

Abstract
In response to DNA damage, p53 activates a G1 cell cycle checkpoint, in part through induction of the cyclin-dependent kinase inhibitor p21(Waf1/Cip1). Here we report the identification of a new direct p53 target, Ptprv (or ESP), encoding a transmembrane tyrosine phosphatase. Ptprv transcription is dramatically and preferentially increased in cultured cells undergoing p53-dependent cell cycle arrest, but not in cells undergoing p53-mediated apoptosis. This observation was further confirmed in vivo using a Ptprv null-reporter mouse line. A p53-responsive element is present in the Ptprv promoter and p53 is recruited to this site in vivo. Importantly, while p53-dependent apoptosis is intact in mice lacking Ptprv, Ptprv-null fibroblasts and epithelial cells of the small intestine are defective in G1 checkpoint control. Thus, Ptprv is a new direct p53 target and a key mediator of p53-induced cell cycle arrest. Finally, Ptprv loss enhances the formation of epidermal papillomas after exposure to chemical carcinogens, suggesting that Ptprv acts to suppress tumor formation in vivo.
Keywords: DNA damage, growth arrest, p53, Ptprv, skin carcinogenesis
Introduction
In response to a variety of stressors, the p53 tumor suppressor functions mainly as a sequence-specific DNA-binding transcription factor to promote antiproliferative responses, including cell cycle checkpoints, cellular senescence, and apoptosis (Lane, 1992). A number of proapoptotic genes, encoding for the mitochondrial proteins Bax, Noxa, Puma, Apaf-1, or proteins that play a role in death receptor-mediated apoptosis, such as Fas, KILLER/DR5, and PIDD, are direct p53 targets (El-Deiry, 1998). Notably, critical importance in vivo has only been established for few of these genes, including puma and noxA (Jeffers et al, 2003; Villunger et al, 2003). p53-mediated cell cycle exit is partly mediated through induction of the cyclin-dependent kinase inhibitor p21 (El-Deiry et al, 1993). p21-null fibroblasts are defective in p53-mediated G1 arrest in response to DNA damage or nucleotide depletion (Brugarolas et al, 1995; Deng et al, 1995). However, this defect is only partial and, in contrast to p53-null cells, fibroblasts derived from p21-null mice enter senescence and have a lifespan similar to wild-type (WT) cells (Pantoja and Serrano, 1999). In addition, these cells respond to oncogenic Ras by accumulating p53, and by decreasing their proliferation rate (Pantoja and Serrano, 1999), suggesting that p21 is not the only critical mediator of G1 arrest.
Phosphorylation of tyrosine residues is a central feature of many cellular signalling pathways, including those affecting apoptosis, growth, differentiation, and cell cycle regulation (Neel and Tonks, 1997). This phosphorylation is antagonistically controlled by protein tyrosine kinases (PTKs) and phosphatases (PTPs). In an intriguing symmetry to receptor tyrosine kinases, many PTPs display a transmembrane topology. In all, 21 genes in the mouse and human genomes encode for members of the receptor PTP family (Alonso et al, 2004). Identifying physiologically relevant ligands or substrates for these RPTPs has proven difficult.
Although a variety of PTKs has been directly linked to tumorigenesis through somatic activating mutations, only a few PTP genes have been implicated in cancer. Recent mutational analysis of the tyrosine phosphatome in colorectal cancers identified multiple inactivating somatic mutations in several PTPs, including three RPTPs. Expression of one of them, PTPRT, in human cancer cells inhibited cell proliferation (Wang et al, 2004). Genetic inactivation of the DEP-1 induces a vascular phenotype characterized by enlarged, oversized vessels with abnormally high endothelial cell proliferation (Takahashi et al, 2003). DEP-1 overexpression in mammary carcinoma cell lines and in thyroid carcinoma cells suppresses the transformed phenotype (Keane et al, 1996; Trapasso et al, 2000). The frequent deletion of the gene in human cancers is another indication that DEP-1 acts as a tumor suppressor (Ruivenkamp et al, 2002).
Here, we report the identification of a new p53 transcription target coding for Ptprv (also known as ESP (Lee et al, 1996)), a protein that is structurally highly related to DEP-1. Both proteins are composed of an extracellular domain containing exclusively multiple type III fibronectin repeats, a transmembrane segment, and either a single or two intracellular classical PTP domains for DEP-1 and Ptprv, respectively. The physiological role of Ptprv and its relevance to tumorigenesis is currently unknown. We show that, under specific conditions, p53 directly regulates transcription of Ptprv, and that Ptprv is a key mediator of p53-mediated cell cycle arrest following DNA damage. In addition, we provide evidences suggesting that Ptprv plays a role in tumor suppression.
Results
Ptprv expression is increased in the mdm4-mutant embryos
We had reported that disruption of the mdm2-related gene, mdm4, leads to a p53-dependent embryonic lethality, mainly caused by an arrest of the mutant cells in the G1 phase of the cell cycle (Migliorini et al, 2002). With the aim to identify new putative mediators of p53-dependent cell cycle arrest, we monitored changes in gene expression in mdm4-mutant embryos using oligonucleotide microarrays (Martoriati et al, 2005). We selected Ptprv for further analysis because it appeared dramatically upregulated in mdm4 mutants. Real-time quantitative PCR, and nonradioactive in situ hybridization (ISH) confirmed the induction of Ptprv expression in mdm4-mutant embryos (Supplementary Figure 1). The level of Ptprv induction (ranging from 70- to 100-fold) is roughly 10 times higher than that of any other known p53-target gene, including p21.
Ptprv is a p53-inducible gene in cultured cells
We next determined Ptprv expression levels after endogenous p53 activation in a series of cellular systems by RNAse protection assays, semiquantitative and quantitative reverse transcription polymerase chain reaction (RT–PCR). Activation of p53 by induction of a conditional allele of p19ARF in NIH-3T3 cells (pMTArf cells; Kuo et al, 2003) increases Ptprv expression (Figure 1A). Ptprv mRNA was also induced in response to DNA damage, such as following doxorubicin treatment (Figure 1B) or UV irradiation (Figure 1C) in WT, but not in the p53-null embryonic stem (ES) cells. In addition, Ptprv mRNA was induced in WT but not p53-null MEFs in response to γ-radiation (γ-IR) (Figure 1D), doxorubicin treatment (Figure 1E), and to a lesser extent UV irradiation (data not shown). Doxorubicin-treated MEFs undergo p53-dependent cell cycle arrest unless otherwise sensitized to undergo apoptosis through the expression of the adenovirus E1A oncogene (Lowe et al, 1994). Notably, induction of Ptprv expression by doxorubicin was significantly suppressed in cells expressing E1A (Figure 1E). Using real-time PCR, we confirmed the more pronounced increase in Ptprv mRNA in E1A-negative cells (27±7.6-fold of induction) compared to E1A-positive cells (2.1±0.5-fold of induction). In addition, Ptprv expression was not induced in p53 WT MEFs by H2O2, which causes oxidative cell death (data not shown). These results suggest that Ptprv is preferentially upregulated by stimuli inducing p53-mediated cell cycle arrest rather than apoptosis. Finally, human Ptprv mRNA levels are also induced in the p53-null osteosarcoma SAOS-2, after expression of a conditional p53 allele (Figure 1F; Nakano and Vousden, 2001). Together, these results demonstrate that activation of p53 in normal or tumor cells leads to upregulation of Ptprv expression. The kinetic of this induction is similar to that of several known p53-inducible target genes, such as p21 (Figure 1A–C and E), suggesting that Ptprv is a direct p53 transcriptional target.
