

Abstract
Multiple sclerosis is mediated by T-cell responses to central nervous system antigens such as myelin basic protein (MBP). To investigate self-peptide/major histocompatibility complex (MHC) recognition and T-cell receptor (TCR) degeneracy, we determined the crystal structure, at 2.8 Å resolution, of an autoimmune TCR (3A6) bound to an MBP self-peptide and the multiple sclerosis-associated MHC class II molecule, human leukocyte antigen (HLA)-DR2a. The complex reveals that 3A6 primarily recognizes the N-terminal portion of MBP, in contrast with antimicrobial and alloreactive TCRs, which focus on the peptide center. Moreover, this binding mode, which may be frequent among autoimmune TCRs, is compatible with a wide range of orientation angles of TCR to peptide/MHC. The interface is characterized by a scarcity of hydrogen bonds between TCR and peptide, and TCR-induced conformational changes in MBP/HLA-DR2a, which likely explain the low observed affinity. Degeneracy of 3A6, manifested by recognition of superagonist peptides bearing substitutions at nearly all TCR-contacting positions, results from the few specific interactions between 3A6 and MBP, allowing optimization of interface complementarity through variations in the peptide.
Keywords: autoimmunity, complex, MHC class II, multiple sclerosis, T-cell receptor
Introduction
Multiple sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system (CNS) believed to be mediated by autoreactive T cells (Steinman, 1996; Sospedra and Martin, 2005). T-cell responses to CNS antigens, such as myelin basic protein (MBP), myelin oligodendrocyte glycoprotein and proteolipid protein, are considered a primary autoimmune event in MS, both during induction and perpetuation of the disease. Similar to other polygenic chronic inflammatory diseases, susceptibility to MS is associated with certain major histocompatibility complex (MHC) class II haplotypes, in particular human leukocyte antigen (HLA)-DR2 (Ebers et al, 1996; Oksenberg et al, 1996). Two HLA-DR β-chains, DRB1*1501 and DR5*0101, are coexpressed in the HLA-DR2 haplotype (Fogdell et al, 1995). Both pair with the nonpolymorphic HLA-DR α-chain (DRA*0101), resulting in formation of two functional heterodimers, HLA-DR2a (DRA*0101, DRB5*0101) and HLA-DR2b (DRA*0101, DRB1*1501). These two alleles, which exist in strong linkage disequilibrium (Fogdell et al, 1995), are thought to act in concert in conferring susceptibility to MS (Lang et al, 2002).
In humans, an immunodominant T-cell epitope has been identified in the middle portion of MBP, corresponding to residues 84–102 (Steinman, 1996; Sospedra and Martin, 2005). The encephalitogenic potential of MBP 84–102 was shown in a clinical trial with an altered peptide ligand, in which several patients developed exacerbations of disease (Bielekova et al, 2000). Moreover, MBP 84–102 can be presented by both DR2a and DR2b to T-cell clones from MS patients (Wucherpfennig et al, 1993; Vergelli et al, 1997; Sospedra and Martin, 2005). We previously determined the crystal structure of HLA-DR2a in complex with MBP 84–102 (Li et al, 2000), revealing that the peptide is bound in a strikingly different conformation by DR2a compared to DR2b (Smith et al, 1998), such that the peptide register is shifted by three residues. This change in alignment causes the two alleles to display completely different MBP residues for interaction with T-cell receptors (TCRs), which explains why T-cell lines specific for MBP 84–102 presented by DR2a fail to recognize this peptide in the context of DR2b, and vice versa (Wucherpfennig et al, 1993; Vergelli et al, 1997). However, understanding the basis for T-cell recognition of self-peptide/MHC ligands requires direct information on the structure of complexes with autoimmune TCRs.
To this end, we have studied the human CD4+ T-cell clone 3A6, which recognizes the immunodominant MBP 84–102 epitope in the context of HLA-DR2a (Vergelli et al, 1997; Hemmer et al, 2000). This autoreactive clone was isolated from a DR2-positive MS patient with a relapsing–remitting disease course. Its antigen specificity has been characterized extensively using peptide analogs and combinatorial peptide libraries, which reveal degenerate recognition (Hemmer et al, 1998, 2000; Wilson et al, 1999; Zhao et al, 2001). We have recently expressed the 3A6 TCR and DR2a molecules in transgenic mice (Quandt et al, in preparation), and these mice develop typical symptoms of experimental autoimmune encephalomyelitis (EAE), an animal disease sharing similarities with MS (Steinman, 1996; Sospedra and Martin, 2005). The resulting CNS autoimmunity attributed to specific TCR recognition of myelin presented by human MHC class II reproduces, in vivo, the peptide/MHC specificity of the T-cell clone from which 3A6 was derived and demonstrates the pathogenic potential of this autoimmune TCR.
To date, structural studies of TCR/peptide/MHC complexes (Rudolph et al, 2002) have been largely restricted to TCRs specific for microbial and other foreign epitopes (Garboczi et al, 1996; Reinherz et al, 1999; Hennecke et al, 2000; Kjer-Nielsen et al, 2003; Stewart et al, 2003), or displaying alloreactivity (Garcia et al, 1998; Reiser et al, 2000). The only exceptions are the mouse autoimmune TCR 172.10, which recognizes MBP 1–11, an encephalitogenic epitope in rodents, presented by I-Au (Maynard et al, 2005) and the human TCR Ob.1A12, which recognizes MBP 85–99 in the context of HLA-DR2b (Hahn et al, 2005). A crucial question raised by these very recent studies is whether autoimmune TCRs generally engage peptide/MHC in the same manner as antimicrobial TCRs, or whether fundamental differences in binding mode exist that may allow autoreactive T cells to escape negative selection (Wilson and Stanfield, 2005). To address this issue, and to gain insights into the basis for TCR degeneracy (Kersh and Allen, 1996; Holler and Kranz, 2004; Wucherpfennig, 2004) and molecular mimicry (Wucherpfennig and Strominger, 1995; Hemmer et al, 1997; Lang et al, 2002), a major model for the development of autoimmune disease, we examined the binding of TCR 3A6 to HLA-DR2a bearing the MBP 89–101 self-peptide, and determined the crystal structure of this autoimmune TCR in complex with MBP/HLA-DR2a.
Results
Interaction of TCR 3A6 with MBP and a superagonist peptide
We used surface plasmon resonance (SPR) to demonstrate specific binding of TCR 3A6 (Vα22/Vβ5.1) to HLA-DR2a bearing MBP 89–101. To permit directional coupling to streptavidin-coated biosensor surfaces, peptide tags containing biotinylation sequences were added to the C-termini of the α-chains of both TCR 3A6 and HLA-DR2a (see Materials and methods). However, no apparent binding could be detected upon injection of relatively high (0.1–100 μM) concentrations of soluble TCR over immobilized peptide/MHC, or by injecting soluble peptide/MHC over immobilized TCR. Indeed, weak affinity for self-ligands is expected for TCRs expressed by autoreactive T cells, like 3A6, that have escaped negative selection (Kappler et al, 1987; Alam et al, 1996; Davis et al, 1998; Stefanova et al, 2002; van der Merwe and Davis, 2003). To increase the avidity of the interaction with MBP/HLA-DR2a, we generated TCR 3A6 tetramers by mixing the biotinylated receptor with streptavidin. In this case, specific binding was readily detected by flowing TCR tetramers over a surface immobilized with peptide/MHC (Figure 1A). No binding was observed when MBP 89–101 was replaced by an irrelevant peptide (Figure 1B). By contrast, 3A6 tetramers bound much tighter to HLA-DR2a bearing a superagonist peptide (K38-27) than to MBP/HLA-DR2a, as indicated by the marked reduction in dissociation rate (Figure 1C). The superagonist, which differs from MBP at all nonanchor positions except P2, was identified previously using synthetic combinatorial peptide libraries (Hemmer et al, 2000). K38-27 stimulates 3A6T cells ∼10 000-fold more efficiently than the MBP self-peptide, consistent with the stronger interaction seen by SPR.
Figure 1.
