

Abstract
The performance of a 5′ nuclease real-time PCR assay was studied to optimize an automated method of detection of preenriched Salmonella enterica cells in buffered peptone water (BPW). The concentrations and interactions of the PCR reagents were evaluated on the basis of two detection responses, the threshold cycle (CT) and the fluorescence intensity by a normalized reporter value (ΔRn). The CT response was identified as the most suitable for detection modeling to describe the PCR performances of different samples. DNA extracted from S. enterica serovar Enteritidis was studied in double-distilled H2O (ddH2O) and in two different enrichment media (brain heart infusion and BPW) with two PCR mixtures based on AmpliTaq Gold or rTth. A descriptive model was proposed and fitted to the available experimental data. Equivalent PCR performances for the two PCR mixtures were obtained when DNA was diluted in ddH2O. However, the level of detection of DNA was affected when BPW was present during amplification. Use of the rTth mixture generated a 1-log-unit wider linear range of amplification, and the DNA detection levels were 2 × 10−13 g/microwell for the rTth mixture and 2 × 10−12 g/microwell for the AmpliTaq Gold mixture. To verify the improved amplification capacity of the rTth mixture, BPW was inoculated with 1 CFU of S. enterica serovar Enteritidis per ml and the mixture was incubated at 30°C. Samples for PCR were withdrawn every 4 h during a 36-h enrichment. Use of the rTth mixture resulted in an earlier PCR detection during enrichment than use of the AmpliTaq Gold mixture. For accurate detection (CT ≤ 30) of S. enterica serovar Enteritidis inoculated in BPW, the rTth mixture required 8.4 h of enrichment, while the AmpliTaq Gold mixture needed 11.6 h. In conclusion, the principle applied can improve the methodology of 5′ nuclease real-time PCR for numerical optimization of sample pretreatment strategies to provide automated diagnostic PCR procedures.
Standard methods for detection of Salmonella spp. in complex biological samples are based on preenrichment in buffered peptone water (BPW) followed by selective enrichment and plating, which can take up to 5 days to complete (26). For this reason rapid methods have been developed, and real-time PCR has become a promising technique. The technique is highly specific and sensitive, but it is also performed in a closed tube with automated detection (6). In addition, because of its quantitative nature, real-time PCR may permit the use of mathematical modeling to describe PCR detection.
However, it has been shown that clinical samples inhibit or interfere with PCR, which makes sample preparation necessary prior to PCR (19). Thus, to improve the robustness of diagnostic real-time PCR, the PCR inhibitors must be characterized (3). One way to overcome PCR inhibition is to use alternative DNA polymerases (1) and buffer systems with PCR facilitators (2), if necessary, combined with a suitable sample preparation method. A common diagnostic sample-processing procedure prior to real-time PCR is the use of enrichment followed by a physical sample preparation method or DNA extraction (14, 30). The enrichment step allows the target bacteria to multiply to detectable concentrations, while it also dilutes dead cells and PCR-inhibitory substances. Furthermore, enrichment facilitates subsequent confirmation by culture.
In the present study, a recently developed 5′ nuclease assay for Salmonella enterica was used (13). However, to adapt the assay for enrichment PCR in order to avoid false-negative results due to PCR inhibition of enrichment media (20), the influences and interactions of various PCR reagents on the detection response were studied. Similar to other real-time PCRs based on SYBR green and hybridization probes (37), the reaction in the 5′ nuclease (TaqMan) PCR (22) undergoes an exponential phase and a plateau phase (10). In an optimized assay, the exponential phase should include no limiting factor and PCR products should accumulate at a steady rate. However, at some point the amplification kinetics change, which leads to the plateau phase. Both phases can be studied by the 5′ nuclease real-time PCR in an ABI Prism 7700 sequence detection system. The software generates two detection responses, i.e., the threshold cycle (CT) and the fluorescence intensity by a normalized reporter value (ΔRn) (protocol P N 4304437, 1998; TaqMan Universal PCR Master Mix; Applied Biosystems). The ΔRn value is obtained by subtracting the value for the reporter signal, R+n (emission intensity of the reporter/emission intensity of a passive reference), from the value for the background signal, R−n (emission intensity of the reporter [no template]/emission intensity of a passive reference [no template]) (protocol P N 4304437, 1998; TaqMan Universal PCR Master Mix; Applied Biosystems). The CT value is the cycle number at which the fluorescence is greater than the fixed threshold fluorescence (protocol P N 4304437, 1998; TaqMan Universal PCR Master Mix; Applied Biosystems). The exponential phase is the region where ΔRn exhibits a linear increase, while the plateau phase is the region where the amplification efficiency drops. Changes in amplification efficiency during the exponential phase will lead to less accurate detection, and several factors have been shown to affect the efficiency of PCR, for instance, the DNA quality (11) and the presence of PCR-inhibitory substances (23). Thus, to estimate the influences of different components of clinical samples on the performance of PCR in terms of detection probability, linear range of amplification, and amplification efficiency, the appropriate detection response must be selected for modeling. A suitable model would provide a better understanding for comparisons of the linear ranges of amplification when the PCR mixture contains different samples with substances originating from complex biological matrices. Furthermore, if biological contaminants originating from clinical samples are going to be present during DNA amplification, it is necessary to establish standard curves for an objective interpretation of the results. The objectives of the present study were (i) to study the influences and interactions of various PCR reagents on CT and ΔRn in order to identify the most suitable response for the description of PCR performance and (ii) to estimate, using a new model, the PCR performances of different DNA polymerases and PCR mixtures in the presence of enrichment medium. The intended use of the proposed model was for numerical optimization of sample pretreatment strategies to facilitate verification to detect preenriched S. enterica cells without any special sample preparation prior to PCR.
