

Abstract
It is not clear how the host initially recognizes and responds to infection by gram-negative pathogenic Brucella spp. It was previously shown (D. S. Weiss, B. Raupach, K. Takeda, S. Akira, and A. Zychlinsky, J. Immunol. 172:4463-4469, 2004) that the early macrophage response against gram-negative bacteria is mediated by Toll-like receptor 4 (TLR4), which signals in response to lipopolysaccharide (LPS). Brucella, however, has a noncanonical LPS which does not have potent immunostimulatory activity. We evaluated the kinetics of TLR4 activation and the cytokine response in murine macrophages after Brucella infection. We found that during infection of macrophages, Brucella avoids activation of TLR4 at 6 h but activates TLR4, TLR2, and myeloid differentiation factor 88 (MyD88) at 24 h postinfection. Interestingly, even though its activation is delayed, MyD88 is important for host defense against Brucella infection in vivo, since MyD88−/− mice do not clear the bacteria as efficiently as wild-type, TLR4−/−, TLR2−/−, or TLR4/TLR2−/− mice.
Toll-like receptors (TLRs) are among the first receptors to detect a microbial infection. They recognize conserved microbial components and signal for inflammation (2, 33). Lipopolysaccharide (LPS) and bacterial lipoproteins (BLP) are two such microbial components and are recognized by TLR4 and TLR2, respectively (3, 5, 35).
TLRs are one-pass transmembrane receptors containing extracellular leucine-rich repeat domains and an intracellular Toll/interleukin-1 receptor homology (TIR) domain. Upon activation, the TIR domain can recruit myeloid differentiation factor 88 (MyD88), one of several adaptor molecules that act directly downstream of TLRs (9, 10, 16, 18, 32, 42, 43). MyD88 is critical for TLR-mediated activation of the transcription factor nuclear factor κB (NF-κB) and thus for the induction of proinflammatory cytokines such as tumor necrosis factor α (TNF-α) (22).
Brucellae are gram-negative bacteria that are found worldwide and cause an inflammatory disease characterized by fever, fatigue, weakness, and weight loss (21). The highest incidence of disease is in developing countries and among people who have close contact with livestock. A chronic generalized infection may follow the initial infection and results in part from the ability of Brucella to reside within macrophages and epithelial cells (15). Brucella spp., as well as some other gram-negative bacteria, contain an LPS with long-chain fatty acids that has low immunostimulatory activity (26, 27, 36). In contrast, enterobacteria, such as Salmonella spp., contain a highly potent LPS. TLR4 is essential for the initial responses of macrophages against Salmonella (24, 37, 41). It is not clear how macrophages initially recognize and respond to gram-negative bacteria that contain a noncanonical LPS, such as Brucella.
Here we show that Brucella signals through TLR4, TLR2, and MyD88 and activates macrophages at 24 but not 6 h postinfection. Salmonella, in contrast, activates TLR4 and macrophages at 6 h. These differences may be due to the low activity of Brucella LPS. Interestingly, even though its activation is delayed, MyD88 is required for inflammation and efficient clearance of Brucella in vivo.
MATERIALS AND METHODS
Reagents.
Highly purified Salmonella enterica serovar Minnesota LPS was from List Biologicals (Campbell, CA). Brucella abortus LPS was prepared from phenol-water extracts of whole bacteria, and the crude material was extensively purified and characterized as described elsewhere (4, 12, 25). The purified Brucella LPS was free of contaminants and displayed the characteristic quantities of N-formyl-perosamine O polysaccharide, 2-keto-3-deoxyoctulosonic acid, and the diaminoglucose backbone acylated with long-chain C28:0 to C30:0 fatty acids (28, 29). Murine TNF-α and murine interleukin-12 (IL-12) p40 enzyme-linked immunosorbent assays (ELISAs) were from R&D Systems (Minneapolis, MN), and the murine IL-10 ELISA was from BD Biosciences (San Diego, CA). The anti-mouse TLR4/MD-2PE antibody was from eBioscience (San Diego, CA). ABTS [2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid)] was from Sigma Chemical Co. (St. Louis, Mo.).
Bacterial strains.
Overnight cultures of Brucella abortus strain 19 (25) were prepared by thawing and adding a glycerol stock containing 1010 bacteria to 150 ml tryptic soy broth. Cultures were grown with shaking overnight at 37°C. Wild-type Salmonella enterica serovar Typhimurium SL1344 (17) was grown standing overnight at 37°C in high-salt Luria broth (LB; 0.3 M NaCl) supplemented with 200 μg/ml streptomycin.
Mice.