Figure 1.
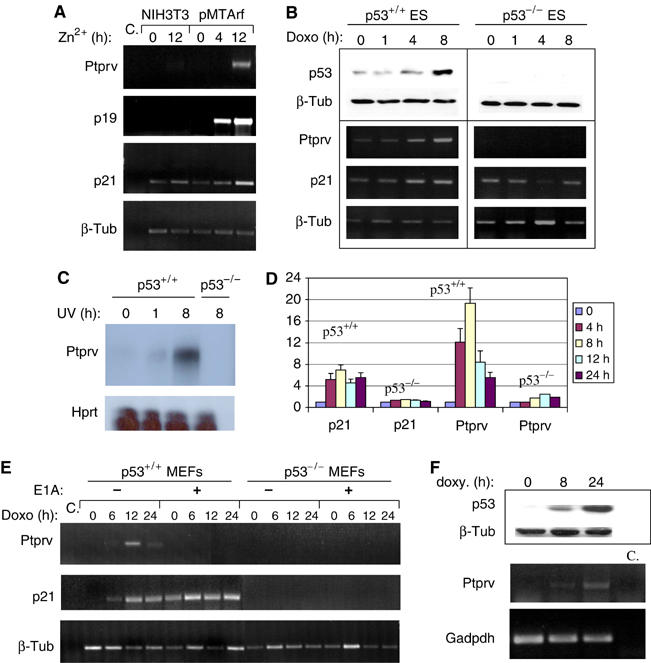
Ptprv expression is p53-inducible. (A) Semiquantitative PCR analysis shows expression of Ptprv in p19ARF-inducible pMTArf cells exposed to zinc for 4 and 12 h. Expression of p19ARF, p21, and Ptprv was detected only in the pMTArf cells, not in the NIH-3T3 parental cell line. β-Tub served here as a normalization control. (B) Western analysis (top panel) and semiquantitative PCR (lower panel) show levels of p53 protein and Ptprv mRNA, respectively, in p53 WT and p53-deficient ES cells, exposed to doxorubicin (Doxo, 0.2 μg/ml). Induction of p21 and Ptprv mRNA was concomitant with p53 protein stabilization, and was detected only in WT cells. β-Tub served here as a normalization control. (C) RNAse protection assay shows induction of Ptprv expression only in p53 WT and not in p53-deficient ES cells, exposed to UVC (30 J/m2). (D) Q-PCR analysis shows the induction of expression of Ptrpv in WT and not in p53-null MEFs following γ-IR (10 Gy). The data represent the mean (±the standard deviation) of three independent experiments. (E) Semiquantitative PCR analysis shows Ptprv expression in p53 WT and p53-deficient E1A-expressing MEFs, exposed to doxorubicin (Doxo, 0.2 μg/ml). Induction of Ptprv expression was only detected in WT E1A-negative cells and not in WT E1A-expressing or p53-deficient cells. β-Tub served here as a normalization control. (F) Western analysis (top panels) and semiquantitative PCR (lower panels) show expression of p53 protein levels and Ptprv mRNA levels, respectively, in p53-inducible SAOS-2 cells following doxycycline (Doxy) treatment.
p53 binds Ptprv promoter in vivo
Other well-characterized p53-responsive genes contain p53-binding sites either upstream of the first exon or within the first intron. Sequence analysis of the murine Ptprv locus reveals the presence of three conserved putative p53-binding sites (p53RE1-3), including perfect-match consensus sequences as defined by El-Deiry et al, 1992 (Figure 2A). To determine whether p53 binds to the candidate response elements in the mouse Ptprv, we carried out band shift assays, comparing the binding activities of double-stranded oligonucleotides containing Ptprv-derived sequences and well-characterized p53-binding sites (PG13) as control. Only one (p53RE2) of the three sites tested efficiently binds p53 after activation of its DNA-binding function with the p53 antibody PAb421 (data not shown). An oligonucleotide with mutations in p53RE2 at the positions strictly conserved in the consensus p53-binding site (positions in bold in Figure 1A) showed no p53-binding activity. Binding to p53RE2 was efficiently competed by excess of unlabelled p53RE2, but not by oligonucleotides harboring mutant p53RE2-binding sites (Figure 2B). We next tested the ability of p53 to bind the mouse p53RE2 in vivo, using chromatin immunoprecipitation (ChIP). pMTArf cells were grown for 24 h in the presence of zinc to induce p19ARF expression and subsequent p53 stabilization. Chromatin was immunoprecipitated using either control IgG- or p53-specific polyclonal antibodies (FL-393), and the Ptprv promoter regions surrounding p53RE2 or p53RE3 as a negative control, amplified by PCR (Figure 2C, left panels). Amplification of a region containing the p53-binding sites in the first intron of the Mdm2 gene served as positive control. Similar experiments were carried out in doxorubicin-treated ES cells (Figure 2C, right panels). Results reveal a specific recruitment of p53 to p53RE2, but not to p53RE3, of the promoter region of Ptprv, thereby proving that Ptprv is a direct p53-transcription target.
Figure 2.
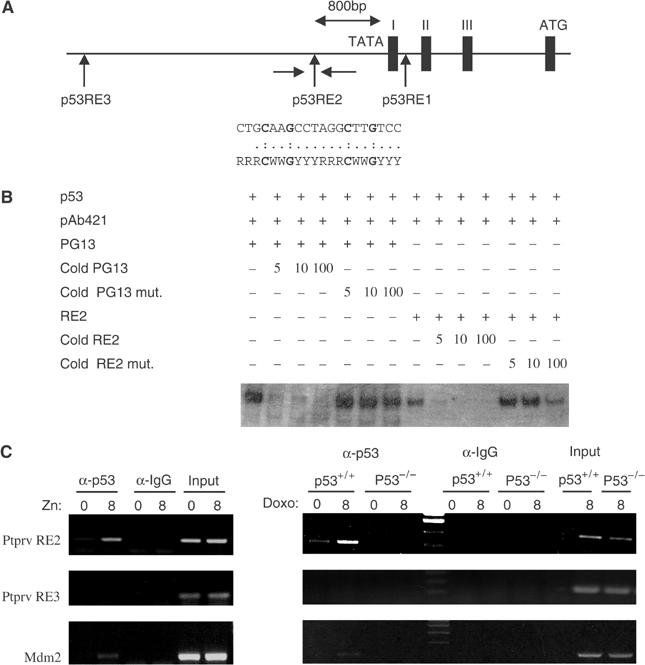
Identification of a p53-binding site in the Ptprv promoter. (A) Partial genomic structure of mouse Ptprv showing exon–intron organization at the 5′ end of the locus. Three potential p53-binding sites were identified (RE1–RE3) in this region. The complete sequence of RE2 is compared to the consensus p53-binding site and positions mutated in p53RE2 mut. indicated in bold. (B) DNA-binding activity of in vitro-translated p53 to oligonucleotides containing the consensus p53-binding site (PG13) and Ptprv RE2. DNA binding was activated using the C-terminal anti-p53 antibody pAb421. Competitive inhibition of p53 binding to RE2 was observed with unlabeled cold RE2, but not mutant p53RE2 (p53RE2 mut.), when added at five-, 10-, and 100-fold molar excess over the labeled RE2 oligonucleotide. (C) ChIP assay of p53 DNA-binding activity in zinc-inducible p19ARF pMTArf cells (left panel) and doxorubicin (Doxo)-treated WT and p53-null ES cells (right panel). A rabbit polyclonal antibody to p53 (FL-393) or control rabbit IgG was used. PCR analysis using primers described in Materials and methods is shown using input DNA (1/20 of ChIP) or DNA after ChIP.