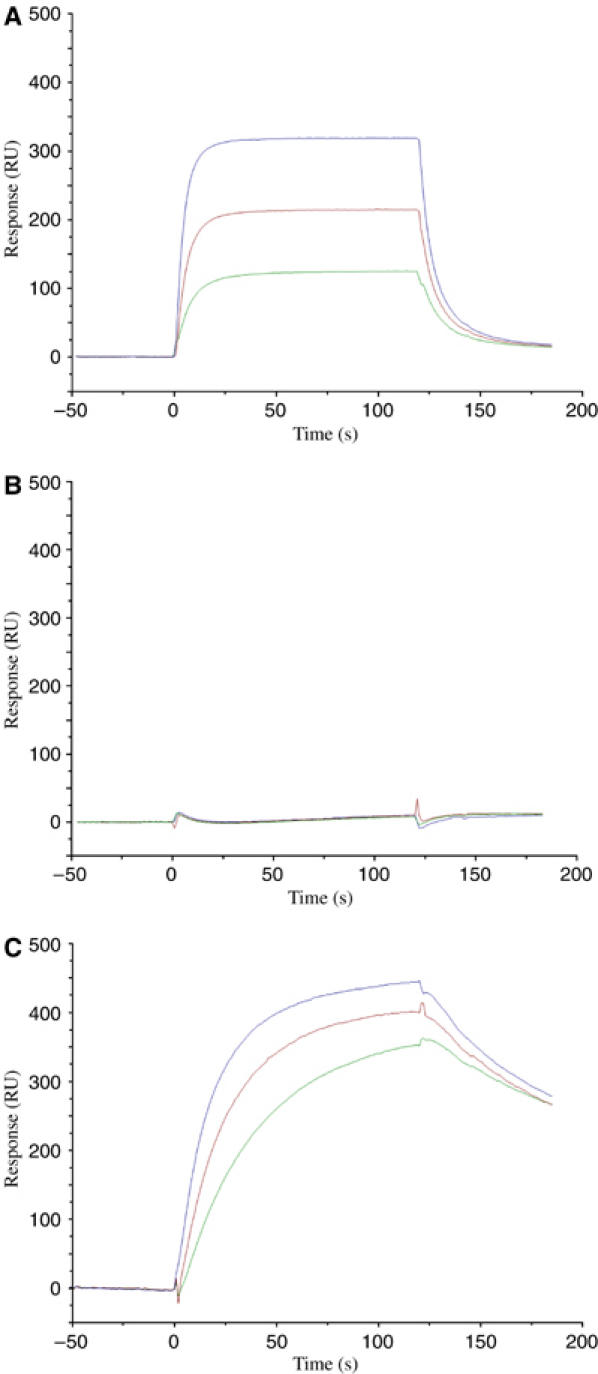
Binding of TCR 3A6 tetramers to MBP/DR2a and K38-27/DR2a complexes. (A) Sensograms showing the binding of 3A6 tetramers at concentrations of 2.3 μM (blue), 1.2 μM (red) and 0.6 μM (green) to HLA-DR2a loaded with MBP 89–101 (VHFFKNIVTPRTP). HLA-DR2a was immobilized on a BIAcore SA chip by injecting 5 μl of biotin-HLA-DR2a (0.1 μM) over the chip. (B) Binding of 3A6 tetramers to HLA-DR2a loaded with an irrelevant peptide. (C) Binding of 3A6 tetramers to HLA-DR2a loaded with K38-27 (WFKLTTTKL), a superagonist peptide (Hemmer et al, 2000).
Although the multivalency of TCR tetramers precludes determination of binding constants (Kd), it is apparent that 3A6 binds MBP/HLA-DR2a with low affinity, probably with a Kd at the upper end of the range for TCR–peptide/MHC interactions (0.1–500 μM) (Davis et al, 1998; van der Merwe and Davis, 2003). Nevertheless, the 3A6T cell clone is activated by low concentrations of MBP peptide (EC50∼30 nM) (Wilson et al, 1999; Hemmer et al, 2000). Reduced affinity may have allowed this autoreactive T cell to escape thymic deletion, without compromising its ability to induce CNS inflammation in 3A6/DR2a transgenic mice (Quandt et al, in preparation).
Overview of the complex structure
Initially, we attempted cocrystallizing TCR 3A6 and MBP/HLA-DR2a as individual molecules. However, no complex crystals were obtained, probably because of the weak interaction between them. To stabilize the 3A6 TCR/MBP/HLA-DR2a complex for crystallization, we covalently attached the MBP peptide to the N-terminus of the TCR Vβ domain via an octapeptide linker, as described for the influenza hemagglutinin (HA)-specific HA1.7 TCR (Hennecke et al, 2000). The fused MBP peptide was then loaded into the binding groove of empty HLA-DR2a molecules using the peptide exchange catalyst HLA-DM to generate a long-lived 3A6 TCR/MBP/HLA-DR2a complex that crystallized readily. The structure was determined by molecular replacement to a resolution of 2.8 Å (Table I). The interface between TCR and peptide/MHC is in unambiguous electron density for each of the four complex molecules in the asymmetric unit of the crystal (Figure 2). Several loops in the MHC β2 and TCR constant domains (mainly Cα) lack electron density; however, these disordered regions are distant from the interface. As also noted for the HA1.7/HA/HLA-DR1 complex (Hennecke et al, 2000), no density could be detected for the octapeptide linker, indicating flexibility. The root-mean-square (r.m.s.) deviation in α-carbon positions for the four complexes in the asymmetric unit ranges between 0.53 and 0.87 Å. For the TCR VαVβ and MHC α1β1 modules alone, including the MBP peptide, the r.m.s. deviation in α-carbon positions varies from 0.28 to 0.51 Å. Based on this close similarity, the following description of TCR–MHC interactions applies to all four complex molecules, unless stated otherwise.
Table 1.
Crystallographic data statistics
| Data collection | |
| Space group | P1 |
| Unit cell | a=96.48 Å, b=97.69 Å, c=124.02 Å, α=74.39°, β=83.19°, γ=61.55° |
| Asymmetric unit | 4 3A6/MBP/HLA-DR2a complexes |
| Resolution limit (Å) | 2.80 |
| Observations | 177 348 |
| Unique reflections | 81 551 |
| Completeness (%)a | 86.3 (86.5) |
| Mean I/σ(I)a | 5.9 (2.5) |
| Rmerge (%)a,b | 8.0 (28.4) |
| Refinement | |
| Resolution range (Å) | 30–2.80 |
| Rwork (%)c | 27.51 |
| Rfree (%)c | 32.55 |
| Non-hydrogen protein atoms | 23 495 |
| Water molecules | 148 |
| R.m.s. deviations from ideal | |
| Bonds (Å) | 0.009 |
| Angles (deg) |
1.7 |
| aValues in parentheses correspond to the highest resolution shell (2.80–2.92). | |
| bRmerge(I)=(∑∣I(i)−〈I(h)〉∣/∑I(i)), where I(i) is the ith observation of the intensity of the hkl reflection and 〈I〉 is the mean intensity from multiple measurements of the hkl reflection. | |
| cRwork (Rfree)= ∑∥Fo∣−∣Fc∥/∑∣Fo∣; 2% of data were used for Rfree. | |
Figure 2.
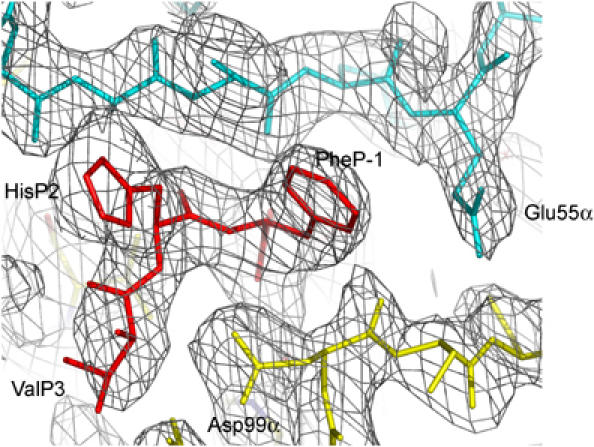
Electron density in the interface of the 3A6/MBP/HLA-DR2a complex. Density from the final 2Fo−Fc map at 2.8 Å resolution showing the N-terminal region of the MBP peptide. TCR is yellow, peptide is red and MHC is cyan. Contours are at 1σ.
For the 3A6/MBP/HLA-DR2a complex (Figure 3A), the orientation angle of TCR to peptide/MHC, defined as the angle between the line formed by the peptide direction and a line between the centers of mass of the Vα and Vβ domains (Reinherz et al, 1999), is 47°, compared to 70 and 80° for the HA1.7/HA/HLA-DR1 and D10/CA/I-Ak complexes, respectively (Reinherz et al, 1999; Hennecke et al, 2000). Thus, the binding angles for MHC class II complexes span as broad a range as those observed for class I complexes (Rudolph et al, 2002). However, 3A6 does not exhibit the highly asymmetrical interaction with MHC seen with Ob.1A12 (Hahn et al, 2005), whose 110° orientation angle lies far outside the range for all reported MHC class I- or class II-restricted TCRs (Rudolph et al, 2002).