Mice were bred under specific-pathogen-free conditions at New York University Medical Center, New York, or at the Bundesinstitut für Risikobewertung, Berlin, Germany. Mice were housed in filter-top cages and provided with sterile water and food ad libitum. TLR4−/− (19), TLR2−/− (40), MyD88−/− (22), IL-1β−/− (44), and IL-18−/− (39) mice have been described previously. We generated TLR4/TLR2−/− mice by crossing TLR4−/− and TLR2−/− mice, and IL-1β/IL-18−/− mice by crossing IL-1β−/− and IL-18−/− mice. All knockout mice were backcrossed at least seven times and were Nramp susceptible (Nramps/s). C57BL/6 mice were from Taconic (Germantown, NY) or the Bundesinstitut für Risikobewertung, Berlin, Germany.
Bone marrow-derived macrophages.
Bone marrow-derived macrophages were prepared as described elsewhere (38). Briefly, bone marrow was collected from the femurs and tibiae of mice. Bone marrow cells were plated on sterile petri dishes and incubated in Dulbecco's modified Eagle medium containing 10% fetal calf serum, 5% horse serum, 10 mM HEPES, 1 mM pyruvate, 10 mM l-glutamine, and 20% macrophage colony-stimulating factor (M-CSF) conditioned medium. M-CSF conditioned medium was collected from an L929 M-CSF cell line. Bone marrow cells were incubated at 37°C under 7% CO2, and macrophages were harvested after 6 days. All assays were performed in standard tissue culture plates at 37°C under 7% CO2 in similar media excluding horse serum and M-CSF conditioned medium.
Macrophage infections.
Twenty-four-well plates were seeded with 200,000 macrophages per well. Cells were allowed to adhere overnight and were then washed the following day. Assays were performed in 500 μl medium. Multiplicities of infection (MOIs) of 50:1 for Brucella and 1:1 for Salmonella were chosen because at these MOIs, similar numbers of each type of bacteria enter macrophages. Therefore, at these MOIs, we can compare the macrophage response to Brucella and Salmonella infection under conditions where bacterial invasion/uptake are similar. After addition of bacteria, plates were spun at 4°C for 15 min at 850 × g. Plates were then incubated at 37°C under 7% CO2 at time zero. After 30 min, the medium was removed, and cells were washed with a medium containing 25 μg/ml gentamicin and then left in a medium containing 10 μg/ml gentamicin.
Measurement of cytokine production.
Cells were allowed to adhere overnight and were then washed the following day. Salmonella or Brucella LPS was added to the cells, and assays were performed in 200 μl medium. Supernatants were collected and frozen until they were assayed by ELISA for determination of TNF-α concentrations according to the manufacturer's specifications. Supernatants from samples in bacterial infection assays were treated similarly and assayed for TNF-α, IL-12p40, or IL-10 concentrations.
TLR4 expression.
Macrophages were either infected with Brucella (MOI, 50:1) or Salmonella (MOI, 1:1) or left uninfected. Cells were harvested 6 or 24 h later. Cells were blocked for 30 min at 4°C with 10% mouse serum-10% horse serum and then stained with an anti-murine TLR4 antibody (1:100 dilution) for 15 min at 4°C. Cells were washed twice, and staining was quantified using a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ). TLR4−/− macrophages were stained as a control for antibody specificity.
Mouse infections and histological studies.
Age- and sex-matched mice were used for all experiments. Mice were injected intraperitoneally (i.p.) with 106 CFU Brucella in 100 μl of sterile phosphate-buffered saline (PBS). For bacterial colonization experiments, spleens were collected on the indicated days, then weighed and homogenized in 1 ml sterile PBS. Serial dilutions were plated on Luria agar plates, and CFU per gram of spleen was calculated. For histological analysis, mouse spleens were collected, fixed with 4% paraformaldehyde, and embedded in paraffin. Sections were made and stained with hematoxylin and eosin according to standard protocols.
Serum levels of anti-Brucella LPS antibodies.
Maxisorp plates (Nunc A/S, Roskilde, Denmark) were coated with 5 μg/ml B. abortus LPS in PBS overnight at 4°C. Nonadsorbed material was removed by washing, and plates were dried, sealed, and stored at −20°C. Serum samples were diluted 1:500 and added to the plates for 1 h at 37°C. Peroxidase-conjugated polyclonal anti-mouse immunoglobulin G (heavy and light chains) (Dianova, Hamburg, Germany) was diluted 1:2,000 in 0.05% Tween 20-PBS-0.1% bovine serum albumin. Samples were developed with ABTS and H2O2. The results are expressed as the optical density at 405 nm (OD405).
Statistics.
Statistical significance was calculated using the Student's t test for TNF-α production assays and the Mann-Whitney test for bacterial colonization experiments. Unless otherwise stated, the analyses compare knockout macrophages or mice to the wild-type control. A P value of <0.05 was considered significant.
RESULTS
Brucella LPS has lower biological activity than Salmonella LPS.
