis control programmes. 28. https://www.who.int/publications/i/item/9789240031104 (2021).
- 9.Agbana, T. et al. Schistoscope: Towards a locally producible smart diagnostic device for schistosomiasis in Nigeria. In IEEE Global Humanitarian Technology Conference (GHTC) (2019).
- 10.Maturana, C. R. et al. Development of an automated artificial intelligence-based system for urogenital schistosomiasis diagnosis using digital image analysis techniques and a robotized microscope. PLoS Negl. Trop. Dis.18, e0012614 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minetti, C. et al. Focusing nucleic acid-based molecular diagnostics and xenomonitoring approaches for human helminthiases amenable to preventive chemotherapy. Parasitol. Open2, (2016).
- 12.Weerakoon, K. G., Gordon, C. A. & McManus, D. P. DNA diagnostics for schistosomiasis control. Trop. Med. Infect. Dis.310.3390/tropicalmed3030081 (2018). [DOI] [PMC free article] [PubMed]
- 13.Keller, D. et al. Performance of a real-time PCR approach for diagnosing Schistosoma haematobium infections of different intensity in urine samples from Zanzibar. Infect. Dis. Poverty9, (2020). [DOI] [PMC free article] [PubMed]
- 14.Esiere, R. K. et al. Detecting Schistosoma haematobium infection by microscopy and polymerase chain reaction (PCR) in school children in three senatorial districts of cross river State, Nigeria. J. Parasitic Dis.46, 272–279 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamburger, J. Molecular tools and schistosomiasis transmission elimination. Am. J. Trop. Med. Hyg.103, 1376–1379. 10.4269/ajtmh.20-0111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abbasi, I., King, C. H., Muchiri, E. M. & Hamburger, J. Detection of Schistosoma mansoni and Schistosoma haematobium DNA by loop-mediated isothermal amplification: identification of infected snails from early prepatency. Am. J. Trop. Med. Hyg.83, 427–432 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gandasegui, J. et al. The rapid-heat LAMPellet method: A potential diagnostic method for human urogenital schistosomiasis. PLoS Negl. Trop. Dis.9, 1–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gandasegui, J. et al. Field and laboratory comparative evaluation of a LAMP assay for the diagnosis of urogenital schistosomiasis in Cubal, central Angola. Trop. Med. Int. Health23, 992–1001 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Salas-Coronas, J. et al. Evaluation of loop-mediated isothermal amplification (LAMP) in urine samples for the diagnosis of imported schistosomiasis. Trop. Med. Infect. Dis.8, (2023). [DOI] [PMC free article] [PubMed]
- 20.Rosser, A., Rollinson, D., Forrest, M. & Webster, B. L. Isothermal recombinase polymerase amplification (RPA) of Schistosoma haematobium DNA and oligochromatographic lateral flow detection. Parasit. Vectors8, (2015). [DOI] [PMC free article] [PubMed]
- 21.Hamburger, J. et al. Polymerase chain reaction assay based on a highly repeated sequence of Schistosoma haematobium: A potential tool for monitoring schistosome-infested water. Am. J. Trop. Med. Hyg.65, 907–911 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Webster, B. L., Southgate, V. R., Timothy, D. & Littlewood, J. A revision of the interrelationships of Schistosoma including the recently described Schistosoma guineensis. Int. J. Parasitol.36, 947–955 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Rostron, P. et al. Development of a recombinase polymerase amplification (RPA) fluorescence assay for the detection of Schistosoma haematobium. Parasit. Vectors12, (2019). [DOI] [PMC free article] [PubMed]
- 24.Archer, J. et al. Analytical and clinical assessment of a portable, isothermal recombinase polymerase amplification (RPA) assay for the molecular diagnosis of urogenital schistosomiasis. Molecules25, (2020). [DOI] [PMC free article] [PubMed]
- 25.Frimpong, M. et al. Evaluation of a real-time recombinase polymerase amplification assay for rapid detection of Schistosoma haematobium infection in resource-limited setting. Acta Trop.216, (2021). [DOI] [PubMed]
- 26.Archer, J. et al. Validation of the isothermal Schistosoma haematobium recombinase polymerase amplification (RPA) assay, coupled with simplified sample preparation, for diagnosing female genital schistosomiasis using cervicovaginal lavage and vaginal self-swab samples. PLoS Negl. Trop. Dis.16, (2022). [DOI] [PMC free article] [PubMed]
- 27.Donnelly, O. et al. Refining the Schistosoma haematobium recombinase polymerase amplification (Sh-RPA) assay: moving towards point-of-care use in endemic settings. Parasit. Vectors17, (2024). [DOI] [PMC free article] [PubMed]
- 28.Özay, B. & McCalla, S. E. A review of reaction enhancement strategies for isothermal nucleic acid amplification reactions. Sens. Actuators Rep.3, (2021).
