

Abstract
Summary: Alzheimer’s disease is a progressive neurodegenerative disorder and the leading cause of dementia in the Western world. Postmortem, it is characterized neuropathologically by the presence of amyloid plaques, neurofibrillary tangles, and a profound gray matter loss. Neurofibrillary tangles are composed of an abnormally hyperphosphorylated intracellular protein called tau, tightly wound into paired helical filaments and thought to impact microtubule assembly and protein trafficking, resulting in the eventual demise of neuronal viability. The extracellular amyloid plaque deposits are composed of a proteinacious core of insoluble aggregated amyloid-β (Aβ) peptide and have led to the foundation of the amyloid hypothesis. This hypothesis postulates that Aβ is one of the principal causative factors of neuronal death in the brains of Alzheimer’s patients. With multiple drugs now moving through clinical development for the treatment of Alzheimer’s disease, we will review current and future treatment strategies aimed at improving both the cognitive deficits associated with the disease, as well as more novel approaches that may potentially slow or halt the deadly neurodegenerative progression of the disease.
Keywords: Alzheimer’s disease, symptomatic, amyloid, immunization, secretase, Aβ
INTRODUCTION
Alzheimer’s disease (AD) is a chronic and progressive neurodegenerative disorder set to become the developed worlds largest socioeconomic healthcare burden over the coming decades. AD is thought to affect 4–8% of the population over 65 years of age, with the incidence continuing to increase with increasing age. Current U.S. estimates on the numbers of patients suffering from the disease range from three to five million, with an annual estimated cost of approximately $100 billion dollars. It is estimated that by 2050 the number of patients with AD could be as high as 25 million.1 Neuropathologically, the disease was first described in 1907 by Alois Alzheimer and is characterized by a progressive loss of neurons and synapses with the presence of large numbers of extracellular amyloid plaques and intracellular neurofibrillary tangles (FIG. 1). Antemortem clinical diagnosis of AD is difficult and requires a recorded decline in cognitive function as well as evidence of progressive deficits in other behavioral areas such as executive function and language skills. Unqualified diagnosis of AD can still only be made neuropathologically postmortem by examination of patients’ brains and the detection of amyloid plaques and tangles.
FIG. 1.

The neuropathological steps of Alzheimer’s disease.
SYMPTOMATIC APPROACHES FOR THE TREATMENT OF AD
In AD, multiple regions of brain gray matter have a profound neuronal loss, including basal forebrain, hippocampus, entorhinal, and temporal cortices. Braak and Braak2 have developed a model of disease progression based on changes in the pattern of neurobrillalry tangles. They suggest that the neurodegenerative process begins with neuronal loss in the glutamatergic pathways of the entorhinal cortex before extending to the hippocampus and amygdala and then more widely to neocortical and subcortical areas. Despite this extensive neurodegenerative process, not all neurons are susceptible to the disease process, with certain populations of neurons being clearly more vulnerable than others. Indeed in the mid-1970s, it was the initial neurochemical discovery of reduced levels of choline acetyltransferase that identified a particularly susceptible neuronal population in the basal forebrain. This population of acetylcholine containing neurons was greatly decreased in the brains of AD patients leading to the development of the cholinergic hypothesis and the first therapeutic agents for the treatment of AD.3,4 Since that time, many approaches to enhance cholinergic function have been tried but only the acetylcholinesterase inhibitors have become a mainstay of AD pharmacotherapy and by far the most successful therapies in current clinical use. They work by inhibiting the hydrolysis of acetylcholine in the synaptic cleft and prolonging the level of acetylcholine at the synapse resulting in at least a partial correction of this neurotransmitter deficit in the brains of patients. Tacrine was the first cholinesterase inhibitor approved by the FDA for the symptomatic treatment of AD but was subsequently withdrawn from the market place due to hepatotoxicity, resulting in an unacceptable risk benefit profile for the drug. Three compounds with similar efficacy but an improved safety profile were subsequently developed and launched. Donepezil and galantamine are both selective acetylcholinesterase inhibitors,5,6 whereas rivastigmine is an inhibitor of both acetylcholinesterase and butyrylcholinesterase.7 Some have argued that cholinesterase inhibitors may also have disease modifying or disease slowing attributes associated with them. One of the best studied examples in the literature is that of phenserine. This molecule was reported to not only inhibit acetylcholinesterase but also modulate the translation and subsequent processing of the amyloid precursor protein, resulting in reduced levels of the toxic Aβ peptide.8 Unfortunately, as is true with many molecules, the preclinical promise has so far not translated in the clinic with the drug not meeting its efficacy end points in late stage clinical development (see Table 1). However, there are other reports suggesting limited disease-modifying effects of cholinesterase inhibitors. For example, in open label studies, patients initially on placebo and then administered donepezil or rivastigmine cognitively never matched patients on cholinesterase inhibitor drugs from the outset of the study.9,10 Although most would question any potentially disease modifying effects for this class of drug, cholinesterase inhibitors do exhibit small and consistent improvements in patient memory and global function. Nevertheless, it is clear that they remain far from ideal therapies given that their effects are neither long lasting nor robustly altering of progression.
TABLE 1.
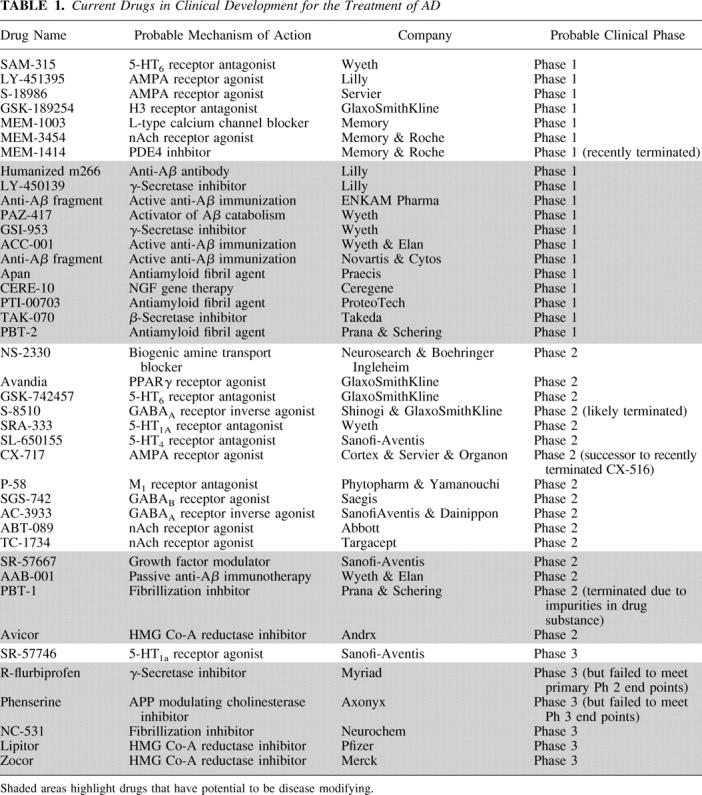
More recently, a noncholinergic agent, memantine, has been approved for the symptomatic treatment of moderate to severe AD.11 Oxidative stress and glutamate induced excitotoxicity are thought to play a critical role in the neurodegenerative process of AD.12,13 As such, blockade of the NMDA receptor, one of the principal excitatory glutamate receptors in the brain, has been shown to have neuroprotective effects in a number of acute preclinical in vitro and in vivo models.14 Memantine, is a noncompetitive, moderate affinity, NMDA antagonist, developed on the basis of the above “excitotoxic glutamate” hypothesis. Unlike most other centrally acting NMDA antagonists that have been abandoned in clinical development because of severe psychomimetic and cardiovascular adverse effects, memantine was surprisingly well tolerated in patients. Clinical trials with memantine demonstrated cognitive improvements in AD patients with a reduction in the number of caregiver hours required.15–17 Despite the fact that memantine’s mechanism of action may be suggestive of a disease modifying potential, thus far the drug has only been tested as a symptomatic agent and the efficacy appears comparable to that of the non disease modifying cholinesterase inhibitors in terms of robustness and duration.18 It remains to be seen if there is synergy to be gained for patients cotreated with both a cholinesterase inhibitor and memantine.