Ptprv is a p53 transcription target in vivo
A Ptprv-null reporter mutant mouse line was generated (Dacquin et al, 2004) and used to confirm in vivo the p53-dependent induction of the Ptprv promoter in response to DNA damage. The knock-in mice express a β-galactosidase reporter under the control of resident Ptprv regulatory elements (Figure 3A). To make sure that this reporter is adequate to assay transactivation by p53, induction of lacZ expression was analyzed in Ptprv-targeted ES cells following exposure to UV-C irradiation. Like endogenous Ptprv (Figure 1D), lacZ expression under the control of Ptprv regulatory elements is detected 8 h after treatment (Figure 3B). Reporter gene expression was then evaluated in 12.5 days post-coitus (dpc) embryos after whole-body γ-irradiation of compound pregnant females. In agreement with the reported histochemical staining of Ptprv-nLacZ mice, β-galactosidase activity is detectable in the foetal gonadal ridge and in the apical ectodermal ridge of the limb buds of nontreated Ptprv heterozygotes (Figure 3C; Dacquin et al, 2004). Following irradiation, β-galactosidase activity was induced in the majority of the cells of Ptprv+/− p53+/− embryos, though to various extents (Figure 3C). The branchial arches, the maxillary areas, the limb buds, and the tail were the sites of strongest expression, whereas no staining was found in the heart. This lacZ expression profile is very similar to the pattern observed in irradiated embryos harboring the p53-dependent promoter of the mdm2 gene, fused to lacZ (Gottlieb et al, 1997). In contrast, however, lacZ staining is barely detectable in gelatin sections of the developing neuroepithelium and in the eye (data not shown), sites of marked p53-dependent apoptosis in irradiated embryos (Wubah et al, 1996). This observation is consistent with a selective upregulation of Ptprv by p53 depending on the cellular context and/or the cells fate. In order to confirm that the induction of lacZ expression is p53-dependent, the Ptprv-null reporter mutation was transferred into a p53-deficient background. Strikingly, while constitutive basal transgene expression was still present in the foetal gonadal ridge and in the apical ectodermal ridge of the limb buds of Ptprv+/− p53−/− embryos, no lacZ induction was found following γ-IR (Figure 3C). Together, these data indicate that Ptprv expression is induced in vivo following DNA damage in a strict p53-dependent manner.
Figure 3.
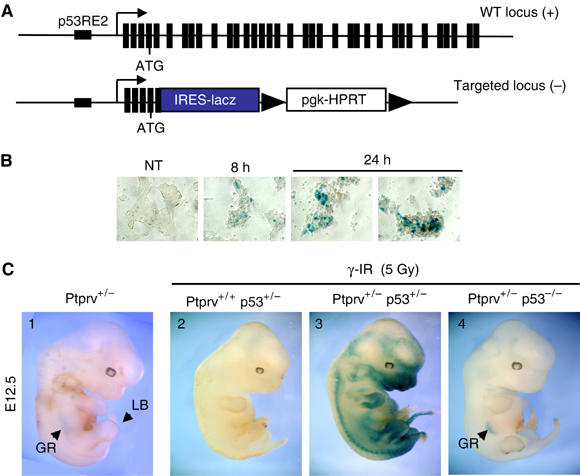
Ptprv induction in vivo. (A) Schematic representation of the WT and the targeted Ptprv loci. An internal robosome entry site-nLacZ casette was knocked in the Ptprv locus, leading to deletion of the majority of the Ptprv coding sequence (Dacquin et al, 2004). (B, C) Cells and embryos of the Ptprv-nLacZ mice were sujected to in situ and whole-mount staining, respectively, with X-gal substrate to visualize the activity of the lacZ reporter gene. (B) Ptprv+/− ES cells were left untreated (NT) or exposed to UVC, staining was performed 8 and 24 h after exposure. (C) Whole-mount lacZ staining of E12.5 embryos performed either without (embryo 1) or with prior exposure to 5 Gy of γ-IR (embryos 2–4). Staining was performed 8 H postirradiation. GR, gonadal ridge (staining as an arc in the center of the embryo); LB, limb bud.
Ptprv is not required for p53-mediated apoptosis in vivo
The poor induction of expression in cells undergoing p53-mediated cell death suggests that Ptprv is dispensable for this biological response. To confirm this view, Ptprv-deficient MEFs were infected with E1A (Lowe et al, 1994) and exposed to doxorubicin or deprived of serum, conditions known to trigger p53-dependent apoptosis. In contrast to p53-null cells, both WT and Ptprv-null MEFs were equally sensitive to apoptosis (Figure 4A). Moreover, sections through the neuroepithelium of irradiated 13.5-day-old embryos were assayed for apoptosis. Consistent with a readily detectable number of pyknotic nuclei evident by H&E staining, apoptosis was widespread in both WT and in Ptprv-deficient embryos, but, as expected, not detected in p53-null embryos (Figure 4B). We next measured apoptosis in freshly isolated thymocytes after γ-IR. Susceptibility to cell death following DNA damage in these cells is also p53-dependent (Clarke et al, 1993; Lowe et al, 1993), and, accordingly, p53−/− thymocytes were resistant to γ-IR (Figure 4C). In contrast, WT and Ptprv-deficient cells were equally sensitive to this treatment. Together, these data indicate that p53-dependent apoptosis following DNA damage does not require Ptprv.
Figure 4.

Ptprv is dispensible for p53-induced apoptosis (A) MEFs with the indicated genotypes were infected with a retrovirus expressing E1A and exposed to doxorubicin (Doxo; left panel) or serum deprived (right panel). Apoptotic cells were measured with annexin V staining and FACS analysis. The graphs represent the average percentages±s.e.m. of viable cells in three independent experiments. (B) E13.5 Embryos with the indicated genotypes were treated in utero with γ-IR (10 Gy). Sections through the neuroepithelium were assayed for apoptosis. The sections were stained with antibody directed against the activated form of caspase-3. (C) Freshly isolated mouse thymocytes were γ-irradiated (IR, 10 Gy) and apoptosis was measured 10 h later by FACS analysis. The graph represents the average percentages±s.e.m. of viable cells in two independent experiments.
Ptprv-deficient MEFs proliferate faster in culture and reach a higher cell density than WT cells
p53-deficient MEFs divide more rapidly and reach a higher saturation density than WT cells (Harvey et al, 1993). We therefore studied the growth rates and saturation densities of early-passage Ptprv-deficient cells (Figure 5A). The Ptprv-null cells initially did not show any significant differences in growth rate compared to the WT cells, but differences became apparent at day 5. The p53 and Ptprv-mutant cells grew more rapidly than WT cells. By day 9, all cell lines appeared to have reached saturation density. p53−/− and Ptprv−/− MEFs reached a much higher saturation density compared to control genotypes (Figure 5A, right panel). Moreover, flow cytometry confirmed that, at day 9, a significant percentage of p53- and Ptprv-null cells continued to synthesize DNA (Figure 5B). Thus, Ptprv loss gives MEFs a growth advantage allowing cells to bypass contact inhibition to an extent similar to p53−/− MEFs. It should be noted, however, that absence of Ptprv does not result in full immortalization of murine fibroblasts.