MATERIALS AND METHODS
Bacterial strain.
S. enterica serovar Enteritidis S83, a swine isolate obtained from the collection of the Department of Microbiology, Danish Veterinary Laboratory, Copenhagen, Denmark, was used. To obtain fresh cells of the strain, it was grown overnight on blood agar plates at 37°C. Twenty-five milliliters of brain heart infusion (BHI; Difco Laboratories, Detroit, Mich.) and 25 ml of BPW (Merck, Darmstadt, Germany) were each inoculated with a colony, and the mixtures were incubated at 37°C overnight. BPW is the preenrichment medium used in the International Organization for Standardization method (32), and BHI is used in serological tests for Salmonella spp. (9). Sterile glycerol (Merck) was added to the overnight cultures, giving a final concentration of 10% (vol/vol), and the concentration (in numbers of CFU per milliliter) was determined by serial dilution followed by plate counting on tryptone glucose extract (TGE) agar (Merck) at 37°C for 24 h. One-milliliter aliquots of the overnight cultures in BPW and BHI were stored at −20°C until testing.
DNA extraction.
DNA was isolated from S. enterica serovar Enteritidis colonies on TGE agar (Merck) with a QIAmp DNA Mini Kit (Qiagen GmbH, Hilden, Germany). Purification was performed by the protocols recommended by the supplier (protocols for bacteria, protocol C, and tissue protocol). The concentration of the extracted DNA was determined by measuring the optical density at 260 nm with a spectrophotometer (GeneQuant RNA/DNA Calculator; Pharmacia, Uppsala, Sweden).
Salmonella 5′ nuclease PCR.
The Salmonella 5′ nuclease PCR assay amplified a 0.12-kb amplicon that originated from the invA gene (13). The oligonucleotides (primers and TaqMan probe) were purchased from DNA Technology Ltd. (Århus, Denmark). The TaqMan probe was labeled with 6-carboxyfluorescein (FAM; reporter dye) and 6-carboxytetramethylrhodamine (TAMRA; quencher dye). The total PCR mixture consisted of 50 μl; and two different PCR mixtures were used since two DNA polymerases, AmpliTaq Gold (Applied Biosystems, Foster City, Calif.) and rTth (Applied Biosystems), were tested with the buffer systems supplied with each of the polymerases. The PCR mixture for AmpliTaq Gold was composed of the following reagents and concentrations: 100 nM TaqMan probe, forward and reverse primers at concentrations of 900 nM each, 1× TaqMan Universal PCR Master Mix (including deoxynucleoside triphosphates [dNTPs; 200 μM dATP, 200 μM dCTP, 200 μM dGTP, and 400 μM dUTP]; Applied Biosystems), 0.01 U of uracil-N-glycosylase per μl, 2.5 mM MgCl2, and 0.025 U of AmpliTaq Gold per μl. The PCR mixture for rTth DNA polymerase had the following reagents and concentrations: 100 nM TaqMan probe, forward and reverse primers at concentrations of 900 nM each, 1× chelating buffer (N808-0098; Applied Biosytems), 0.05 U of rTth DNA polymerase per μl, dNTPs (dATP, dCTP, dGTP, and dTTP, each at a concentration of 200 μM; Applied Biosystems), 2.5 mM MgCl2, and 8% (vol/vol) glycerol (Merck). Double-distilled H2O (ddH2O) was used in both PCR mixtures, and the sample volume was 5 μl. The samples and PCR mixture were distributed over a 96-well microwell plate (MicroAmp; Applied Bisosytems), and the microwell plates were sealed with MicroAmp optical caps (Applied Biosystems). Two nontemplate controls were added to each microwell plate. Amplification took place in an ABI Prism 7700 (TaqMan) sequence detection system (Applied Biosystems). Each amplification was run online and started with a denaturation step at 94°C for 10 min, followed by 40 cycles at 94°C for 15 s and 55°C for 60 s. The fluorescence measurements were made online, and at the end of the PCR program these were analyzed with the sequence detection systems software (version 1.6.3; Applied Biosystems). The reference dye for the rTth reaction mixture was TAMRA, while the reference dye for the AmpliTaq Gold mixture was 6-carboxy-X-rhodamine (ROX) dye. All calculations of the CT value were performed with the threshold set at 0.1.
Factorial design experiment.
To study the interactions of the PCR reagents in the PCR mixture, a factorial design experiment (central composite 25 factorial design) was used, and the experimental design is given in Table 1. The factors, in terms of the PCR reagents, tested were the MgCl2 concentration (in millimolar), the probe concentration (in nanomolar), the primer concentration (in nanomolar), the DNA polymerase concentration (in units per microliter), and the amount of Salmonella DNA (in grams). Two DNA polymerases with their corresponding buffer systems, AmpliTaq Gold and rTth, were tested in two different experiments. Center points were included, which made it possible to test if curvature was present (24). Axial points were included in the design to make it possible to further evaluate the curvature. The center points were chosen so that they were at the same concentrations as those used for the PCR reagents for the Salmonella 5′ nuclease assay, as described previously (13). The results generated were used for statistical data analysis with the statistical software S-Plus (version 4.0; Math Soft, Seattle, Wash.).