Although there are now a number of symptomatic therapies available to patients, there is still a clear need for improved symptomatic therapies that not only improve on current treatment standards with regard to cognitive deficits but also address the variety of other behavioral disturbances associated with disease (psychosis, depression, aggression, etc.). A number of alternate symptomatic approaches are actively being investigated and may have the potential to add to and perhaps surpass current treatment approaches either in terms of their efficacy or tolerability (see Table 1). Some of the mechanisms being investigated include modulation of cholinergic receptors using selective M1 receptor agonists or α7 nicotinic receptor agonists, blockade of selective serotonergic receptors using 5-HT6 or 5-HT1A receptor antagonists,19,20 activation of AMPA receptors using selective ampakines,21 and blockade of histamine H3 receptors.22,23 Each of these approaches may have a variety of potential mechanistic advantages over current therapies. Some such as the 5-HT6 or 5-HT1A antagonists exhibit robust preclinical efficacy in rodent and nonhuman primate models of cognition and have the potential to modulate multiple neurotransmitter systems such as acetylcholine, glutamate, and serotonin exclusively in the brain, suggestive of broader and improved efficacy and a reduced peripheral side effect profile. Others such as the M1 and α7 selective compounds have the ability to modulate neurotransmitter systems while simultaneously having the potential to block disease progression by modulating levels of Aβ, allowing for the possibility of improved efficacy with the additional benefits of disease modification.24 Although each of these pharmacological approaches are exciting and have the potential to achieve superiority over existing therapies, it should be noted that none have yet been validated, in the clinic treating AD patients. Indeed, M1 receptor agonists have been previously tested in clinical trials without much success.25 However, it is likely that these first generation compounds lacked sufficient muscarinic receptor selectivity resulting in dose-limiting side effects and preventing adequate drug exposure and M1 receptor occupancy of the drug.26,27
DISEASE-MODIFYING APPROACHES FOR THE TREATMENT OF AD
Much attention is now being directed at the development of approaches that counteract the fundamental pathological processes of the disease. Such approaches may effectively slow or halt the progression of the disease and be used in conjunction with existing symptomatic and/or cognitive enhancing therapies. The amyloid hypothesis has arisen from a focus on one of the key pathological features of AD, the amyloid plaque (FIG. 1). These extracellular deposits are composed of a proteinacious core of insoluble aggregated amyloid-β (Aβ) peptide,28 surrounded by a halo of dystrophic neurites, activated microglia, and reactive astrocytes. Aβ is a hydrophobic 39- to 42-amino acid peptide, found in all biological fluids, and derived from the enzymatic cleavage of a larger type I membrane protein, the amyloid precursor protein (APP)29,30 (FIG. 2). A number of key findings have led people to postulate a central role for this peptide in the etiology and pathogenesis of the disease. Linkage studies of familial AD patients identified a number of mutations in two genes, APP and presenilin, associated with aberrant metabolism of APP and an increased production of aggregating forms of Aβ. Furthermore, Down syndrome (trisomy 21) patients who have high levels of Aβ deposits in their brains and dementia from an early age have three copies of the APP gene.31,32 Individuals carrying the apoliprotein E4 (ApoE4) genotype also have an increased risk of developing AD compared with ApoE2 or ApoE3 individuals. Importantly, ApoE4 has been shown to modulate both the aggregation of Aβ, as well as its clearance from the brain.33–36 Finally, multiple reports have highlighted the neurotoxic effects of aggregated and soluble oligomeric species of Aβ in vitro and in vivo, suggesting that excessive production of this peptide is detrimental to the viability of neurons.37,38 Irrespective of the cause or toxic Aβ species, it is likely that a delicate equilibrium between Aβ production and catabolism exists in the brains of normal aging individuals. When this equilibrium is perturbed by either increased Aβ production and/or reduced activity of Aβ catabolic pathways, the consequence is likely a slow accumulation of synapotoxic Aβ species, amyloid deposition, and subsequent neuronal dysfunction and cell death. In this section, we will focus on approaches being investigated to inhibit the production or enhance clearance of Aβ peptides and amyloid deposits.
FIG. 2.

The amyloid cascade hypothesis. The amyloid precursor protein APP is processed by β and γ-secretases via the amyloidogenic pathway to yield a variety of toxic Aβ-containing species, ultimately resulting in neuronal cell death. These amyloidogenic species can be degraded by a number of catabolic proteases such as neprilysin, IDE, and plasmin, thereby clearing Aβ and preventing cell death. The nonamyloidogenic pathway results from α-secretase cleavage within the Aβ sequence of APP.
PROCESSING OF APP TO GENERATE Aβ PEPTIDES
To date, over 150 mutations in three autosomal dominant genes, APP, presenilin (PS)-1 and -2, are known to cause familial Alzheimer’s disease. Surprisingly, the function of APP is still unknown, although the protein is highly conserved throughout evolution, and expressed widely in many different cell types.39 These mutations result in the increased production of certain Aβ peptides by altering the processing of APP by the tandem action of two proteases, β secretase, and γ secretase.40 This clustering of mutations within the APP processing pathway forms the cornerstone of the amyloid hypothesis and has highlighted a number of potential targets for disease-modifying therapeutic intervention.41
One of these targets is the soluble pool of Aβ itself. This is the species of Aβ most closely correlated with disease progression, and postulated mechanisms include the activation of membrane receptors, and the permeabilization of membranes by the binding of Aβ to the cell surface42 A number of strategies to reduce soluble Aβ are currently being employed both preclinically as well as clinically, and include antibody based approaches (discussed below), and reagents that disrupt the structure of Aβ, such as metal chelators.43
DECREASING Aβ PEPTIDE PRODUCTION BY BLOCKING β-SECRETASE
The amiloydogenic pathway involves the sequential proteolysis of APP by β-secretase (BACE) followed by γ-secretase (FIG. 2). Although this is a minor APP processing route, it is this pathway that generates Aβ fragments believed to give rise to AD.44,45 In humans, two β-secretase genes have been identified, referred to as BACE-1 and BACE-2, colocalized with APP in the endosomal compartment.46 Whereas both can process APP at the same site, only BACE-1 is significantly expressed in brain, particularly in neurons, indicating that neurons are the major source of β-amyloid peptides in brain. Because BACE-2 is expressed in heart, kidney, and placenta, drugs developed as β-secretase inhibitors may need to be selective against BACE-2 to prevent unwanted peripheral side effects in the clinic.
Nevertheless, inhibition of β-secretase is a promising strategy. This therapeutic potential was demonstrated by the findings that BACE-1 knockout mice develop normally, and appear to have completely abolished the production of Aβ, suggesting that BACE-1 is the principal β-secretase in neurons.47–49 Such inhibition does not preclude normal processing of APP by the nonamyloidogenic major pathway, and is the first step in the amyloidogenic cascade. Specific BACE-1 inhibitors should therefore have therapeutic potential to slow or halt the progression of this debilitating and ultimately fatal disease, and a number of preclinical candidates are about to enter clinical trials (see Table 1).
Developing specific β-secretase inhibitors has been difficult, in part because there appears to be a nonlinear relationship between decrease of β-secretase activity in vivo, and a reduction of Aβ peptides in brain. Studies using heterozygous BACE-1 knockout animals have shown that a 50% decrease in BACE activity leads to a much smaller decrease (∼15%) of brain Aβ levels. A further difficulty is the low brain penetration of most inhibitors, likely due to the fact that many of these are substrates for P-glycoprotein, plasma membrane proteins that actively extrude a wide range of amphiphilic and hydrophobic drugs from cells, and important in preventing the accumulation of several drugs in brain.50 Finally, crystallographic analyses of BACE-1 monomers have revealed a large catalytic domain, making it more difficult to identify small molecule transition-state analogs. This problem is further exacerbated if active BACE is a dimer, with potentially an even larger substrate-binding pocket. Nevertheless, a number of small molecule inhibitors are close to entering clinical trials.
Other than small molecule inhibitors, a novel approach to regulate production of Aβ based on intracellular expression of single chain antibodies (intrabodies) raised to an epitope adjacent to the β-secretase cleavage site of human APP.51 Such intrabodies are potentially of therapeutic significance particularly if appropriate delivery mechanisms such as by intranasal administration of phage expressing anti-β site-directed antibodies, are shown to be safe in humans.52
DECREASING Aβ PEPTIDE PRODUCTION BY BLOCKING γ-SECRETASE
The products of either α-secretase or β-secretase cleavage of APP become a substrate for the site-specific proteolysis by γ-secretase,31 generating two predominant Aβ peptides either 40 or 42 amino acids in length, and a short intracellular fragment (APP intracellular domain or AICD) that may function as a transcriptional activator in a complex with the adapter protein Fe65 and the nuclear protein Tip60.53 Because this processing step is proximal to the generation of Aβ peptides, the identification of specific γ-secretase inhibitors must be considered one of the most promising strategies for a disease modifying treatment of AD. However, a potential liability of this target is that a number of other proteins are also substrates of this enzyme complex, and in particular the processing of the Notch receptor may be inhibited by γ-secretase blockers.54 Hence, such inhibitors will likely need to be selective against Notch and against other γ-secretase targets.
γ-Secretase is now known to be a hetero-oligomer containing at least four protein components, PS-1/PS-2, nicastrin, anterior pharynx defective-1 (APH-1), and presenilin enhancer-2 (PEN-2), in a high-molecular-weight complex of unknown stoichiometry.55 This complex assembled in endoplasmic reticulum (ER) rapidly moves to the plasma membrane, but the contributions made by each of the subunits are only starting to be unraveled. It is likely that each of the subunits may be a target for therapeutic intervention.