Figure 3.
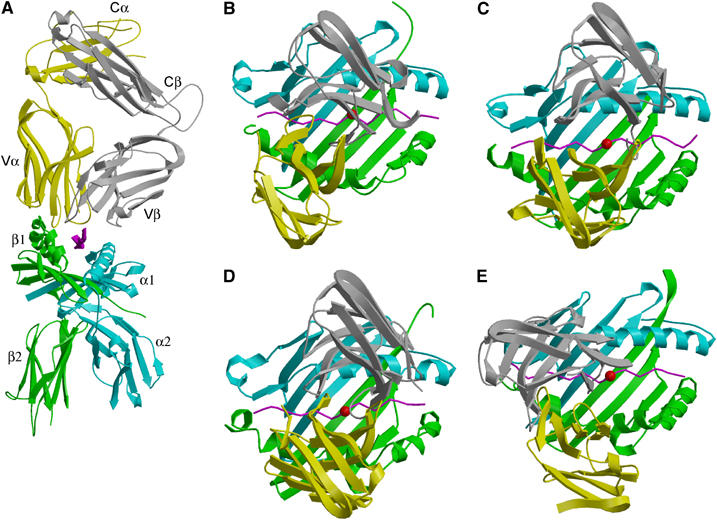
3A6/MBP/HLA-DR2a complex and comparison with other TCR/peptide/MHC class II complexes. (A) Side view of the 3A6/MBP/DR2a complex. TCR α-chain is yellow and β-chain is gray; peptide is magenta; MHC α-chain is cyan and β-chain is green. (B) Top view of the 3A6/MBP/DR2a complex, compared with (C) the HA1.7/HA/DR1 complex (Hennecke et al, 2000), (D) 172.10/MBP/I-Au complex (Maynard et al, 2005) and (E) Ob.1A12/MBP/DR2b complex (Hahn et al, 2005). Color codes are the same as (A). The central P5 residue of the peptide is represented as a red sphere.
The CDR footprint of 3A6 on MBP/HLA-DR2a (Figure 3B) is shifted toward the N-terminus of the bound peptide and toward the MHC β1 α-helix, compared to the CDR footprints of HA1.7 and 172.10 on their class II ligands (Figure 3C and D). In these other complexes (Hennecke et al, 2000; Maynard et al, 2005), CDR2β contacts Lys39α, an invariant residue located in a loop outside the peptide-binding site of the class II molecule. In the 3A6/MBP/HLA-DR2a complex, by contrast, the CDR2β loop is positioned directly over the α1 α-helix (Figure 4A), thereby precluding an interaction with Lys39α. Hence, this interaction does not constitute an obligatory anchor point for docking TCR onto MHC class II, expanding the range of possible docking solutions. Although the 3A6/MBP/DR2a and Ob.1A12/MBP/DR2b complexes (Figure 3B and E) display very different orientation angles (47 and 110°, respectively), it is significant that, in both cases, the CDR footprints are displaced toward the N-terminus of the peptide, a feature that may characterize many autoimmune TCRs (see Discussion).
Figure 4.
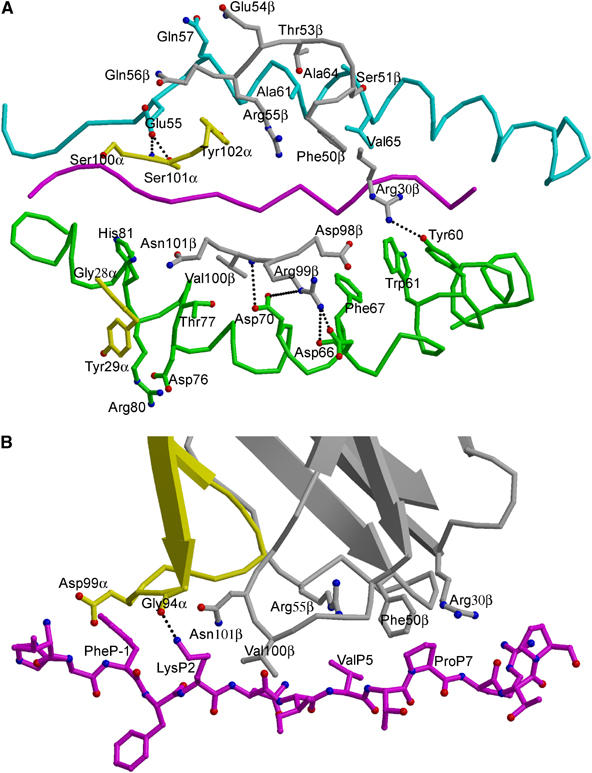
Interactions of TCR 3A6 with HLA-DR2a and the MBP peptide. (A) Interactions of the HLA-DR2a α-chains (cyan) and β-chains (green) with the TCR 3A6 α-chains (yellow) and β-chains (gray). The contact residues are drawn and labeled. Hydrogen bonds are indicated by broken black lines and the MBP peptide is magenta. (B) Interactions of the TCR 3A6 α-chains (yellow) and β-chains (gray) with the MBP peptide (magenta) drawn in a ball-and-stick representation. Contact residues are labeled. The P2 Lys Nζ–O CDR3α Gly94 hydrogen bond is present in only two of the four complex molecules in the asymmetric unit of the crystal.
Interaction of TCR 3A6 with MHC
The 3A6/MBP/DR2a complex buries a total solvent-accessible surface of 2002 Å2, comparable to that in other class II complexes (Reinherz et al, 1999; Hennecke et al, 2000; Maynard et al, 2005), but substantially greater than the surface buried in the Ob.1A12/MBP/DR2b complex (∼1600 Å2) (Hahn et al, 2005). In common with the HA1.7/HA/DR1 complex, Vβ buries significantly more surface on peptide/MHC than Vα (59 and 41%, respectively). This ratio is reversed in the D10/CA/I-Ak and 172.10/MBP/I-Au complexes, as well as in all but one complex involving MHC class I molecules (Reiser et al, 2000; Rudolph et al, 2002). Overall, 16 Vβ and eight Vα residues interact with MHC (Table II).
Table 2.
Interactions between TCR 3A6 and DR2a/MBP in the 3A6/DR2a/MBP complex
| Hydrogen bondsa | van der Waals contactsb | |||
|---|---|---|---|---|
| 3A6 | DR2a | |||
| Gly28α | His81β | |||
| Tyr29α | Asp76β, Thr77β, Arg80β, His81β | |||
| Ser100α | Glu22α | |||
| Ser101α | N, Oγ | Glu55α | Oɛ2 | Glu55α |
| Tyr102α | Glu55α, Gly58α | |||
| Arg30β | Nη2 | Tyr60β | Oη | Tyr60β, Try61β |
| Phe50β | Val65α | |||
| Ser51β | Ala64α, Val65α | |||
| Thr53β | Ala61α | |||
| Gln54β | Gln57α | |||
| Arg55β | Glu55α, Gln57α, Gly58α | |||
| Asn56β | Glu55α | |||
| Asp98β | Phe67β | |||
| Arg99β | Nη2 | Asp66β | O, Oδ2 | Asp77β, Phe67β, Asp66β |
| Val100β | N | Asp70β | Oδ1 | Asp70β, Thr77β |
| Nɛ | Asp70β | Oδ2 | ||
| Asn101β | Thr77β | |||
| 3A6 | MBP | |||
| Thr27α | ValP3 | |||
| Gly94α | O | Lys P2 | Nζ | LysP2 |
| Asp99α | ValP3, HisP2, PheP-1 | |||
| Arg30β | ProP7 | |||
| Phe50β | ValP5, ProP7 | |||
| Arg55β | ValP5 | |||
| Val100β | LysP2 | |||
| Asn101β |
|
|
|
LysP2 |
| aHydrogen bond distance is 2.5–3.4 Å. Listed are all hydrogen bonds present in two or more of the four complex molecules in the asymmetric unit of the crystal. | ||||
| bVan der Waals contacts are <4.0 Å. | ||||
Because of the overall shift in the 3A6 footprint on MHC, compared to the footprints of HA1.7, D10 and 172.10, the CDR2β loop of 3A6, from residue 51 to 56, completely overlays the N-terminal portion of the α1 α-helix of HLA-DR2a (Figure 4A), making numerous van der Waals contacts with residues 55–65 (Table II). Residues 98–101 of CDR3β interact extensively with MHC β1 α-helical residues Asp66β, Phe67β, Asp70β and Thr77β. Most notably, CDR3β Arg99 forms two hydrogen bonds, via its side-chain Nη2 atom, to the main-chain oxygen and side-chain Oδ2 atoms of Asp66β. In addition, CDR3β Val100 is hydrogen bonded, through its main-chain nitrogen, to the side-chain Oδ1 atom of Asp70β. By contrast, the CDR1β loop, which is oriented away from the MHC β1 α-helix, interacts with HLA-DR2a only through Arg30β, whose long side chain contacts Tyr60β and Trp61β (Figure 4A). In other TCR/MHC class II complexes, CDR1β is positioned directly over the MHC α1 α-helix.