TABLE 1.
Experimental design and results from the factorial design experiment with the two PCR reaction mixtures based on AmpliTaq Gold and rTtha
| PCR mixture | Factor |
P value by ANOVAb
|
Experimental design
|
PCR results
|
|||
|---|---|---|---|---|---|---|---|
| CT | ΔRn | Factor level | Concn | Mean CT | Mean ΔRn | ||
| AmpliTaq Gold | MgCl2 | 0.000 | 0.000 | High | 3.5 mM | 23.1 | 1.7 |
| Low | 1.5 mM | 36.2 | 0.2 | ||||
| Center | 2.5 mM | 24.6 | 1.2 | ||||
| Axial high | 4.7 mM | 22.3 | 1.9 | ||||
| Axial low | 0.1 mM | 40.0 | 0.0 | ||||
| Probe | 0.192 | 0.050 | High | 125 nM | 30.6 | 1.1 | |
| Low | 75 nM | 28.6 | 0.8 | ||||
| Center | 100 nM | 26.2 | 1.2 | ||||
| Axial high | 160 nM | 22.5 | 2.1 | ||||
| Axial low | 40 nM | 24.5 | 0.6 | ||||
| Primer | 0.260 | 0.089 | High | 1,200 nM | 30.9 | 0.9 | |
| Low | 600 nM | 28.3 | 1.0 | ||||
| Center | 900 nM | 26.3 | 1.1 | ||||
| Axial high | 1,600 nM | 23.3 | 1.4 | ||||
| Axial low | 185 nM | 23.7 | 1.2 | ||||
| AmpliTaq Gold | 0.218 | 0.536 | High | 0.07 U/μl | 29.1 | 1.0 | |
| Low | 0.03 U/μl | 30.1 | 0.9 | ||||
| Center | 0.05 U/μl | 27.4 | 1.1 | ||||
| Axial high | 0.10 U/μl | 23.6 | 1.4 | ||||
| Axial low | 0.02 U/μl | 24.5 | 1.2 | ||||
| DNA | 0.000 | 0.474 | High | 5.0 × 10−8 g | 27.4 | 1.0 | |
| Low | 5.0 × 10−10 g | 31.9 | 0.9 | ||||
| Central | 5.0 × 10−9 g | 26.2 | 1.2 | ||||
| Axial high | 2.0 × 10−6 g | 15.7 | 1.2 | ||||
| Axial low | 2.0 × 10−11 g | 31.5 | 1.1 | ||||
| rTth | MgCl2 | 0.235 | 0.082 | High | 3.5 mM | 24.5 | 5.5 |
| Low | 1.5 mM | 35.0 | 0.5 | ||||
| Center | 2.5 mM | 22.3 | 5.7 | ||||
| Axial high | 4.7 mM | 21.9 | 7.5 | ||||
| Axial low | 0.1 mM | 36.8 | 0.1 | ||||
| Probe | 0.682 | 0.415 | High | 125 nM | 31.3 | 2.8 | |
| Low | 75 nM | 28.2 | 2.8 | ||||
| Center | 100 nM | 23.2 | 5.5 | ||||
| Axial high | 160 nM | 22.4 | 4.1 | ||||
| Axial low | 40 nM | 24.1 | 2.3 | ||||
| Primer | 0.681 | 0.204 | High | 1,200 nM | 28.1 | 3.2 | |
| Low | 600 nM | 31.4 | 2.5 | ||||
| Center | 900 nM | 23.4 | 5.0 | ||||
| Axial high | 1,600 nM | 22.3 | 5.8 | ||||
| Axial low | 185 nM | 22.0 | 8.6 | ||||
| rTth | 0.758 | 0.769 | High | 0.035 U/μl | 29.6 | 3.2 | |
| Low | 0.015 U/μl | 29.8 | 2.4 | ||||
| Central | 0.025 U/μl | 23.6 | 5.8 | ||||
| Axial high | 0.043 U/μl | 23.1 | 3.6 | ||||
| Axial low | 0.007 U/μl | 21.5 | 2.5 | ||||
| DNA | 0.003 | 0.015 | High | 5.0 × 10−8 g | 27.1 | 3.4 | |
| Low | 5.0 × 10−10 g | 32.4 | 2.3 | ||||
| Central | 5.0 × 10−9 g | 23.3 | 5.1 | ||||
| Axial high | 2.0 × 10−6 g | 14.5 | 9.1 | ||||
| Axial low | 2.0 × 10−11 g | 30.5 | 3.8 | ||||
The different factors and their concentrations are given. The experiment with the AmpliTaq Gold mixture used a basic 25 factorial design with 2-by-5 axial points and one observation at the center point, giving a total of 86 observations. The experiment with the rTth mixture used a basic 25 factorial design augmented with 2-by-5 axial points and five observations at the center point, giving a total of 47 observations.
ANOVA, one-way analysis of variance for main effects in studying the significance of the different factors. Significant factor interactions are not presented here but are given in the text (see Results and Discussion).
Modeling of 5′ nuclease PCR performance.