A number of strategies for decreasing β-amyloid peptide by interference at the level of γ-secretase present themselves. The most direct pathway is the inhibition of the holoenzyme complex by brain-penetrant small molecule inhibitors. The potential usefulness of such inhibition has been demonstrated in a number of animal models and also in early stage clinical trials. A second strategy for lowering Aβ peptides is to modulate γ-secretase to shift bias away from the generation of the species of Aβ believed to be most toxic—Aβ42. A number of modulators [including NSAID (nonsteroidal anti-inflammatory drug)-like molecules] have the ability to increase the production of shorter Aβ species such as Aβ38, and decrease the production of Aβ42. For example, a subset of NSAIDs has been shown to reduce secretion of the highly amyloidogenic Aβ42. A correlation has been found between Rho and its effector, Rho-associated kinase, preferentially regulated the amount of Aβ42 produced in vitro and that only those NSAIDs effective as Rho inhibitors lowered Aβ42. Selective Rock inhibitors also lowered brain levels of Aβ42 in a transgenic mouse model of Alzheimer’s disease. Thus, the Rho-Rock pathway may regulate amyloid precursor protein processing, and a subset of NSAIDs can reduce Aβ42 through inhibition of Rho activity.51
If such specific modulators can be identified then they present an intriguing drug class that reduces the amyloid burden by altering the specificity of γ secretase. Further points of intervention include altering the maturation of the γ secretase complex, either by interfering with the protein trafficking or assembly of the four γ-secretase components.
MODULATION OF Aβ PEPTIDE PRODUCTION BY α-SECRETASES
The predominant pathway by which APP is processed does not give rise to Aβ fragments, and hence is referred to as the nonamyloidogenic pathway.31 The initial APP processing involves the cleavage of APP by α-secretase. The identification of proteins with α-secretase activity is ongoing, and currently includes a constitutive activity [a disintegrin and metalloproteinase (ADAM)-10],56 as well as a PKC-regulated activity (ADAM-17).57 Because the α-secretase cleavage site is within the Aβ sequence of APP, and none of these proteolytic fragments have been associated with the generation of AD, enhanced cleavage at this site may represent a disease modifying strategy for AD as first postulated by Nitsch and colleagues.58 The expectation is that an elevation of α-secretase activity will compete with β-secretase activity, and hence result in decreased levels of Aβ peptides. However, such a strategy requires both substrates to be in the same compartment, at the same time, and whether approaches targeting the elevation of α-secretase activity will be fruitful for identifying therapies remains to be established.59
A related strategy to increase the fraction of APP cleaved by α-secretase is to modulate the trafficking of APP in such a way as to increase the likelihood that α-secretase will cleave APP. There are preliminary data that members of the sortin nexin family of proteins can reduce the rate of APP endocytosis, and increase sAPPα production, possibly by exposing the APP substrate to ADAM-10 for an extended period of time. Similarly, strategies that increase the production of ADAM-10 by inhibiting protease inhibitors such as tissue inhibitor of matrix metalloproteinase (TIMP)1 and TIMP3, may represent further therapeutically tractable approaches to further shift the bias of APP processing from the amiloydogenic to the nonamilyoidogenic pathway. An analogous mechanism was identified by Li et al.60 who screened 100,000 sequences from a human brain-derived cDNA library to identify cDNA sequences that can decrease β-secretase cleavage and elevate α-cleavage. This group found that small ubiquitin-related modifier (SUMO)-2 significantly modulates APP processing to decrease Aβ secretion from cells by 80%. Biological implications of SUMOylation include alterations in protein stability or subcellular location.61–63 Hence, the activation of SUMO-2 is a potential therapeutic target for a disease-modifying strategy in AD.
Finally, it is of interest that cholinesterase inhibitors have also been shown to elevate the production of sAPPα in a dose-dependent manner. The mechanism by which this occurs is not well defined, but in part appear to involve elevation of PKCε. If cholinesterase inhibitors can be shown to lower Aβ42 in brain, it is possible that this class of drug may have both symptomatic, as well as disease-modifying properties within a single molecule.8,24
ACTIVE AND PASSIVE IMMUNIZATION LOWERS BRAIN Aβ LEVELS AND IMPROVES MEMORY
One of the most interesting, unexpected, and novel findings over the past decade of AD research with regard to therapeutic approaches aimed at slowing or halting Aβ mediated pathology, were those made by Schenk and colleagues at Elan.64 In this study, young PDAPP transgenic mice were immunized, before they had amyloid plaque deposits, with an intraperitoneal injection of aggregated Aβ1–42 once a month for 11 months. This led to a polyclonal antibody response directed toward Aβ, resulting in significantly reduced amyloid deposits and neuritic pathology in the brains of the animals. More importantly and of relevance to testing the approach in patients, Aβ immunization of older PDAPP mice with significant levels of preexisting plaques, also resulted in a clear reduction in plaque pathology, suggesting this approach was able to not only slow the progression of amyloid deposition but perhaps even reverse it.64 The excitement garnered around this potential concept of being able to immunize Alzheimer’s patients to halt or reverse the disease process has led to a rapid confirmation and extension of the original studies from a multitude of academic and industrial groups, building further enthusiasm and impetus to the approach.65–71 Bard and colleagues66 were the first to demonstrate that one could circumvent the immune response (i.e., not rely on the animals’ ability to generate anti-Aβ antibodies after active immunization with Aβ peptide) by direct administration of anti-Aβ antibodies into transgenic APP mice. This passive immunization approach was found to be very effective at clearing amyloid plaques and reversing neuritic pathology to a degree similar to that seen in the original active peptide-immunization experiments of Schenk and colleagues. Importantly, only antibodies binding aggregated Aβ in vitro reduced amyloid pathology, in contrast to antibodies unable to bind to plaques and recognizing only soluble forms of Aβ.72 Numerous studies continue to be published on a variety of active and passive immunization strategies as well as on alternate routes of drug administration. Weiner and colleagues73 successfully lowered central Aβ levels and pathology using an intranasal administration of Aβ peptide in a mouse model of AD, a finding repeated by others.74 Other groups have treated APP transgenic mice with Aβ peptide sequences expressed on recombinant adeno-associated virus (via several different routes of administration) or using phage display and demonstrated significant reductions in plaque burden and neuroinflammation, as well as improved cognitive performance.75,76 An interesting proof of principle in humans has also been reported by Dodel and colleagues77,78 based on the fact that a small percentage of antibodies in a human Ig preparations are directed against Aβ peptide sequences. Intravenous infusion of Igs in five AD patients over a 6-month period prevented further cognitive decline suggesting this approach could potentially act like a passive Aβ directed immunotherapy approach. Irrespective of the approach taken, results have consistently demonstrated that active or passive immunization strategies, targeting sequences within the Aβ peptide, are able to slow disease pathology and reverse memory deficits in preclinical models of AD.65,70–72,79–83
Although there is a general agreement as to the preclinical effectiveness of both active and passive immunization approaches, several hypotheses exist for how these approaches elicit their effects. None are mutually exclusive and it is quite possible that several of are correct and important in mediating the observed benefits in preclinical models. Microglial-mediated phagocytosis is one potential mechanism by which amyloid deposits may be cleared from the brain. In this instance, anti-Aβ antibodies are proposed to enter the brain after treatment with an active or passive immunization protocol, bind to aggregated Aβ, and subsequently recruit phagocytosing microglia, via their cell surface expressed Fc-receptors, to sites of amyloid deposition.66 Another potential mechanism of action centers on the ability of anti-Aβ antibodies, recognizing N-terminal Aβ epitopes, to inhibit the formation of toxic Aβ fibrils as well as dissolve pre-existing fibrils in vitro.84,85 Subsequent studies identified residues 3–6 Gln-Phe-Arg-His (EFRH) of Aβ as the minimally effective epitope for this activity.86 In support of this hypothesis, antibodies directed to N-terminal-specific Aβ epitopes inhibit fibrillogenesis and cell death in transgenic TgCRND8 mice80 and are capable of attenuating amyloid deposition and neuritic dystrophy.72,87 These data are also consistent with reports of non-Fc-mediated clearance of amyloid81,83 and suggest two phases for amyloid clearance—a microglial-dependent phase followed by a microglial independent process in which diffuse Aβ deposits are cleared.88 More recently, data from the laboratories of Selkoe and Rowan have demonstrated that neutralizing antibodies able to bind synaptotoxic oligomeric species of Aβ can acutely reverse deficits in LTP in vitro and in vivo.89 These findings suggest such antibodies may therefore have a rapid effect on learning and memory in addition to the more chronic and prolonged disease slowing or reversing effects. These finding are consistent with data generated in our own laboratories showing that treatment with a variety of amyloid-lowering drugs, whether they be protease inhibitors or anti-Aβ antibodies, rapidly and robustly reverse cognitive deficits observed in tgAPP animals.