With respect to MHC recognition by Vα, the CDR2α loop of 3A6 is too short to interact with DR2a or MBP. Residue Gly28 of CDR1α contacts the imidazole ring of HLA-DR2a His81β, while CDR1α Tyr29 packs tightly against Asp76β, Thr77β, Arg80β and His81β, forming numerous, predominantly hydrophobic, interactions with both main- and side-chain atoms of these MHC β1 α-helical residues (Figure 4A). Residues Ser100 and Ser101 of CDR3α both contact HLA-DR2a Glu55α, which is located in an extended strand immediately preceding the α1 α-helix. This strand, which replaces two turns of the corresponding α-helix in MHC class I, is a distinctive feature of MHC class II; however, it is not contacted by TCRs HA1.7, D10 or 172.10 (Reinherz et al, 1999; Hennecke et al, 2000; Maynard et al, 2005). A hydrogen bond also links the main-chain nitrogen of CDR3α Ser101 to the side-chain Oɛ2 atom of HLA-DR2a Gln55α. This unique engagement of a nonhelical MHC α1 residue is attributable to the displacement of 3A6 toward the N-terminus of the bound peptide, relative to other TCRs (Figure 5A).
Figure 5.
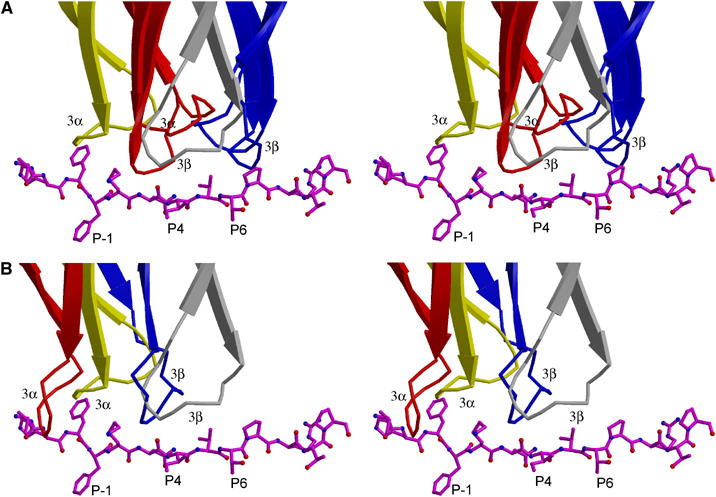
Position of TCRs along peptide antigen. (A) Ribbon diagram (stereo view) showing the position of the CDR3α (yellow) and CDR3β (gray) loops of 3A6 along the MBP peptide, compared to the position of the corresponding loops of TCRs HA1.7 and 172.10 (CDR3α loops are red; CDR3β loops are blue), upon superposition of the MHC α1β1 modules in the three complexes. The MBP peptide is drawn in a ball-and-stick format; anchor residues are labeled. Carbon atoms are magenta, oxygen atoms are red and nitrogen atoms are blue. (B) Position of the 3A6 CDR3α (yellow) and CDR3β (gray) loops along the MBP peptide, compared to the position of the corresponding loops of TCR Ob.1A12 (CDR3α loop is red; CDR3β loop is blue).
Five of the six residues contacted by 3A6 on the nonpolymorphic HLA-DR α-chain are also contacted by HA1.7 (Hennecke et al, 2000). Similarly, four of the eight β-chain residues contacted by 3A6 are contacted as well by HA1.7. All four (Asp66β, Asp70β, Thr77β, His81β) are polymorphic in human MHC class II alleles, but identical in DR2a and DR1. Despite this, the footprint of 3A6 on HLA-DR2a differs much more from that of HA1.7 on DR1 than does the latter from the footprint of 172.10 on I-Au (Figure 3B–D). In the 3A6/MBP/DR2a complex, CDR1α Tyr29 and CDR3β Arg99 are the major MHC-contacting residues: CDR1α Tyr29 alone accounts for half the contacts made by Vα, while CDR3β Arg99 contributes a third of those from Vβ. On the MHC side of the interface, the DR2a β1 domain, which interacts with CDR1α and CDR3β, makes twice as many contacts with TCR as the α1 domain. Therefore, the specific orientation of 3A6 on the MHC molecule, characterized by a tilt toward the β1 α-helix and a translation to its C-terminal end, may be dictated primarily by residues CDR1α Tyr29 and CDR3β Arg99, serving as anchor points.
Peptide recognition by TCR 3A6
Remarkably, few hydrogen bonds (and no salt bridges) are observed between the CDR loops of 3A6 and MBP 89–101, involving either main- or side-chain atoms of the TCR or peptide, in contrast to other TCR/peptide/MHC complexes (Rudolph et al, 2002). Moreover, none of the hydrogen bonds exist in all four complex molecules in the asymmetric unit of the crystal, suggesting that they do not constitute stable, energetically important interactions. Indeed, only one hydrogen bond, linking the amino group of P2 Lys to the main-chain oxygen in CDR3α Gly94, is found in more than one complex molecule; this particular bond is present in two of four complexes in the asymmetric unit (Table II). Interactions between TCR and peptide are mainly restricted to van der Waals contacts, with limited juxtaposition of hydrophobic surfaces. Thus, 3A6 appears at least as structurally degenerate in its recognition of the MBP peptide as 172.10, which forms several hydrogen bonds to MBP 1–11, one of which is specific for a side chain of the bound peptide (Maynard et al, 2005). The functional degeneracy of the 3A6 interface is supported by the success of combinatorial libraries in isolating peptides, such as K38-27 (Figure 1C), with multiple substitutions at TCR-contacting positions, that stimulate 3A6 T cells far more efficiently than MBP itself (Hemmer et al, 2000) (see Discussion).
TCR 3A6 mainly recognizes four MBP residues: P-1 Phe, P2 Lys, P5 Val and P7 Pro (Figure 4B). On the TCR side of the interface, CDR3α interacts with the N-terminal portion of the peptide, whose central and C-terminal portions engage all three CDR loops of Vβ. Notably, P-1 Phe is enveloped by the CDR3α loop, whose tip is 12.7 Å from that of CDR3β in the 3A6 binding site. Compared to HA1.7 or 172.10, large differences are observed in the position of both CDR3 loops along the peptide (Figure 5A). Thus, the difference in location of the tip of CDR3α between 3A6 and HA1.7 is 12.6 Å and the difference for CDR3β is 11.5 Å (the corresponding differences are 8.8 and 8.6 Å between 3A6 and 172.10). As a result, the P-1 residue in these other complexes is situated outside the pocket formed by CDR3α and CDR3β. Indeed, this pocket, which accommodates a single peptide side chain in other TCRs, including Ob.1A12 (Hahn et al, 2005), irrespective of MHC class I or II restriction (Rudolph et al, 2002), accommodates residues P-1 and P2 in 3A6 (Figure 4B). However, both 3A6 and Ob.1A12 mainly recognize the N-terminal portion of the peptide (Figure 5B).
In agreement with the crystal structure, combinatorial peptide libraries have shown that replacement of P-1 Phe by tryptophan markedly augments the stimulatory capacity of the MBP peptide (Hemmer et al, 2000). By contrast, replacement (or truncation) of the N-terminal P-3 Val of MBP 89–101, located at the periphery of the interface with TCR, has minimal effect on T-cell activation (Hemmer et al, 1998, 2000). Besides position P-1, substitutions at TCR-contacting positions P2, P5 and P7 also significantly affect T-cell activation. In the crystal structure, the P2 Lys side chain contacts CDR3α Gly94 and CDR3β Asn101, at the interface between Vα and Vβ (Figure 4B). The residue P5 Val contacts CDR2β Phe50 and Arg55 through its side chain, while the pyrrolidine ring of P7 Pro stacks against CDR1β Arg30 and CDR2β Phe50.