We incubated bone marrow-derived macrophages with LPS from Brucella or Salmonella to compare their activities. At 10 ng/ml, Salmonella LPS induced TNF-α release from wild-type macrophages (Fig. 1). In contrast, 50 μg/ml of Brucella LPS was required to induce a similar TNF-α response. Smaller amounts of Brucella LPS were insufficient to induce a comparable level of TNF-α secretion. These data indicate that Brucella LPS is 5,000-fold less active than Salmonella LPS as measured by TNF-α secretion. These results agree with previous observations (8, 14).
FIG. 1.

Brucella LPS is less active than Salmonella LPS. Macrophages were treated with the indicated concentrations of Salmonella or Brucella LPS. Supernatants were collected at 24 h and assayed for TNF-α by ELISA. Statistical comparison of Salmonella LPS (10 ng/ml) and Brucella LPS (10 μg/ml) showed a significant difference (P < 0.0001). Data are representative of three independent experiments. Error bars represent, standard deviations.
Slow induction of cytokines by Brucella-infected macrophages.
To determine the timing of the macrophage response to Brucella infection, we infected macrophages with Brucella and measured cytokine production. Macrophages produced relatively low levels of TNF-α 24 h after Brucella infection but no detectable TNF-α at the earlier time points tested (Fig. 2A). This was in striking contrast to the strong induction of TNF-α seen at 2, 6, and 24 h after Salmonella infection (Fig. 2A). IL-12p40 and IL-10, two other cytokines induced upon TLR activation, were produced 24 and 48 h after Brucella infection, respectively (Fig. 2B and C). However, both cytokines were induced 6 h after Salmonella infection, and higher levels were produced during the experiment (Fig. 2B and C). These results show that the macrophage response to Brucella infection is delayed and less robust compared to that to Salmonella infection.
FIG. 2.

Slow induction of cytokine production by Brucella-infected macrophages. Macrophages were infected with Brucella (MOI, 50:1) or Salmonella (MOI, 1:1). Supernatants were collected at the indicated time points postinfection and assayed by ELISA for levels of (A) TNF-α, (B) IL-12p40, or (C) IL-10. Data are representative of four independent experiments. Error bars represent standard deviations.
Down-regulation of TLR4 is delayed after Brucella infection.
Activation of TLR4 down-regulates its own cell surface expression (31, 41). As an independent assay for TLR4 function, we tested the timing of TLR4 down-regulation after Brucella and Salmonella infection. Brucella infection induced TLR4 down-regulation at 24 h but not at 6 h (Fig. 3). Salmonella infection, however, down-regulated TLR4 by 6 h (Fig. 3). These results show that in addition to slow induction of cytokines, Brucella induces the down-regulation of TLR4 more slowly than Salmonella. We next investigated whether TLRs mediate the activation of macrophages by Brucella.
FIG. 3.
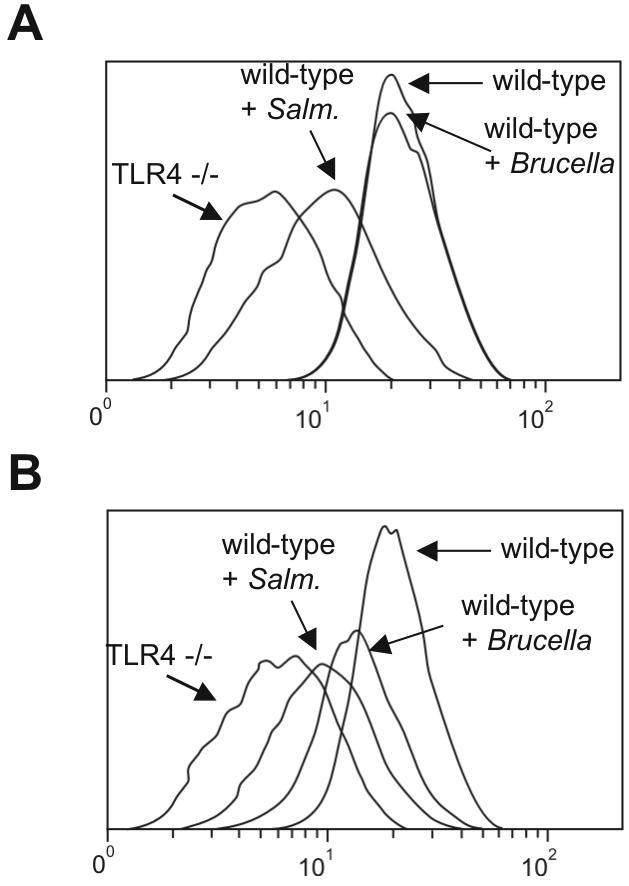
TLR4 down-regulation is delayed after Brucella infection. Macrophages were infected with Brucella (MOI, 50:1) or Salmonella (MOI, 1:1), collected 6 (A) or 24 (B) hours postinfection, and stained with an anti-TLR4 antibody. Expression levels were quantified by fluorescence-activated cell sorting. Histograms for unstimulated wild-type and TLR4−/− macrophages, as well as Brucella- and Salmonella-infected macrophages, are shown. Data are representative of three independent experiments.