The final mechanism proposed is based on the “peripheral amyloid sink” hypothesis70,71,90 and was developed using a monoclonal antibody named m266.91 This antibody is directed to epitopes within the central domain of Aβ and binds only to soluble forms of this peptide. Chronic administration of m266 resulted in rapid increase in plasma Aβ and reduction in total brain Aβ. The hypothesis states that sequestration of peripheral plasma Aβ shifts the equilibrium between central and peripheral Aβ pools, resulting in a net efflux of peptide from the CNS and into the periphery from where it can be degraded by normal proteolytic processes.70,90,92 Acute administration of m266 has also been shown to improve cognitive behavior in APP transgenic mice, likely by altering brain levels of soluble species of Aβ.93
Results from immunotherapy with aggregated Aβ1-42 led to the development of the first human active immunization trial with a synthetic Aβ peptide called AN1792 in combination with a QS-21 adjuvant. Despite extensive safety and tolerability studies in animals, this trial was halted in early phase 2a after reports of an acute meningoencephalitis in 18 of 300 treated patients.94 Analysis of all AN1792-treated subjects showed that approximately 20% developed robust antibody responses to Aβ, and there was no correlation between severity of encephalitis and the antibody titer produced. The first analysis of efficacy in this interrupted AN1792 trial was reported for a small subset of AN1792-treated patients and suggestive of a slowing cognitive decline, as measured by Alzheimer’s Disease Assessment Scale-cognition (ADAS-COG), and mini-mental state examination (MMSE), particularly in those patients generating the highest antibody titers.95 In contrast, a more recent and complete analysis of all patients treated in the AN1792 phase 2 trial demonstrated no significant effects on exploratory measures of cognition or disability [ADAS-COG, Disability Assessment for Dementia (DAD) Clinical Dementia Rating (CDR), or MMSE].96 However, significant improvements were observed in a nine component neuropsychological test battery (NTB), indicating less worsening of performance in antibody responders. Furthermore, improvement in memory components of the NTB, including immediate and delayed memory, were associated with an increased antibody response, suggestive of a dose response effect. In addition, measures of tau were also decreased in a small subset of patients undergoing CSF analysis, suggesting a reduction of degenerating neurons, although no differences in Aβ levels were observed.96 In a second recent report, magnetic resonance imaging (MRI) was also used to examine cerebral volume changes in patients treated with AN1792. A comparison of predose MRI scans with scans 12 months after dosing of AN1792, surprisingly demonstrated that antibody responders to AN1792 had increased brain volume loss, greater ventricular enlargement, and greater hippocampal volume loss. However, increased brain volume loss did not result in cognitive decline and indeed was suggestive of cognitive improvement using the NTB.97 It remains unclear what these change in brain volume reflect mechanistically. It is possible that clearance of Aβ deposits from the brains of AD patients or indeed changes in plaque composition or associated inflammatory components could result in changes in brain water content and a concomitant apparent reduction in brain volume. In support of this, studies have suggested that amyloid deposits can occupy approximately 10% of cerebral areas such as the entorhinal cortex.98,99 Alternatively, the observed reduction in brain volume and ventricular enlargement could reflect a continuing of the neurodegenerative process. This, however, does not seem likely given that patients with good antibody response to AN1792 did not show any increase in cognitive decline but instead a reduction in decline as measured by the NTB.97
Additional support for a positive effect of AN1792 via amyloid clearance has come from three post mortem cases (two with encephalitis and one without). The brains of these patients had clear evidence of Alzheimer’s like pathology, but interestingly also had brain regions, particularly in the neocortex, almost completely devoid of amyloid plaques and with clear evidence of Aβ phagocytosing microglia.100–102 No effects were observed on either vascular amyloid deposits or neurofibrillary tangles, the latter despite preclinical evidence suggesting that passive immunization can attenuate early tau pathology in transgenic animals.103
Examination of the two encephalitis cases postmortem revealed a marked CD4 positive T-cell infiltration suggestive of a T-cell response to Aβ.100–102 Given that T-cell epitopes have been mapped to the carboxy terminus of Aβ104,105 and that efficacy in preclinical studies appears to be driven largely by amino terminal epitopes of Aβ, it may be possible to create an immunotherapy with a reduced risk of encephalitis by specifically targeting the amino terminal domain of Aβ, thereby circumventing potentially harmful T-cell responses. Although active immunization is likely to be easier to administer to AD patients, passive immunotherapy using humanized monoclonal anti-Aβ antibodies does confer some potential advantages. In addition to eliminating potentially toxic T-cell-mediated responses to Aβ, antibody therapy will be easier to control and stop, should any adverse events be observed during the course of a clinical trial. This is, of course, more difficult to achieve with active immunization where individuals treated with the Aβ immunogen may continue to generate an immune response to the drug months after the last dose. Concerns have also recently been raised about the potential for active and passive immunotherapy approaches to cause micro hemorrhages. In preclinical studies, passive immunization with antibodies recognizing a variety of Aβ epitopes in transgenic APP mice with pre-existing evidence of cerebral amyloid angiopathy (CAA) resulted in an increased severity and/or incidence of CAA-associated microhemorrhages.106–108 The physiological implications of these findings remain unclear given the doses of antibody used were in some instances extremely high and the animals used had pre-existing cerebral amyloid angiopathy. Furthermore, these findings have not been observed by others or reported in clinical trials to date.109
As active and passive immunization approaches continue to bring together minds from two of the most complex and poorly understood fields of science, neuroscience and immunology, there is real hope that further improvements to our understanding will enable the successful clinical implementation of these approaches for the treatment of this devastating neurodegenerative disorder. If a safe and well-tolerated immunization strategy can be successfully developed, one can envisage a scenario where improving diagnosis of the disease will allow patients to be treated earlier and earlier, preventing progression of neuropathology and the onset of memory impairment.
ENHANCED PROTEOLYTIC DEGRADATION OF Aβ AS AN APPROACH TO DIMINISH STEADY-STATE SOLUBLE AND AGGREGATED Aβ LEVELS
Insufficient clearance of brain Aβ has been proposed to account for elevated Aβ levels and the accumulation of pathogenic amyloid deposits in sporadic AD as the balance between production and degradation determines steady-state levels of Aβ.110 Several proteases involved in Aβ degradation have been identified that contribute to the regulation of Aβ levels under normal physiological conditions,111–114 and may potentially be targeted for therapeutic strategies to enhance Aβ clearance by catabolism (FIG. 2).115–117
Insulin-degrading enzyme (IDE) is a cytosolic metalloendopeptidase that hydrolyzes numerous peptides with poor substrate selectivity and specificity and was the first protease to be implicated in the proteolytic degradation of Aβ.118 IDE isolated from human brain extracts was demonstrated to cleave Αβ40 and Aβ42 preventing aggregation and neurotoxicity of Aβ in vitro.119 In contrast to the reduction of soluble and insoluble Aβ levels, the reduction of amyloid burden and the improved survival of rates of transgenic mice overexpressing IDE,120 IDE knockout mice demonstrate a clear elevation of brain Aβ levels.121 Genetic association with late-onset AD122 and the correlation of high steady-state enzyme levels in brain areas less vulnerable to amyloid pathology in AD123 support the involvement of IDE in Aβ degradation.
Neprilysin (NEP) is a 90- to 100-kDa plasma membrane-bound, extracellular, metalloendopeptidase that preferentially hydrolyzes oligopepetides on the amino terminal of hydrophobic amino acid residues.124 NEP is expressed in brain and has been demonstrated to hydrolyze Aβ42 in vitro and in vivo.124,125 Correlations achieved with chronic overexpression experiments in transgenic mice,120 and evaluation of knockout animals126 suggest that NEP is a physiologically relevant protease and contributes to the degradation of brain Aβ. A 50% reduction of cortical amyloid deposits in transgenic APP mice, after an intracerebral injection of a viral construct expressing NEP, provides further compelling evidence for a potential NEP-mediated Aβ-clearance mechanism in vivo.127 Interestingly, Sisodia and colleagues128 demonstrated that exposure of transgenic mice to an “enriched environment” in combination with exercise results in an elevation of brain NEP activity, and that this is correlated with a pronounced reduction in cerebral Aβ levels and amyloid deposits.
Mutation screening analysis and association studies suggest that NEP might influence the susceptibility to sporadic AD,129 and a decrease of NEP immunoreactivity is observed in the brain of AD patients.130 The recent observation that somatostatin regulates brain Aβ42 levels through the modulation of proteolytic degradation by NEP suggests a potential therapeutic strategy by targeting somatostatin receptors.131
Plasmin, a serine protease released after cleavage of the zymogen plasminogen, can also modulate the clearance of Aβ.132 Kinetic studies measuring the turnover rates of soluble and aggregated Aβ, evaluation of Aβ fibrils by electron microscopy, and Aβ neuroprotection assays in rat cortical cultures, indicate that Aβ is a plasmin substrate in vitro.133,134 Aggregated Aβ also up-regulates the expression of tissue plasminogen activator (tPA) in plaque-bearing transgenic APP mice135 and can activate the generation of plasmin by cleavage of plasminogen.136 Plasminogen is expressed in brain,137 although plasmin activity appears reduced in brain138 and plasma139 of AD patients. Urokinase plasminogen activator (uPA), a functional analog to tPA, has been mapped to a locus140 previously linked to a familial AD locus on chromosome 10.141 Decreased plasmin activity may explain reduced Aβ degradation and accumulation of amyloid pathology in AD,138 and strategies to elevate plasmin activity may be of therapeutic relevance.
Other Aβ42-cleaving peptidases including endothelin converting enzyme-1, matrix metallopeptidase-9 and angiotensin-converting enzyme, have all been implicated in Aβ degradation in vitro, although in vivo evidence thus far is less compelling.114,115,142
It is probable that several peptidases contribute to the degradation of Aβ in vivo and may participate in regulating both normal steady-state brain Aβ levels with an appropriate balance of Αβ formation and catabolism, and pathology with the accumulation of amyloid plaques in AD. Further understanding of Αβ catabolism may lead to the discovery of novel strategies involving the therapeutically regulated Αβ degradation.