Conformational changes in self-peptide/MHC induced by TCR binding
Superposition of the α1β1 domains of free MBP/HLA-DR2a (Li et al, 2000) onto those of MBP/HLA-DR2a in the complex with 3A6 revealed small, but significant, structural differences in both MHC and peptide (Figure 6A). Overall, the peptide-binding groove of HLA-DR2a widens upon TCR engagement. In the MHC β1 domain, residues 60–74, located at the bend of the α-helix, undergo an average displacement of 1.5 Å in the position of their Cα atoms, away from the MBP peptide; Asp66β shows the largest individual displacement (2.7 Å in its Cα position). The average shift for helical residues 54–63 in the MHC α1 domain is 0.5 Å, also away from the peptide. The net result of these concerted movements is that P6 Thr and P9 Thr are drawn toward the β1 α-helix, with a concomitant loss of contacts to the α1 α-helix. Regarding conformational changes in the peptide, the r.m.s. deviation of Cα atoms of MBP 89–101 before and after TCR binding is 1.4 Å, mainly because of rearrangements in residues P5 through P10, which bring P7 Pro into contact with 3A6 (Figure 6B). In other TCR/peptide/MHC class II complexes, no significant changes in MHC or peptide conformation are associated with TCR engagement (Reinherz et al, 1999; Hennecke et al, 2000; Hahn et al, 2005; Maynard et al, 2005). As evidence that structural differences between free and TCR-bound forms of MBP/HLA-DR2a are induced by 3A6, no such differences were observed in the complex between MBP/DR2a and the superantigen SPE-C, which also contacts both peptide and MHC (Li et al, 2001) (Figure 6C). This implies that the superantigen, unlike the TCR, has evolved to optimize its fit to the ligand. Interestingly, SPE-C, like 3A6, interacts with the N-terminal portion of MBP 89–101, but at positions P2, P-1 and P3.
Figure 6.
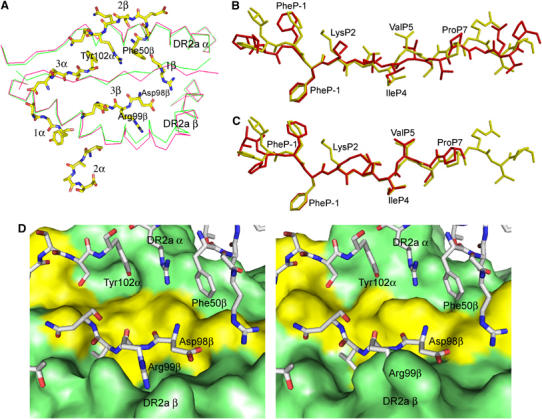
Conformational changes in MBP/HLA-DR2a upon binding TCR 3A6. (A) Top view of the superposition of MBP/HLA-DR2a in unliganded form (green) and in complex with 3A6 (red). 3A6 CDR loops are shown in a stick format. Carbon atoms are yellow, oxygen atoms are red and nitrogen atoms are blue. (B) Comparison of the MBP peptide conformation before (yellow) and after (red) binding to TCR 3A6. (C) Comparison of the MBP peptide conformation before (yellow) and after (red) binding to the superantigen SPE-C (Li et al, 2001). The three C-terminal residues of MBP 89–101 are disordered in the SPE-C/MBP/DR2a structure. (D) Comparison of the MBP/HLA-DR2a surface after (left panel) and before (right panel) binding to 3A6. TCR residues, in particular Asp98β and Arg99β, fit better into pockets on the bound MBP/HLA-DR2a surface (left) than into pockets on the free MBP/HLA-DR2a surface (right). 3A6 residues are shown in a stick format and labeled.
The main consequence of TCR-induced adjustments in MBP/HLA-DR2a is to create two shallow grooves on the surface of the ligand for accommodating the CDR3α and CDR3β loops of 3A6, in particular Tyr102α and Arg99β (Figure 6D). In this way, the geometrical fit between TCR and peptide/MHC is improved, based on calculations of the shape correlation statistic (Sc) for this interface (Lawrence and Colman, 1993). Thus, the Sc value for the experimentally determined complex is 0.62 (Sc=1.0 for interfaces with perfect fits), but only 0.50 for a hypothetical complex constructed using the free MBP/HLA-DR2a structure that assumes no conformational changes (Li et al, 2000). Although improved shape complementarity is expected to result in increased affinity, any enhancement would be partially offset by the energetic cost of distorting MBP/DR2a (and possibly 3A6) from its ground state conformation.
Discussion
Escape from negative selection
Several mechanisms have been proposed to explain why some autoreactive T cells, such as 3A6, escape deletion in the thymus. Thymocytes undergo selection based on recognition of self-peptide/MHC complexes, such that weak interactions with self-peptide/MHC permit T-cell survival (positive selection), whereas strong interactions induce apoptosis (negative selection) (Kappler et al, 1987; Alam et al, 1996). In some cases, failure of negative selection could result from unusually weak binding of the self-peptide to MHC, which would effectively destabilize the complex with TCR. Indeed, the very short half-life of the MBP 1–11/I-Au complex could explain the incomplete deletion of T cells like 172.10 (Garcia et al, 2001). Although the affinity of TCR 172.10 for MBP 1–11/I-Au (Kd∼5 μM) is at the high end of the range for TCR–peptide/MHC interactions (Davis et al, 1998; van der Merwe and Davis, 2003), SPR binding measurements were conducted using an engineered version of MBP 1–11/I-Au in order to stabilize the interaction with TCR (Garcia et al, 2001). By contrast, low affinity of self-peptide for MHC cannot account for the existence of T cells recognizing MBP 89–101 in MS patients, as this peptide binds tightly to HLA-DR molecules (Wucherpfennig et al, 1993; Vogt et al, 1994). Another mechanism for escaping negative selection involves expression of splice variants of self-proteins in the thymus that do not contain the relevant T-cell epitope (Klein et al, 2000). However, it has been demonstrated that MBP T-cell epitopes are displayed by antigen-presenting cells in active MS lesions (Krogsgaard et al, 2000). Based on the highly unusual topology of the Ob.1A12/MBP/DR2b complex, it has also been proposed that escape from negative selection involves an alteration in the geometry of interaction with the CD4 coreceptor (Hahn et al, 2005). However, the more conventional docking mode of TCR 3A6 makes this possibility unlikely.
A more plausible explanation for the survival of T cells reactive with MBP in humans is reduced TCR affinity for MBP/HLA-DR complexes. Indeed, we have shown by SPR that 3A6 binds weakly to MBP/DR2a. Despite this, 3A6 T cells are activated by nanomolar concentrations of MBP peptide (Hemmer et al, 2000) and mice transgenic for 3A6 and DR2a develop CNS autoimmunity (Quandt et al, in preparation). Two main structural features likely explain the low affinity of 3A6 for MBP/DR2a. First, interactions between 3A6 and MBP are limited to van der Waals contacts, with no hydrogen bonds or salt bridges linking TCR and peptide. Second, 3A6 binding is associated with conformational changes in the relatively rigid MBP/DR2a molecule, and perhaps also in the more flexible CDR loops of the TCR, which would effectively reduce the binding free energy.
Structural characteristics of autoimmune TCR/peptide/MHC complexes
The 3A6/MBP/DR2a complex allows us to address whether TCR recognition of self-peptide/MHC may exhibit certain common elements that distinguish autoimmune from antimicrobial or alloreactive TCRs. Overall, the diagonal orientation of TCR 3A6 relative to MBP/DR2a resembles that in other TCR/peptide/MHC complexes (Rudolph et al, 2002; Maynard et al, 2005), in marked contrast to Ob.1A12, which engages MBP/DR2b at an orientation angle far outside the normal range (Hahn et al, 2005). Despite this, and the fact that DR2a and DR2b present completely different MBP residues to TCR as a result of a three-residue shift in peptide register (Li et al, 2000), it is striking that both 3A6 and Ob.1A12 primarily recognize the N-terminal, rather than central, portion of the peptide, as do all other TCRs (Rudolph et al, 2002), including 172.10. In the 172.10/MBP/I-Au complex, the CDR3 loops overlay the central region of the peptide-binding groove in the classical manner (Maynard et al, 2005). However, the MBP 1–11/I-Au ligand is unusual in that the N-terminal third of the binding groove is empty, as a result of a shift of the peptide (He et al, 2002).