Figure 5.
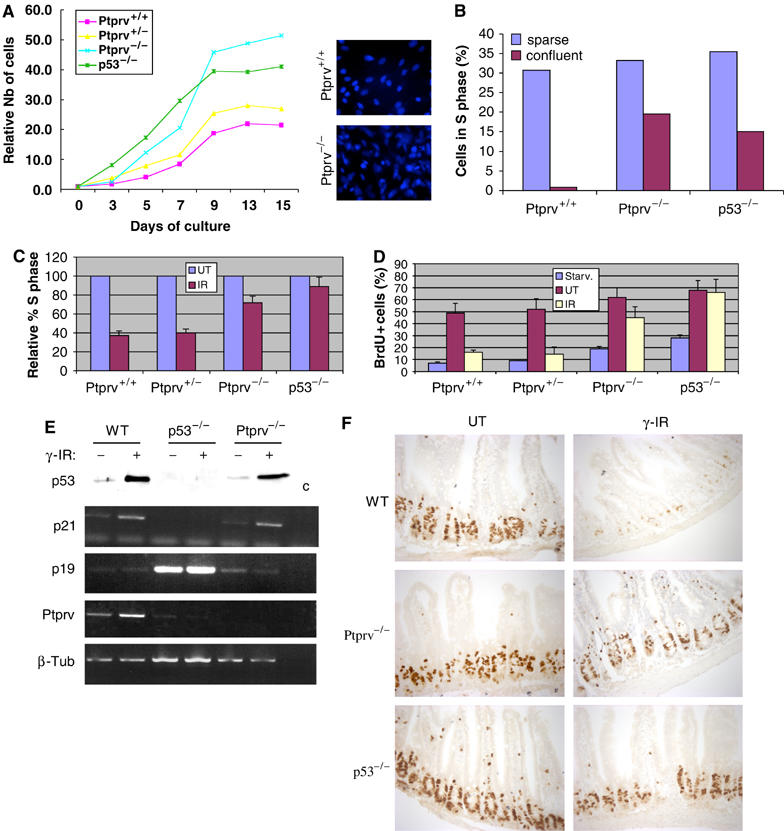
Loss of Ptprv results in defective G1 checkpoint control. (A) To measure cell proliferation, passage 2 MEFs of different genotypes were plated at day 0. Cultures were harvested at daily intervals, and the total number of cells were determined and normalized to the number of cells at day 0 (4 h after plating) (relative number of cells). The right panel shows a Hoechst staining (5 μg/ml) of WT and Ptprv-null cultures at day 9. (B) Sparse or confluent MEF cultures were labeled with BrdU and analyzed by FACS. The graph displays the relative reduction in the number of S-phase cells (BrdU-positive) compared with sparse cultures of the same genotype. FACS analysis also revealed that the WT and Ptprv-null cells have equivalent cell volumes (data not shown). (C) Effect of γ-IR (10 Gy) on the cell cycle of asynchronously growing MEFs of different genotypes. The percentage of S-phase cells is shown relative to the percentage of S-phase cells in UT cultures. Three independent experiments were performed, and the mean values with standard deviations (error bars) are presented. (D) S-phase entry following serum stimulation and γ-IR of synchronized MEFs. The percentages of BrdU-positive cells immediately after starvation (Starv.), cells grown for 24 h in growth medium (UT), or cells that had undergone γ-IR are shown. Three independent experiments were performed, and the mean values with standard deviations (error bars) are presented. (E) Western analysis (top panel) and semiquantitative PCR (lower panel) show expression of p53 protein levels and p21 mRNA levels, respectively, in p53 WT, p53-deficient, and Ptprv-null MEFs, exposed to γ-IR. Induction of p21 mRNA was concomitant with p53 protein stabilization, and was detected in WT and in Ptprv-null cells. β-Tub served here as a normalization control. (F) Cell proliferation in the epithelium of small intestine of WT, Ptprv-null and p53-null mice after γ-IR (15 Gy). Comparison of BrdU incorporation in UT mice and irradiated mice (γ-IR) 8 h after irradiation.
DNA-damage-induced G1/S arrest is partly Ptprv-dependent
WT MEFs undergo a well-characterized arrest in G1 when treated with γ-IR, while p53-null MEFs fail to do so (Kastan et al, 1992). To decipher the functional importance of Ptprv to this checkpoint, subconfluent asynchronously growing MEFs were γ-irradiated and the number of cells in S phase after 24 h evaluated. As indicated in Figure 5C, WT cells show a 60% reduction in the number of S-phase cells relative to unirradiated samples averaged over several experiments. By contrast, p53-defective cells were almost completely deficient in the ability to arrest in G1 (Figure 5C). Ptprv-null cells showed an intermediate phenotype with, on average, a 30% reduction in cells entering S phase. As interpretation of these pulse-labeling results on asynchronous cultures is complicated by the ability of the cells to arrest in G1, G2, or M, we measured more precisely the effect of irradiation on the G1- to S-phase transition using synchronized cells. MEFs were synchronized at G0 by serum starvation (0.1% serum for 96 h), and treated with 0 or 10 Gy of γ-IR. Cells were then stimulated to enter the cell cycle in the presence of BrdU by the addition of serum. Cells were harvested 24 h after irradiation, and the number of S-phase cells quantified by bivariate FACS analysis (Figure 5D). p53−/− and, to a lesser extent, Ptprv−/− cells show a defect in G1 arrest relative to WT and Ptprv heterozygous cells. As p21-null cells are also significantly deficient in their ability to arrest in G1 in response to DNA damage (Brugarolas et al, 1995; Deng et al, 1995), impairment of the G1 checkpoint in Ptprv-null cells may be due to an inability of these cells to upregulate p21 expression. However, p53 stabilization and concomitant induction of p21 transcription were observed in both WT and Ptprv-null cells following DNA damage, excluding this possibility (Figure 5E). In conclusion, these results clearly indicate that induction of Ptprv contributes to the p53-mediated G1 arrest response to γ-IR.
Cell proliferation in epithelia of the guts is limited to the crypts, in which stem cells and early progenitors are located. WT epithelia block proliferation as early as 8 h after exposure to a high dose of whole-body irradiation (15 Gy), while in p53-null mice DNA replication continues (Komarova et al, 2004). We monitored cell proliferation in the epithelium of the small intestine of Ptprv-null mice after γ-IR. As expected, virtually no BrdU incorporation was seen in the small intestine of irradiated WT animals, while proliferation was largely unaffected in the crypts of p53-null and Ptprv-deficient animals (Figure 5F). These results further underline a key role for Ptprv as a mediator of p53-induced cell cycle arrest in vivo.