MODULATION OF TAU PHOSPHORYLATION AS A THERAPEUTIC TARGET
Neurofibrillary tangles (NFTs) in the brain of Alzheimer’s disease patients are recognized as the other principal pathological hallmark at autopsy. Tangles are generated after the aggregation and assembly of a hyperphosphorylated microtubule binding, tau, into insoluble intracellular paired helical filaments (PHFs). Abnormal accumulations of hyperphosphorylated tau are also seen in the swollen, tortuous, neuritic processes often found in association with senile plaques. The phosphorylation state of tau regulates its ability to stimulate microtubule assembly.143 Indeed, excessive tau phosphorylation in brain extracts from AD cases is thought to contribute to the observed impairment in microtubule assembly.144
Tau is primarily, although not exclusively, a neuronal protein. In adult human brain, there are six major isoforms of tau generated by alternative mRNA splicing. Tau has zero, one, or two N-terminal inserts (resulting from the splicing in or out of exons 2 and 3) and three or four microtubule-binding domains (resulting from the splicing in or out of exon 10).145 The splicing of tau is developmentally regulated, as is its phosphorylation state. In fetal brain, only the shortest tau isoform is present (minus exons 2, 3, and 10)146 and fetal tau is more extensively phosphorylated than adult tau.147 Tau from fetal brain promotes microtubule assembly less efficiently than tau from adult brain148 and elevated levels of phosphorylated tau correlate with the presence of dynamic microtubules during periods of high plasticity in the developing mammalian brain.149 The longest form of adult human brain tau has eight Ser or Thr residues and five Tyr residues; therefore, almost 20% of the molecule has the potential to be phosphorylated.150 In vitro, tau is a substrate for over 20 protein kinases. However, the number of protein kinases that actually phosphorylate tau in vivo is likely to be lower. Site-specific phosphorylation of tau is essential for its normal function and there is increasing evidence that inappropriate phosphorylation of tau leads to tau dysfunction, resulting in decreased cell viability. Indeed, all neurodegenerative diseases in which tau pathology has been observed contain high levels of abnormally phosphorylated tau.151 These diseases include a group of rare autosomal dominant neurodegenerative diseases collectively known as frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), which are caused by mutations in the tau gene located on chromosome 17q21.151 In Alzheimer’s disease, whereas the number and density of NFTs are strongly correlated with the degree of cognitive impairment, developing tauopathy is thought to occur secondary to Aβ multimerization,117,152,153 although prior to the formation of β-amyloid plaques.154 Irrespective of the precise location of tau hyperphosphorylation and NFTs in the pathogenic cascade of AD (FIG. 1), aberrant tau phosphorylation is thought to be play a significant role in the pathogenesis in AD, and therefore inhibition of this process should slow or halt the neurodegenerative disease progression.
Nearly 20 kinases are reported to phosphorylate tau in vitro, therefore exact identification of the relevant kinase(s) responsible for pathology has proven difficult. The rationale for developing therapeutic inhibitors for cyclin-dependent kinase-5 (cdk-5)155 and glycogen synthase kinase-3 (GSK-3)156 activities has been reviewed elsewhere, but to our knowledge drug discovery studies remain preclinical at this time. Aβ can induce tau phosphorylation by the progressive and sustained activation of a number of kinase pathways. For example, application of Aβ42 induces the conversion of p35 to p25 in primary cortical neurons, leading to activation of cdk-5 and subsequent tau hyperphosphorylation In addition, high levels of p25 have been found in the brains of AD patients.157,158 In vitro, Aβ can also activate the Src family tyrosine kinases resulting in phosphorylation of numerous neuronal proteins such as tau and the microtubule-associated protein 2c.159 Recent studies evaluating the cascade of events leading to neurofibrillary pathology suggest that hyperphosphorylation of tau by kinases such as cdk-5 and GSK-3 is preceded by phosphorylation of the tau microtubule binding domain by microtubule affinity regulating kinase (MARK). It is suggested that inhibition of MARK may block the event(s) triggering microtubule disruption, tau hyperphosphorylation, aggregation, formation of neurofibrillary tangles and neurodegeneration.160 Alternative exploratory strategies for reducing tau hyperphosphorylation include increasing the activity phosphatases such as protein phosphatase-2A, thereby promoting the enzymatic dephosphorylation of tau.161
This is an exciting time for AD research, as there is little doubt that our understanding of the disease has increased significantly over the past 20 years. Many promising compounds are now moving through clinical development and over the next 3–5 years it is conceivable that we may have not only improved symptomatic AD therapies but also the first of the disease-modifying agents. Importantly, if these therapies are safe and effective in slowing or halting the underlying pathological progression of the disease then it will be critical for us to understand how best to bring these medicines to patients as early on in the disease process as possible (i.e., before significant cognitive deficits have occurred). As such, it is essential that improved diagnostic markers for AD are identified, more sensitive than current diagnostic tests. Many groups are making progress in this area, with perhaps some of the most promising innovation coming from the development of new amyloid imaging agents such as Pittsburgh Compound-B.162 It is still too early to tell whether this or any of the other diagnostic markers being developed will allow clinicians to diagnose patients early. Nevertheless, with the advent of disease-modifying therapies a realistic vision for the future, the holy grail for patients and their families is the ability to diagnose and treat not just patients with significant cognitive impairment, but presymptomatic AD patients who are able to live and function normally on therapy.
REFERENCES
- 1.Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am J Public Health 88: 1337–1342, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 82: 239–259, 1991. [DOI] [PubMed] [Google Scholar]
- 3.Bowen DM, Smith CB, White P, Davison AN. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 99: 459–496, 1976. [DOI] [PubMed] [Google Scholar]
- 4.Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 2: 1403, 1976. [DOI] [PubMed] [Google Scholar]
- 5.Kryger G, Silman I, Sussman JL. Three-dimensional structure of a complex of E2020 with acetylcholinesterase from Torpedo californica. J Physiol (Lond) Paris 92: 191–194, 1998. [DOI] [PubMed] [Google Scholar]
- 6.Greenblatt HM, Kryger G, Lewis T, Silman I, Sussman JL. Structure of acetylcholinesterase complexed with (-)-galanthamine at 2.3 A resolution. FEBS Lett 463: 321–326, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Farlow M. A clinical overview of cholinesterase inhibitors in Alzheimer’s disease. Int Psychogeriatrics 1: 93–126, 2002. [DOI] [PubMed] [Google Scholar]
- 8.Lahiri DK, Farlow MR, Hintz N, Utsuki T, Greig NH. Cholinesterase inhibitors, β-amyloid precursor protein and amyloid β-peptides in Alzheimer’s disease. Acta Neurol Scand Suppl 176: 60–67, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Doody RS, Geldmacher DS, Gordon B, Perdomo CA, Pratt RD. Open-label, multicenter, phase 3 extension study of the safety and efficacy of donepezil in patients with Alzheimer disease. Arch Neurol 58: 427–433, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Farlow M, Anand R, Messina J Jr, Hartman R, Veach J. A 52-week study of the efficacy of rivastigmine in patients with mild to moderately severe Alzheimer’s disease. Eur Neurol 44: 236–241, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Bullock R. New drugs for Alzheimer’s disease and other dementias. Br J Psychiatry 180: 135–139, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Greenamyre JT, Young AB. Excitatory amino acids and Alzheimer’s disease. Neurobiol Aging 10: 593–602, 1989. [DOI] [PubMed] [Google Scholar]
- 13.Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S. Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer’s and Parkinson’s diseases. Ann N Y Acad Sci 893: 154–175, 1999. [DOI] [PubMed] [Google Scholar]
- 14.Buchan AM, Slivka A, Xue D. The effect of the NMDA receptor antagonist MK-801 on cerebral blood flow and infarct volume in experimental focal stroke. Brain Res 574: 171–177, 1992. [DOI] [PubMed] [Google Scholar]
- 15.Winblad B, Poritis N. Memantine in severe dementia: results of the 9M-Best Study (benefit and efficacy in severely demented patients during treatment with memantine). Int J Geriatr Psychiatry 14: 135–146, 1999. [DOI] [PubMed] [Google Scholar]
- 16.Winblad B, Jelic V. Treating the full spectrum of dementia with memantine. Int J Geriatr Psychiatry 18: S41–S46, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Winblad B, Mobius HJ, Stoffler A. Glutamate receptors as a target for Alzheimer’s disease—are clinical results supporting the hope? J Neural Transm Suppl 217–225, 2002. [DOI] [PubMed]