Brucella-induced TNF-α production is dependent on TLRs and MyD88.
We tested if TLRs are involved in the induction of TNF-α by Brucella. Brucella-induced TNF-α production was dependent on both TLR4 and TLR2, since at 24 h postinfection, TLR4−/− and TLR2−/− macrophages produced lower levels of TNF-α than wild-type macrophages (Fig. 4). TLR4/TLR2−/− and MyD88−/− cells did not produce significant amounts of TNF-α. These results demonstrate that the induction of TNF-α in response to Brucella is completely dependent on the combination of TLR4 and TLR2, as well as MyD88.
FIG. 4.

TLR4, TLR2, and MyD88 are required for Brucella-induced TNF-α production. Macrophages were infected with Brucella at an MOI of 50:1; supernatants were collected 24 h postinfection and assayed for TNF-α by ELISA. **, P < 0.005 compared to the wild type. Differences between TLR/TLR2−/− and TLR4−/− (P = 0.0032) or TLR2−/− (P = 0.0002) mice are significant. Data are representative of four independent experiments. Error bars represent standard deviations.
MyD88 is required to control Brucella infection in vivo.
Since TLR4, TLR2, and MyD88 are involved in macrophage TNF-α production in response to Brucella infection in vitro, we tested their roles in host defense against Brucella in vivo. We infected mice intraperitoneally with 106 CFU Brucella and quantified the number of bacteria in the spleen. Spleens from wild-type mice contained >107 CFU/g on day 4 postinfection (Fig. 5A). On day 14 postinfection, the mice harbored 106 CFU/g spleen, and on day 42 postinfection, they contained between 104 and 105 CFU/g spleen, showing that they were progressively clearing the infection (Fig. 5B and C). TLR4−/−, TLR2−/−, and TLR4/TLR2−/− mice were also able to effectively fight the infection and contained similar levels of Brucella as wild-type mice on each day tested (Fig. 5).
FIG. 5.

MyD88 is required for efficient clearance of Brucella in vivo. Mice were infected i.p. with 106 CFU Brucella, and spleens were collected on (A) day 4, (B) day 14, and (C) day 42. Bacteria were quantified, and the number of bacteria per gram of tissue was calculated. **, P < 0.005 for comparison with wild-type mice; ***, P < 0.0005. Data are representative of five independent experiments.
MyD88−/− mice harbored similar numbers of Brucella as wild-type mice on day 4 postinfection but, strikingly, did not begin to clear the infection by day 14, when their spleens contained 60 times more bacteria than wild-type spleens (Fig. 5A and B). On day 42 postinfection, MyD88−/− spleens contained fewer bacteria than on day 14 but still harbored 10 times more bacteria than wild-type spleens (Fig. 5C). Taken together, these results show that MyD88, but not TLR4 or TLR2, is required for efficient host defense against Brucella infection in vivo.
In addition to its role in the TLR pathway, MyD88 is required for IL-1β and IL-18 signaling (1). To test if these two cytokines contribute to the phenotype of the MyD88−/− mice, we infected IL-1β, IL-18, and IL-1β/IL-18 knockout mice with Brucella. On day 14, when the difference in bacterial burden between wild-type and MyD88−/− mice was most pronounced, IL-1β−/−, IL-18−/−, and IL-1β/IL-18−/− mice harbored similar levels of bacteria as wild-type mice (Fig. 6). These results show that IL-1β and IL-18 are not required for host defense against Brucella infection. The phenotype of the MyD88−/− mice is therefore likely due to defects in signaling from TLRs other than TLR4 and TLR2 or from another, unknown receptor.
FIG. 6.

IL-1β and IL-18 are not required for host defense against Brucella. Mice were infected i.p. with 106 CFU Brucella, and spleens were collected on day 14. Bacteria were quantified, and the number of bacteria per gram of tissue was calculated. **, statistically significant in comparison to wild-type mice (P < 0.005).
MyD88−/− mice have decreased splenic inflammation after Brucella infection.
To further understand the phenotype of the MyD88−/− mice, we examined sections of spleens from mice on day 14 postinfection. Infected wild-type spleens showed significant inflammation, with large numbers of infiltrating cells, and the normal splenic architecture was disrupted (Fig. 7A). In contrast, infected MyD88−/− spleens were less inflamed and contained fewer infiltrating cells, and the splenic architecture was preserved (Fig. 7B). These results suggest that the delayed clearance of Brucella by MyD88−/− mice can in part be explained by decreased inflammation.
FIG. 7.