Loss of Ptprv enhances papilloma formation in experimental mouse skin carcinogenesis
In WT MEFs and primary epidermal keratinocytes, constitutive signaling by oncogenic Ras provokes a p19ARF and p53-dependent cell cycle arrest (Serrano et al, 1997; Palmero et al, 1998; Lin and Lowe, 2001). As Ptprv is a p53 target, its expression is induced in WT MEFs expressing activated Ras (not shown). Mutations in endogenous Hras, leading to constitutively active Ras protein, can be achieved in vivo by treatment of the mouse skin with the carcinogen 7,12-dimethylbenzanthracene (DMBA). This treatment induces benign squamous cell papillomas, nearly 100% of which have sustained an AT mutation in codon 61 of Hras (Quintanilla et al, 1986). Interestingly, the expression levels of both p19ARF and p53 were increased in such papillomas (Kelly-Spratt et al, 2004), indicating that p19ARF regulates p53 in response to Ras in vivo. Using the Ptprv-nlacZ mice, we first determined whether Ptprv expression is induced in the skin of DMBA-treated mice. In untreated (UT) mice, lacZ staining was only evident in hair follicles and was absent from the interfollicular epidermis (Figure 6A). The hair follicle is composed of concentric epithelial layers, an external outer root sheath (ORS) attached to the basal lamina, an internal layer, the inner root sheath (IRS), and the hair shaft (Panteleyev et al, 2001). Follicles periodically undergo cycles of growth (anagen), destruction (catagen), and rest (telogen). The zone between the noncycling and cycling segments is a stem cell niche, the ORS ‘bulge' (Alonso and Fuchs, 2003). Expression of the reporter gene within the hair follicle is confined to the bulge, which contains infrequently cycling, label-retaining cells (LRCs) and related keratinocyte progeny in the basal layer (BL) and ORS, and is absent from the IRS. LacZ expression was particularly evident in resting hair follicle, at the end of the telogen (hair club) (Figure 6A). Expression of the reporter in these structures is p53-independent because it is also observed in a p53-null background (not shown). Strikingly, upon treatment of the Ptprv-null reporter mice with the tumour-initiating agent DMBA, a strong induction of expression was observed in the hyperplastic epidermis and in the majority of the epithelial tumor cells (Figure 6A). We next assessed the significance of Ptprv induction for tumor suppression. Ptprv+/+ and Ptprv−/− littermates were treated with DMBA and scored each week for papilloma and carcinoma formation. Papillomas began to appear in both genotypes 8 weeks after the first application of DMBA, indicating that the latency period for tumor appearance was the same in both groups. By 21 weeks, the average number of papillomas (more than 2 mm in diameter) was 2.53-fold significantly higher (P<0.025) in mice homozygous for a null mutation in the Ptprv gene compared with WT control mice (Figure 6B and C). There seemed to be an increased mean volume of tumors with decreasing Ptprv gene dosage (data not shown); however, due to the large-size variability among tumors, the median tumor size was not statistically significantly different across genotypes (Kruskal–Wallis test). Mortality in Ptprv-deficient mice was significantly higher (P<0.001; Logrank test) than in age-matched WT littermates (Figure 6D), and the most common cause of death in Ptprv−/− mice appeared to be lymphoma. Seven out of the eight mutant mice that died during the course of the experiment had indeed developed lymphomas, whereas only one of the two WT mice that died showed signs of lymphoma. Other nonepithelial tumors, like hemangiosarcomas, were also found in mutant mice (two of eight mice), while none were found in WT animals. Importantly, the skin tumors in both groups were exclusively benign squamous tumors, with no or very few frank carcinomas, indicating that the rate of malignant conversion of papillomas to carcinomas is not increased in the absence of Ptprv. Of note, Ptprv-null animals did not develop tumors spontaneously at an appreciable frequency in their first year of life (not shown).
Figure 6.
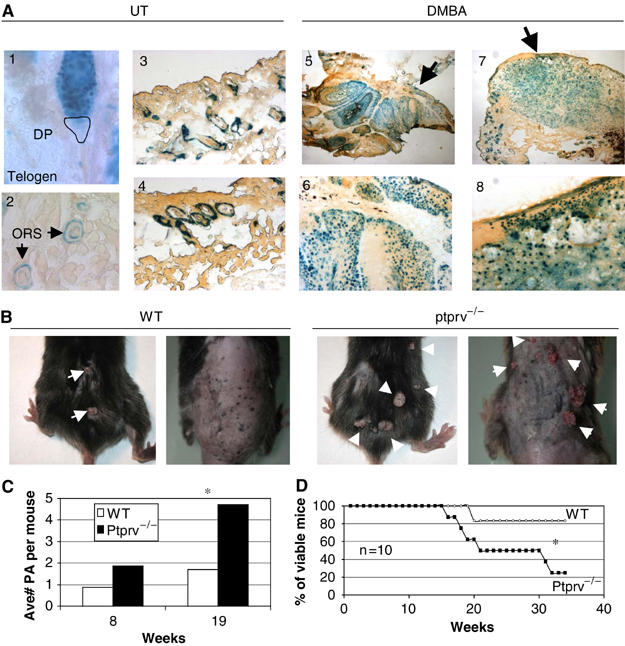
Ptprv-deficient mice display increased papilloma yield after chemical carcinogenesis. (A) Expression of Ptprv in the skin of UT or DMBA-treated Ptprv-null/reporter mice as measured by X-gal staining on cryosections. LacZ expression in UT mice is only detected in hair follicles, particularly in resting hair follicles (telogen, see 1). Within the hair follicles, expression is confined to the ORS (see 2–4), and is excluded from the dermal papilla (DP). No expression was detected in the normal epidermis. In contrast, strong staining was observed in the hyperplastic epidermis (see 6 and 8) of DMBA-treated mice. Two different lesions isolated from DMBA-treated mice are shown (see images 5 and 7, acquired with an × 2 objective). Images 6 and 8 are the same lesions showed at higher magnification (acquired with an × 10 objective). Strong expression was evident in most tumor cells of epithelial origin in both lesions (see 5 and 7). (B) Gross morphology of DMBA-induced skin tumors at 16 weeks of treatment. Two representative WT and Ptprv-null mice are shown. (C) Average number of papillomas (more than 2 mm in diameter) per mouse after 8 and 19 weeks of treatment. (D) Survival curves of DMBA-treated WT (n=10) and Ptprv-null mice (n=10) with respect to the number of weeks. In (C) and (D), asterisks denote statistical significance.
Discussion
Our data indicate that Ptprv is a transcriptional target of p53. Ptprv is induced in a p53-dependent manner following DNA damage, culture shock, ARF-dependent stabilization of p53, and expression of oncogenic Ras. Consistent with these findings, an oligonucleotide microarray-based, gene expression profile analysis had previously identified Ptprv as a putative p53-inducible gene (Kannan et al, 2001). We show here that induction of Ptprv expression is dramatic in cells undergoing p53-mediated cell cycle arrest, and thus in all in vitro and in vivo settings tested (see Figures 1A and 2Cfor examples). In these conditions, Ptprv induction was consistently more pronounced than that of p21, which is widely used as a marker of p53 activation. This is most likely due to a very low constitutive basal expression level in most tissues (Dacquin et al, 2004). We therefore propose the use of Ptprv as a sensitive marker of p53 activation, and the Ptprv-nLacZ mouse model as a new valuable tool for monitoring p53 transcriptional activity in vivo.
Notably, Ptprv is only poorly induced in response to apoptotic stimuli or when cells are sensitized to death by oncogenes, such as E1A. The molecular basis for the selective induction of the Ptprv promoter by p53 is currently unknown, but this observation makes this promoter unique and therefore worth studying further.