- 18.Koch HJ, Szecsey A, Haen E. NMDA-antagonism (memantine): an alternative pharmacological therapeutic principle in Alzheimer’s and vascular dementia. Curr Pharm Des 10: 253–259, 2004. [DOI] [PubMed] [Google Scholar]
- 19.King MV, Sleight AJ, Woolley ML, Topham IA, Marsden CA, Fone KC. 5-HT6 receptor antagonists reverse delay-dependent deficits in novel object discrimination by enhancing consolidation—an effect sensitive to NMDA receptor antagonism. Neuropharmacology 47: 195–204, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Schechter LE, Smith D, Rosenzweig-Lipson S, Sukoff S, Dawson L, Marquis K, et al. Lecozotan (Sra-333): a selective serotonin1a receptor antagonist that enhances the stimulated release of glutamate and acetylcholine in the hippocampus and possesses cognitive-enhancing properties. J Pharmacol Exp Ther 10.1124/jpet.105.086363, 13 June 2005. [DOI] [PubMed]
- 21.Danysz W. CX-516 Cortex pharmaceuticals. Curr Opin Investig Drugs 3: 1081–1088, 2002. [PubMed] [Google Scholar]
- 22.Komater VA, Browman KE, Curzon P, Hancock AA, Decker MW, Fox GB. H3 receptor blockade by thioperamide enhances cognition in rats without inducing locomotor sensitization. Psychopharmacology (Berl) 167: 363–372, 2003. [DOI] [PubMed] [Google Scholar]
- 23.Fox GB, Esbenshade TA, Pan JB, Radek RJ, Krueger KM, Yao BB, et al. Pharmacological properties of ABT-239 [4-(2-{2-[(2R)-2-methylpyrrolidinyl]ethyl}-benzofuran-5-yl)benzonitrile]: II. Neurophysiological characterization and broad preclinical efficacy in cognition and schizophrenia of a potent and selective histamine H3 receptor antagonist. J Pharmacol Exp Ther 313: 176–190, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Fisher A, Pittel Z, Haring R, Bar-Ner N, Kliger-Spatz M, Natan N, et al. M1 muscarinic agonists can modulate some of the hallmarks in Alzheimer’s disease: implications in future therapy. J Mol Neurosci 20: 349–356, 2003. [DOI] [PubMed] [Google Scholar]
- 25.Thal LJ, Forrest M, Loft H, Mengel H. Lu 25-109, a muscarinic agonist, fails to improve cognition in Alzheimer’s disease. Lu25-109 Study Group. Neurology 54: 421–426, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Loudon JM, Bromidge SM, Brown F, Clark MS, Hatcher JP, Hawkins J, et al. SB 202026: a novel muscarinic partial agonist with functional selectivity for M1 receptors. J Pharmacol Exp Ther 283: 1059–1068, 1997. [PubMed] [Google Scholar]
- 27.Bartolomeo AC, Morris H, Buccafusco JJ, Kille N, Rosenzweig-Lipson S, Husbands MG, et al. The preclinical pharmacological profile of WAY-132983, a potent M1 preferring agonist. J Pharmacol Exp Ther 292: 584–596, 2000. [PubMed] [Google Scholar]
- 28.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120: 885–890, 1984. [DOI] [PubMed] [Google Scholar]
- 29.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297: 353–356, 2002. [DOI] [PubMed] [Google Scholar]
- 30.Sisodia SS, St George-Hyslop PH. γ-Secretase, Notch, Aβ and Alzheimer’s disease: where do the presenilins fit in? Nat Rev Neurosci 3: 281–290, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Selkoe DJ, Schenk D. Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol 43: 545–584, 2003. [DOI] [PubMed] [Google Scholar]
- 32.Robakis NK, Pangalos MN. Involvement of amyloid as a central step in the development of Alzheimer’s disease. Neurobiol Aging 15 [Suppl 2]: S127–S129, 1994. [DOI] [PubMed] [Google Scholar]
- 33.Kaplitt M, Gouras GK, Makimura H, Jovanovic J, Sweeney D, Greengard P, et al. Apolipoprotein E, A β-amyloid, and the molecular pathology of Alzheimer’s disease. Therapeutic implications. Ann N Y Acad Sci 802: 42–49, 1996. [DOI] [PubMed] [Google Scholar]
- 34.Carter DB, Dunn E, McKinley DD, Stratman NC, Boyle TP, Kuiper SL, et al. Human apolipoprotein E4 accelerates β-amyloid deposition in APPsw transgenic mouse brain. Ann Neurol 50: 468–475, 2001. [DOI] [PubMed] [Google Scholar]
- 35.Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, et al. Binding of human apolipoprotein E to synthetic amyloid β peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA 90: 8098–8102, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 97: 2892–2897, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, et al. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 46: 860–866, 1999. [DOI] [PubMed] [Google Scholar]
- 38.Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-β oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans 30: 552–557, 2002. [DOI] [PubMed] [Google Scholar]
- 39.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, et al. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 359: 322–325, 1992. [DOI] [PubMed] [Google Scholar]
- 40.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349: 704–706, 1991. [DOI] [PubMed] [Google Scholar]
- 41.Suh YH, Checler F. Amyloid precursor protein, presenilins, and α-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer’s disease. Pharmacol Rev 54: 469–525, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Verdier Y, Penke B. Binding sites of amyloid β-peptide in cell plasma membrane and implications for Alzheimer’s disease. Curr Protein Peptide Sci 5: 19–31, 2004. [DOI] [PubMed] [Google Scholar]
- 43.Curtain CC, Ali FE, Smith DG, Bush AI, Masters CL, Barnham KJ. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer’s disease amyloid-β peptide with membrane lipid. J Biol Chem 278: 2977–2982, 2003. [DOI] [PubMed] [Google Scholar]
- 44.Sinha S, Anderson J, John V, McConlogue L, Basi G, Thorsett E, et al. Recent advances in the understanding of the processing of APP to β amyloid peptide. Ann N Y Acad Sci 920: 206–208, 2000. [DOI] [PubMed] [Google Scholar]
- 45.Gruninger-Leitch F, Schlatter D, Kung E, Nelbock P, Dobeli H. Substrate and inhibitor profile of BACE (β-secretase) and comparison with other mammalian aspartic proteases. J Biol Chem 277: 4687–4693, 2002. [DOI] [PubMed] [Google Scholar]
- 46.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286: 735–741, 1999. [DOI] [PubMed] [Google Scholar]
- 47.Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, et al. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat Neurosci 4: 233–234, 2001. [DOI] [PubMed] [Google Scholar]
- 48.Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, et al. Mice deficient in BACE1, the Alzheimer’s β-secretase, have normal phenotype and abolished β-amyloid generation. Nat Neurosci 4: 231–232, 2001. [DOI] [PubMed] [Google Scholar]
- 49.Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, et al. BACE knockout mice are healthy despite lacking the primary β-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum Mol Genet 10: 1317–1324, 2001. [DOI] [PubMed] [Google Scholar]
- 50.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophy Acta 455: 152–162, 1976. [DOI] [PubMed] [Google Scholar]
- 51.Paganetti P, Calanca V, Galli C, Stefani M, Molinari M. β-Site specific intrabodies to decrease and prevent generation of Alzheimer’s Aβ peptide. J Cell Biol 168: 863–868, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arbel M, Yacoby I, Solomon B. Inhibition of amyloid precursor protein processing by β-secretase through site-directed antibodies. Proc Natl Acad Sci USA 102: 7718–7723, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Q, Sudhof TC. Cleavage of amyloid-β precursor protein and amyloid-β precursor-like protein by BACE 1. J Biol Chem 279: 10542–10550, 2004. [DOI] [PubMed] [Google Scholar]
- 54.Selkoe D, Kopan R. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci 26: 565–597, 2003. [DOI] [PubMed] [Google Scholar]
- 55.Shirotani K, Edbauer D, Prokop S, Haass C, Steiner H. Identification of distinct γ-secretase complexes with different APH-1 variants. J Biol Chem 279: 41340–41345, 2004. [DOI] [PubMed] [Google Scholar]
- 56.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, et al. Constitutive and regulated α-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA 96: 3922–3927, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng Y, Saftig P, Hartmann D, Blobel C. Evaluation of the contribution of different ADAMs to tumor necrosis factor α (TNFα) shedding and of the function of the TNFα ectodomain in ensuring selective stimulated shedding by the TNFα convertase (TACE/ADAM17). J Biol Chem 279: 42898–42906, 2004. [DOI] [PubMed] [Google Scholar]
- 58.Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science 258: 304–307, 1992. [DOI] [PubMed] [Google Scholar]
- 59.Lichtenthaler SF, Haass C. Amyloid at the cutting edge: activation of α-secretase prevents amyloidogenesis in an Alzheimer disease mouse model. J Clin Invest 113: 1384–1387, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y, Wang H, Wang S, Quon D, Liu YW, Cordell B. Positive and negative regulation of APP amyloidogenesis by sumoylation. Proc Natl Acad Sci USA 100: 259–264, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Desterro JM, Rodriguez MS, Hay RT. SUMO-1 modification of IκBα inhibits NF-κB activation. Mol Cell 2: 233–239, 1998. [DOI] [PubMed] [Google Scholar]
- 62.Verger A, Perdomo J, Crossley M. Modification with SUMO. A role in transcriptional regulation. EMBO Rep 4: 137–142, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gill G. Post-translational modification by the small ubiquitin-related modifier SUMO has big effects on transcription factor activity. Curr Opin Genet Dev 13: 108–113, 2003. [DOI] [PubMed] [Google Scholar]