MyD88−/− mice have decreased splenic inflammation after Brucella infection. Mice were infected i.p. with 106 CFU Brucella, and spleens were collected on day 14. Sections were made and stained with hematoxylin and eosin. Representative spleen sections from (A) wild-type and (B) MyD88−/− mice are shown at a magnification of ×10. Arrows highlight areas of white pulp (B), showing that the normal splenic architecture is preserved in MyD88−/− mice, while this is not observed in wild-type mice (A).
MyD88 is not required to induce anti-Brucella LPS antibodies.
Antibodies against Brucella LPS contribute to host defense in vivo (23, 30). We tested the levels of anti-Brucella LPS antibodies in the sera of infected mice on day 14 postinfection by ELISA. Wild-type mice had varying levels of anti-Brucella LPS antibodies, within a range of OD405s of 0.5 to 2.0 (Fig. 8). Similar results were seen for infected TLR4−/−, TLR2−/−, TLR4/TLR2−/−, and MyD88−/− mice. Uninfected mice did not contain anti-Brucella LPS antibodies. These results demonstrate that TLR4, TLR2, and MyD88 are not required for the induction of anti-Brucella LPS antibodies in vivo.
FIG. 8.

TLR4, TLR2, and MyD88 are not required for production of anti-Brucella LPS antibodies. Serum was collected from Brucella-infected mice (three mice per group) on day 14 and assayed for anti-LPS antibodies by ELISA. Each bar represents the level of anti-LPS antibodies from one mouse. Error bars represent variations between triplicate samples. Data are representative of two independent experiments.
DISCUSSION
Slow activation of macrophages by Brucella.
We show that Brucella activates macrophages 24 h postinfection but not 6 h postinfection, as measured by cytokine production and TLR4 down-regulation (Fig. 2 and 3). This is in contrast to Salmonella, which induces a strong macrophage response at both 6 and 24 h and induces TNF-α production as early as 2 h postinfection (Fig. 2 and 3). TLR4, which responds to LPS, is required for early macrophage activation during Salmonella infection (24, 37, 41). The lack of early macrophage activation by Brucella may be due to the 5,000-fold-lower activity of its LPS compared to that of Salmonella LPS (Fig. 1). Indeed, the structure of Brucella LPS strikingly departs from that of Salmonella, a fact consistent with its lower biological activity (29).
Interestingly, the combination of TLR4 and TLR2, as well as MyD88, is necessary and sufficient for induction of TNF-α production by Brucella 24 h postinfection (Fig. 4). It is not known which Brucella components activate TLR4 and TLR2. Brucella LPS may signal through TLR4. It is still an open question whether there is enough Brucella LPS during an infection to signal through TLR4, or if another Brucella component activates TLR4. If indeed Brucella LPS signals through TLR4, it is not clear why it signals at 24 h but not at 6 h, since TLR4 is expressed prior to and at 6 h. Brucella LPS may signal differently than classical enterobacterial LPS. Brucella LPS, in contrast to enterobacterial LPS, is not degraded by macrophages and instead accumulates in lysosomes up to 24 h postinfection and later recycles to the plasma membrane (11). Accumulation may be required for signaling, either from an intracellular location or from a plasma membrane location. These properties may explain the slow kinetics of Brucella LPS signaling in the context of an infection.
Brucella BLP may activate TLR2. Giambartolomei et al. described Brucella BLP as the active components in heat-killed Brucella, and Huang et al. showed that TLR2 is involved in the recognition of heat-killed Brucella (13, 20). Whichever Brucella components signal through TLR4 and TLR2, it is clear that both receptors are involved in the recognition of live Brucella.
MyD88, but not TLR4, TLR2, IL-1β, or IL-18, is required for efficient clearance of Brucella in vivo.
Efficient clearance of Brucella infection requires MyD88, as evidenced by the fact that MyD88−/− mice harbor more bacteria than wild-type mice at days 14 and 42 postinfection (Fig. 5). These mice have reduced splenic inflammation, which may explain their delay in bacterial clearance (Fig. 7). Despite their roles in macrophage induction of TNF-α in vitro, TLR4 and TLR2 are not required for clearance of Brucella in vivo (Fig. 5). The lack of a role for TLR4 is in agreement with data from Huang et al., who showed that TLR4 is not required for TNF-α production in response to heat-killed Brucella in vivo (20). TLR4 is activated quickly by bacteria containing classical LPS, and its primary role in host defense may be in the initial recognition of microbes. The lack of early TLR4 activation by Brucella may explain why TLR4 is not required for host defense against the infection. We verified the lack of a role for TLR4 in another host genetic background by using C3H/HeJ mice, which have a naturally occurring, inactivating mutation in TLR4 (35). Consistent with our observations for TLR4 knockout mice, infected C3H/HeJ mice contained similar numbers of Brucella as infected wild-type C3H/HeN mice (data not shown).
MyD88 is required for TLR signaling, as well as for IL-1β and IL-18 signaling. We find that IL-1β and IL-18 are not necessary for host defense against Brucella infection (Fig. 6). Taken together, these results suggest that the phenotype of the MyD88 knockout mice is likely due to a lack of signaling from TLRs other than TLR4 and TLR2, or from an as yet uncharacterized receptor.