The amount of product at cycle n (P) is dependent on the initial amount of target template (T), the amplification efficiency (E), and the number of cycles (n) (10):
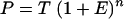 |
(1) |
The efficiency can be determined by a standard curve by serial dilution of template in ddH2O. Different methods have been used for calculation of the amplification efficiency (17, 29). In the present study, the following equation was used to calculate the amplification efficiency (17):
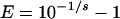 |
(2) |
where s is the slope of the log-linear range. The optimal PCR condition is obtained when E is equal to 1, giving the slope = −1/log2 = −3.3219. However, for the diagnostic purposes of real-time PCR, it is needed to describe the practical operating range of the assay when clinical samples are analyzed. Therefore, a model that describes the PCR performance in terms of detection probability, linear range of amplification, and amplification efficiency is proposed. It is known that in a certain region, the relationship between the CT value and the DNA concentration in the sample is essentially linear (protocol P N 4304437, 1998; TaqMan Universal PCR Master Mix; Applied Biosystems). However, since the PCR program is usually run for a fixed number of cycles, this puts a limit on the largest possible CT value. When the concentration of target DNA is below a certain level, the response will simply be this maximal CT value (in the assay described here, the value is 40), and the linear relationship mentioned above will no longer be valid. A value corresponding to the maximal value of CT can be defined to be no detection. The borderline between these two regions is not a clear cutoff and needs to be determined. A model commonly used to describe the detection probability is a logistic regression model (4), where the detection probability of the target template can be estimated. From this we propose a new combined model that weights the models of the two ranges according to a concentration-dependent detection probability:
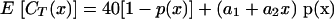 |
(3) |
where x is the concentration of target template in the sample, p is concentration-dependent detection probability, and a1 and a2 are the intercept and slope, respectively, of the linear response model. This means that the expected response is modeled as a weighted average of the CT value when there is no detection (CT = 40) and the expected CT value in the region of linear response (CT = a1 + a2x). The weights are determined by the detection probability, which is assumed to depend on the DNA concentration according to a logistic regression model:
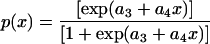 |
(4) |
where a3 and a4 are the intercept and slope, respectively, of the logit model. From the model, given in equation 3, the linear range of amplification and slope (a2) can be estimated. By using the slope the amplification efficiency can be determined by equation 2, where s is equal to a2. The parameters a1 to a4 of the model were estimated numerically by the method of least squares (Matlab Optimization Toolbox; The Math Works, Inc., Natick, Mass.).
The model was evaluated on the basis of the available experimental data from studies on the PCR performances of the two DNA polymerases (AmpliTaq Gold and rTth). The experiment was performed three times with 10-fold dilutions of extracted DNA. The range of the DNA dilution was 2 × 10−14 to 2 × 10−8 g. To study the PCR performance when enrichment media were present in the PCR mixture, 5 μl of ddH2O in each microwell was replaced by 5 μl of BHI (Difco Laboratories) or BPW (Merck). This experiment was performed three times. In addition, the effect of changing the medium concentration at a fixed concentration of DNA (2 × 10−9 g/microwell) was studied in a rough screening experiment, but the data obtained were not used for modeling. The concentrations tested were 74, 37, 29, 22, 15, and 4 g/liter for BHI and 76, 51, 25, 21, 15, 10, and 3 g/liter for BPW. The standard concentration of BHI is 37 g/liter, and that of BPW is 25 g/liter; the other concentrations tested were 3 (only BPW), 2, 0.8, 0.6, 0.4, and 0.2 times the standard concentration of each medium. The experiment was performed by adding 5 μl of the prepared medium with each concentration to the microwell.
Enrichment PCR.
A total of 10 ml of BPW was inoculated with 1 CFU of S. enterica serovar Enteritidis per ml, and the mixture was incubated in a water bath with orbital motion at 30°C for 36 h. The culture was enriched for 36 h at ambient temperature to allow study of the PCR performance of cells withdrawn in the lag phase, exponential phase, and stationary phase of growth. The growth was followed by determination of the numbers of CFU by plating on TGE agar (Merck) every 4 h. Samples of 100 μl for PCR were also withdrawn every 4 h. The samples for PCR were frozen at −20°C and stored until all samples could be analyzed at the same time by the described PCR assay.
RESULTS
Factorial design experiment.
It was demonstrated that the quantitative CT response and the end-point response, ΔRn, in the 5′ nuclease real-time PCR were affected differently by the various PCR reagents (Fig. 1). The differences were related to variations in the amplification kinetics rather than interference of fluorescence measurements. When no PCR reagents were limited, the amplification efficiency was sufficient and the two distinct phases were identified, as illustrated in Fig. 1E. In addition, a third phase, a lag phase (i.e., the cycle period required to reach the threshold for the CT response) can also be seen in the amplification plot. The assumed quadratic model did not fit the observations of the axial points well, although this does not influence the general interpretation of the results. It was found that the CT response was significantly (P < 0.001) dependent on the DNA concentration (Table 1). To obtain the optimal performance of the Salmonella PCR assay studied, the concentration of the center point of the factorial design experiment for all except one of the PCR reagents (MgCl2) should be used in the AmpliTaq Gold mixture. The concentration of MgCl2 in the AmpliTaq Gold mixture-based PCR was found to be the factor that most significantly affected the CT value (P < 0.001), and for this response, MgCl2 was found to interact with most of the PCR reagents, especially the primers (P = 0.005) and the probe (P = 0.04). Regarding the value of ΔRn, the MgCl2 (P < 0.001) and the probe (P = 0.05) concentrations were found to be significantly involved in the response (Table 1).
FIG. 1.