- 64.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400: 173–177, 1999. [DOI] [PubMed] [Google Scholar]
- 65.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, et al. A β peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 408: 979–982, 2000. [DOI] [PubMed] [Google Scholar]
- 66.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: 916–919, 2000. [DOI] [PubMed] [Google Scholar]
- 67.Lemere CA, Maron R, Selkoe DJ, Weiner HL. Nasal vaccination with β-amyloid peptide for the treatment of Alzheimer’s disease. DNA Cell Biol 20: 705–711, 2001. [DOI] [PubMed] [Google Scholar]
- 68.Weiner HL, Selkoe DJ. Inflammation and therapeutic vaccination in CNS diseases. Nature 420: 879–884, 2002. [DOI] [PubMed] [Google Scholar]
- 69.DeMattos RB, Brendza RP, Heuser JE, Kierson M, Cirrito JR, Fryer J, et al. Purification and characterization of astrocyte-secreted apolipoprotein E and J-containing lipoproteins from wild-type and human apoE transgenic mice. Neurochem Int 39: 415–425, 2001. [DOI] [PubMed] [Google Scholar]
- 70.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A β antibody alters CNS and plasma A β clearance and decreases brain A β burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 98: 8850–8855, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-β efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science 295: 2264–2267, 2002. [DOI] [PubMed] [Google Scholar]
- 72.Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D, et al. Epitope and isotype specificities of antibodies to β-amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proc Natl Acad Sci USA 100: 2023–2028, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weiner HL, Lemere CA, Maron R, Spooner ET, Grenfell TJ, Mori C, et al. Nasal administration of amyloid-β peptide decreases cerebral amyloid burden in a mouse model of Alzheimer’s disease. Ann Neurol 48: 567–579, 2000. [PubMed] [Google Scholar]
- 74.Leverone JF, Spooner ET, Lehman HK, Clements JD, Lemere CA. Aβ1–15 is less immunogenic than Aβ1–40/42 for intranasal immunization of wild-type mice but may be effective for “boosting.” Vaccine 21: 2197–2206, 2003. [DOI] [PubMed] [Google Scholar]
- 75.Zhang J, Wu X, Qin C, Qi J, Ma S, Zhang H, et al. A novel recombinant adeno-associated virus vaccine reduces behavioral impairment and β-amyloid plaques in a mouse model of Alzheimer’s disease. Neurobiol Dis 14: 365–379, 2003. [DOI] [PubMed] [Google Scholar]
- 76.Lavie V, Becker M, Cohen-Kupiec R, Yacoby I, Koppel R, Wedenig M, et al. EFRH-phage immunization of Alzheimer’s disease animal model improves behavioral performance in Morris water maze trials. J Mol Neurosci 24: 105–113, 2004. [DOI] [PubMed] [Google Scholar]
- 77.Dodel R, Hampel H, Depboylu C, Lin S, Gao F, Schock S, et al. Human antibodies against amyloid β peptide: a potential treatment for Alzheimer’s disease. Ann Neurol 52: 253–256, 2002. [DOI] [PubMed] [Google Scholar]
- 78.Dodel RC, Du Y, Depboylu C, Hampel H, Frolich L, Haag A, et al. Intravenous immunoglobulins containing antibodies against β-amyloid for the treatment of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 75: 1472–1474, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, et al. A β peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 408: 982–985, 2000. [DOI] [PubMed] [Google Scholar]
- 80.McLaurin J, Cecal R, Kierstead ME, Tian X, Phinney AL, Manea M, et al. Therapeutically effective antibodies against amyloid-β peptide target amyloid-β residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat Med 8: 1263–1269, 2002. [DOI] [PubMed] [Google Scholar]
- 81.Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, et al. Non-Fc-mediated mechanisms are involved in clearance of amyloid-β in vivo by immunotherapy. J Neurosci 22: 7873–7878, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, et al. Imaging of amyloid-β deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med 7: 369–372, 2001. [DOI] [PubMed] [Google Scholar]
- 83.Das P, Howard V, Loosbrock N, Dickson D, Murphy MP, Golde TE. Amyloid-β immunization effectively reduces amyloid deposition in FcRγ−/− knock-out mice. J Neurosci 23: 8532–8538, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Solomon B, Koppel R, Hanan E, Katzav T. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer β-amyloid peptide. Proc Natl Acad Sci USA 93: 452–455, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer β-amyloid by site-directed mAb. Proc Natl Acad Sci USA 94: 4109–4112, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Frenkel D, Balass M, Solomon B. N-terminal EFRH sequence of Alzheimer’s β-amyloid peptide represents the epitope of its anti-aggregating antibodies. J Neuroimmunol 88: 85–90, 1998. [DOI] [PubMed] [Google Scholar]
- 87.Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, et al. Amyloid-β antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J Neurosci 23: 10879–10883, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, et al. Intracranially administered anti-Aβ antibodies reduce β-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci 23: 3745–3751, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, et al. Amyloid β protein immunotherapy neutralizes Aβ oligomers that disrupt synaptic plasticity in vivo. Nat Med 11: 556–561, 2005. [DOI] [PubMed] [Google Scholar]
- 90.DeMattos RB, Bales KR, Parsadanian M, O’Dell MA, Foss EM, Paul SM, et al. Plaque-associated disruption of CSF and plasma amyloid-β (Aβ) equilibrium in a mouse model of Alzheimer’s disease. J Neurochem 81: 229–236, 2002. [DOI] [PubMed] [Google Scholar]
- 91.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, et al. Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids. Nature 359: 325–327, 1992. [DOI] [PubMed] [Google Scholar]
- 92.Lemere CA, Spooner ET, LaFrancois J, Malester B, Mori C, Leverone JF, et al. Evidence for peripheral clearance of cerebral Aβ protein following chronic, active Aβ immunization in PSAPP mice. Neurobiol Dis 14: 10–18, 2003. [DOI] [PubMed] [Google Scholar]
- 93.Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, et al. Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer’s disease model. Nat Neurosci 5: 452–457, 2002. [DOI] [PubMed] [Google Scholar]
- 94.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology 61: 46–54, 2003. [DOI] [PubMed] [Google Scholar]
- 95.Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, et al. Antibodies against β-amyloid slow cognitive decline in Alzheimer’s disease. Neuron 38: 547–554, 2003. [DOI] [PubMed] [Google Scholar]
- 96.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, et al. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64: 1553–1562, 2005. [DOI] [PubMed] [Google Scholar]
- 97.Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, et al. Effects of Aβ immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 64: 1563–1572, 2005. [DOI] [PubMed] [Google Scholar]
- 98.Bussiere T, Friend PD, Sadeghi N, Wicinski B, Lin GI, Bouras C, et al. Stereologic assessment of the total cortical volume occupied by amyloid deposits and its relationship with cognitive status in aging and Alzheimer’s disease. Neuroscience 112: 75–91, 2002. [DOI] [PubMed] [Google Scholar]
- 99.Colle MA, Duyckaerts C, Laquerriere A, Pradier L, Czech C, Checler F, et al. Laminar specific loss of isocortical presenilin 1 immunoreactivity in Alzheimer’s disease. Correlations with the amyloid load and the density of tau-positive neurofibrillary tangles. Neuropathol Appl Neurobiol 26: 117–123, 2000. [DOI] [PubMed] [Google Scholar]
- 100.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nat Med 9: 448–452, 2003. [DOI] [PubMed] [Google Scholar]
- 101.Ferrer I, Boada Rovira M, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-β immunization in Alzheimer’s disease. Brain Pathol 14: 11–20, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, et al. Aβ vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology 64: 129–131, 2005. [DOI] [PubMed] [Google Scholar]
- 103.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43: 321–332, 2004. [DOI] [PubMed] [Google Scholar]
- 104.Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, et al. Increased T cell reactivity to amyloid β protein in older humans and patients with Alzheimer disease. J Clin Invest 112: 415–422, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Glaser R, Kiecolt-Glaser JK, Malarkey WB, Sheridan JF. The influence of psychological stress on the immune response to vaccines. Ann N Y Acad Sci 840: 649–655, 1998. [DOI] [PubMed] [Google Scholar]
- 106.Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, et al. Cerebral hemorrhage after passive anti-Aβ immunotherapy. Science 298: 1379, 2002. [DOI] [PubMed] [Google Scholar]
- 107.Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, et al. Passive immunotherapy against Aβ in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflamm 1: 24, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β. J Neurosci 25: 629–636, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Goni F, Sigurdsson EM. New directions towards safer and effective vaccines for Alzheimer’s disease. Curr Opin Mol Ther 7: 17–23, 2005. [PubMed] [Google Scholar]
- 110.Selkoe DJ. Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid β-protein. Ann N Y Acad Sci 924: 17–25, 2000. [DOI] [PubMed] [Google Scholar]