Our data agree with the results of Campos et al., who showed that TLR2 does not play a role in host defense against Brucella (6). However, using C3H/HeJ mice, Campos et al. observed that TLR4 might play a role in clearance of Brucella (6). The discrepancy with our results may be due to the fact that Campos et al. used a different strain of Brucella displaying lipid A fatty acid profiles that depart considerably from that of the Brucella strain we used. Indeed, the characteristic long-chain fatty acids (C28:0 to C30:0), which are a hallmark of many alpha-2 proteobacterial LPSs, including that of Brucella (28, 29), were not detected in their preparation (6).
Host defense against Brucella infection depends in part on the antibody response (23, 30). We investigated whether the MyD88−/− phenotype was due in part to defects in antibody production. On day 14 postinfection, when spleens from MyD88 knockout mice contained more bacteria than spleens from wild-type mice, both groups of mice had similar levels of anti-Brucella LPS antibodies (Fig. 8). This result suggests that the phenotype of the MyD88−/− mice is due to defects in innate immune function rather than to defects in antibody production. It will be interesting to test CD8 T-cell responses in Brucella-infected MyD88 knockout mice, which also contribute to the host response against Brucella (23).
Does Brucella manipulate TLR function to find its niche?
To live in host cells, Brucella redirects the intracellular trafficking pathway, avoiding late endosomes and lysosomes, and localizing to membrane-bound compartments resembling the endoplasmic reticulum (ER) 6 to 8 h postinfection (7, 34). This is a mandatory step in the pathogenesis of Brucella, since mutants that are defective in localization to the ER-like compartments are avirulent (7). The lack of early TLR4 signaling may buy time for the bacteria to redirect the intracellular trafficking pathway and localize to the ER-like compartments before the macrophage is effectively armed to fight the bacteria. By the time TLRs signal and the macrophage is activated, Brucella is already in its intracellular niche. Therefore, the low activity of Brucella LPS and subsequent evasion of early TLR4 signaling may be critical aspects of Brucella pathogenesis.
Acknowledgments
We thank Björn Eilers for help with the FACS analysis, Beatrix Fauler and Volker Brinkmann for help with the histological analysis, and Molly Ingersoll and Bärbel Raupach for critical reading of the manuscript.
Edgardo Moreno was a research fellow of the Deutscher Akademischer Austauschdienst (DAAD) during the course of this investigation. This work was partially supported by the Network for Research and Training in Tropical Diseases in Central America (NeTropica).
Editor: J. N. Weiser
REFERENCES
- 1.Adachi, O., T. Kawai, K. Takeda, M. Matsumoto, H. Tsutsui, M. Sakagami, K. Nakanishi, and S. Akira. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143-150. [DOI] [PubMed] [Google Scholar]
- 2.Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499-511. [DOI] [PubMed] [Google Scholar]
- 3.Aliprantis, A. O., R. B. Yang, M. R. Mark, S. Suggett, B. Devaux, J. D. Radolf, G. R. Klimpel, P. Godowski, and A. Zychlinsky. 1999. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285:736-739. [DOI] [PubMed] [Google Scholar]
- 4.Aragon, V., R. Diaz, E. Moreno, and I. Moriyon. 1996. Characterization of Brucella abortus and Brucella melitensis native haptens as outer membrane O-type polysaccharides independent from the smooth lipopolysaccharide. J. Bacteriol. 178:1070-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brightbill, H. D., D. H. Libraty, S. R. Krutzik, R. B. Yang, J. T. Belisle, J. R. Bleharski, M. Maitland, M. V. Norgard, S. E. Plevy, S. T. Smale, P. J. Brennan, B. R. Bloom, P. J. Godowski, and R. L. Modlin. 1999. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science 285:732-736. [DOI] [PubMed] [Google Scholar]
- 6.Campos, M. A., G. M. Rosinha, I. C. Almeida, X. S. Salgueiro, B. W. Jarvis, G. A. Splitter, N. Qureshi, O. Bruna-Romero, R. T. Gazzinelli, and S. C. Oliveira. 2004. Role of Toll-like receptor 4 in induction of cell-mediated immunity and resistance to Brucella abortus infection in mice. Infect. Immun. 72:176-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Celli, J., C. de Chastellier, D. M. Franchini, J. Pizarro-Cerda, E. Moreno, and J. P. Gorvel. 2003. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J. Exp. Med. 198:545-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duenas, A. I., A. Orduna, M. S. Crespo, and C. Garcia-Rodriguez. 2004. Interaction of endotoxins with Toll-like receptor 4 correlates with their endotoxic potential and may explain the proinflammatory effect of Brucella spp. LPS. Int. Immunol. 16:1467-1475. [DOI] [PubMed] [Google Scholar]