Could this focus on the N-terminal half of the self-peptide by 3A6 and Ob.1A12 be broadly characteristic of autoimmune TCRs? Although additional structures are clearly required to fully assess this possibility, it is tempting to speculate that the N-terminal site is intrinsically suboptimal for TCR binding, favoring escape from negative selection. Conversely, antimicrobial TCRs achieve higher affinities by focusing on the central portion of peptides, presumably a more favorable site for TCR engagement. Importantly, however, both binding modes are compatible with a wide range of orientation angles of TCR to peptide/MHC: 47–110° (at least) for autoimmune TCRs and 45–80° for antimicrobial or alloreactive TCRs (Rudolph et al, 2002).
Basis for TCR degeneracy
The concept of TCR degeneracy has been widely evoked to explain the ability of a single TCR to recognize the diverse peptides it encounters during thymic education and peripheral surveillance (Kersh and Allen, 1996; Holler and Kranz, 2004; Wucherpfennig, 2004). TCR degeneracy also underlies the molecular mimicry hypothesis for autoimmune diseases, whereby autoimmunity can be triggered by bacterial or viral peptides that mimic self-peptides (Wucherpfennig and Strominger, 1995; Hemmer et al, 1997). However, the structural basis for TCR degeneracy has remained elusive. For example, a T-cell clone from a DR2-positive MS patient was shown to recognize both a DR2b-restricted MBP and a DR2a-restricted EBV peptide (Lang et al, 2002). However, crystal structures of the MBP/DR2b and EBV/DR2a complexes revealed topologically equivalent surfaces that would be presented for TCR recognition. Hence, crossreactivity in this case appears to be achieved by strict conservation of putative TCR-contacting residues (no complex structures are available), without recourse to TCR degeneracy.
An unbiased way to study T-cell degeneracy makes use of positional scanning synthetic combinatorial peptide libraries (PS-SCL) (Hemmer et al, 1997; Pinilla et al, 1999). Accordingly, decamer PS-SCL containing all possible combinations (199) of 10-mers in systematically arranged mixtures were screened to identify sequences that stimulate the 3A6T cell clone (Wilson et al, 1999; Hemmer et al, 2000). The observed proliferative responses to these mixtures indicate that many peptides are recognized by 3A6, since individual peptides are present at concentrations of only ∼10−16 M, or 108-fold lower than typically required for activating CD4+ T-cell clones. These results, and similar ones for other autoreactive T-cell clones (Hemmer et al, 1997), imply considerable TCR degeneracy. On the basis of information obtained using PS-SCL, optimal ligands have been identified for 3A6 (Hemmer et al, 2000). Remarkably, some of these superagonists, for example K38-27, stimulate this T-cell clone with EC50 values in the pico- to femtomolar range. The 3A6/MBP/DR2a structure provides a basis for understanding TCR degeneracy in this system, as it enables us to distinguish superagonists with differences in TCR-binding residues (which would indicate true crossreactivity) from superagonists that differ simply in MHC-binding residues.
In principle, superagonism may be explained by improved binding of superagonist peptides to MHC, increased affinity of superagonist/MHC complexes for TCR, or a combination of the two. Since the MBP peptide already exhibits tight binding to HLA-DR2a (Vogt et al, 1994), it is unlikely that enhanced affinity for MHC can explain the greatly increased stimulatory capacity of superagonist peptides. In support of this conclusion, all identified superagonists, including K38-27, retain three out of four anchor residues: P-1 Phe (the primary anchor), P4 Ile and P6 Thr (both secondary anchors) (Hemmer et al, 2000; Li et al, 2000). The sole exception is P9 Thr in the MBP peptide, which is replaced by leucine or glycine in the superagonists. However, HLA-DR2a is tolerant of substitutions at the P9 position (Vogt et al, 1994). Consequently, superagonist activity most likely arises from increased affinity of superagonist/MHC complexes for TCR, as demonstrated by SPR analysis of 3A6 binding to HLA-DR2a loaded with MBP versus K38-27.
In the 3A6/MBP/HLA-DR2a complex, 3A6 interacts directly with MBP residues P-1 Phe, P2 Lys, P5 Val and P7 Pro. With the exception of P2 Lys, superagonist peptides contain substitutions at each of the three other TCR-contacting positions, in addition to an asparagine-to-leucine substitution at P3 (Hemmer et al, 2000). Although P3 Asn does not itself contact 3A6, this residue is nevertheless in the interface and could indirectly affect TCR binding. The consensus motif for superagonists is P-1WFKLIXTYKZP9, where MHC anchor residues are in bold, P5 X is Leu/Thr/Pro, P7 Y is Thr/Lys/Pro and P9 Z is Leu/Gly (the corresponding MBP sequence is P-1FFKNIVTPRTP9). We propose that substitutions at P-1, P3, P5 and P7 increase affinity for 3A6 by optimizing shape and chemical complementary with TCR. For example, since the volume of the interfacial cavity occupied by MBP residue P-1 Phe appears sufficient to accommodate the larger tryptophan side chain found in superagonists, the stimulatory effect of this replacement likely results from additional hydrophobic or van der Waals interactions with 3A6, augmenting affinity.
In the case of TCR 172.10, PS-SCL did not identify stimulatory peptides with TCR-contacting residues different from MBP 1–11, despite the apparent paucity of specific interactions between 172.10 and MBP in the crystal structure (Maynard et al, 2005). By contrast, the degeneracy of 3A6 is demonstrated by the isolation of superagonist mimics of MBP 89–101 containing substitutions at nearly all TCR-contacting positions. This degeneracy most likely reflects the imperfect fit and scarcity of hydrogen bonds between 3A6 and MBP observed in the crystal structure, which offer ample opportunity for optimizing the TCR–peptide/MHC interface through variations of the peptide. Finally, it is conceivable that TCRs like 3A6, which primarily recognize the N-terminal portion of peptides, are more inherently crossreactive than TCRs targeting the central portion, since the overall conformation of peptides bound to MHC class II molecules is much more highly conserved for residues P-1–P4 than P5–P9 (Ghosh et al, 1995). Such crossreactivity could enhance the pathogenic potential of T cells bearing these TCRs, as it would increase the probability of self-peptide/MHC recognition, resulting in autoimmunity.
Materials and methods
Protein expression and purification
Empty HLA-DR2a was folded in vitro from inclusion bodies produced in Escherichia coli as described (Li et al, 2000), except that no peptide was added during folding. The gene encoding residues Asp1–Cys211 of the TCR 3A6 α-chain was amplified by PCR and inserted into the pET-22b expression vector (Novagen). The β-chain construct, which encoded a fusion protein comprising the MBP 89–101 peptide (VHFFKNIVTPRTP) connected by an octapeptide linker (GGSGGGGG) to residues Gly3–Cys248 of the 3A6 β-chain (Hennecke et al, 2000), was cloned into the same vector. The TCR α- and β-chains were expressed separately as inclusion bodies in BL21(DE3)pLys E. coli cells (Novagen), as described for antibody light and heavy chains (Li et al, 2003). The inclusion bodies were dissolved in 8 M urea, 50 mM Tris–HCl (pH 8.0) and 10 mM DTT. For in vitro folding, the 3A6 α- and β-chains were mixed in a 1.2:1 molar ratio and diluted into ice-cold folding buffer containing 1 M L-arginine–HCl, 50 mM Tris–HCl (pH 8.5), 1 mM EDTA, 3 mM reduced glutathione, 0.9 mM oxidized glutathione, 60 mg/l PMSF, 2 mg/l pepstatin A, 3 mg/l benzamidine, 2 mg/l leupeptin and 2 mg/l aprotin to a final protein concentration of 150 mg/l. After 72 h at 4°C, the folding mixture was concentrated 50-fold, dialyzed against 50 mM MES (pH 6.0) and centrifuged to remove aggregates. Following dialysis against 50 mM Tris–HCl (pH 8.0), the sample was applied to a POROS 20 HQ (PerSeptive Biosystems) anion exchange FPLC column equilibrated with the same buffer; disulfide-linked 3A6 heterodimer was eluted using a linear NaCl gradient.
Assembly of the 3A6/MBP/HLA-DR2a complex
The 3A6/MBP/DR2a complex was formed by loading the MBP peptide, fused to TCR 3A6, into empty HLA-DR2a using soluble HLA-DM (Hennecke et al, 2000). 3A6, DR2a and HLA-DM were incubated at a molar ratio of 6:3:1 for 15 h at 37°C in 50 mM sodium citrate (pH 5.8) containing 2 mg/l leupeptin and 2 mg/l aprotinin. The complex was separated from aggregates, HLA-DM, and uncomplexed MHC and TCR with a Superdex S-200 column (Pharmacia). Further purification was carried out using a Mono Q anion exchange column in 50 mM Tris–HCl (pH 8.0).