Consistent with its preferential induction by p53, Ptprv is dispensable for p53-mediated apoptosis but necessary for efficient p53-dependent cell cycle arrest in response to DNA damage. A role of p21 in the p53-dependent G1 checkpoint had already been clearly established (Brugarolas et al, 1995; Deng et al, 1995). The similarities between the biological consequences of p21 loss and Ptprv deficiency are remarkable. The growth properties of p21−/− and Ptprv−/− cultured MEFs are virtually identical. These cells grow faster and to higher saturation densities than control cells. They are both compromised, to similar extents, in their G1 cell cycle checkpoint following γ-IR. However, they both still enter replicative senescence, even if they do so, with a slight delay as compared to WT cells, and decrease their proliferation in response to overexpression of oncogenic Ras (Pantoja and Serrano, 1999). In addition, Ptprv- and p21-deficient mice display an increased predisposition to tumour formation after exposure to chemical carcinogens (Weinberg et al, 1999). Loss of both genes indeed enhanced the formation of benign epidermal papillomas after exposure to DMBA and, thus, even if slightly different protocols were used. While a classical DMBA/TPA two-step protocol was used for the p21-null mice, we did not expose the Ptprv-null mice to TPA. Notably, none of these mutants recapitulate the increased rate of malignant conversion of papillomas to carcinomas observed in the absence of p53 function (Kemp et al, 1993). It therefore appears that both p21 and Ptprv act as mediators of p53-induced cell cycle arrest in response to DNA damage or activated Ras. However, loss of either Ptprv or p21 does not recapitulate the phenotypes observed in cells and mice lacking p53. Our data raise the possibility that Ptprv and p21 activate nonoverlapping pathways, contributing to a full p53-dependent response. It would therefore be of interest to investigate the DNA-damage response in cells in which both Ptprv and p21 are inactivated, the ability of these cells to proliferate following Ras expression, and the incidence and malignant conversion of epidermal papillomas in Ptprv/p21 double knockout mice after carcinogens exposure.
The molecular mechanism through which increased Ptprv expression triggers a cell proliferation block remains to be elucidated. Recent experiments have shown that RPTPs can bind to themselves (homophilic) as well as to other proteins (heterophilic). Interestingly, several RPTPs, including RPTPμ, DEP-1, RPTPκ, and RPTPλ, interact with and regulate the tyrosine phosphorylation level of catenins (Fuchs et al, 1996; Cheng et al, 1997; Zondag et al, 2000; Holsinger et al, 2002), which are critical in physiological and pathological events such as cell proliferation, migration, and adhesion. DEP-1 colocalizes with the junctional protein VE-cadherin at cell borders in endothelial cells. Interestingly, members of the cadherin family of cell–cell adhesion molecules function in the suppression of cell growth and tumor invasion. Junctional components such catenins, however, can promote cell growth by inducing the transcription of genes involved in proliferation and cancer progression (reviewed in Ben-Ze'ev et al, 2000). The growth-inhibitory effects of the cadherin complex may involve binding and sequestration of the signaling pool of the catenins. Furthermore, inducible DEP-1 expression in fibroblasts antagonizes platelet-derived growth factor-stimulated tyrosine phosphorylation of a number of cellular proteins, including the PDGF receptor itself, components of the MAPK pathway (Erk1/2, p21Ras), Src, and activation of the receptor-associated phosphoinositide-3 kinase (Kellie et al, 2003). Thus, the implication of Ptprv in the regulation of the cadherin-catenin and MAPK pathways should therefore be investigated.
Several lines of evidence, albeit preliminary, support the role of RPTPs in tumor suppression (see introduction). The data presented herein provide yet another link between RPTPs and cancer. The identification of Ptprv as a key downstream p53 target suggests that it might be required for tumor suppression by p53. In agreement, we show here that, even if Ptprv-null animals did not develop tumors spontaneously at an appreciable frequency in their first year of life, these mice show a significant acceleration of skin tumour formation compared to WT littermates when treated with carcinogens (DMBA). Since, this treatment induces Hras mutations at high frequency (Quintanilla et al, 1986), it is tempting to speculate that mutated Ras can cooperate with Ptprv loss to accelerate tumor formation in vivo. Intriguingly, most of the lethal tumors found in DMBA-treated Ptrpv-null mice were aggressive lymphomas that infiltrated thymus, lymph nodes, and vital organs such as liver and lungs. In contrast, DMBA-treated ARF-null or p53-null animals primarily develop invasive skin carcinomas (Kemp et al, 1993). Notably, mice deficient for DMP1, an ARF transcriptional activator, develop tumors with a spectrum very similar to the one observed in mice lacking Ptprv when treated with DMBA (Inoue et al, 2000). DMP1 was originally isolated from a T-lymphocyte cDNA library and DMP1-null T-cells in long-term culture have a proliferative advantage when stimulated by IL-2 and antibody to CD3. By analogy, it will be interesting to assess the relevance of Ptprv for the development and function of cells of lymphoid origin.
Materials and methods
Cell culture and cell cycle analysis
MEFs, Saos-2 (p53-inducible), NIH-3T3, or NIH-3T3-derived pMTarf and phoenix packaging cell lines were prepared and/or grown as described (Martoriati et al, 2005). p53 activity was induced as follows: NIH-3T3 or pMTarf cells were treated with ZnSO4 (100 μM); Saos-2 p53-inducible cells were treated with doxycycline (2 μg/ml); ES cells were treated with doxorubicin (0.2 μg/ml); MEFs were treated with doxorubicin (0.2 μg/ml), exposed to UV (30 J/m2), H2O2 (50 μM), and γ-IR (10 Gy). BrdU staining on coverslips and flow cytometry analysis were as previously described (Migliorini et al, 2002).
Retroviral infection and apoptosis assays
pBABE(Puro) and pBABE(Puro)-E1A 12S were transfected into phoenix packaging cells and the supernatants used to infect MEFs as previously described (Danovi et al, 2004). After 2–4 days of selection, MEFs were replated for apoptosis assays. We treated MEFs with 0.2 μg/ml doxorubicin (Sigma) or 0.1% fetal serum for 24 and 48 h. Cells were trypsinized and pooled with floating cells and apoptosis measured using annexin-V detection kit (Roche Applied Science #1858777) and FACS analysis or a Trypan blue exclusion assay. We treated pregnant females at 13.5 dpc with 10 Gy γ-IR, and 8 h later collected the embryos. Embryonic heads were fixed, paraffin-embedded, and apoptosis assayed using an antibody recognizing the activated form of caspase 3 (Cell Signaling), as previously described (Migliorini et al, 2002). Freshly isolated thymocytes from 4-weeks-old mice were treated with γ-IR and apoptosis assayed by annexin V staining 16 h later.
Semiquantitative RT–PCR expression analysis
Total RNA was prepared using RNeasy miniKit (QIAGEN) according to the manufacturer's protocol. In all, 1 μg of total RNA was reverse-transcribed in a final volume of 20 μl using a SuperScript kit (Invitrogen). PCR primers were as follows: p19ARF: sense 5′-AGGGATCCTTGGTCACTGTGAGGATTC-3′; antisense: 5′-GCAAAGCTTGAGGCCGGATTTAGCTCTGCT-3′; p21: sense 5′-GGAGCAAAGTGTGCCGTTGTC-3′; antisense 5′-GAGGAAGTACTGGGCCTCTTG-3′; Ptprv: sense 5′-GTTGATGCCTTACAACCTGTGGCG-3′; antisense 5′-AGCTGCTTCACGCGCCTCTGTT-3′; β-tubulin sense: 5′-CAACGTCAAGACGGCCGTGTG-3′, antisense: 5′-GACAGAGGCAAACTGAGCACC-3′.
TaqMan real-time quantitative RT–PCR assays
These assays were performed following the manufacturer's specifications (PE Applied Biosystems). Primer pairs and TaqMan probes were designed by Applied Biosystems (Assays on demand). Each sample was analyzed in triplicate.