- 9.Fitzgerald, K. A., E. M. Palsson-McDermott, A. G. Bowie, C. A. Jefferies, A. S. Mansell, G. Brady, E. Brint, A. Dunne, P. Gray, M. T. Harte, D. McMurray, D. E. Smith, J. E. Sims, T. A. Bird, and L. A. O'Neill. 2001. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 413:78-83. [DOI] [PubMed] [Google Scholar]
- 10.Fitzgerald, K. A., D. C. Rowe, B. J. Barnes, D. R. Caffrey, A. Visintin, E. Latz, B. Monks, P. M. Pitha, and D. T. Golenbock. 2003. LPS-TLR4 signaling to IRF-3/7 and NF-κB involves the toll adapters TRAM and TRIF. J. Exp. Med. 198:1043-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forestier, C., E. Moreno, J. Pizarro-Cerda, and J. P. Gorvel. 1999. Lysosomal accumulation and recycling of lipopolysaccharide to the cell surface of murine macrophages, an in vitro and in vivo study. J. Immunol. 162:6784-6791. [PubMed] [Google Scholar]
- 12.Freer, E., N. Rojas, A. Weintraub, A. A. Lindberg, and E. Moreno. 1995. Heterogeneity of Brucella abortus lipopolysaccharides. Res. Microbiol. 146:569-578. [DOI] [PubMed] [Google Scholar]
- 13.Giambartolomei, G. H., A. Zwerdling, J. Cassataro, L. Bruno, C. A. Fossati, and M. T. Philipp. 2004. Lipoproteins, not lipopolysaccharide, are the key mediators of the proinflammatory response elicited by heat-killed Brucella abortus. J. Immunol. 173:4635-4642. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein, J., T. Hoffman, C. Frasch, E. F. Lizzio, P. R. Beining, D. Hochstein, Y. L. Lee, R. D. Angus, and B. Golding. 1992. Lipopolysaccharide (LPS) from Brucella abortus is less toxic than that from Escherichia coli, suggesting the possible use of B. abortus or LPS from B. abortus as a carrier in vaccines. Infect. Immun. 60:1385-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorvel, J. P., and E. Moreno. 2002. Brucella intracellular life: from invasion to intracellular replication. Vet. Microbiol. 90:281-297. [DOI] [PubMed] [Google Scholar]
- 16.Hoebe, K., X. Du, P. Georgel, E. Janssen, K. Tabeta, S. O. Kim, J. Goode, P. Lin, N. Mann, S. Mudd, K. Crozat, S. Sovath, J. Han, and B. Beutler. 2003. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424:743-748. [DOI] [PubMed] [Google Scholar]
- 17.Hoiseth, S. K., and B. A. Stocker. 1981. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291:238-239. [DOI] [PubMed] [Google Scholar]
- 18.Horng, T., G. M. Barton, R. A. Flavell, and R. Medzhitov. 2002. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 420:329-333. [DOI] [PubMed] [Google Scholar]
- 19.Hoshino, K., O. Takeuchi, T. Kawai, H. Sanjo, T. Ogawa, Y. Takeda, K. Takeda, and S. Akira. 1999. Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162:3749-3752. [PubMed] [Google Scholar]
- 20.Huang, L. Y., J. Aliberti, C. A. Leifer, D. M. Segal, A. Sher, D. T. Golenbock, and B. Golding. 2003. Heat-killed Brucella abortus induces TNF and IL-12p40 by distinct MyD88-dependent pathways: TNF, unlike IL-12p40 secretion, is Toll-like receptor 2 dependent. J. Immunol. 171:1441-1446. [DOI] [PubMed] [Google Scholar]
- 21.Joklik, W. K., H. P. Willett, D. B. Amos, and C. M. Wilfert. 1992. Brucella, p. 609. In Zinsser Microbiology, 20th ed. Appleton and Lange, Norwalk, Conn.
- 22.Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11:115-122. [DOI] [PubMed] [Google Scholar]
- 23.Ko, J., and G. A. Splitter. 2003. Molecular host-pathogen interaction in brucellosis: current understanding and future approaches to vaccine development for mice and humans. Clin. Microbiol. Rev. 16:65-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li, Q., and B. J. Cherayil. 2003. Role of Toll-like receptor 4 in macrophage activation and tolerance during Salmonella enterica serovar Typhimurium infection. Infect. Immun. 71:4873-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreno, E., M. W. Pitt, L. M. Jones, G. G. Schurig, and D. T. Berman. 1979. Purification and characterization of smooth and rough lipopolysaccharides from Brucella abortus. J. Bacteriol. 138:361-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreno, E., D. T. Berman, and L. A. Boettcher. 1981. Biological activities of Brucella abortus lipopolysaccharides. Infect. Immun. 31:362-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreno, E., R. S. Kurtz, and D. T. Berman. 1984. Induction of immune and adjuvant immunoglobulin G responses in mice by Brucella lipopolysaccharide. Infect. Immun. 46:74-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreno, E., E. Stackebrandt, M. Dorsch, J. Wolters, M. Busch, and H. Mayer. 1990. Brucella abortus 16S rRNA and lipid A reveal a phylogenetic relationship with members of the alpha-2 subdivision of the class Proteobacteria. J. Bacteriol. 172:3569-3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moriyon, I. 2003. Against gram-negative bacteria: the LPS case, p. 204. In J. P. Gorvel (ed.), Intracellular pathogens in membrane interactions and vacuole biogenesis. Landes Bioscience/Eurekah.com, Georgetown, Tex.