Graphical illustration of Salmonella DNA amplification kinetics of the 5′ nuclease PCR assay (13) for the low and high axial levels used in the factorial design experiment. In all five graphs (A to E) the highest final ΔRn was obtained for the high axial concentrations of each PCR reagent. All PCR reagents except the PCR reagent being studied are at the concentration of the center level (Table 1). (A) High (4.7 mM) and low (0.1 mM) MgCl2 concentrations; (B) high (160 nM) and low (40 nM) probe concentrations; (C) high (1,600 nM) and low (185 nM) primer (both forward and reverse primers) concentrations; (D) high (0.10 U/μl) and low (0.02 U/μl) AmpliTaq Gold concentrations; (E) high (2 × 10−6 g/microwell) and low (2 × 10−11 g/microwell) DNA concentrations.
The results suggest that for accurate positive detection by the 5′ nuclease Salmonella assay studied, the detection probability should be >95%, the CT value should be <30 (Fig. 2), and ΔRn should be >1.0 (Table 1). The CT value was found to be the most appropriate response for estimation of the DNA concentration by plotting the CT value against the DNA concentration. The ΔRn response seemed to be a more uncertain measure, and the overall precision of ΔRn was insufficient. Therefore, the CT response was used in the remainder of the study as the main response for the description and estimation of the performance of the PCR, i.e., detection probability, linear range of amplification, and amplification efficiency.
FIG. 2.

Graphical appearance of the model used to estimate PCR performance. The experiment was performed three times with 10-fold dilutions of DNA, and the model (the line connecting the datum points) fits the experimental data (✻) that were obtained well. Salmonella DNA amplification took place in the presence of ddH2O and different media with two amplification mixtures: (A) AmpliTaq Gold with ddH2O; (B) rTth with ddH2O; (C) AmpliTaq Gold with BPW; (D) rTth with BPW; (E) AmpliTaq Gold with BHI; (F) rTth with BHI. The vertical dashed lines in all graphs are the log DNA concentration at a detection probability of 0.95 from the estimated model. The horizontal dashed lines at CT equal to 30 marks the proposed upper limit for sufficient PCR performance. From the model the slope was determined and all graphs (A to F) gave close to optimal amplification efficiencies.
Modeling of 5′ nuclease PCR performance.
The proposed model was developed on the basis of the available data generated from the experiment on the PCR performances of the two PCR mixtures based on AmpliTaq Gold and rTth. As can be seen from Fig. 2, the model fit the experimental data well. However, the estimates of parameters a3 and a4 were not very reliable in the experiments in which there were few observations in the region between the regions of constant CT and linear response. From the experiment it was observed that the PCR performances of the two PCR mixtures were similar for samples with no enrichment medium. The model allowed the accuracy of the assay to be studied since it could be used to compare the performances of PCR with DNA in ddH2O and PCR with DNA in enrichment medium. The presence of BPW (10% [vol/vol]) and BHI (10% [vol/vol]) resulted in different PCR performances. The presence of BPW decreased the practical operating range for the AmpliTaq Gold system, as indicated by the lower detection probability at lower DNA concentrations (Fig. 2C), while rTth was affected in the same way in the presence of BHI (Fig. 2F).
When the DNA concentration was constant (2 × 10−9 g/microwell) and the concentrations of the different media were changed, it was observed that the rTth mixture provided more robust detection than the AmpliTaq Gold mixture (Fig. 3). The use of rTth permitted PCR amplification of S. enterica cells when 5 μl of BPW was present in a total PCR sample volume of 50 μl without the necessity for special sample preparation prior to PCR.
FIG. 3.

DNA amplification by the Salmonella 5′ nuclease PCR assay in the presence of 5 μl of BHI and 5 μl of BPW. The graphs show the results of the CT response at a constant Salmonella DNA concentration (2 × 10−9 g/microwell) while the concentrations of BHI and BPW in the media were changed. The experiment was performed with both the AmpliTaq Gold mixture and the rTth mixture. The PCR results correspond to the following DNA amplification combinations: AmpliTaq Gold mixture and BPW medium (⧫), AmpliTaq Gold mixture and BHI medium (▴), rTth mixture and BPW medium (⋄), and rTth mixture and BHI medium (▵).
Enrichment PCR.
For accurate detection (CT ≤ 30), the experiment showed that it was possible to detect an inoculation concentration of 1 CFU of S. enterica serovar Enteritidis per ml after 8.4 h of enrichment with the rTth mixture instead of the AmpliTaq Gold mixture, which required 11.6 h of enrichment, as estimated by linear regression (Fig. 4). The exponential growth (determined as the numbers of CFU per milliliter) corresponded to a linear decrease in the CT value between 8 and 16 h of incubation. It was noted that the detectable linear range was reached 3.2 h more quickly with the rTth mixture than with the AmpliTaq Gold mixture. To avoid bias it was found that sample withdrawal should be made during the exponential growth phase of the bacteria.
FIG. 4.

Enrichment PCR for S. enterica serovar Enteritidis. The graph illustrates the dynamic detection range, i.e., the time during which positive detection is possible, by plotting the CT value against the incubation time. BPW was inoculated with S. enterica serovar Enteritidis at a concentration of 1 CFU/ml, and the mixture was incubated at 37°C. Samples for PCR analysis were withdrawn every 4 h. •, numbers of CFU per milliliter; ▪, results for AmpliTaq Gold mixture; ▴, results for rTth mixture.