- 29.Lee, P. L. M. DNA amplification in the field: move over PCR, here comes LAMP. Mol. Ecol. Resour.17, 138–141. 10.1111/1755-0998.12548 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Skerra, A. Phosphorothioate primers improve the amplification of DNA sequences by DNA polymerases with proofreading activity. Nucleic Acids Res.20http://nar.oxfordjournals.org/ (1992). [DOI] [PMC free article] [PubMed]
- 31.Cai, S., Jung, C., Bhadra, S. & Ellington, A. D. Phosphorothioated primers lead to loop-mediated isothermal amplification at low temperatures. Anal. Chem.90, 8290–8294 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Gong, J. et al. An enhanced method for nucleic acid detection with CRISPR-Cas12a using phosphorothioate modified primers and optimized gold-nanopaticle strip. Bioact. Mater.6, 4580–4590 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang, Y., Zhang, L., Shen, Y., Yu, E. Y. W. & Ding, X. Nested phosphorothioated hybrid primer-mediated isothermal amplification for specific and dye-based subattomolar nucleic acid detection at low temperatures. ACS Sens.8, 1261–1271 (2023). [DOI] [PubMed] [Google Scholar]
- 34.El Wahed, A. A. et al. Suitcase lab for rapid detection of SARS-CoV-2 based on recombinase polymerase amplification assay. Anal. Chem.93, 2627–2634 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mesquita, S. G. et al. Development of real-time and lateral flow recombinase polymerase amplification assays for rapid detection of Schistosoma mansoni. Front. Microbiol.13, (2022). [DOI] [PMC free article] [PubMed]
- 36.Nawrot, B., Paul, N., Rebowska, B. & Stec, W. J. Significance of stereochemistry of 3′-terminal phosphorothioate- modified primer in DNA polymerase-mediated chain extension. Mol. Biotechnol.40, 119–126 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Noble, J. E., Wang, L., Cole, K. D. & Gaigalas, A. K. The effect of overhanging nucleotides on fluorescence properties of hybridising oligonucleotides labelled with Alexa-488 and FAM fluorophores. Biophys. Chem.113, 255–263 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Leger, E. & P Webster, J. Hybridizations within the genus schistosoma: implications for evolution, epidemiology and control. Parasitology144, 65–80. 10.1017/S0031182016001190 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Nogueira, R. A. et al. Praziquantel: an update on the mechanism of its action against schistosomiasis and new therapeutic perspectives. Mol. Biochem. Parasitol.25210.1016/j.molbiopara.2022.111531 (2022). [DOI] [PubMed]
- 40.Ullah, H. et al. Schistosomiasis related circulating cell-free DNA: A useful biomarker in diagnostics. Mol. Biochem. Parasitol.25110.1016/j.molbiopara.2022.111495 (2022). [DOI] [PubMed]
- 41.Daher, R. K., Stewart, G., Boissinot, M. & Bergeron, M. G. Recombinase polymerase amplification for diagnostic applications. Clin. Chem.62, 947–958 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Land, K. J., Boeras, D. I., Chen, X. S., Ramsay, A. R. & Peeling, R. W. REASSURED diagnostics to inform disease control strategies, strengthen health systems and improve patient outcomes. Nat. Microbiol.4, 46–54 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emery, A., Allan, F., Rabone, M. & Rollinson, D. Schistosomiasis collection at the natural history museum (SCAN). Parasit. Vectors5, 185 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Obeng, B. B. et al. Application of a circulating-cathodic-antigen (CCA) strip test and real-time PCR, in comparison with microscopy, for the detection of Schistosoma haematobium in urine samples from Ghana. Ann. Trop. Med. Parasitol.102, 625–633 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Vinkeles Melchers, N. V. S. et al. Diagnostic performance of schistosoma real-time PCR in urine samples from Kenyan children infected with Schistosoma haematobium: Day-to-day variation and follow-up after praziquantel treatment. PLoS Negl. Trop. Dis.8, e2807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated and analysed during this study are included in this publication (and its Supplementary Information file).