- 111.Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer’s Aβ peptide: the many roads to perdition. Neuron 43: 605–608, 2004. [DOI] [PubMed] [Google Scholar]
- 112.Selkoe DJ. Clearing the brain’s amyloid cobwebs. Neuron 32: 177–180, 2001. [DOI] [PubMed] [Google Scholar]
- 113.Mukherjee A, Hersh LB. Regulation of amyloid β-peptide levels by enzymatic degradation. J Alzheimer’s Dis 4: 341–348, 2002. [DOI] [PubMed] [Google Scholar]
- 114.Morelli L, Bulloj A, Leal MC, Castano EM. Amyloid β degradation: a challenging task for brain peptidases. Subcell Biochem 38: 129–145, 2005. [DOI] [PubMed] [Google Scholar]
- 115.Turner AJ, Fisk L, Nalivaeva NN. Targeting amyloid-degrading enzymes as therapeutic strategies in neurodegeneration. Ann N Y Acad Sci 1035: 1–20, 2004. [DOI] [PubMed] [Google Scholar]
- 116.Ling Y, Morgan K, Kalsheker N. Amyloid precursor protein (APP) and the biology of proteolytic processing: relevance to Alzheimer’s disease. Int J Biochem Cell Biol 35: 1505–1535, 2003. [DOI] [PubMed] [Google Scholar]
- 117.Walker LC, Ibegbu CC, Todd CW, Robinson HL, Jucker M, LeVine H 3rd, et al. Emerging prospects for the disease-modifying treatment of Alzheimer’s disease. Biochem Pharmacol 69: 1001–1008, 2005. [DOI] [PubMed] [Google Scholar]
- 118.Kurochkin IV, Goto S. Alzheimer’s β-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett 345: 33–37, 1994. [DOI] [PubMed] [Google Scholar]
- 119.Mukherjee A, Song E, Kihiko-Ehmann M, Goodman JP Jr, Pyrek JS, Estus S, et al. Insulysin hydrolyzes amyloid β peptides to products that are neither neurotoxic nor deposit on amyloid plaques. J Neurosci 20: 8745–8749, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, et al. Enhanced proteolysis of β-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40: 1087–1093, 2003. [DOI] [PubMed] [Google Scholar]
- 121.Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA 100: 4162–4167, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bertram L, Tanzi RE. Alzheimer’s disease: one disorder, too many genes? Hum Mol Genet R135–R141, 2004. [DOI] [PubMed]
- 123.Caccamo A, Oddo S, Sugarman MC, Akbari Y, LaFerla FM. Age- and region-dependent alterations in Aβ-degrading enzymes: implications for Aβ-induced disorders. Neurobiol Aging 26: 645–654, 2005. [DOI] [PubMed] [Google Scholar]
- 124.Carson JA, Turner AJ. β-Amyloid catabolism: roles for neprilysin (NEP) and other metallopeptidases? J Neurochem 81: 1–8, 2002. [DOI] [PubMed] [Google Scholar]
- 125.Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, et al. Identification of the major Aβ1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med 6: 143–150, 2000. [DOI] [PubMed] [Google Scholar]
- 126.Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, et al. Metabolic regulation of brain Aβ by neprilysin. Science 292: 1550–1552, 2001. [DOI] [PubMed] [Google Scholar]
- 127.Marr RA, Rockenstein E, Mukherjee A, Kindy MS, Hersh LB, Gage FH, et al. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J Neurosci 23: 1992–1996, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lazarov O, Robinson J, Tang YP, Hairston IS, Korade-Mirnics Z, Lee VM, et al. Environmental enrichment reduces Aβ levels and amyloid deposition in transgenic mice. Cell 120: 701–713, 2005. [DOI] [PubMed] [Google Scholar]
- 129.Shi J, Zhang S, Tang M, Ma C, Zhao J, Li T, et al. Mutation screening and association study of the neprilysin gene in sporadic Alzheimer’s disease in Chinese persons. J Gerontol A Biol Sci Med Sci 60: 301–306, 2005. [DOI] [PubMed] [Google Scholar]
- 130.Wang DS, Lipton RB, Katz MJ, Davies P, Buschke H, Kuslansky G, et al. Decreased neprilysin immunoreactivity in Alzheimer disease, but not in pathological aging. J Neuropathol Exp Neurol 64: 378–385, 2005. [DOI] [PubMed] [Google Scholar]
- 131.Saito T, Iwata N, Tsubuki S, Takaki Y, Takano J, Huang SM, et al. Somatostatin regulates brain amyloid β peptide Aβ42 through modulation of proteolytic degradation. Nat Med 11: 434–439, 2005. [DOI] [PubMed] [Google Scholar]
- 132.Periz G, Fortini ME. Proteolysis in Alzheimer’s disease. Can plasmin tip the balance? EMBO Rep 1: 477–478, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tucker HM, Kihiko M, Caldwell JN, Wright S, Kawarabayashi T, Price D, et al. The plasmin system is induced by and degrades amyloid-β aggregates. J Neurosci 20: 3937–3946, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tucker HM, Kihiko-Ehmann M, Wright S, Rydel RE, Estus S. Tissue plasminogen activator requires plasminogen to modulate amyloid-β neurotoxicity and deposition. J Neurochem 75: 2172–2177, 2000. [DOI] [PubMed] [Google Scholar]
- 135.Kingston IB, Castro MJ, Anderson S. In vitro stimulation of tissue-type plasminogen activator by Alzheimer amyloid β-peptide analogues. Nat Med 1: 138–142, 1995. [DOI] [PubMed] [Google Scholar]
- 136.Van Nostrand WE, Porter M. Plasmin cleavage of the amyloid β-protein: alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry 38: 11570–11576, 1999. [DOI] [PubMed] [Google Scholar]
- 137.Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci 17: 543–552, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B, Dotti CG. Brain plasmin enhances APP α-cleavage and Aβ degradation and is reduced in Alzheimer’s disease brains. EMBO Rep 1: 530–535, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Aoyagi T, Wada T, Kojima F, Nagai M, Harada S, Takeuchi T, et al. Deficiency of fibrinolytic enzyme activities in the serum of patients with Alzheimer-type dementia. Experientia 48: 656–659, 1992. [DOI] [PubMed] [Google Scholar]
- 140.Tucker HM, Kihiko-Ehmann M, Estus S. Urokinase-type plasminogen activator inhibits amyloid-β neurotoxicity and fibrillogenesis via plasminogen. J Neurosci Res 70: 249–255, 2002. [DOI] [PubMed] [Google Scholar]
- 141.Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, et al. Linkage of plasma Aβ42 to a quantitative locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Science 290: 2303–2304, 2000. [DOI] [PubMed] [Google Scholar]
- 142.Hersh LB. Peptidases, proteases and amyloid β-peptide catabolism. Cur Pharm Des 9: 449–454, 2003. [DOI] [PubMed] [Google Scholar]
- 143.Selkoe DJ. Defining molecular targets to prevent Alzheimer disease. Arch Neurol 62: 192–195, 2005. [DOI] [PubMed] [Google Scholar]
- 144.Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, et al. Defective brain microtubule assembly in Alzheimer’s disease. Lancet 2: 421–426, 1986. [DOI] [PubMed] [Google Scholar]
- 145.Jenkins SM, Johnson GV. Modulation of tau phosphorylation within its microtubule-binding domain by cellular thiols. J Neurochem 73: 1843–1850, 1999. [PubMed] [Google Scholar]
- 146.Kosik KS, Orecchio LD, Bakalis S, Neve RL. Developmentally regulated expression of specific tau sequences. Neuron 2: 1389–1397, 1989. [DOI] [PubMed] [Google Scholar]
- 147.Watanabe A, Hasegawa M, Suzuki M, Takio K, Morishima-Kawashima M, Titani K et al. In vivo phosphorylation sites in fetal and adult rat tau. J Biol Chem 268: 25712–25717, 1993. [PubMed] [Google Scholar]
- 148.Yoshida H, Ihara Y. Tau in paired helical filaments is functionally distinct from fetal tau: assembly incompetence of paired helical filament-tau. J Neurochem 61: 1183–1186, 1993. [DOI] [PubMed] [Google Scholar]
- 149.Brion JP, Octave JN, Couck AM. Distribution of the phosphorylated microtubule-associated protein tau in developing cortical neurons. Neuroscience 63: 895–909, 1994. [DOI] [PubMed] [Google Scholar]
- 150.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3: 519–526, 1989. [DOI] [PubMed] [Google Scholar]
- 151.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci 24: 1121–1159, 2001. [DOI] [PubMed] [Google Scholar]
- 152.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293: 1487–1491, 2001. [DOI] [PubMed] [Google Scholar]
- 153.Hutton M, McGowan E. Clearing tau pathology with Aβ immunotherapy-reversible and irreversible stages revealed. Neuron 43: 293–294, 2004. [DOI] [PubMed] [Google Scholar]
- 154.Price JL, Morris JC. So what if tangles precede plaques? Neurobiol Aging 25: 721–723, 2004. [DOI] [PubMed] [Google Scholar]
- 155.Tsai LH, Lee MS, Cruz J. Cdk5, a therapeutic target for Alzheimer’s disease? Biochimica et Biophysica Acta 1697: 137–142, 2004. [DOI] [PubMed] [Google Scholar]
- 156.Bhat RV, Budd Haeberlein SL, Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem 89: 1313–1317, 2004. [DOI] [PubMed] [Google Scholar]
- 157.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: 615–622, 1999. [DOI] [PubMed] [Google Scholar]
- 158.Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405: 360–364, 2000. [DOI] [PubMed] [Google Scholar]
- 159.Williamson R, Scales T, Clark BR, Gibb G, Reynolds CH, Kellie S, et al. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-β peptide exposure: involvement of Src family protein kinases. J Neurosci 22: 10–20, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Drewes G. MARKing tau for tangles and toxicity. Trends Biochem Sci 29: 548–555, 2004. [DOI] [PubMed] [Google Scholar]
- 161.Iqbal K, Grundke-Iqbal I. Inhibition of neurofibrillary degeneration: a promising approach to Alzheimer’s disease and other tauopathies. Curr Drug Targets 5: 495–502, 2004. [DOI] [PubMed] [Google Scholar]
- 162.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 55: 306–319, 2004. [DOI] [PubMed] [Google Scholar]