DISCUSSION
A number of 5′ nuclease PCR assays have been developed and evaluated for the detection of Salmonella spp. (7, 13, 16, 25), and most of them have used ΔRn as the main response (7, 13, 16). In the present study the ΔRn response was found to be an uncertain variable for estimation of the concentration of the target bacteria, although other investigators have most often applied it to the detection of pathogenic bacteria by the 5′ nuclease PCR (5, 27, 35). Determination of ΔRn may be adequate as a qualitative method of analysis, but the CT value should be used to monitor the accuracy. However, expression of results by use of CT values in the range of 35 to 40 are not as accurate and precise as expression of results by use of CT values in the range of 20 to 30 (10), making it necessary to reevaluate positive samples with CT values in the range of 35 to 40 (15). Thus, it is appropriate to present the raw data for CT and ΔRn independently in order to use the power of the TaqMan technique (ABI Prism 7700 sequence detection system) for diagnostic applications. The CT value can be used to describe PCR performance, while the ΔRn value can verify the presence of the PCR product.
As illustrated by the amplification phases in Fig. 1E, the amplification efficiency is not constant during the PCR. Factors that may change during DNA amplification are, for instance, the concentrations of PCR reagents, melting temperatures, and the functionality of the DNA polymerase. Two of the most important PCR reagents that affect the amplification conditions are the DNA polymerase with its supplied buffer (especially salts) and MgCl2 (8, 12). The divalent Mg ion not only acts as a DNA polymerase cofactor (21, 28) but is also involved in many other mechanisms during the PCR. No significant interactions (P > 0.05) between the ion and the enzymes were observed in these experiments, which indicated that a higher concentration did not improve the functionality of the enzyme. Others have shown that MgCl2 facilitates the annealing of primers and the TaqMan probe (36). Also, in our experiment it was observed that higher concentrations of MgCl2 increased the amount of hydrolyzed probe, which led to a stronger fluorescence signal. However, as for many other alkaline metals, the divalent Mg ion binds primarily to phosphates on the DNA backbone (18). Therefore, the ion probably binds to primers, probes, and dNTPs; and the balance between dNTP and MgCl2 has been determined in PCR optimization protocols (12). From the factorial design experiment with the AmpliTaq Gold mixture, it was found that the interaction between MgCl2, the primer, and the probe was significant (P < 0.05) and that a higher concentration of MgCl2 gave a stronger fluorescence signal. The experiment confirmed that the amplification efficiency decreased when the highest primer concentration (concentration of each primer, 1,200 nM) was used. When MgCl2 was present at a low concentration (1.5 mM), it was indicated that the ion probably became trapped and amplification conditions were not favorable. As a result, to optimize the performance of the 5′ nuclease PCR, it is necessary to maintain free Mg ions in the PCR mixture. It is also important to consider the proportions of MgCl2 and components of nucleic acids (including templates, primers, probes, and dNTPs). However, no significant interactions were observed in the rTth amplification mixture (P > 0.05); this might have been due to the buffer composition, the fewer numbers of observations in comparison to those for the AmpliTaq Gold mixture, or the interference of some combinations of PCR reagents with the fluorescence measurements. This should be evaluated in more detail in future studies, but the concentrations of the PCR reagents used in the center point of the factorial design experiment for the rTth mixture resulted in appropriate detection responses (Table 1). Furthermore, for future studies of 5′ nuclease real-time PCRs, the stabilities of the different fluorescent dyes (FAM, ROX, and TAMRA) should be considered.
The PCR performances of the two PCR mixtures were similar when DNA was diluted in ddH2O and the lowest amplifiable DNA concentration was about 10−13 g/microwell (Fig. 2A and B). At DNA concentrations of about 10−13 to 10−11 g/microwell, the PCR performance was negatively affected for both the AmpliTaq Gold mixture and the rTth mixture, but the AmpliTaq Gold mixture was more prone to be negatively affected (Fig. 2C). However, at higher DNA concentrations (10−10 to 10−8 g/microwell), the PCR performance was more accurate. It is suggested that the model proposed for estimation of PCR performance can be applied when complex biological samples are present in the PCR sample. It was demonstrated that the presence of BPW (10% [vol/vol]) and BHI (10% [vol/vol]) affected the performance of the PCR. The results from the model showed that the rTth mixture was less influenced by the presence of BPW than the AmpliTaq Gold mixture was (Fig. 2C and D). It has been shown that rTth is more salt tolerant and resistant to other ions than some other DNA polymerases (1). Phosphates are present at rather high concentrations (10.5 g/liter) in BPW (Merck), and this might affect the conditions for PCR amplification. In comparison to the AmpliTaq Gold buffer (composed of 150 mM Tris-HCl and 500 mM KCl) the buffer system of rTth contains a higher concentration of KCl (1,000 mM) and contains PCR facilitators such as Tween 20 and glycerol (2). The higher concentration of KCl, the presence of PCR facilitators, and the characteristics of the DNA polymerase in the rTth mixture may improve the DNA amplification of the system to maintain the practical operating range of the assay when BPW is present during PCR. This means that amplification can take place at a lower concentration of target DNA, implying faster detection at low initial concentrations of the target organism.