Crystallization, structure determination and refinement
Small, clustered crystals of 3A6/MBP/DR2a were grown in hanging drops by mixing 1 μl of protein solution at 12 mg/ml with 1 μl of reservoir solution containing 10% (w/v) PEG 8000, 0.2 M ammonium sulfate and 100 mM sodium citrate (pH 5.8–6.2). Large, single crystals were obtained after 2–3 rounds of seeding. For data collection, crystals were transferred to a cryoprotectant solution (mother liquor containing 20% (v/v) glycerol), prior to flash cooling in a nitrogen stream. X-ray diffraction data were collected in-house at 100 K using a Siemens HI-STAR area detector. The crystals are triclinic with four complex molecules per asymmetric unit (Table I). Diffraction data were processed and scaled using CrystalClear (Pflugrath, 1999). The data set is 86.3% complete to 2.8 Å resolution with Rmerge=8.0%. Data collection statistics are shown in Table I.
The structure of the 3A6/DR2a/MBP complex was solved by molecular replacement using the program Phaser (Read, 2001; Storoni et al, 2004). The free MBP/HLA-DR2a structure (PDB accession code 1FV1) (Li et al, 2000), the CαCβ module of TCR HA1.7 (1FYT) (Hennecke et al, 2000) and the VαVβ module of TCR N15 (1NFD) (Wang et al, 1998) were used as search models. The four MHC molecules were located first by automatic rotation and translation search. After fixing the four MHC molecules, one TCR CαCβ module was found with a Z-score of 25.06. The positions of the other three CαCβ modules were obtained by superposition according to the positions of the four MHC molecules. By fixing the four MHC and CαCβ, the four VαVβ modules were located in a straightforward manner; Z-scores exceeded 20 for all four solutions. Rigid-body refinement gave an initial Rfree of 39.1% and Rwork of 38.0%, with all nonconserved residues in the VαVβ domains mutated to alanine. Refinement was carried out using CNS1.1 (Brünger et al, 1998), including iterative cycles of simulated annealing, positional refinement and B-factor refinement, interspersed with model rebuilding into σA-weighted Fo−Fc and 2Fo−Fc electron density maps using TURBO-FRODO (Roussel and Cambillau, 1989). The final model includes 23 495 non-hydrogen protein atoms and 148 water molecules, resulting in a final Rvalue of 27.51% and Rfree of 32.55 at 2.80 Å resolution. Refinement statistics are summarized in Table I.
Production of biotin-HLA-DR2a, biotin-3A6 and 3A6 tetramer
For production of biotin-DR2a and biotin-3A6, 17-amino-acid tags were added to the C-termini of the α-chains of both TCR and MHC (Cull and Schatz, 2000): GGGLNDIFEAQKIEWHE for DR2a and GSLHHILDAQKMVWNHR for 3A6. The lysine residue in both tags can be recognized by biotin-protein ligase and specifically biotinylated. The tagged proteins were produced and purified in the same way as the wild-type proteins. Biotinylation was performed using a Biotinylation Kit (Avidity, LLC). Briefly, protein at a concentration of 2 mg/ml in 10 mM Tris–HCl (pH 8.0) was mixed with 1/8 volume of Biomix-A and 1/8 volume of Biomix-B; birA enzyme was added to a final concentration of 1 μg/100 μl. The reaction mixture was incubated at 30°C for 30 min; biotinylated protein was separated from excess biotin with a Superdex S-200 column. To produce TCR tetramers, streptavidin dissolved in PBS was added dropwise into a biotin-3A6 solution at the final molar ratio of 1:4; 3A6 tetramers were purified using a Superdex S-200 column.
SPR analysis
The interaction of TCR 3A6 with HLA-DR2a was assessed by SPR using a BIAcore 1000 biosensor. Typically, biotin-DR2a was immobilized on the sensor chip by flowing a biotin-DR2a solution (∼20 μg/ml) containing 0.5 M NaCl over a BIAcore SA chip preimmobilized with streptavidin at high surface density. After immobilizing 1000–2000 RU, 20 μl of a 20 μM biotin solution was injected to block the remaining streptavidin sites. As the blank, biotin alone was injected. To assess TCR binding, solutions containing different concentrations of 3A6 (monomer or tetramer) were injected sequentially over flow cells immobilized with DR2a.
Protein Data Bank accession code
Atomic coordinates and structure factors for the TCR 3A6/MBP/HLA-DR2a complex have been deposited under accession code 1ZGL.
Acknowledgments
We thank L Deng and R Langley for critical reading of the manuscript and LJ Stern for advice on HLA-DR expression. We are grateful to D Zaller for the gift of Drosophila cells producing HLA-DM. This work was supported by grants from the National Multiple Sclerosis Society (RG2747) and the National Institutes of Health (AI36900) to RAM.
References
- Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, Jameson SC, Gascoigne NR (1996) T-cell receptor affinity and thymocyte positive selection. Nature 381: 616–620 [DOI] [PubMed] [Google Scholar]
- Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, Gran B, Eaton J, Antel J, Frank JA, McFarland HF, Martin R (2000) Encephalitogenic potential of myelin basic protein peptide (83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med 6: 1167–1175 [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Gross-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography and NMR systems: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Cull MG, Schatz PJ (2000) Biotinylation of proteins in vivo and in vitro using small peptide tags. Methods Enzymol 326: 430–440 [DOI] [PubMed] [Google Scholar]
- Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, Chien Y (1998) Ligand recognition by αβ T cell receptors. Annu Rev Immunol 16: 523–544 [DOI] [PubMed] [Google Scholar]
- Ebers GC, Kukay K, Bulman DE, Sadovnick AD, Rice G, Anderson C, Armstrong H, Cousin K, Bell RB, Hader W, Paty DW, Hashimoto S, Oger J, Duquette P, Warren S, Gray T, O'Connor P, Nath A, Auty A, Metz L, Francis G, Paulseth JE, Murray TJ, Pryse-Phillips W, Nelson R, Freedman M, Brunet D, Bouchard J-P, Hinds D, Risch N (1996) A full genome search in multiple sclerosis. Nat Genet 13: 472–476 [DOI] [PubMed] [Google Scholar]
- Fogdell A, Hillert J, Sachs C, Ollerup O (1995) The multiple sclerosis- and narcolepsy-associated HLA class II haplotype includes the DRB5*0101 allele. Tissue Antigens 46: 333–336 [DOI] [PubMed] [Google Scholar]
- Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC (1996) Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature 384: 134–141 [DOI] [PubMed] [Google Scholar]
- Garcia KC, Degano M, Pease LR, Huang M, Peterson PA, Teyton L, Wilson IA (1998) Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science 279: 1166–1172 [DOI] [PubMed] [Google Scholar]
- Garcia KC, Radu CG, Ho J, Ober RJ, Ward ES (2001) Kinetics and thermodynamics of T cell receptor–autoantigen interactions in murine experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 98: 6818–6823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P, Amaya M, Mellins E, Wiley DC (1995) The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature 378: 457–462 [DOI] [PubMed] [Google Scholar]
- Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW (2005) Unconventional topology of self peptide–major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol 6: 490–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XL, Radu C, Sidney J, Sette A, Ward ES, Garcia KC (2002) Structural snapshot of aberrant antigen presentation linked to autoimmunity: the immunodominant epitope of MBP complexed with I-Au. Immunity 17: 83–94 [DOI] [PubMed] [Google Scholar]
- Hemmer B, Fleckenstein BT, Vergelli M, Jung G, McFarland H, Martin R, Wiesmuller KH (1997) Identification of high potency microbial and self ligands for a human autoreactive class II-restricted T cell clone. J Exp Med 185: 1651–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmer B, Pinilla C, Gran B, Vergelli M, Ling N, Conlon P, McFarland HF, Houghten R, Martin R (2000) Contribution of individual amino acids within MHC molecule or antigenic peptide to TCR ligand potency. J Immunol 164: 861–871 [DOI] [PubMed] [Google Scholar]