- 30.Moriyon, I., M. J. Grillo, D. Monreal, D. Gonzalez, C. Marin, I. Lopez-Goni, R. C. Mainar-Jaime, E. Moreno, and J. M. Blasco. 2004. Rough vaccines in animal brucellosis: structural and genetic basis and present status. Vet. Res. 35:1-38. [DOI] [PubMed] [Google Scholar]
- 31.Nomura, F., S. Akashi, Y. Sakao, S. Sato, T. Kawai, M. Matsumoto, K. Nakanishi, M. Kimoto, K. Miyake, K. Takeda, and S. Akira. 2000. Endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J. Immunol. 164:3476-3479. [DOI] [PubMed] [Google Scholar]
- 32.Oshiumi, H., M. Matsumoto, K. Funami, T. Akazawa, and T. Seya. 2003. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 4:161-167. [DOI] [PubMed] [Google Scholar]
- 33.Pasare, C., and R. Medzhitov. 2004. Toll-like receptors and acquired immunity. Semin. Immunol. 16:23-26. [DOI] [PubMed] [Google Scholar]
- 34.Pizarro-Cerda, J., S. Meresse, R. G. Parton, G. van der Goot, A. Sola-Landa, I. Lopez-Goni, E. Moreno, and J. P. Gorvel. 1998. Brucella abortus transits through the autophagic pathway and replicates in the endoplasmic reticulum of nonprofessional phagocytes. Infect. Immun. 66:5711-5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poltorak, A., X. He, I. Smirnova, M. Y. Liu, C. Van Huffel, X. Du, D. Birdwell, E. Alejos, M. Silva, C. Galanos, M. Freudenberg, P. Ricciardi-Castagnoli, B. Layton, and B. Beutler. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085-2088. [DOI] [PubMed] [Google Scholar]
- 36.Rasool, O., E. Freer, E. Moreno, and C. Jarstrand. 1992. Effect of Brucella abortus lipopolysaccharide on oxidative metabolism and lysozyme release by human neutrophils. Infect. Immun. 60:1699-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Royle, M. C., S. Totemeyer, L. C. Alldridge, D. J. Maskell, and C. E. Bryant. 2003. Stimulation of Toll-like receptor 4 by lipopolysaccharide during cellular invasion by live Salmonella typhimurium is a critical but not exclusive event leading to macrophage responses. J. Immunol. 170:5445-5454. [DOI] [PubMed] [Google Scholar]
- 38.Schaible, U. E., and S. H. E. Kaufmann. 2002. Studying trafficking of intracellular pathogens in antigen-presenting cells. Methods Microbiol. 31:343-360. [Google Scholar]
- 39.Takeda, K., H. Tsutsui, T. Yoshimoto, O. Adachi, N. Yoshida, T. Kishimoto, H. Okamura, K. Nakanishi, and S. Akira. 1998. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 8:383-390. [DOI] [PubMed] [Google Scholar]
- 40.Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K. Takeda, and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443-451. [DOI] [PubMed] [Google Scholar]
- 41.Weiss, D. S., B. Raupach, K. Takeda, S. Akira, and A. Zychlinsky. 2004. Toll-like receptors are temporally involved in host defense. J. Immunol. 172:4463-4469. [DOI] [PubMed] [Google Scholar]
- 42.Yamamoto, M., S. Sato, K. Mori, K. Hoshino, O. Takeuchi, K. Takeda, and S. Akira. 2002. A novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-β promoter in the Toll-like receptor signaling. J. Immunol. 169:6668-6672. [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto, M., S. Sato, H. Hemmi, S. Uematsu, K. Hoshino, T. Kaisho, O. Takeuchi, K. Takeda, and S. Akira. 2003. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat. Immunol. 4:1144-1150. [DOI] [PubMed] [Google Scholar]
- 44.Zheng, H., D. Fletcher, W. Kozak, M. Jiang, K. J. Hofmann, C. A. Conn, D. Soszynski, C. Grabiec, M. E. Trumbauer, A. Shaw, et al. 1995. Resistance to fever induction and impaired acute-phase response in interleukin-1 beta-deficient mice. Immunity 3:9-19. [DOI] [PubMed] [Google Scholar]