To carry out enrichment PCR, the physiological state of the cell must be known. Recent studies have claimed that environmental factors, including growth phase, pH, osmotic conditions, temperature, and nutritional stress, can influence the amount of nucleic acid (33, 34). The physiological state of a cell may also influence the ability of the cell to be lysed and thereby contribute to the template available for DNA amplification (31). In enrichment PCR it is of importance to define the optimal point in time for sample withdrawal. In the present study it was demonstrated that the detection accuracy was optimal when the bacteria were in the exponential growth phase (at 8 to 16 h of incubation) (Fig. 4). However, if the aim is to attain qualitative PCR detection, then sample withdrawal during the stationary phase would also provide an appropriate detection response. The results obtained from the enrichment PCR experiment are only for cells inoculated in broths, and caution should be taken when clinical samples are present during enrichment, which needs to be evaluated in future studies. However, before clinical samples are studied in detail it is necessary to establish standard curves for real-time PCR, which are at the moment rare. In addition, since real-time PCR is a new technology in terms of its diagnostic application for the detection of pathogenic bacteria, unambiguous definitions of the detection limit and the linear range of amplification are needed for an objective interpretation of the data.
In conclusion, for optimization of real-time PCR the effects of different PCR reagents on the detection responses must be evaluated for modeling of the PCR performance. For cells inoculated in a broth (BPW), it can be concluded that by optimization of PCR mixtures that maintained the practical operating range of the Salmonella 5′ nuclease assay studied, it was possible to obtain an automated high-throughput method. For accurate detection (CT < 30), it was found that use of the rTth mixture reduced the enrichment time required by 3.2 h in comparison to that required by use of the AmpliTaq Gold mixture.
Acknowledgments
This work was financially supported by VINNOVA (The Swedish Agency for Innovation Systems), the Commission of the European Communities within the program Quality of Life and Management of Living Resources (grant QLRT-1999-00226), and a scholarship (FS-147) from KSLA, The Royal Swedish Academy of Agriculture and Forestry.
REFERENCES
- 1.Abu Al-Soud, W., and P. Rådström. 1998. Capacity of nine thermostable DNA polymerases to mediate DNA amplification in the presence of PCR-inhibiting samples. Appl. Environ. Microbiol. 64:3748–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abu Al-Soud, W., and P. Rådström. 2000. Effects of amplification facilitators on diagnostic PCR in the presence of blood, feces, and meat. J. Clin. Microbiol. 38:4463–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abu Al-Soud, W., and P. Rådström. 2001. Purification and characterization of PCR-inhibitory components in blood cells. J. Clin. Microbiol. 39:485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agresti, A. 1996. An introduction to categorical data analysis. John Wiley & Sons, Inc., New York, N.Y.
- 5.Bassler, H. A., S. J. Flood, K. J. Livak, J. Marmaro, R. Knorr, and C. A. Batt. 1995. Use of a fluorogenic probe in a PCR-based assay for the detection of Listeria monocytogenes. Appl. Environ. Microbiol. 61:3724–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Batt, C. A. 1997. Molecular diagnostics for dairy-borne pathogens. J. Dairy Sci. 80:220–229. [DOI] [PubMed] [Google Scholar]
- 7.Chen, S., A. Yee, M. Griffiths, C. Larkin, C. T. Yamashiro, R. Behari, C. Paszko-Kolva, K. Rahn, and S. A. De Grandis. 1997. The evaluation of a fluorogenic polymerase chain reaction assay for the detection of Salmonella species in food commodities. Int. J. Food Microbiol. 35:239–250. [DOI] [PubMed] [Google Scholar]
- 8.Favre, N., and W. Rudin. 1996. Salt-dependent performance variation of DNA polymerases in coamplification PCR. BioTechniques 21:28–30. [DOI] [PubMed] [Google Scholar]
- 9.Food and Drug Administration. 1998. Bacterial analytical manual, 8th ed., revision A. AOAC International, Gaithersburg, Md.
- 10.Freeman, W. M., S. J. Walker, and K. E. Vrana. 1999. Quantitative RT-PCR: pitfalls and potential. BioTechniques 26:112–122, 124–125. [DOI] [PubMed] [Google Scholar]
- 11.Golenberg, E. M., A. Bickel, and P. Weihs. 1996. Effect of highly fragmented DNA on PCR. Nucleic Acids Res. 24:5026–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henegariu, O., N. A. Heerema, S. R. Dlouhy, G. H. Vance, and P. H. Vogt. 1997. Multiplex PCR: critical parameters and step-by-step protocol. BioTechniques 23:504–511. [DOI] [PubMed] [Google Scholar]
- 13.Hoorfar, J., P. Ahrens, and P. Rådström. 2000. Automated 5′ nuclease PCR assay for identification of Salmonella enterica. J. Clin. Microbiol. 38:3429–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jourdan, A. D., S. C. Johnson, and I. V. Wesley. 2000. Development of a fluorogenic 5′ nuclease PCR assay for detection of the ail gene of pathogenic Yersinia enterocolitica. Appl. Environ. Microbiol. 66:3750–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Killgore, G. E., B. Holloway, and F. C. Tenover. 2000. A 5′ nuclease PCR (TaqMan) high-throughput assay for detection of the mecA gene in staphylococci. J. Clin. Microbiol. 38:2516–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimura, B., S. Kawasaki, T. Fujii, J. Kusunoki, T. Itoh, and S. J. Flood. 1999. Evaluation of TaqMan PCR assay for detecting Salmonella in raw meat and shrimp. J. Food Prot. 62:329–335. [DOI] [PubMed] [Google Scholar]