- Hemmer B, Vergelli M, Gran B, Ling N, Conlon P, Pinilla C, Houghten R, McFarland HF, Martin R (1998) Predictable TCR antigen recognition based on peptide scans leads to identification of agonist ligands with no sequence homology. J Immunol 160: 3631–3636 [PubMed] [Google Scholar]
- Hennecke J, Carfi A, Wiley DC (2000) Structure of a covalently stabilized complex of a human αβ T-cell receptor, influenza HA peptide and MHC class II molecule, HLA-DR1. EMBO J 19: 5611–5624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler PD, Kranz DM (2004) T cell receptors: affinities, cross-reactivities, and a conformer model. Mol Immunol 40: 1027–1031 [DOI] [PubMed] [Google Scholar]
- Kappler JW, Roehm N, Marrack P (1987) T cell tolerance by clonal elimination in the thymus. Cell 49: 273–280 [DOI] [PubMed] [Google Scholar]
- Kersh GJ, Allen PM (1996) Essential flexibility in the T-cell recognition of antigen. Nature 380: 495–498 [DOI] [PubMed] [Google Scholar]
- Kjer-Nielsen L, Clements CS, Purcell AW, Brooks AG, Whisstock JC, Burrows SR, McCluskey J, Rossjohn J (2003) A structural basis for the selection of dominant αβ T cell receptors in antiviral immunity. Immunity 18: 53–64 [DOI] [PubMed] [Google Scholar]
- Klein L, Klugmann M, Nave KA, Tuohy VK, Kyewski B (2000) Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat Med 6: 56–61 [DOI] [PubMed] [Google Scholar]
- Krogsgaard M, Wucherpfennig KW, Cannella B, Hansen BE, Svejgaard A, Pyrdol J, Ditzel H, Raine C, Engberg J, Fugger L (2000) Visualization of myelin basic protein (MBP) T cell epitopes in multiple sclerosis lesions using a monoclonal antibody specific for the human histocompatibility leukocyte antigen (HLA)-DR2–MBP 85–99 complex. J Exp Med 191: 1395–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang HLE, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, Hjorth P, Sondergaard L, Svejgaard A, Wucherpfennig K, Stuart DI, Bell JI, Jones EY, Fugger L (2002) A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol 3: 940–943 [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Colman PM (1993) Shape complementarity at protein–protein interfaces. J Mol Biol 234: 946–950 [DOI] [PubMed] [Google Scholar]
- Li Y, Dimasi N, McCormick JK, Martin R, Schuck P, Schlievert PM, Mariuzza RA (2001) Crystal structure of a superantigen bound to the high-affinity, zinc-dependent site on MHC class II. Immunity 14: 93–104 [DOI] [PubMed] [Google Scholar]
- Li Y, Li H, Martin R, Mariuzza RA (2000) Structural basis for the binding of an immunodominant peptide from myelin basic protein in different registers by two HLA-DR2 proteins. J Mol Biol 304: 177–188 [DOI] [PubMed] [Google Scholar]
- Li Y, Li H, Yang F, Smith-Gill SJ, Mariuzza RA (2003) X-ray snapshots of the maturation of an antibody response to a protein antigen. Nat Struct Biol 10: 482–488 [DOI] [PubMed] [Google Scholar]
- Maynard J, Petersson K, Wilson DH, Adams EJ, Blondelle SE, Boulanger MJ, Wilson DB, Garcia KC (2005) Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity 22: 81–92 [DOI] [PubMed] [Google Scholar]
- Oksenberg JR, Seboun E, Hauser SL (1996) Genetics of demyelinating diseases. Brain Pathol 6: 289–302 [DOI] [PubMed] [Google Scholar]
- Pflugrath JW (1999) The finer things in X-ray data collection. Acta Crystallogr D 55: 1718–1725 [DOI] [PubMed] [Google Scholar]
- Pinilla C, Martin R, Gran B, Appel JR, Boggiano C, Wilson DB, Houghten RA (1999) Exploring immunological specificity using synthetic peptide combinatorial libraries. Curr Opin Immunol 11: 193–202 [DOI] [PubMed] [Google Scholar]
- Read RJ (2001) Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D 57: 1373–1382 [DOI] [PubMed] [Google Scholar]
- Reinherz EL, Tan K, Tang L, Kern P, Liu J, Xiong Y, Hussey RE, Smolyar A, Hare B, Zhang R, Joachimiak A, Chang HC, Wagner G, Wang J (1999) The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science 286: 1913–1921 [DOI] [PubMed] [Google Scholar]
- Reiser JB, Darnault C, Guimezanes A, Gregoire C, Mosser T, Schmitt-Verhulst AM, Fontecilla-Camps JC, Malissen B, Housset D, Mazza G (2000) Crystal structure of a T cell receptor bound to an allogeneic MHC molecule. Nat Immunol 1: 291–297 [DOI] [PubMed] [Google Scholar]
- Roussel A, Cambillau C (1989) TURBO-FRODO. In Silicon Graphics Partners Directory, pp 77–78. Mountain View, CA, USA: Silicon Graphics [Google Scholar]
- Rudolph MG, Luz JG, Wilson IA (2002) Structural and thermodynamic correlates of T cell signaling. Annu Rev Biophys Biomol Struct 31: 121–149 [DOI] [PubMed] [Google Scholar]
- Smith KJ, Pyrdol J, Gauthier Wiley DC, Wucherpfennig KW (1998) Crystal structure of HLA-DR2 (DRA*0101, DRB1*1501) complexed with a peptide from human myelin basic protein. J Exp Med 188: 1511–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sospedra M, Martin R (2005) Immunology of multiple sclerosis. Annu Rev Immunol 23: 683–747 [DOI] [PubMed] [Google Scholar]
- Stefanova I, Dorfman JR, Germain RN (2002) Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes. Nature 420: 429–434 [DOI] [PubMed] [Google Scholar]
- Steinman L (1996) Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell 85: 299–302 [DOI] [PubMed] [Google Scholar]
- Stewart GB, McMichael AJ, Bell JI, Stuart DI, Jones EY (2003) A structural basis for immunodominant T cell receptor recognition. Nat Immunol 4: 657–663 [DOI] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr D 60: 432–438 [DOI] [PubMed] [Google Scholar]
- van der Merwe PA, Davis SJ (2003) Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol 21: 659–684 [DOI] [PubMed] [Google Scholar]
- Vergelli M, Kalbus M, Rojo SC, Hemmer B, Kalbacher H, Tranquill L, Beck H, McFarland HF, De Mars R, Long EO, Martin R (1997) T cell response to myelin basic protein in the context of the multiple sclerosis-associated HLA-DR15 haplotype: peptide binding, immunodominance and effector functions of T cells. J Neuroimunol 77: 195–203 [DOI] [PubMed] [Google Scholar]
- Vogt AB, Kropshofer H, Kalbacher H, Kalbus M, Rammensee H-G, Coligan JE, Martin R (1994) Ligand motifs of HLA-DRB5*0101 and DRB1*1501 molecules delineated from self-peptides. J Immunol 153: 1665–1673 [PubMed] [Google Scholar]
- Wang J, Lim K, Smolyar A, Teng M, Liu J, Tse AG, Liu J, Hussey RE, Chishti Y, Thomson CT, Sweet RM, Nathenson SG, Chang HC, Sacchettini JC, Reinherz EL (1998) Atomic structure of an αβ T cell receptor (TCR) heterodimer in complex with an anti-TCR Fab fragment derived from a mitogenic antibody. EMBO J 17: 10–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DB, Pinilla C, Wilson DH, Schroder K, Boggiano C, Judkowski V, Kaye J, Hemmer B, Martin R, Houghten RA (1999) Immunogenicity. I. Use of peptide libraries to identify epitopes that activate clonotypic CD4+ T cells and induce T cell responses to native peptide ligands. J Immunol 163: 6424–6434 [PubMed] [Google Scholar]
- Wilson IA, Stanfield RL (2005) MHC restriction: slip-sliding away. Nat Immunol 6: 434–435 [DOI] [PubMed] [Google Scholar]
- Wucherpfennig KW (2004) T cell receptor crossreactivity as a general property of T cell recognition. Mol Immunol 40: 1009–1017 [DOI] [PubMed] [Google Scholar]
- Wucherpfennig KW, Sette A, Southwood S, Oseroff C, Matsui M, Strominger JL, Hafler DA (1993) Structural requirements for binding of an immunodominant myelin basic protein peptide to DR2 isotypes and for its recognition by human T cell clones. J Exp Med 179: 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucherpfennig KW, Strominger JL (1995) Molecular mimicry in T-cell mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 80: 695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Gran B, Pinilla C, Markovic-Plese S, Hemmer B, Tzou A, Whitney LW, Biddison WE, Martin R, Simon R (2001) Combinatorial peptide libraries and biometric score matrices permit the quantitative analysis of specific and degenerate interactions between clonotypic TCR and MHC peptide ligands. J Immunol 167: 2130–2141 [DOI] [PubMed] [Google Scholar]