Electromobility shift assay (EMSA)
Oligonucleotides encoding the p53-binding sites (p53RE2) located in Ptprv (5′-CTGCAAGCCTAGGCTTGTCC-3′), and the consensus p53-binding site (PG13: 5′-GGGCAAGCCCGGGCAAGCCC-3′) were 32P end-labeled with T4 kinase (10 U/μl, Invitrogen) and hybridized with the complementary oligonucleotide. Unlabeled p53RE2 and mutant p53RE2 (5′-CTGAAAACCTAGGATTATCC-3′) probes were used as specific competitor and unspecific competitor, respectively. Probes were incubated with an in vitro transcribed and translated p53 protein (Promega) and the mouse antibody pAb421 (Oncogene). The DNA–protein complexes were separated from the nonbound probes by electrophoresis.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were carried out as described (Martoriati et al, 2005). Ptprv p53RE2-, p53RE3-, and mdm2-specific products were amplified using primers: 5′-CTGGGATGCTCTGGGTGGATA-3′, 5′-TCCGCCTTTGTGCCATGGCTT-3′; 5′-AAGACGAGTGTCTCAAGCATTG-3′, 5′-GTGTGCAGAGGATGGAATGG-3′, and 5′-GGTGCCTGGTCCCGGACTCGCCGGG-3′, 5′-CCGAGAGGGTCCCCCAGGGGTGTCC-3′, respectively.
Western blot analysis
Western analyses were performed as described (Martoriati et al, 2005). The primary antibodies used were: polyclonal anti-p53 (FL-393, Santa Cruz) and monoclonal anti-β-tubulin (Sigma).
RNAse protection assays
32P-labeled antisense probe for Ptprv was generated using T7 and by a BamHI-linearized pcR2.1-Ptprv plasmid (Invitrogen) containing a 575 bp fragment of mouse Ptprv ORF. A 249 bp HPRT probe, obtained using T7 and BamHI-linearized pCR2.1-HPRT containing a 249 bp fragment of mouse HPRT ORF, was used as loading control.
β-galactosidase assays
E12.5 embryos were isolated 8 h after exposure to γ-IR (10 Gy). Embryos, frozen sections or PBS-washed cultured cells on plates were fixed for 10 min in a 0.2% gluteraldehyde solution, rinsed in a 0.02% NP40 solution and stained overnight at 37°C in a X-gal staining solution (1 mg/ml X-gal, 5 mM K3Fe(CN)6, and 5 mM K4Fe(CN)6).
In situ quantitation of BrdU-positive cells in the small intestine
BrdU (50 mg/kg) was injected intraperitoneally 6 h after irradiation (15 Gy). At 2 h after injection, mice were killed and a part of small intestine (ileum) was fixed for 15 h in paraformaldehyde 4% and paraffin embedded. Sections were prepared, permeabilized, and DNA was denatured at 37°C for 1 h in HCl 2 N. An anti-BrdU rat monoclonal antibody was used as primary antibody (1 h incubation, diluted 1/60) and an anti-rat polyclonal antibody, HRP-conjugated, was used as secondary antibody (10 m incubation, 1/250).
Tumor induction protocol
Skin papillomas were induced as previously described (Capozza et al, 2003). The backs of 10 WT and Ptprv−/− mice were shaved and treated weekly (for 10 weeks) with a solution of 0.5% DMBA (Fluka) in acetone. The number and the size of tumors were evaluated each week. Statistical significance for the difference in the number of tumors across genotypes was determined using Student's t-test, and P<0.05 was defined as significant. When moribund, the mice were killed and a complete necropsy was performed. Skin tumors, lymph nodes, lungs, liver, thymus, spleen, and sternum were collected, fixed in 4% neutral buffered formalin, and further processed for histopathological evaluation.
Supplementary Material
Supplementary Figure 1
Acknowledgments
We thank Ines Bonk, Dieter Defever, and Ludivine Wacheul for excellent technical help. We thank Austin Smith and Christian Dani for providing us with the Ptprv-nlacZ mice, Simone Minardi, Loris Bernard, and Laura Tizzoni for technical help with the Affymetrix and Q-PCR technologies. We also thank M Rouselle and C Sherr (St Jude Children's Research Hospital, Memphis, TN, USA) for the pMTArf cell line, R Jaenisch (Whitehead Institute for Biomedical research, Cambridge, USA) for the p53-deficient ES cells, and K Vousden for the SAOS-2-p53 cell line. We thank Aart Jochemsen, Geert Berx, and Jody Haigh for helpful discussions and comments on the manuscript. A Martoriati, G Doumont, and P Mee were supported by grants from the FNRS, Télévie, and BBSRC, respectively. This work was supported in part by grants from FB Insurance, AICR, Belgium Federation against cancer (nonprofit organization), and by EC FP6 funding. This publication reflects only our views. The commission is not liable for any use that may be made of the information herein.
References
- Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T (2004) Protein tyrosine phosphatases in the human genome. Cell 117: 699–711 [DOI] [PubMed] [Google Scholar]
- Alonso L, Fuchs E (2003) Stem cells of the skin epithelium. Proc Natl Acad Sci USA 100 (Suppl 1): 11830–11835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ze'ev A, Shtutman M, Zhurinsky J (2000) The integration of cell adhesion with gene expression: the role of beta-catenin. Exp Cell Res 261: 75–82 [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ (1995) Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 377: 552–557 [DOI] [PubMed] [Google Scholar]
- Capozza F, Williams TM, Schubert W, McClain S, Bouzahzah B, Sotgia F, Lisanti MP (2003) Absence of caveolin-1 sensitizes mouse skin to carcinogen-induced epidermal hyperplasia and tumor formation. Am J Pathol 162: 2029–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Wu K, Armanini M, O'Rourke N, Dowbenko D, Lasky LA (1997) A novel protein-tyrosine phosphatase related to the homotypically adhering kappa and mu receptors. J Biol Chem 272: 7264–7277 [DOI] [PubMed] [Google Scholar]
- Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH (1993) Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 362: 849–852 [DOI] [PubMed] [Google Scholar]
- Dacquin R, Mee PJ, Kawaguchi J, Olmsted-Davis EA, Gallagher JA, Nichols J, Lee K, Karsenty G, Smith A (2004) Knock-in of nuclear localised beta-galactosidase reveals that the tyrosine phosphatase Ptprv is specifically expressed in cells of the bone collar. Dev Dyn 229: 826–834 [DOI] [PubMed] [Google Scholar]
- Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P, Gobbi A, Helin K, Pelicci P-G, Jochemsen A, Marine J-C (2004) Amplification of Mdmx (Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol 24: 5835–5843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P (1995) Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82: 675–684 [DOI] [PubMed] [Google Scholar]
- El-Deiry WS (1998) Regulation of p53 downstream genes. Semin Cancer Biol 8: 345–357 [DOI] [PubMed] [Google Scholar]
- El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B (1992) Definition of a consensus binding site for p53. Nat Genet 1: 45–49 [DOI] [PubMed] [Google Scholar]
- El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825 [DOI] [PubMed] [Google Scholar]
- Fuchs M, Muller T, Lerch MM, Ullrich A (1996) Association of human protein-tyrosine phosphatase kappa with members of the armadillo family. J Biol Chem 271: 16712–16719 [DOI] [PubMed] [Google Scholar]