- 17.Klein, D., P. Janda, R. Steinborn, M. Muller, B. Salmons, and W. H. Unzburg. 1999. Proviral load determination of different feline immunodeficiency virus isolates using real-time polymerase chain reaction: influence of mismatches on quantification. Electrophoresis 20:291–299. [DOI] [PubMed] [Google Scholar]
- 18.Koltover, I., K. Wagner, and C. R. Safinya. 2000. DNA condensation in two dimensions. Proc. Natl. Acad. Sci. USA 97:14046–14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lantz, P. G., W. Abu Al-Soud, R. Knutsson, B. Hahn-Hägerdal, and P. Rådström. 2000. Biotechnical use of polymerase chain reaction for microbiological analysis of biological samples. Biotechnol. Annu. Rev. 5:87–130. [DOI] [PubMed] [Google Scholar]
- 20.Lantz, P. G., R. Knutsson, Y. Blixt, W. Abu Al-Soud, E. Borch, and P. Rådström. 1998. Detection of pathogenic Yersinia enterocolitica in enrichment media and pork by a multiplex PCR: a study of sample preparation and PCR-inhibitory components. Int. J. Food Microbiol. 45:93–105. [DOI] [PubMed] [Google Scholar]
- 21.Lawyer, F. C., S. Stoffel, R. K. Saiki, S. Y. Chang, P. A. Landre, R. D. Abramson, and D. H. Gelfand. 1993. High-level expression, purification, and enzymatic characterization of full-length Thermus aquaticus DNA polymerase and a truncated form deficient in 5′ to 3′ exonuclease activity. PCR Methods Appl. 2:275–287. [DOI] [PubMed] [Google Scholar]
- 22.Lyamichev, V., M. A. Brow, and J. E. Dahlberg. 1993. Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science 260:778–783. [DOI] [PubMed] [Google Scholar]
- 23.Meijerink, J., C. Mandigers, L. van de Locht, E. Tonnissen, F. Goodsaid, and J. Raemaekers. 2001. A novel method to compensate for different amplification efficiencies between patient DNA samples in quantitative real-time PCR. J. Mol. Diagn. 3:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montgomery, D. C. 1997. Design and analysis of experiments. John Wiley & Sons Inc., New York, N.Y.
- 25.Nogva, H. K., A. Bergh, A. Holck, and K. Rudi. 2000. Application of the 5′-nuclease PCR assay in evaluation and development of methods for quantitative detection of Campylobacter jejuni. Appl. Environ. Microbiol. 66:4029–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nordic Committee on Food Analysis. 1999. Salmonella detection in food, method 71, 5th ed. Nordic Committee on Food Analysis, Esbo, Finland.
- 27.Oberst, R. D., M. P. Hays, L. K. Bohra, R. K. Phebus, C. T. Yamashiro, C. Paszko-Kolva, S. J. Flood, J. M. Sargeant, and J. R. Gillespie. 1998. PCR-based DNA amplification and presumptive detection of Escherichia coli O157:H7 with an internal fluorogenic probe and the 5′ nuclease (TaqMan) assay. Appl. Environ. Microbiol. 64:3389–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rüttimann, C., M. Cotoras, J. Zaldivar, and R. Vicuna. 1985. DNA polymerases from the extremely thermophilic bacterium Thermus thermophilus HB-8. Eur. J. Biochem. 149:41–46. [DOI] [PubMed] [Google Scholar]
- 29.Schnell, S., and C. Mendoza. 1997. Enzymological considerations for a theoretical description of the quantitative competitive polymerase chain reaction (QC-PCR). J. Theor. Biol. 184:433–440. [DOI] [PubMed] [Google Scholar]
- 30.Sharma, V. K., and S. A. Carlson. 2000. Simultaneous detection of Salmonella strains and Escherichia coli O157:H7 with fluorogenic PCR and single-enrichment-broth culture. Appl. Environ. Microbiol. 66:5472–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silva, M. C., and C. A. Batt. 1995. Effect of cellular physiology on PCR amplification efficiency. Mol. Ecol. 4:11–16. [DOI] [PubMed] [Google Scholar]
- 32.Tietjen, M., and D. Y. Fung. 1995. Salmonellae and food safety. Crit. Rev. Microbiol. 21:53–83. [DOI] [PubMed] [Google Scholar]
- 33.Tolker-Nielsen, T., M. H. Larsen, H. Kyed, and S. Molin. 1997. Effects of stress treatments on the detection of Salmonella typhimurium by in situ hybridization. Int. J. Food Microbiol. 35:251–258. [DOI] [PubMed] [Google Scholar]
- 34.Uyttendaele, M., R. Schukkink, B. von Gemen, and J. Debevere. 1996. Influence of bacterial age and pH of lysis buffer on type of nucleic acid isolated. J. Microbiol. Methods 26:133–138. [Google Scholar]
- 35.Vishnubhatla, A., D. Y. Fung, R. D. Oberst, M. P. Hays, T. G. Nagaraja, and S. J. Flood. 2000. Rapid 5′ nuclease (TaqMan) assay for detection of virulent strains of Yersinia enterocolitica. Appl. Environ. Microbiol. 66:4131–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Witham, P. K., C. T. Yamashiro, K. J. Livak, and C. A. Batt. 1996. A PCR-based assay for the detection of Escherichia coli Shiga-like toxin genes in ground beef. Appl. Environ. Microbiol. 62:1347–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wittwer, C. T., M. G. Herrmann, A. A. Moss, and R. P. Rasmussen. 1997. Continuous fluorescence monitoring of rapid cycle DNA amplification. BioTechniques 22:130–131, 134–138. [DOI] [PubMed] [Google Scholar]