- Gottlieb E, Haffner R, King A, Asher G, Gruss P, Lonai P, Oren M (1997) Transgenic mouse model for studying the transcriptional activity of the p53 protein: age- and tissue-dependent changes in radiation-induced activation during embryogenesis. EMBO J 16: 1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey M, Sands AT, Weiss RS, Hegi ME, Wiseman RW, Pantazis P, Giovanella BC, Tainsky MA, Bradley A, Donehower LA (1993) In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene 8: 2457–2467 [PubMed] [Google Scholar]
- Holsinger LJ, Ward K, Duffield B, Zachwieja J, Jallal B (2002) The transmembrane receptor protein tyrosine phosphatase DEP1 interacts with p120(ctn). Oncogene 21: 7067–7076 [DOI] [PubMed] [Google Scholar]
- Inoue K, Wen R, Rehg JE, Adachi M, Cleveland JL, Roussel MF, Sherr CJ (2000) Disruption of the ARF transcriptional activator DMP1 facilitates cell immortalization, Ras transformation, and tumorigenesis. Genes Dev 14: 1797–1809 [PMC free article] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4: 321–328 [DOI] [PubMed] [Google Scholar]
- Kannan K, Kaminski N, Rechavi G, Jakob-Hirsch J, Amariglio N, Givol D (2001) DNA microarray analysis of genes involved in p53 mediated apoptosis: activation of Apaf-1. Oncogene 20: 3449–3455 [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ Jr (1992) A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71: 587–597 [DOI] [PubMed] [Google Scholar]
- Keane MM, Lowrey GA, Ettenberg SA, Dayton MA, Lipkowitz S (1996) The protein tyrosine phosphatase DEP-1 is induced during differentiation and inhibits growth of breast cancer cells. Cancer Res 56: 4236–4243 [PubMed] [Google Scholar]
- Kellie S, Craggs G, Bird IN, Jones GE (2003) The tyrosine phosphatase DEP-1 induces cytoskeletal rearrangements, aberrant cell–substratum interactions and a reduction in cell proliferation. J Cell Sci 117: 609–618 [DOI] [PubMed] [Google Scholar]
- Kelly-Spratt KS, Gurley KE, Yasui Y, Kemp CJ (2004) p19Arf suppresses growth, progression, and metastasis of Hras-driven carcinomas through p53-dependent and -independent pathways. PLoS Biol 2: 1138–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp CJ, Donehower LA, Bradley A, Balmain A (1993) Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 74: 813–822 [DOI] [PubMed] [Google Scholar]
- Komarova EA, Kondratov RV, Wang K, Christov K, Golovkina TV, Goldblum JR, Gudkov AV (2004) Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene 23: 3265–3271 [DOI] [PubMed] [Google Scholar]
- Kuo ML, Duncavage EJ, Mathew R, den Besten W, Pei D, Naeve D, Yamamoto T, Cheng C, Sherr CJ, Roussel MF (2003) Arf induces p53-dependent and -independent antiproliferative genes. Cancer Res 63: 1046–1053 [PubMed] [Google Scholar]
- Lane DP (1992) Cancer. p53, guardian of the genome. Nature 358: 15–16 [DOI] [PubMed] [Google Scholar]
- Lee K, Nichols J, Smith A (1996) Identification of a developmentally regulated protein tyrosine phosphatase in embryonic stem cells that is a marker of pluripotential epiblast and early mesoderm. Mech Dev 59: 153–164 [DOI] [PubMed] [Google Scholar]
- Lin AW, Lowe SW (2001) Oncogenic ras activates the ARF-p53 pathway to suppress epithelial cell transformation. Proc Natl Acad Sci USA 98: 5025–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Jacks T, Houstman DE, Ruley HE (1994) Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proc Natl Acad Sci USA 91: 2026–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T (1993) p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 362: 847–849 [DOI] [PubMed] [Google Scholar]
- Martoriati A, Doumont G, Alcalay M, Bellefroid E, Pelicci PG, Marine JC (2005) dapk1, encoding an activator of a p19ARF-p53 mediated apoptotic checkpoint, is a transcription target of p53. Oncogene 24: 1461–1466 [DOI] [PubMed] [Google Scholar]
- Migliorini D, Denchi EL, Danovi D, Jochemsen A, Capillo M, Gobbi A, Helin K, Pelicci PG, Marine JC (2002) Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol 22: 5527–5538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683–694 [DOI] [PubMed] [Google Scholar]
- Neel BG, Tonks NK (1997) Protein tyrosine phosphatases in signal transduction. Curr Opin Cell Biol 9: 193–204 [DOI] [PubMed] [Google Scholar]
- Palmero I, Pantoja C, Serrano M (1998) p19ARF links the tumour suppressor p53 to Ras. Nature 395: 125–126 [DOI] [PubMed] [Google Scholar]
- Panteleyev AA, Jahoda CA, Christiano AM (2001) Hair follicle predetermination. J Cell Sci 114 (Part 19): 3419–3431 [DOI] [PubMed] [Google Scholar]
- Pantoja C, Serrano M (1999) Murine fibroblasts lacking p21 undergo senescence and are resistant to transformation by oncogenic Ras. Oncogene 18: 4974–4982 [DOI] [PubMed] [Google Scholar]
- Quintanilla M, Brown K, Ramsden M, Balmain A (1986) Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 322: 78–80 [DOI] [PubMed] [Google Scholar]
- Ruivenkamp CA, van Wezel T, Zanon C, Stassen AP, Vlcek C, Csikos T, Klous AM, Tripodis N, Perrakis A, Boerrigter L, Groot PC, Lindeman J, Mooi WJ, Meijjer GA, Scholten G, Dauwerse H, Paces V, van Zandwijk N, van Ommen GJ, Demant P (2002) Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat Genet 31: 295–300 [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602 [DOI] [PubMed] [Google Scholar]
- Takahashi T, Takahashi K, St John PL, Fleming PA, Tomemori T, Watanabe T, Abrahamson DR, Drake CJ, Shirasawa T, Daniel TO (2003) A mutant receptor tyrosine phosphatase, CD148, causes defects in vascular development. Mol Cell Biol 23: 1817–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapasso F, Iuliano R, Boccia A, Stella A, Visconti R, Bruni P, Baldassarre G, Santoro M, Viglietto G, Fusco A (2000) Rat protein tyrosine phosphatase eta suppresses the neoplastic phenotype of retrovirally transformed thyroid cells through the stabilization of p27(Kip1). Mol Cell Biol 20: 9236–9246 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302: 1036–1038 [DOI] [PubMed] [Google Scholar]
- Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, van der Heijden MS, Parmigiani G, Yan H, Wang TL, Riggins G, Powell SM, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE (2004) Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 304: 1164–1166 [DOI] [PubMed] [Google Scholar]
- Weinberg WC, Fernandez-Salas E, Morgan DL, Shalizi A, Mirosh E, Stanulis E, Deng C, Hennings H, Yuspa SH (1999) Genetic deletion of p21WAF1 enhances papilloma formation but not malignant conversion in experimental mouse skin carcinogenesis. Cancer Res 59: 2050–2054 [PubMed] [Google Scholar]
- Wubah JA, Ibrahim MM, Gao X, Nguyen D, Pisano MM, Knudsen TB (1996) Teratogen-induced eye defects mediated by p53-dependent apoptosis. Curr Biol 6: 60–69 [DOI] [PubMed] [Google Scholar]
- Zondag GC, Reynolds AB, Moolenaar WH (2000) Receptor protein-tyrosine phosphatase RPTPmu binds to and dephosphorylates the catenin p120(ctn). J Biol Chem 275: 11264–11269 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1